

Abstract
Background
Oculopharyngeal muscular dystrophy (OPMD) is a late‐onset muscle disease affecting one per 80 000 of the general population characterized by profound dysphagia and ptosis, and limb weakness at later stages. Affected muscles are characterized by increased fibrosis and atrophy. Myostatin is a negative regulator of muscle mass, and inhibition of myostatin has been demonstrated to ameliorate symptoms in dystrophic muscles.
Methods
In this study, we performed a systemic delivery of a monoclonal antibody to immunologically block myostatin in the A17 mouse model of OPMD. The mice were administered a weekly dose of 10 mg/kg RK35 intraperitonially for 10 weeks, following which histological analyses were performed on the samples.
Results
This treatment significantly (P < 0.01) improved body mass (11%) and muscle mass (for the tibialis anterior and extensor digitorum longus by 19% and 41%) in the A17 mice treated with RK35 when compared to saline controls. Similarly, a significantly (P < 0.01) increased muscle strength (18% increase in maximal tetanic force) and myofibre diameter (17% and 44% for the tibialis anterior and extensor digitorum longus), and reduced expression of markers of muscle fibrosis (40% reduction in area of expression), was also observed. No change in the density of intranuclear inclusions (a hallmark of disease progression of OPMD) was however observed.
Conclusions
Our study supports the clinical translation of such antibody‐mediated inhibition of myostatin as a treatment of OPMD. This strategy has implications to be used as adjuvant therapies with gene therapy based approaches, or to stabilize the muscle prior to myoblast transplantation.
Keywords: Anti‐myostatin antibody, RK35, OPMD, Muscle atrophy
Introduction
Oculopharyngeal muscular dystrophy (OPMD) is a rare late‐onset muscular dystrophy characterized by progressive weakening of the extraocular and pharyngeal muscles, manifesting as ptosis and dysphagia, and at later stages of the disease as a weakening of proximal limb musculature.1 The disease is caused by mutations in the poly(A)‐binding protein nuclear 1 (PABPN1) gene that introduces an abnormal expansion of polyalanine‐encoding (GCN)n trinucleotide repeats in the coding region of exon 1. PABPN1 plays crucial roles in regulating the poly(A) tail length on mRNAs,2, 3 controlling the use of alternative polyadenylation sites,4, 5 influencing pre‐mRNA splicing,6, 7 and also processing of long non‐coding RNA (lncRNAs)8 and small nucleolar RNA (snoRNA).9 It also has an important role in poly(A)‐mediated RNA decay or export from the nucleus2 and in RNA hyperadenylation.10
Repeat alanine expansions with 11–18 repeats lead to a mutant expanded PABPN1 protein (expPABPN1), which causes misfolding, and subsequent accumulation of intranuclear aggregates in muscles.11 The aggregates sequester wild‐type PABPN1 among other essential components of the cell, thereby dysregulating essential functions of the cell.12, 13, 14, 15, 16, 17, 18, 19, 20 Interestingly, it has been shown that PABPN1 expression in muscles declines with age in healthy individuals, with OPMD patients presenting molecular signatures of premature ageing.12, 21 Global approaches have generated a body of evidence—including mitochondrial defects, altered RNA metabolism, specific oxidative fibre atrophy, and increased proteasome activity—suggesting a premature ageing in OPMD muscles. This has also been implicated in the comparison of the molecular signature of OPMD patients and models of muscle ageing.12 Altogether these data strongly suggest common features between OPMD and ageing. No cure currently exists for the disease; to correct ptosis and dysphagia, patients undergo surgical procedures that partially alleviate the symptoms. Recent clinical efforts have however shown promise with myoblast transplantation22 and small molecule drug therapies.23, 24
Myostatin acts as a negative regulator of muscle mass, and naturally occurring myostatin null mutants (such as the Piedmontese cattle, Texel sheep, and the bully whippet dogs) display a remarkable hypermuscular phenotype.25 Myostatin signalling primarily occurs via the interaction of the activin receptor II B and bone morphogenetic protein pathways, with other pathways such as the IGF‐1, p21/Cdk, and Wnt signalling are known to interact with myostatin signalling.25, 26, 27 Inhibition of myostatin has been reported to increase muscle mass and myofibre size (by cytoplasmic growth27). A large number of strategies (involving propeptide, gene therapy, gene editing, ligand traps, and monoclonal antibodies) have been investigated to attenuate myostatin activity in both preclinical and clinical settings,28, 29, 30, 31, 32, 33, 34 especially in cases of cachexia, sarcopenia, muscular atrophy, and other muscular dystrophies such as Duchenne muscular dystrophy (DMD).35, 36, 37, 38, 39, 40 An anti‐myostatin strategy, while not targeting the underlying genetic cause of the disease, may lead to an improvement in the quality of life of patients by delaying the onset of locomotion and atrophy related disability. Though early clinical trials in DMD, or Becker's muscular dystrophy with the first generation anti‐myostatin antibody MYO‐029 displayed limited therapeutic potential,29 other therapeutic candidates have been since developed. The anti‐myostatin antibody RK35 developed by Pfizer presents a next generation hybridoma and has been previously tested in models of amyotrophic lateral sclerosis, nemaline myopathy, and DMD, where it demonstrated efficacy in improving muscle mass and function.32, 41, 42, 43 The humanized equivalent of RK35 (Domagrozumab) is currently in clinical testing in DMD patients following a Phase 1 study in healthy volunteers.32 To our knowledge, this is the first report to examine the effect of inhibiting myostatin in a model of OPMD.
The A17 transgenic mouse over‐expresses bovine expPABPN1 specifically in skeletal muscle,44 and it is the most commonly used murine model of OPMD. This mouse exhibits loss of body mass, muscle atrophy, decreased muscle strength, transcriptomic dysregulation, and accumulation of insoluble aggregates in myonuclei.13, 17 Here, we show that treatment of OPMD mice with the murine anti‐myostatin monoclonal antibody RK35, while not affecting accumulation of intranuclear aggregates, prevents loss of body mass, muscle atrophy, and muscle strength and reduces deposition of fibrotic collagen proteins, which are relevant endpoints for a therapy targeting OPMD. Overall, our data support the clinical translation of myostatin inhibition to ameliorate symptoms of OPMD.
Material and methods
Animal handling
A17 and FvB mice were bred in‐house, and all mice were housed individually with food and water ad libitum in a minimal disease facility at Royal Holloway, University of London. Individual mice were identified by ear‐notching at about 4 weeks of age, and each mouse was monitored as per the recommendations of Animals (Scientific Procedures) Act (1986). Due to the heterozygous nature of the disease model, the OPMD mice were analysed to confirm the genotype by PCR, with primers directed against the bovine PABPN1 insert (5′‐GAACCAACAGACCAGGCATC‐3′ and 5′‐GTGATGGTGATGATGACCGG‐3′). The PCR cycle implemented initial denaturation at 95°C for 2 min, followed by 40 cycles of 95°C denaturation, 60°C for annealing, and 72°C for extension with each step lasting for 30 s. The final extension was conducted at 72°C for 10 min.17
Male 12‐week‐old mice were weighed prior to each injection. Initial body weights were used to evenly distribute animals among cohorts to ensure equivalent average body weights prior to the commencement of experimental protocols. In this experiment, we administered the anti‐myostatin blocking antibody RK35 [Pfizer, USA; diluted in sterile saline (Sigma Aldrich, UK) for a final volume of 200 μL] which was injected at 10 mg/kg weekly i.p. for 10 weeks into 9 A17 mice (disease model for OPMD) and 8 FvB (littermate strain control) mice. A further 10 A17 mice and 9 FvB mice were administered the vehicle (sterile saline) as a control in the experiment.
