

Abstract
Background
Pancreatic ductal adenocarcinoma (PDAC) is a particularly lethal malignancy partly due to frequent, severe cachexia. Serum activin correlates with cachexia and mortality, while exogenous activin causes cachexia in mice.
Methods
Isoform‐specific activin expression and activities were queried in human and murine tumours and PDAC models. Activin inhibition was by administration of soluble activin type IIB receptor (ACVR2B/Fc) and by use of skeletal muscle specific dominant negative ACVR2B expressing transgenic mice. Feed‐forward activin expression and muscle wasting activity were tested in vivo and in vitro on myotubes.
Results
Murine PDAC tumour‐derived cell lines expressed activin‐βA but not activin‐βB. Cachexia severity increased with activin expression. Orthotopic PDAC tumours expressed activins, induced activin expression by distant organs, and produced elevated serum activins. Soluble factors from PDAC elicited activin because conditioned medium from PDAC cells induced activin expression, activation of p38 MAP kinase, and atrophy of myotubes. The activin trap ACVR2B/Fc reduced tumour growth, prevented weight loss and muscle wasting, and prolonged survival in mice with orthotopic tumours made from activin‐low cell lines. ACVR2B/Fc also reduced cachexia in mice with activin‐high tumours. Activin inhibition did not affect activin expression in organs. Hypermuscular mice expressing dominant negative ACVR2B in muscle were protected for weight loss but not mortality when implanted with orthotopic tumours. Human tumours displayed staining for activin, and expression of the gene encoding activin‐βA (INHBA) correlated with mortality in patients with PDAC, while INHBB and other related factors did not.
Conclusions
Pancreatic adenocarcinoma tumours are a source of activin and elicit a systemic activin response in hosts. Human tumours express activins and related factors, while mortality correlates with tumour activin A expression. PDAC tumours also choreograph a systemic activin response that induces organ‐specific and gene‐specific expression of activin isoforms and muscle wasting. Systemic blockade of activin signalling could preserve muscle and prolong survival, while skeletal muscle‐specific activin blockade was only protective for weight loss. Our findings suggest the potential and need for gene‐specific and organ‐specific interventions. Finally, development of more effective cancer cachexia therapy might require identifying agents that effectively and/or selectively inhibit autocrine vs. paracrine activin signalling.
Keywords: Pancreatic cancer, Cachexia, Activins, Activin receptor type IIb, Weight loss
Introduction
Cancer cachexia is a multifactorial syndrome with progressive weight loss mainly resulting from reduction of skeletal muscle and fat mass; other cachexia‐associated clinical manifestations include chronic inflammation, anorexia, and fatigue.1, 2, 3 Simply increasing food intake or nutritional support is inadequate to reverse the cachectic state. Patients with cancer cachexia respond poorly to anti‐cancer treatments including chemotherapy and radiotherapy and experience increased treatment toxicity. Lethality of pancreatic ductal adenocarcinoma (PDAC) is due in large part to the frequent occurrence of severe cachexia in patients with PDAC. Currently, there are no approved, effective therapeutics to treat cachexia, despite many promising pre‐clinical studies.
Activin proteins have been implicated in cancer cachexia. Activins are members of the transforming growth factor‐beta (TGF‐β) superfamily and play multiple biological roles including regulation of development, homeostasis, tissue repair, and inflammation.4, 5, 6, 7, 8 Activins are composed of two polypeptide subunits, each being encoded by a separate gene, subunit βA (gene Inhba), βB (Inhbb), βC (Inhbc), and βE (Inhbe). Two βA or two βB form activin A or activin B, respectively, while one βA and one βB form activin AB.9 Activin βC10, 11, 12 and βE13 were discovered more recently and have been linked with cachexia,14, 15 although their roles are less understood. Deregulated activin A is often observed in various malignancies including pancreatic cancer,6, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28 showing overexpression in tumours or elevation in blood. This suggests that activin A has potential endocrine effects on the host organs. Elevated plasma activin A level is also documented with cachexia and mortality in PDAC.29, 30 Activin B has received much less attention regarding its role in cancer or cancer cachexia.31, 32, 33 Experimentally, exogenous activin induces weight loss and muscle wasting in mice,27, 28 while injection of a soluble activin receptor or activin‐receptor blocking antibody induces skeletal muscle hypertrophy and prevents or slows cachexia in mice with cancer or other muscle wasting conditions.34, 35
Given activin A deregulation in cancer and likely its systemic impact on host biology, we aimed to investigate activin expression and cachexia in murine models of PDAC and in patients and to evaluate the therapeutic utility of blocking the activin receptor ACVR2B‐mediated signalling pathway.
Materials and methods
Ethics
The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle.36 All mouse studies were approved by the Institutional Animal Care and Use Committee at Indiana University School of Medicine and in compliance with the National Institutes of Health Guidelines for Use and Care of Laboratory Animals. Human tumour specimens were obtained under an IRB approved protocol number 1312105608 from patients undergoing surgery for pancreatic cancer at Indiana University.
Pancreatic cancer cell lines
Pancreatic cancer cells, KPC32043, KPC32047, and KPC32908 (gifted by David Tuveson, Cold Spring Harbor Laboratories) derived from the genetically engineered KrasG12D;Trp53R172H;Pdx1::Cre mouse were maintained in Dulbecco's modified Eagle's medium (DMEM) medium. All media were supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Conditioned medium (CM) was collected from confluent cultures.
Effects on myotube size
C2C12 cells (ATCC, Manassas, VA, USA) were maintained in DMEM with 10% fetal bovine serum and 1% penicillin/streptomycin. Myoblast differentiation to myotubes was induced by shifting confluent cultures to differentiation medium (DM), consisting of DMEM with 2% horse serum. DM was replaced every day for 6 days. KPC cell CM was added to C2C12 myoblast or 4‐day‐old differentiated myotube cultures at 25%, 50%, or 75% in DM for 24 or 48 h. Cultures were fixed in 4% paraformaldehyde and permeabilized in 1% Triton X‐100 at room temperature, followed by blocking in Sea Block Blocking Buffer (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 0.2% Tween 20. Fixed myotubes were incubated with myosin heavy chain (MyHC) primary antibody (1:100, Developmental Studies Hybridoma Bank, Iowa City, IA, USA) and then with Alexa Fluor 488‐conjugated anti‐mouse secondary antibody (1:1000, Invitrogen, Carlsbad, CA, USA). Nuclei were stained with 1 μg/mL 4′,6‐diamidino‐2‐phenylinole (Calbiochem, Millipore Sigma, Burlington, MA, USA). Images were acquired at 10× using a monochrome (MRm) Zeiss AxioCam camera mounted on a Zeiss Axio Observer.Z1 inverted microscope. Fifteen fields from each well were randomly selected at imaging acquisition, and three wells per experimental condition gave 45 fields from which >200 qualified myotubes per well and condition were measured at or near the middle of isolated, long, multinucleate myotubes avoiding regions of clustered nuclei, using Fiji (https://fiji.sc). The average diameter of myotubes per condition was calculated and expressed as mean ± SD.
Mice
Mice were housed up to five per cage in a pathogen‐free facility on a 12 h light cycle, with ad libitum access to autoclaved food and sterile water. At the end of experiments, mice were euthanized under isoflurane general anaesthesia with cardiac puncture for collection of platelet‐poor plasma. Tissues were collected and weighed, then snap frozen in liquid nitrogen for protein or RNA extraction, and muscle frozen in cold 2‐methylbutane for cryosectioning or fixed in 10% neutral buffered formalin solution. Frozen plasma and tissue were stored at −80°C.