Forelimb grip strength analysis
To assess the forelimb grip strength of the animals, mice were allowed to rest on an angled mesh assembly, facing away from the meter (Linton Instrumentation, Norfolk, UK) and with its hind limbs not in contact with the mesh. A pulling force was exerted on the tail of the mouse parallel to the angle of the mesh, causing the animal to resist with both forelimbs. The force was recorded as the maximal force over a period of 30‐s, with five observations recorded per day. To prevent habituation of the mice, the test was performed over 3 days in 30‐s intervals of data collection and rest, over five observations per day resulting in 15 observations per mouse. Procedures were performed in accordance with Treat‐NMD protocols.
In situ muscle electrophysiology
At the end of the experiment, muscle function was assessed using the right tibialis anterior (TA) muscle. A17 and FvB mice were deeply anaesthetized by using a mix of Hypnorm/Hypnovel solution (i.p.) and were carefully monitored throughout the experiment to ensure that there was no reflex response to toe pinch. The distal tendon of the TA muscle was dissected from surrounding tissue and tied with 4.0 braided surgical silk (Interfocus, Cambridge, UK). The sciatic nerve was exposed and superfluous branches axotomized, leaving the TA motor innervation via the common peroneal nerve intact. The foot was secured to a platform and the ankle and knee immobilized using stainless steel pins. The TA tendon was attached to the lever arm of a 305B dual‐mode servomotor transducer (Aurora Scientific, Ontario, Canada) via a custom‐made steel s‐hook. TA muscle contractions were elicited by stimulating the distal part of common peroneal nerve via bipolar platinum electrodes, using supramaximal square‐wave pulses of 0.02 ms (701A stimulator; Aurora Scientific). Data acquisition and control of the servomotors were conducted using a Lab‐View‐based DMC programme (Dynamic muscle control and Data Acquisition; Aurora Scientific). Optimal muscle length (Lo) was determined by incrementally stretching the muscle using micromanipulators until the maximum isometric twitch force was achieved. Maximum isometric tetanic force (Po) was determined from the plateau of the force–frequency relationship following a series of stimulations at 10, 30, 40, 50, 80, 100, 120, and 150 Hz. The specific force (N/cm2) was calculated by dividing Po by TA muscle cross‐sectional area. Overall cross‐sectional area was estimated using previously established protocols.13
Sample collection and processing
Mice were euthanized 1 week after the last injection of RK35, and TA, extensor digitorum longus (EDL), and soleus muscles were harvested, weighed and mounted in O.C.T. compound (Thermo Fisher Scientific, Leicestershire, UK), frozen in 2‐methylbutane (isopentane) chilled with liquid nitrogen, and stored at −80°C.
Histological and immunohistochemical analysis
Transverse sections of the tissue were sectioned at 10 to 12 different intervals at a thickness of 10 μm, along the length of the muscle, allowing the maximal cross‐sectional area to be determined, and the sections mounted on coated slides (VWR International, Leicestershire, UK), and stored at −80°C. Transverse sections of the TA and EDL were air‐dried, fixed, and stained with anti‐PABPN1 (rabbit monoclonal, diluted 1:100, Abcam ab75855, overnight at 4°C), anti‐laminin (rat monoclonal, diluted 1:800, Sigma‐Aldrich L0663, 1 h at room temperature), and anti‐collagen VI (rabbit polyclonal, Abcam ab6588, 1:200, 1 h room temperature) antibodies or with picosirius red using previously established protocols.13 Slides were stained with DAPI (4′,6‐diamidino‐2‐phenylindole, 1μg/ml) to visualize nuclei, and coverslips were mounted using mounting medium (Vector Labs, California, USA). Whole muscle images (for laminin based fibre morphometry) or random fields (for PABPN1 and collagen immunostaining) were captured using a microscope (Zeiss, Cambridge, UK). For analysis of fibre morphometry, the median diameter from 1000 or 400 randomly selected fibres were determined respectively for TA and EDL muscles for each individual animal analysed. For analysis of succinate dehydrogenase (SDH) activity, tissue sections were incubated in the dark for an hour, in a solution of 0.1 M Na2HPO4, 0.1 M NaH2PO4, 0.1 M sodium succinate, and 2.4 mM nitro‐blue tetrazolium (Sigma Aldrich, UK), and random fields were analysed for the percentage of SDH positive fibres.
Quantitative PCR analysis
Total RNA was extracted from frozen skeletal muscles biopsies using Trizol (Invitrogen) according to the manufacturer's instructions. RNA samples were quantified using a ND‐1000 NanoDrop spectrophotometer (NanoDrop Technologies). RNA (50–250 ng) was reverse transcribed using M‐MLV reverse transcriptase (Invitrogen) according to the manufacturer's instructions. cDNA was used for quantitative PCR reaction using SYBR green mix buffer (LightCycler® 480 Sybr green I Master) in a total of 9 uL reaction volume. PCR reaction was carried out as follows: 8 min at 95°C followed by 50 cycles: 15 s at 95°C, 15 s at 60°C, and 15 s at 72°C. Specificity of the PCR products was checked by melting curve analysis using the following programme: 65°C increasing by 0.11°C/s to 97°C. The expression level of each mRNA was normalized to that of murine RPLP0 mRNA (large ribosomal protein, subunit P0) expression. Expression levels were calculated according to the DDCt method. Primers were designed to target Myh2 (5′‐ACTTTGGCACTACGGGGAAAC‐3′ and 5′‐CAGCAGCATTTCGATCAGCTC‐3′), Myh4 (5′‐CTTTGCTTACGTCAGTCAAGGT‐3′ and 5′‐AGCGCCTGTGAGCTTGTAAA‐3′), and Rplp0 as a housekeeping control (5′‐GAGGACCTCACTGAGATTCGG‐3′ and 5′‐TTCTGAGCTGGCACAGTGAC‐3′).
Statistical analyses
After checking for the conditions of normality and homoscedasticity, a one way analysis of variance (ANOVA) was performed to compare multiple groups. Multiple comparison were performed with the Benjamini, Hochberg, and Yekutieli correction for the false discovery rate (FDR) to correct for multiple testing used. All descriptive statistical techniques were performed using GraphPad Prism v7.00 (GraphPad Software, California, USA).
Results
Anti‐myostatin antibody RK35 treatment significantly reduces loss of body mass and muscle atrophy in A17 mice
Twelve‐week‐old A17 (OPMD mice) and FvB (WT; wild‐type strain control) mice were treated by weekly intraperitoneal (i.p.) injection of RK35 antibody (10 mg/kg in saline) or saline over a 10‐week period. Body mass, which has been correlated to muscle strength and is considered a rapid and longitudinal method to follow disease progression,45 was measured weekly. The treatment was well tolerated, and all mice grew during the 10‐week time treatment (Figure 1A). At the end of the experiment, saline‐treated OPMD mice had significantly lower body mass than saline‐treated WT mice (8% reduction; P < 0.05). Administration of RK35 antibody resulted in an increase of 11% (P < 0.01) in the final body mass of OPMD mice when compared to saline‐treated OPMD mice, normalizing it to that of saline‐treated WT control mice (Figure 1B). In this A17 OPMD model, myopathy induces muscle atrophy.17, 44 As different mammalian muscle fibre types possess varying sensitivities to this pathophysiological atrophy, three distinct muscles (TA, EDL, and soleus which are composed of mixed fibre type, a majority of type IIb fibres, and a majority of type I fibres, respectively46) were examined. The muscle mass of both TA and EDL increased after treatment by about 19% (P < 0.01) and 41% (P < 0.01), respectively (Figure 1C–D), in A17 mice treated with RK35 compared with mice treated with saline. The muscle mass of TA and EDL of RK35 treated A17 mice was either not significantly different to those of wild‐type FvB controls (TA muscles, P > 0.05) or significantly heavier (EDL muscles, 14% heavier, P < 0.05) (Figure 1C–D). Similar changes were observed for TA and EDL muscles of treated FvB mice; TA and EDL muscles of RK35 treated wild‐type FvB mice where 18% (P < 0.01) and 20% (P < 0.01) heavier than muscles of saline‐treated FvB mice, respectively (Figure 1C–D). Muscle mass of soleus was not affected by the treatment with RK35 in either A17 or FvB mice (Figure 1E). This was not unexpected because myostatin null mutants display minimal change in the contractile properties of the soleus.47 These data demonstrate that a systemic treatment with clinically relevant doses of RK35 antibody significantly increases the body and the muscle masses of the TA and EDL in OPMD mice.