Pancreatic ductal adenocarcinoma models
For the orthotopic cancer models, KPC pancreatic cancer cells (500 000 or 5 million as indicated) were injected into the pancreas of ~10‐week‐old wild‐type (WT) C57BL/6 male mice (Jackson Laboratory, Bar Harbor, ME, USA). Briefly, mice were anaesthetized by inhalation of isoflurane, and a 1.2 cm lengthwise incision into the upper left quadrant was made. The cell suspension (40 μL) was injected into the pancreas, and the cavity was closed with suture and clips. Controls were similarly treated with the exception of cell injection. Mice were treated twice daily for 48 h with buprenorphine for pain, and wound clips were removed at 7 days. Mice were weighed daily and euthanized under isoflurane anaesthesia when they reached the pre‐designated endpoint. Body composition, mouse activities, and muscle strength were monitored weekly or at the end of experiments. The genetically engineered mouse model of PDAC, KPC (KrasG12D;Trp53R172H;Pdx1‐Cre) was generated by crossing LSL‐KrasG12D mice with LSL‐p53R172H mice (National Cancer Institute), producing LSL‐KrasG12D;LSL‐p53R172H (i.e. KP), which was then crossed with Pdx1‐Cre (Jackson Laboratory) that expresses a pancreas‐specific Cre recombinase, giving rise to KPC mice that develop tumours in the pancreas. The KPC genetically engineered mouse model mimics human PDAC by introducing a high degree of genomic instability due to point mutations in the oncogene Kras and the tumour suppressor gene p53.37 For activin inhibition, C57BL/6J mice bearing orthotopic KPC tumours were treated with either phosphate buffered saline (vehicle control) or ACVR2B/Fc (10 mg/kg) at the designated intervals. Hypermuscular MLC‐dnACVR2B mice were a gift from Se‐Jin Lee. These over‐express in skeletal muscle a dominant negative ACVR2B lacking the intracellular domain. Male and female mice and WT littermates were orthotopically implanted with murine KPC cells (5 × 105 per mouse) at age of 10 weeks.
Activity was assessed by counting horizontal and vertical movements of individual mice in a fresh cage placed within a VersaMax AccuScan activity monitor (Columbus Instruments, Columbus, OH, USA) recording for 30 min. Grip strength was assessed using a wire mesh grid and all four limbs, repeated three times on each mouse with peak force recorded in grams using an attached force transducer (Columbus Instruments). Activity on an inverted grid was used to assess combined forepaw and hind paw strength. Mice were placed on a wire grid that was then inverted over a foam pad, resulting in mice hanging upside‐down; the number of times the mice crossed into marked quadrants was recorded (Figure 5B). To evaluate treatment effects on indicators of wellness/distress, an 11‐point scoring system based upon Paster et al.38 plus body condition scoring was used, where 11 was normal and <3 consistent with imminent death. Factors assessed included appearance (2 = bright eyes, well‐groomed shiny hair; 1 = dull fur, lack of grooming; 0 = pilorerection, hunched back, rough coat, abnormal posture), behaviour (2 = active, interactive, alert; 1 = isolated, decreased activity, decreased alertness; 0 = immobile, weak, vocalizations), provoked behaviour (3 = quickly moves away; 2 = slow to move; 1 = moves after a short period; 0 = does not move), clinical signs (2 = normal body temperature/respiratory rate; 1 = decreased body temperature/respiratory rate; 0 = hypothermia, markedly reduced respiratory rate), and body habitus (2 = normal; 1 = thin, 0 = emaciated).
Figure 5.