Figure 1.
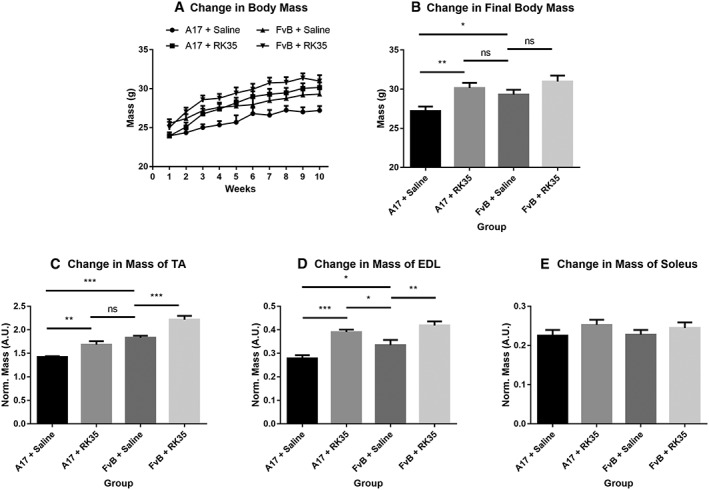
Treatment with RK35 antibody significantly increases body mass in A17 mice: Mice (n = 8–10) were weighed and administered a weekly regimen of either saline or the RK35 antibody (10 mg/kg i.p.) for 10 weeks from the 12th week of age. (A) The average body mass per group plotted against weeks shows that body mass increases in all groups of mice. (B) The average body mass at the final week shows that A17 mice treated with RK35 antibody are heavier than A17 mice treated with saline. TA (C), EDL (D), and soleus (E) were sampled, and mean mass of all muscles were normalized to the initial body mass. Muscle masses of the TA and EDL were increased in both the treated oculopharyngeal muscular dystrophy and control mice, with no change observed in the soleus. Data presented as mean ± standard error of the mean, with P‐values obtained by analysis of variance after a false discovery rate correction (*P < 0.05 and **P < 0.01). EDL, extensor digitorum longus; TA, tibialis anterior.
RK35 treatment improves the histopathology of oculopharyngeal muscular dystrophy
To further characterize the increase in muscle mass observed in treated mice, average myofibre size was measured in TA and EDL muscles. Whole muscle cross sections were stained with anti‐laminin antibodies, blinded, and analysed for myofibre size assessing the minimum Feret's diameter. The average myofibre cross‐sectional area of TA and EDL muscles of A17 mice were 28% (P < 0.001) and 22% (P < 0.001) smaller compared to FvB mice (Figure 2). Administration of the RK35 antibody in A17 mice resulted in a significant increase in the average diameter of TA (17%, P < 0.01) and EDL myofibres (44%, P < 0.001) respectively, when compared to saline‐treated A17 mice (Figure 2A–D).
Figure 2.
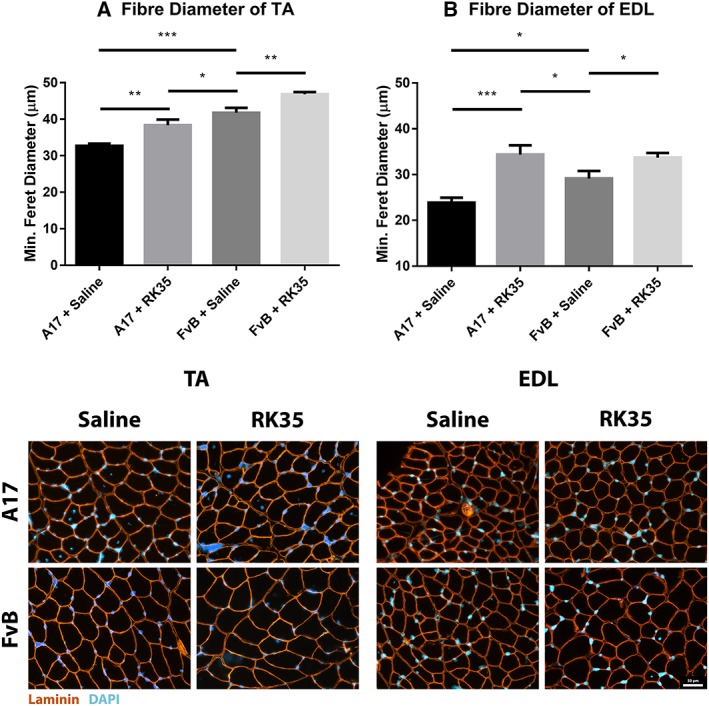
Treatment with RK35 antibody increases myofibre diameter in oculopharyngeal muscular dystrophy mice: Mice were administered with a weekly regimen of either saline or the RK35 antibody (10 mg/kg IP) for 10 weeks from 12‐week of age. Muscle samples from five randomly selected mice per group were sectioned and immunostained for laminin. (A) 1000 fibres were randomly selected for the TA, whereas (B) 400 fibres were randomly selected for the EDL, and the median minimum Feret diameter was analysed. The mice treated with the antibody consistently displayed an increased myofiber diameter, with the muscle mass of the disease model either normalized to or exceeding the FvB controls. The average median minimum feret diameter per group was plotted, with bars representing standard error of the mean, with P‐values obtained by analysis of variance after a false discovery rate correction (*P < 0.05 and **P < 0.01). EDL, extensor digitorum longus; TA, tibialis anterior.
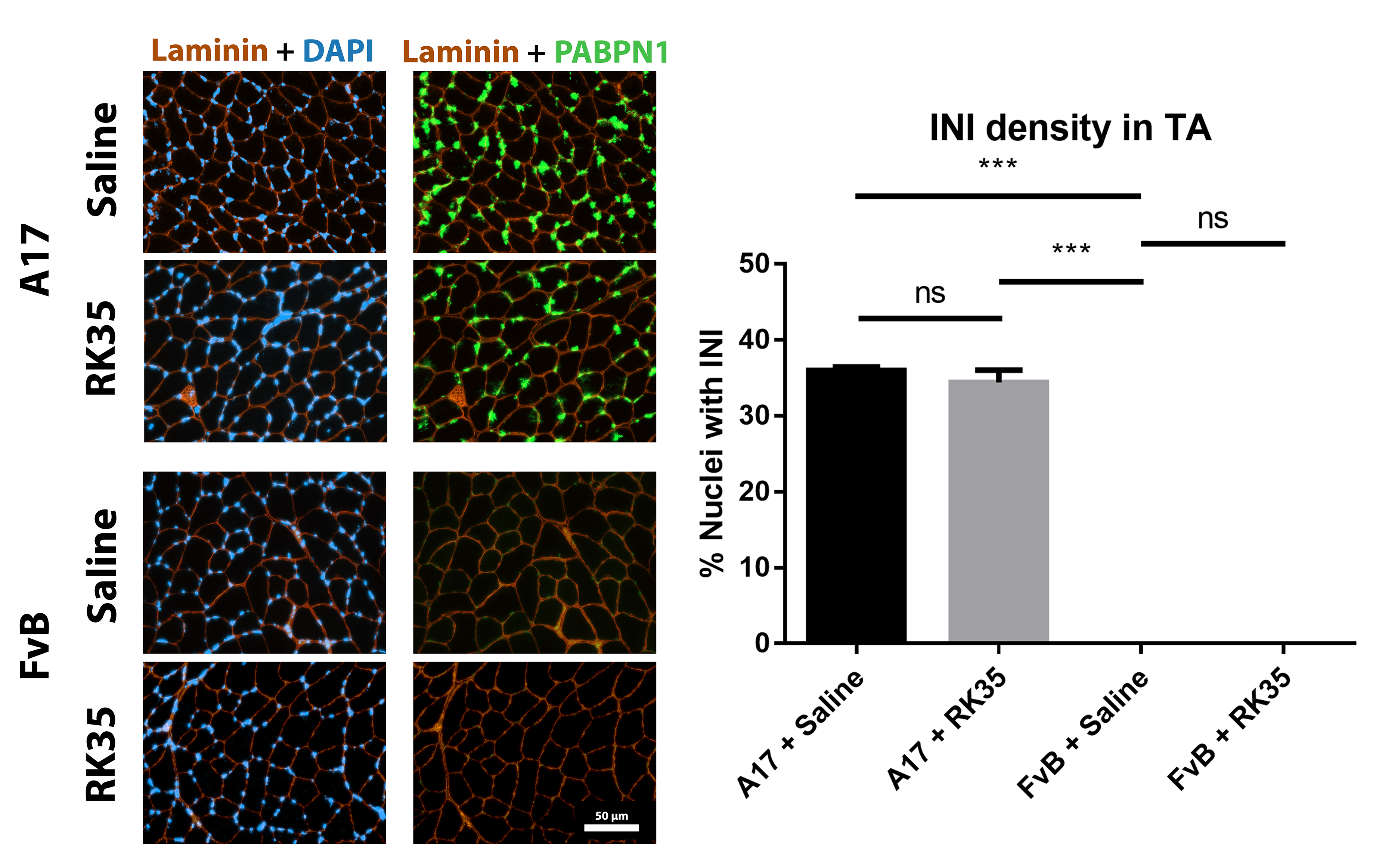
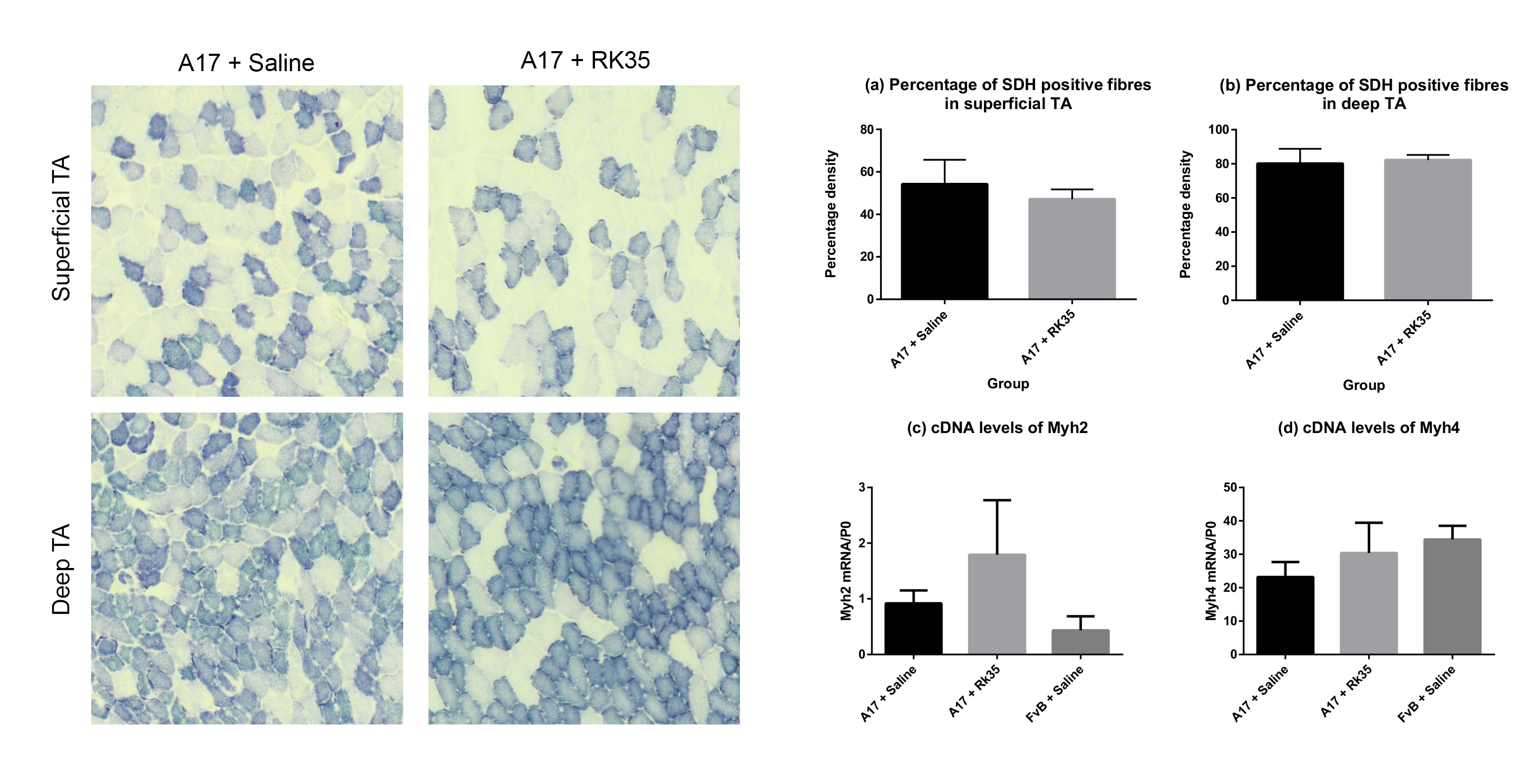
Muscle fibrosis is a distinct histopathological feature of affected OPMD muscles in humans. To study the effect of a RK35‐mediated myostatin inhibition on collagen protein deposition and fibrosis in the A17 mouse, TA muscle sections from RK35 or saline‐treated mice were stained with either picosirius red (to detect collagen proteins I and III) or with an antibody against collagen VI. TA muscles of saline‐treated A17 have 56% (P < 0.001) increased area of collagen VI deposition and 51% (P < 0.01) increased deposition of collagens I and III compared to muscles of age‐matched saline‐treated FvB mice (Figure 3A,B). Inhibition of myostatin in A17 mice reduced deposition of collagen VI in the TA muscle by 40% (P < 0.001), and of collagens I and III by 44% (P < 0.01) when compared to saline‐treated A17 mice normalizing the area covered by collagen proteins to the level observed in wild type mice (Figure 3A–B). One of the characteristic histopathological hallmarks of OPMD is the aggregation of expPABPN1 in muscle tissues.1, 48 To evaluate if inhibition of myostatin in A17 mice resulted in any change in the aggregate content, TA muscle sections from mice treated with RK35 were stained with antibody to PABPN1 and percentage of myonuclei with insoluble aggregates calculated. As expected, the percentage of myonuclei containing aggregates in wild‐type FvB mice was negligible (<1% of observed nuclei) and significantly higher in the saline‐treated A17 mice (36% ± 0.79%, P < 0.001; Figure S1A–F). Administration of RK35 antibody did not change the percentage of myonuclei containing aggregates in treated A17 (Figure S1A–F). We additionally analysed the effect of inhibition of myostatin by the antibody RK35 on myofibre metabolism and fibre type, using two methodologies: measuring change in expression levels of SDH and expression of two transcripts related to myofibre type: Myh2 and Myh4 (coding for myosin type IIa and type IIb respectively; cDNA generated from a muscle extract of TA). We report no significant differences observed between the groups studied (Figure S2A–D). These data show that the anti‐myostatin treatment significantly reduces myofibre atrophy and muscle fibrosis by altering biological pathways that do not affect the formation of aggregates in muscles or fibre type of A17 mice.