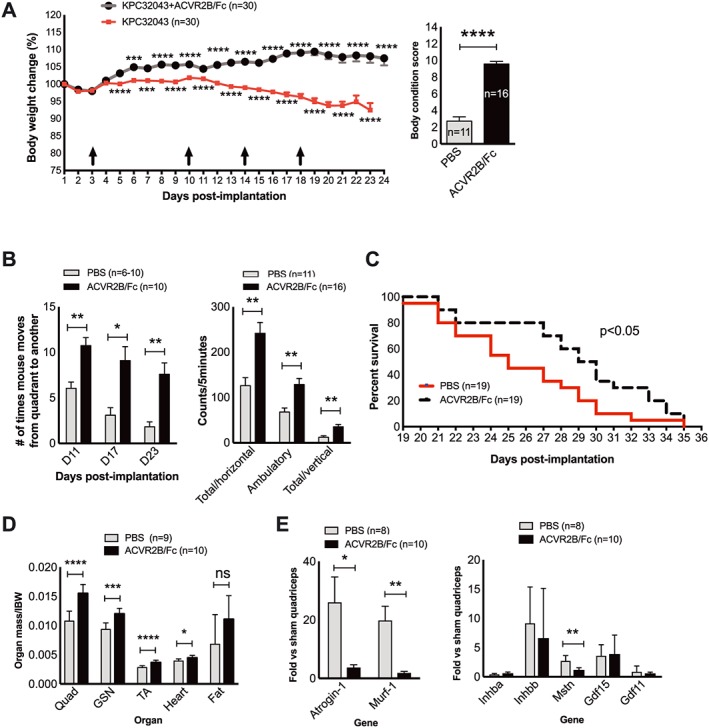
Activin blockade in mice with activinlow PDAC cells via soluble activin receptor reduces weight loss and prolongs survival. (A) Body weights after KPC32043 cell implantation into the pancreas and time points (arrows) of ACVR2B/Fc administration (10 mg/kg body weight via i.p.) (left) and body condition scores at the end of the experiment (right). IBW, initial body weight measured on the day of cell implantation. (B) Mouse activity measured at the indicated time points (left) or at the end of experiment (right) showing a therapeutic effect of activin blockade. (C) Increased survival in mice with KPC32043 tumours treated with ACVR2B/Fc (log–rank test). (D) Effect of activin blockade on organ mass. (E) Effect of activin blockade on the expression of atrogenes (left) or activins and related genes (right) in quadriceps. Data are presented as mean ± SEM. * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001.
The endpoint for euthanasia was dependent on the aims of studies. For tissue endpoints to study response to tumour/therapeutic intervention, all mice were euthanized when either group reached an average score of 3 on the 11‐point scale. For studies of therapeutic intervention effects on survival (Figures 5C and 6B), mice were euthanized when moribund.
Figure 6.
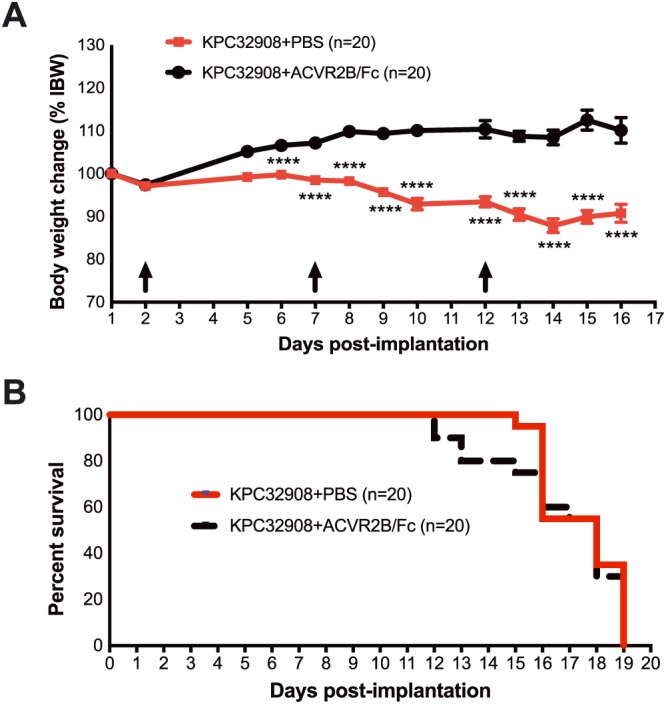
Activin blockade in mice with activinhigh PDAC cells via soluble activin receptor reduces weight loss without improving survival. (A) Body weights after KPC32908 cell implantation into the pancreas and time points (arrows) of ACVR2B/Fc administration (10 mg/kg body weight via i.p.) (left) and body condition scores at the end of the experiment (right). IBW, initial body weight measured on the day of cell implantation. (B) Survival in mice with KPC32908 tumours treated with ACVR2B/Fc (log–rank test). **** P < 0.0001.
Quantitative real‐time polymerase chain reaction
Total RNA was extracted using the QIAzol Lysis Reagent and miRNeasy Mini Kit (217004, Qiagen) and reverse‐transcribed into complementary DNA (cDNA) using the TaqMan Reverse Transcriptase Reagents (4366596, Life Technologies, Carlsbad, CA, USA) or the Verso cDNA Synthesis Kit (AB1453B, Thermo Scientific), following the manufacturer's instructions. Equal amounts of cDNA were subjected to quantitative real‐time polymerase chain reaction (RT–qPCR) performed using the TaqMan Universal Master Mix II with UNG on the LightCycler Instrument (Roche Life Science, Indianapolis, IN, USA). Gene‐specific TaqMan PCR‐based gene expression assays were from Life Technologies, including Inhba (Mm00434339_m1), Inhbb (Mm03023992_m1), Trim63/MuRF‐1 (Mm01185221_m1), Fbxo32/MAFbx/Atrogin1 (Mm00499523_m1), Mstn (Mm01254559_m1), Gdf15 (Mm00442228_m1), Gdf11 (Mm1159973_m1), and Rn18s (Mm03928990_g1). Ct values for target and housekeeping genes were used to calculate relative transcript abundance using the ΔΔCt method. Data were presented as the mean fold change.
Immunohistochemistry
Immunohistochemistry (IHC) was performed on formalin‐fixed, paraffin‐embedded 5 μm tissue sections. Sections were deparaffinized and rehydrated, then heated in 0.01 M citrate buffer (pH 6.0) to retrieve antigens. Endogenous peroxidase was quenched in 3% H2O2. Sections were incubated with the specific primary antibodies to activin A/INHBA from R&D Systems (AF338) or to activin B/INHBB from antibodies‐online (ABIN952903) at 1:100 in Odyssey Blocking Buffer (927‐50010) and then with peroxidase conjugated secondary antibodies, ImmPRESS anti‐goat (MP740550) or ImmPRESS anti‐rabbit (MP‐7401) from Vector Laboratories, Burlingame, CA, USA. The immunostaining was visualized with ImmPACT DAB (SK‐4105) from Vector Laboratories. Thereafter, the sections were counterstained with haematoxylin.
Immunohistochemistry stain imaging and quantification
Images shown were acquired at 20× using a colour Zeiss AxioCam camera mounted on a Zeiss Axio Observer.Z1 inverted microscope. For image quantification, whole slide images were taken using Aperio Scan Scope CS at 20×. The Positive Pixel Count algorithm was used to quantify the amount of a specific stain present in a scanned slide image. A range of colour (range of hues and saturation) and three intensity ranges (weak, positive, and strong) were masked and evaluated. The algorithm counted the number and intensity sum in each intensity range, along with three additional quantities: average intensity, ratio of strong/total number, and average intensity of weak positive pixels. The algorithm had a set of default input parameters when first selected—these inputs have been pre‐configured for brown colour quantification in the three intensity ranges (220‐175, 175‐100, and 100‐0). Pixels, which were stained but did not fall into the positive‐colour specification, were considered negative‐stained pixels—these pixels were counted as well, so that the fraction of positive to total stained pixels was determined. For the tissue microarray IHC, three pathologists scored all biopsies for staining intensity and histopathology diagnosis, blinded to each other's scores and supplier diagnosis.