Figure 3.
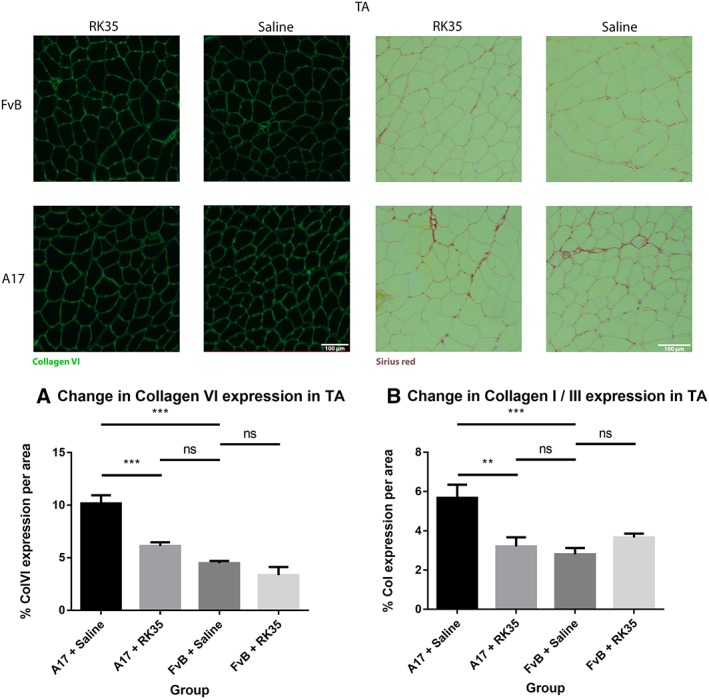
Treatment with RK35 antibody reduces collagen deposition in muscles of oculopharyngeal muscular dystrophy mice: Mice were administered with a weekly regimen of either saline or the RK35 antibody (10 mg/kg i.p.) for 10 weeks from 12‐week of age. Five randomly selected whole TA muscle sections from all groups were stained for (A) collagen VI and (B) picosirius red, and five random fields were imaged and analysed for the percentage area of collagen staining. The mice treated with the antibody consistently displayed a reduced collagen deposition, with the area of collagen in the disease model normalized to wild‐type levels. The average area per group is plotted, bars representing standard error of the mean, with P‐values obtained by analysis of variance after a false discovery rate correction (*P < 0.05 and **P < 0.01). TA, tibialis anterior.
RK35 mediated increase in muscle mass is accompanied by an increase in muscle strength
Forelimb grip strength was measured in A17 and FvB mice treated with the RK35 antibody or saline. The forelimb strength of the A17 mice was 13% lower (P < 0.05) than that of FvB control mice which likely reflects the atrophic and fibrotic condition of OPMD affected muscles (Figure 4A). The administration of RK35 in A17 mice significantly increased the forelimb strength by 23% (P < 0.001) when compared to the saline‐treated A17 mice. This increase in grip strength resulted in mice being equally strong as saline‐treated FvB control mice (Figure 4A). On the contrary, the treatment with RK35 antibody did not modify the forelimb strength of treated FvB mice (Figure 4A). Because this in‐life measure is not a pure readout of muscle strength, as it is dependent upon neurological, anatomical, and behavioural factors, the direct effect of myostatin inhibition on skeletal muscle force production was assessed by measuring maximal tetanic force generated in response to a series of increasing stimulation frequencies by in situ muscle electrophysiology. The maximal tetanic force generated by TA muscles of A17 mice was 55% (P < 0.001) lower than that of FvB controls (Figure 4B,C). Administration of RK35 antibody to A17 mice significantly increased the maximum tetanic force of TA muscles compared to saline‐treated A17 mice by 18% (P < 0.05) while no change in maximum tetanic force was observed between TA of RK35 treated and saline‐treated FvB mice. Normalizing the maximum tetanic force with the calculated muscle cross‐sectional area yields specific maximal force, a measurement that expresses the force of the muscle per unit of area. No difference was observed in specific maximal force for TA muscles between any of the groups analysed (data not shown). Finally, as the treatment with RK35 affected the body weight and the muscle mass of A17 mice, we evaluated the locomotor behaviour to exclude any detrimental effect of such treatment. No change in voluntary locomotion between the saline‐treated and RK35 treated mice was detected, nor a change was observed between the A17 and FvB mice (data not shown). Overall, these data show that in RK35 treated A17 mice the muscle force increases with the hypertrophic effect and this is reflected in an improved limb strength.
Figure 4.

Treatment with RK35 increases muscle strength in oculopharyngeal muscular dystrophy mice: Mice (n = 8–10) were administered with a weekly regimen of either saline or the RK35 antibody (10 mg/kg IP) for 10 weeks from 12‐week of age. (A) Forelimb grip strength was analysed and the average of 15 measurements per mouse, per group is plotted, bars representing standard error of the mean, with P‐values obtained by analysis of variance after a false discovery rate correction. (B–D) The left TA from all groups were mounted on a mechanotransducer and the sciatic nerve was excited (B) average max. force generated at increasing frequencies is plotted. (C) The average max. force generated at 180 Hz is plotted, with bars representing standard error of the mean. The gross muscle strength of the disease model mice treated with the antibody displayed an increased grip strength, although a similar effect was not observed in the control mice. (D) Maximal force values were normalized with the muscle cross‐sectional area to obtain specific force of TA at 180 Hz, and no significant differences observed between the groups. Data presented as mean ± standard error of the mean, with P‐values obtained by analysis of variance after a false discovery rate correction (*P < 0.05 and **P < 0.01). TA, tibialis anterior.
Discussion
Myostatin is a negative regulator of muscle mass. The efficacy of myostatin blockade by the monoclonal antibody RK35 has been examined in the murine models of amyotrophic lateral sclerosis, DMD, and nemaline myopathy,41, 42, 43 and this antibody is currently in phase II clinical trial for DMD (NCT02907619 and NCT02310763). In this study, we tested the effect of myostatin inhibition to ameliorate atrophic symptoms in the A17 OPMD mouse model system using this antibody. The A17 mice co‐express a pathological expPABPN1 transgene in skeletal muscles to mimic the disease at a tissue level. Early studies in the A17 model have reported the onset of loss of muscle force by 18 weeks of age. To evaluate the efficacy of myostatin inhibition before the pathological onset of disease in terms of delaying disease progression, we treated the A17 disease model with the anti‐myostatin antibody RK35 for 10 weeks, from the 12th week of age. As previously shown,17 our study confirmed that A17 mouse model at 22 weeks of age (i.e. at the end of the treatment) has lower body and muscle mass as compared to the FvB age‐matched controls. The lower muscle mass was accompanied by an atrophy of the muscles, and an increased level of observed fibrosis markers resulting in a lower functional quality of muscles as compared to the saline‐treated wild‐type FvB mice. The treatment with RK35 antibody resulted in the reversal of loss of body mass in the OPMD model. The increase in body mass was most evident during the first 5 weeks of the 10‐week treatment regimen, after which the difference was maintained. Using the same antibody, a similar effect in DMD and nemaline myopathy models was observed by St. Andre et al.43 and Tinklenberg et al.,41 respectively. The increased response to the antibody (in terms of change in muscle mass) we observed in the EDL compared to the TA and soleus muscles is likely due to the myofibrillar metabolic profile of glycolytic fast‐twitch fibres that cause an increased production of myostatin transcripts and an increased density of the activin receptor II B receptors to be present in the EDL.47 This observation is particularly interesting as OPMD disease progression is reported to be more specific in fast‐twitch muscle fibres, such as those found in the EDL.17The increase in muscle size observed in muscles of the A17 mice treated with RK35 is promising, as it indicates that inhibition of myostatin may be a valid strategy to counteract the loss of muscle mass in OPMD human patients. Skeletal muscles form the most abundant tissue in the body and maintain the energy balance of the organism by regulating glucose availability in the blood serum. Indeed, muscles absorb glucose which are then stored or oxidized subsequent to a contraction induced/(postprandial secreted) insulin induced signals. We speculate that this increase in muscle mass may assist to further attenuate any secondary effects in systemic metabolism that may arise from muscular atrophy, by perhaps increasing insulin sensitivity of the organism.
Muscle fibrosis forms an essential step in both myogenesis and in muscle regeneration where it helps reforming the contractile unit from myofibre stumps after damage.49, 50 However, in OPMD, the fibrotic deposition is mainly pathological and it is associated with a defective muscle functionality perhaps contributing to dysphagia and ptosis in OPMD subjects. The reduction in the area of fibrotic collagen deposition in the TA is promising and indicates a prevention of loss of structural integrity of skeletal muscles. This might be related to the effect of myostatin inhibition in inducing apoptosis of fibroblasts.51 Again, the effect of RK35 on fibrosis may be crucially important to preserve and possibly restore the functionality of affected muscles of human OPMD subjects.
The increase in muscle mass and the reduction in pathological fibrosis is correlated to an increase in grip strength and maximal tetanic force, corresponding to an improvement in gross muscle function in the treated A17 mice. No such effect was evident in the treated wild‐type FvB mice despite of an increase in muscle mass. This indicates that the antibody treatment may be more effective in atrophic muscles than in healthy muscles most likely because healthy muscles may have already attained the maximal possible biological myofibre diameter.
Among the other therapeutic strategies under clinical and preclinical development for muscular dystrophies, the anti‐myostatin antibody strategy has some advantages. Firstly, the systemic and non‐invasive method of drug administration can ameliorate symptoms in all affected muscles as opposed to specific muscles that are targeted by other locally administered agents (i.e. gene therapy vectors and myoblast transplantation). Furthermore, monoclonal antibodies are less costly to produce, easier to attain regulatory approval, and translate into clinical trials than other gene and cell therapy approaches.52 This strategy has the potential to be used as an adjuvant therapy53 of pharmacological treatments currently in preclinical and clinical development (i.e. trehalose) or gene therapy13 that target aggregate formation without addressing the issue of pre‐existing muscle atrophy or fibrosis. An interesting observation relating to the disease condition itself revolves around the fact that an improvement in muscle force and rescue of muscle atrophy was evident without affecting the number of intranuclear aggregates. While the exact mechanisms associated with disease progression with respect to aggregate formation remains to be elucidated, our results may suggest that the presence of aggregates does not preclude an increase in muscle hypertrophy. Notably, this demonstrates that a treatment strategy that does not target the affect the aggregate formation in muscle might be clinically viable in OPMD. Future experiments can be conducted in order to elucidate the effect of an anti‐myostatin treatment regimen on the signalling pathways of the disease model. In conclusion, we report that a systemic administration of an anti‐myostatin monoclonal antibody delays disease progression in the OPMD mouse model and, as such, is a promising therapeutic strategy to be explored.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Figure S1. Treatment with RK35 does not affect the amount of intranuclear aggregates: Mice were subject to a weekly regimen of either saline or the anti‐myostatin RK35 antibody i.p. for 10 weeks from the 12th week of age till the 22nd week of age. Five muscle samples from all groups were stained for bovine PABPN1 after a 1M KCl treatment, and 5 random fields were imaged and analysed for the percentage number of positive PABPN1 stains to all myonuclei. The administration of the treatment regimen did not change the number of intranuclear aggregates observed. The average density is plotted, bars representing SEM, with p‐values obtained by ANOVA after a FDR correction (*p<0.05; **p<0.01).
{kind=link}
Figure S2. Treatment with RK35 does not affect myofibre type or myofibre oxidative capacity in the OPMD model mice: Mice were subject to a weekly regimen of either saline or the anti‐myostatin RK35 antibody i.p. for 10 weeks from the 12th week of age till the 22nd week of age. Five muscle samples from the A17 mice treated with saline or RK35 were stained for SDH activity. Random fields from different regions of the TA; namely the (a) superficial TA (region around the periphery of the muscle wherein a higher density of fast‐twitch fibres are found) and (b) deep TA (the central portion of the muscle having a higher density of slow fibres) were analysed separately. The administration of the treatment regimen did not change the number of SDH positive fibres observed. The percentage SDH positive fibres is plotted, bars representing SEM, with p‐values obtained by a t‐test (no significant differences observed between groups). Additionally, a qPCR was performed in order to investigate the transcript levels of the myosin heavy chains (c) IIa (encoded by myh2) and (d) IIb (encoded by myh4), with values normalised to the levels of RPLP0. The normalised mean abundance of transcripts are plotted, bars representing SEM, with p‐values obtained by ANOVA after a FDR correction (no significant changes observed between groups).
{kind=link}
Acknowledgements
The authors wish to thank Jane Owens, Michael St‐Andre, Carl Morris, and other colleagues at Pfizer for providing the antibody RK35 and for support designing the study. The authors also wish to acknowledge Martine Oloko for technical assistance during the course of the study. P.H. also wishes to thank S. Harish and H. Parameshwaran for their support. The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle.54 All data supporting this study is provided either in the Results section of the manuscript or as supplementary information accompanying this paper. P.H., A.M., C.T., L.P., and G.D. designed the study. P.H., A.M., L.F., N.L., O.C., and F.R. collected, analysed and interpreted data. P.H. and A.M. participated in manuscript preparation. All authors provided manuscript review and revisions.
Harish, P. , Malerba, A. , Lu‐Nguyen, N. , Forrest, L. , Cappellari, O. , Roth, F. , Trollet, C. , Popplewell, L. , and Dickson, G. (2019) Inhibition of myostatin improves muscle atrophy in oculopharyngeal muscular dystrophy (OPMD). Journal of Cachexia, Sarcopenia and Muscle, 10: 1016–1026. 10.1002/jcsm.12438.