Enzyme‐linked immunosorbent assay
Activin A enzyme‐linked immunosorbent assay (ELISA) was performed using the activin A DuoSet (DY338, R&D Systems, Minneapolis, MN, USA), per manufacturer's protocol. In brief, 96‐well plates were coated with the capture anti‐activin A antibody overnight at room temperature, and non‐specific binding was blocked with diluent followed by sample (CM or mouse plasma) addition and incubation. Biotinylated anti‐activin A detection antibody was added, which were subsequently incubated with streptavidin–horseradish peroxidase. Substrate solution was added to produce a visible signal, and the optical density of the signal was determined using a microplate reader (BioTek, Winooski, VT, USA).
Western blotting analysis of C2C12 myotubes
C2C12 myotubes were lysed in RIPA buffer. Extracted cell lysates were subjected to 4–15% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) and transferred onto nitracellulose membranes (#1620112, Bio‐Rad, Hercules, CA, USA), followed by immunoblotting using primary antibodies: phospho‐p38 MAPK (Thr180/Tyr182) (4511) and total p38 MAPK (8690) from Cell Signaling Technology, Danvers, MA, USA; Smad3 (S423 + S425) (ab52903), total Smad3 (ab40854), and GAPDH (ab9484) from Abcam, Cambridge, UK, USA, and tubulin from the Development Studies Hybridoma Bank at the University of Iowa. Secondary antibodies were as follows: IRDye 800CW goat anti‐rabbit IgG (926‐32211), IRDye 680RD goat anti‐rabbit IgG (925‐68071), IRDye 680LT goat anti‐mouse IgG2b‐specific (926‐68052), and IRDye 680LT goat anti‐mouse IgG1‐Specific (926‐68050) from LI‐COR Biosciences, Lincoln, NE, USA. Signal was detected using a LI‐COR Odyssey system.
ACVR2B/Fc preparation and purification
ACVR2B/Fc fusion protein was purified from CM of Chinese hamster ovary cells (a gift from Se‐Jin Lee, Johns Hopkins University School of Medicine) on Protein A Sepharose (GE Healthcare, Chicago, IL, USA) and dialysed against 1× phosphate buffered saline using a Slide‐A‐Lyzer dialysis kit. The dialysed protein was confirmed by western blotting using recombinant ACVR2B/Fc (R&D Systems) as a positive control. Biological activity of the protein was assessed by confirming a hypertrophic effect in muscle in normal C57BL/6J mice (not shown).
Database analysis of human pancreatic cancer and myostatin gene expression
Oncomine was accessed, and pancreatic cancer data sets with normal controls were analysed.39 Clinical and RNAseq data were downloaded from The Cancer Genome Atlas (TCGA).40 For each of the genes of interest, the association between abundance of gene expression and overall survival was fitted by a Cox proportional hazards regression model. Age, which significantly associated with overall survival (0.0054), was adjusted in the regression. Statistical analysis was performed in R version 3.1.2. UALCAN was used for visualization of gene association with survival.41 Expression of myostatin (MSTN) was queried for human tissues in the GTEx Portal (gtexportal.org) and across other cells and tissues via Illumina BaseSpace Correlation Engine (illumina.com). The data used for the analysis described in this manuscript (Supporting Information, Figure S2) were all obtained from the GTEx Portal and Illumina BaseSpace on 04/04/2019.
Statistical analysis
Two group comparisons were by unpaired Student's t‐test. Comparison of multiple groups was by analysis of variance followed by Tukey's post‐hoc test. For all analyses, the level of significance was set at P < 0.05.