References
- 1. Harish P, Malerba A, Dickson G, Bachtarzi H. progress on gene therapy, cell therapy, and pharmacological strategies toward the treatment of oculopharyngeal muscular dystrophy. Hum Gene Ther 2015;26:286–292. [DOI] [PubMed] [Google Scholar]
- 2. Apponi LH, Leung SW, Williams KR, Valentini SR, Corbett AH, Pavlath GK. Loss of nuclear poly(A)‐binding protein 1 causes defects in myogenesis and mRNA biogenesis. Hum Mol Genet 2010;19:1058–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Benoit B, Mitou G, Chartier A, Temme C, Zaessinger S, Wahle E, et al. An essential cytoplasmic function for the nuclear poly(A)‐binding protein, PABP2, in poly(A) tail length control and early development in Drosophila. Dev Cell 2005;9:511–522. [DOI] [PubMed] [Google Scholar]
- 4. Jenal M, Elkon R, Loayza‐Puch F, Van Haaften G, Kühn U, Menzies FM, et al. The poly(A)‐binding protein nuclear 1 suppresses alternative cleavage and polyadenylation sites. Cell 2012;149:538–553. [DOI] [PubMed] [Google Scholar]
- 5. de Klerk E, Venema A, Anvar SY, Goeman JJ, Hu O, Trollet C, et al. Poly(A) binding protein nuclear 1 levels affect alternative polyadenylation. Nucleic Acids Res 2012;40:9089–9101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bergeron D, Pal G, Beaulieu YB, Chabot B, Bachand F. Regulated intron retention and nuclear pre‐mRNA decay contribute to PABPN1 autoregulation. Mol Cell Biol 2015;35:2503–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Muniz L, Davidson L, West S. Poly(A) Polymerase and the nuclear poly(A) binding protein, PABPN1, coordinate the splicing and degradation of a subset of human pre‐mRNAs. Mol Cell Biol 2015;35:2218–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beaulieu YB, Kleinman CL, Landry‐Voyer AM, Majewski J, Bachand F. Polyadenylation‐dependent control of long noncoding RNA expression by the poly(A)‐binding protein nuclear 1. PLoS Genet 2012;8:e1003078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lemay J‐F, Lemieux C, St‐André O, Bachand F. Crossing the borders: poly(A)‐binding proteins working on both sides of the fence. RNA Biol 2010;7:291–295. [DOI] [PubMed] [Google Scholar]
- 10. Bresson SM, Conrad NK. The human nuclear poly(A)‐binding protein promotes RNA hyperadenylation and decay. PLoS Genet 2013;9:e1003893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Calado A, Tome FM, Brais B, Rouleau GA, Kuhn U, Wahle E, et al. Nuclear inclusions in oculopharyngeal muscular dystrophy consist of poly(A) binding protein 2 aggregates which sequester poly(A) RNA. Hum Mol Genet 2000;9:2321–2328. [DOI] [PubMed] [Google Scholar]
- 12. Anvar SY, Raz Y, Verwey N, Van der Sluijs B, Venema A, Goeman JJ, et al. A decline in PABPN1 induces progressive muscle weakness in oculopharyngeal muscle dystrophy and in muscle aging. Aging (Albany NY) 2013;5:412–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Malerba A, Klein P, Bachtarzi H, Jarmin SA, Cordova G, Ferry A, et al. PABPN1 gene therapy for oculopharyngeal muscular dystrophy. Nat Commun 2017;8:14848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vest KE, Phillips BL, Banerjee A, Apponi LH, Dammer EB, Xu W, et al. Novel mouse models of oculopharyngeal muscular dystrophy (OPMD) reveal early onset mitochondrial defects and suggest loss of PABPN1 may contribute to pathology. Hum Mol Genet 2017;26:3235–3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Apponi LH, Corbett AH, Pavlath GK. Control of mRNA stability contributes to low levels of nuclear poly(A) binding protein 1 (PABPN1) in skeletal muscle. Skelet Muscle 2013;3:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Banerjee A, Vest KE, Pavlath GK, Corbett AH. Nuclear poly(A) binding protein 1 (PABPN1) and Matrin3 interact in muscle cells and regulate RNA processing. Nucleic Acids Res 2017;45:10706–10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Trollet C, Anvar SY, Venema A, Hargreaves IP, Foster K, Vignaud A, et al. Molecular and phenotypic characterization of a mouse model of oculopharyngeal muscular dystrophy reveals severe muscular atrophy restricted to fast glycolytic fibres. Hum Mol Genet 2010;19:2191–2207. [DOI] [PubMed] [Google Scholar]
- 18. Anvar S, 't Hoen PA, Venema A, van der Sluijs B, van Engelen B, Snoeck M, et al. Deregulation of the ubiquitin‐proteasome system is the predominant molecular pathology in OPMD animal models and patients. Skelet Muscle 2011;1:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Davies JE, Rubinsztein DC. Over‐expression of BCL2 rescues muscle weakness in a mouse model of oculopharyngeal muscular dystrophy. Hum Mol Genet 2011;20:1154–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Davies JE, Sarkar S, Rubinsztein DC. Wild‐type PABPN1 is anti‐apoptotic and reduces toxicity of the oculopharyngeal muscular dystrophy mutation. Hum Mol Genet 2008;17:1097–1108. [DOI] [PubMed] [Google Scholar]
- 21. Raz Y, Raz V. Oculopharyngeal muscular dystrophy as a paradigm for muscle aging. Front Aging Neurosci 2014;6:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Perie S, Trollet C, Mouly V, Vanneaux V, Mamchaoui K, Bouazza B, et al. Autologous myoblast transplantation for oculopharyngeal muscular dystrophy: a phase I/IIa clinical study. Mol Ther 2014;22:219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Argov Z, Gliko‐Kabir I, Brais B, Caraco Y, Megiddo D. Intravenous trehalose improves dysphagia and muscle function in oculopharyngeal muscular dystrophy (OPMD): preliminary results of 24 weeks open label phase 2 trial (S28. 004). Neurology 2016;86:S28–S004. [Google Scholar]
- 24. Argov Z, Vornovitsky H, Blumen S, Caraco Y. First human use of high dose IV trehalose: safety, tolerability and pharmacokinetic results from the oculopharyngeal muscular dystrophy (OPMD) therapy trial (P7. 068). Neurology 2015;84:P7–P068. [Google Scholar]
- 25. Lee S‐J. Regulation of muscle mass by myostatin. Annu Rev Cell Dev Biol 2004;20:61–86. [DOI] [PubMed] [Google Scholar]
- 26. Rodgers BD, Garikipati DK. Clinical, agricultural, and evolutionary biology of myostatin: a comparative review. Endocr Rev 2008;29:513–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sartori R, Schirwis E, Blaauw B, Bortolanza S, Zhao J, Enzo E, et al. BMP signaling controls muscle mass. Nat Genet 2013;45:1309–1318. [DOI] [PubMed] [Google Scholar]
- 28. Wagner KR, McPherron AC, Winik N, Lee SJ. Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann Neurol 2002;52:832–836. [DOI] [PubMed] [Google Scholar]
- 29. Wagner KR, Fleckenstein JL, Amato AA, Barohn RJ, Bushby K, Escolar DM, et al. A phase I/IItrial of MYO‐029 in adult subjects with muscular dystrophy. Ann Neurol 2008;63:561–571. [DOI] [PubMed] [Google Scholar]
- 30. Mendell JR, Sahenk Z, Malik V, Gomez AM, Flanigan KM, Lowes LP, et al. A phase 1/2a follistatin gene therapy trial for Becker muscular dystrophy. Mol Ther 2015;23:192–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mendell JR, Sahenk Z, Al‐Zaidy S, Rodino‐Klapac LR, Lowes LP, Alfano LN, et al. Follistatin gene therapy for sporadic inclusion body myositis improves functional outcomes. Mol Ther 2017;25:870–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bhattacharya I, Pawlak S, Marraffino S, Christensen J, Sherlock SP, Alvey C, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of domagrozumab (PF‐06252616), an antimyostatin monoclonal antibody, in healthy subjects. Clin Pharmacol drug Dev 2017;7:484–497. [DOI] [PubMed] [Google Scholar]
- 33. Campbell C, McMillan HJ, Mah JK, Tarnopolsky M, Selby K, McClure T, et al. Myostatin inhibitor ACE‐031 treatment of ambulatory boys with Duchenne muscular dystrophy: results of a randomized, placebo‐controlled clinical trial. Muscle Nerve 2017;55:458–464. [DOI] [PubMed] [Google Scholar]
- 34. Bogdanovich S, Perkins KJ, Krag TOB, Whittemore L‐A, Khurana TS. Myostatin propeptide‐mediated amelioration of dystrophic pathophysiology. FASEB J 2005;19:543–549. [DOI] [PubMed] [Google Scholar]
- 35. Siriett V, Salerno MS, Berry C, Nicholas G, Bower R, Kambadur R, et al. Antagonism of myostatin enhances muscle regeneration during sarcopenia. Mol Ther 2007;15:1463–1470. [DOI] [PubMed] [Google Scholar]
- 36. Gilson H, Schakman O, Combaret L, Lause P, Grobet L, Attaix D, et al. Myostatin gene deletion prevents glucocorticoid‐induced muscle atrophy. Endocrinology 2007;148:452–460. [DOI] [PubMed] [Google Scholar]
- 37. Zhang L, Rajan V, Lin E, Hu Z, Han HQ, Zhou X, et al. Pharmacological inhibition of myostatin suppresses systemic inflammation and muscle atrophy in mice with chronic kidney disease. FASEB J 2011;25:1653–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bogdanovich S, Krag TOB, Barton ER, Morris LD, Whittemore L‐A, Ahima RS, et al. Functional improvement of dystrophic muscle by myostatin blockade. Nature 2002;420:418–421. [DOI] [PubMed] [Google Scholar]
- 39. Whittemore L‐A, Song K, Li X, Aghajanian J, Davies M, Girgenrath S, et al. Inhibition of myostatin in adult mice increases skeletal muscle mass and strength. Biochem Biophys Res Commun 2003;300:965–971. [DOI] [PubMed] [Google Scholar]
- 40. Klimek MEB, Aydogdu T, Link MJ, Pons M, Koniaris LG, Zimmers TA. Acute inhibition of myostatin‐family proteins preserves skeletal muscle in mouse models of cancer cachexia. Biochem Biophys Res Commun 2010;391:1548–1554. [DOI] [PubMed] [Google Scholar]
- 41. Tinklenberg JA, Siebers EM, Beatka MJ, Meng H, Yang L, Zhang Z, et al. Myostatin inhibition using mRK35 produces skeletal muscle growth and tubular aggregate formation in wild type and TgACTA1D286G nemaline myopathy mice. Hum Mol Genet 2017;27:638–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Holzbaur ELF, Howland DS, Weber N, Wallace K, She Y, Kwak S, et al. Myostatin inhibition slows muscle atrophy in rodent models of amyotrophic lateral sclerosis. Neurobiol Dis 2006;23:697–707. [DOI] [PubMed] [Google Scholar]
- 43. St. Andre M, Johnson M, Bansal PN, Wellen J, Robertson A, Opsahl A, et al. A mouse anti‐myostatin antibody increases muscle mass and improves muscle strength and contractility in the mdx mouse model of Duchenne muscular dystrophy and its humanized equivalent, domagrozumab (PF‐06252616), increases muscle volume in cynomolgus monk. Skelet Muscle 2017;7:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Davies JE, Wang L, Garcia‐Oroz L, Cook LJ, Vacher C, O'Donovan DG, et al. Doxycycline attenuates and delays toxicity of the oculopharyngeal muscular dystrophy mutation in transgenic mice. Nat Med 2005;11:672–677. [DOI] [PubMed] [Google Scholar]
- 45. Hasan NAKAK, Kamal HM, Hussein ZA. Relation between body mass index percentile and muscle strength and endurance. Egypt J Med Hum Genet 2016;17:367–372. [Google Scholar]
- 46. Schiaffino S, Reggiani C. Fiber types in mammalian skeletal muscles. Physiol Rev 2011;91:1447–1531. [DOI] [PubMed] [Google Scholar]
- 47. Mendias CL. Contractile properties of EDL and soleus muscles of myostatin‐deficient mice. J Appl Physiol 2006;101:898–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tomé FM, Fardeau M. Nuclear inclusions in oculopharyngeal dystrophy. Acta Neuropathol 1980;49:85–87. [DOI] [PubMed] [Google Scholar]
- 49. Li ZB, Kollias HD, Wagner KR. Myostatin directly regulates skeletal muscle fibrosis. J Biol Chem 2008;283:19371–19378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. March J, Golshirazi G, Cernisova V, Carr H, Leong Y, Lu‐Nguyen N, et al. Targeting TGFβ signaling to address fibrosis using antisense oligonucleotides. Biomedicines 2018;6:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li ZB, Zhang J, Wagner KR. Inhibition of myostatin reverses muscle fibrosis through apoptosis. J Cell Sci 2012;125:3957–3965. [DOI] [PubMed] [Google Scholar]
- 52. da Silva FA, Corte‐Real S, Goncalves J. Recombinant antibodies as therapeutic agents. BioDrugs 2008;22:301–314. [DOI] [PubMed] [Google Scholar]
- 53. Cordova G, Negroni E, Cabello‐Verrugio C, Mouly V, Trollet C. Combined therapies for Duchenne muscular dystrophy to optimize treatment efficacy. Front Genet 2018;9:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Treatment with RK35 does not affect the amount of intranuclear aggregates: Mice were subject to a weekly regimen of either saline or the anti‐myostatin RK35 antibody i.p. for 10 weeks from the 12th week of age till the 22nd week of age. Five muscle samples from all groups were stained for bovine PABPN1 after a 1M KCl treatment, and 5 random fields were imaged and analysed for the percentage number of positive PABPN1 stains to all myonuclei. The administration of the treatment regimen did not change the number of intranuclear aggregates observed. The average density is plotted, bars representing SEM, with p‐values obtained by ANOVA after a FDR correction (*p<0.05; **p<0.01).
Figure S2. Treatment with RK35 does not affect myofibre type or myofibre oxidative capacity in the OPMD model mice: Mice were subject to a weekly regimen of either saline or the anti‐myostatin RK35 antibody i.p. for 10 weeks from the 12th week of age till the 22nd week of age. Five muscle samples from the A17 mice treated with saline or RK35 were stained for SDH activity. Random fields from different regions of the TA; namely the (a) superficial TA (region around the periphery of the muscle wherein a higher density of fast‐twitch fibres are found) and (b) deep TA (the central portion of the muscle having a higher density of slow fibres) were analysed separately. The administration of the treatment regimen did not change the number of SDH positive fibres observed. The percentage SDH positive fibres is plotted, bars representing SEM, with p‐values obtained by a t‐test (no significant differences observed between groups). Additionally, a qPCR was performed in order to investigate the transcript levels of the myosin heavy chains (c) IIa (encoded by myh2) and (d) IIb (encoded by myh4), with values normalised to the levels of RPLP0. The normalised mean abundance of transcripts are plotted, bars representing SEM, with p‐values obtained by ANOVA after a FDR correction (no significant changes observed between groups).