Results
Murine pancreatic ductal adenocarcinoma tumour‐derived cells expressed activin A, but not activin B
Three PDAC cell lines, KPC32043, KPC32047, and KPC32908, were analysed by RT–qPCR for expression of two activin β subunit genes, Inhba and Inhbb. Compared with normal mouse pancreas, Inhba mRNA increased by 52‐fold (P < 0.001), 132‐fold, and 551‐fold (P < 0.0001), respectively (Figure 1A), with highest expression in KPC32908 cells. Inhbb was undetectable (data not shown). To determine whether the increased Inhba expression leads to increase in βA subunit secretion, we collected CM from KPC cell line cultures and measured activin A by ELISA. Indeed, KPC cells secreted activin A with KPC32908 levels being 6.6‐fold higher than KPC32047 (P < 0.05) vs. undetectable levels in KPC32043 CM (Figure 1B). We subsequently refer to these as activinhigh‐KPC32908 and activinlow‐KPC32043 lines.
Figure 1.
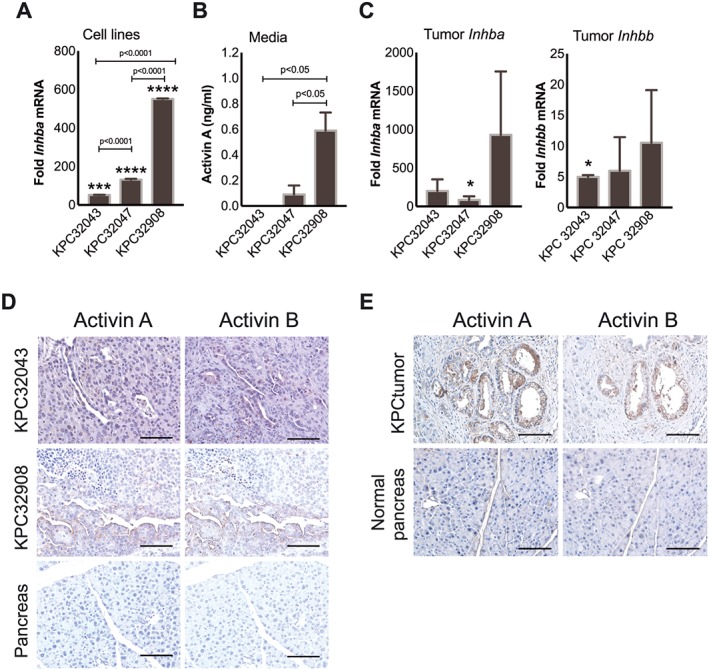
Murine PDAC tumour‐derived cells express activin A but not activin B, while tumours express both activins A and B. (A) Quantitative RT–PCR demonstrating varying levels of Inhba mRNA in cell lines: KPC32043, KPC32047, and KPC32908. Inhbb was undetectable (not shown). Data are presented as the mean fold change relative to normal mouse pancreas ± SD. (B) ELISA demonstrating activin A protein levels in the conditioned medium (CM) collected from the culture of the three KPC cell lines. Data are presented as mean ± SD. (C) Quantitative RT–PCR demonstrating levels of Inhba and Inhbb mRNA in pancreatic tumours generated by injection of the KPC cell lines into the pancreas of C57BL/6J mice. Triplicate replicate data are compared with normal pancreas of sham‐operated mice and presented as mean ± SEM (n = 3 tissues per condition). (D) Immunohistochemistry (IHC) of activin A or activin B in pancreatic tumours from two orthotopic PDAC models (KPC32043 and KPC32908). (E) IHC of pancreatic tumours from mice with genetic PDAC (KPC mice). Normal pancreas tissues were from sex‐matched and age‐matched mice with wild‐type (WT) Kras and WT p53 alleles. Scale bar: 100 μm. * P < 0.05, *** P < 0.001, **** P < 0.0001.
Pancreatic ductal adenocarcinoma cell lines elicited expression of activin B from host stromal cells in an orthotopic tumour model
We examined Inhba and Inhbb mRNA expression in PDAC cachexia developing from orthotopic implantation of 5 million KPC tumour cells into C57BL/6J male mice. Mice were euthanized when they met specific veterinarian body condition criteria of activity and appearance. In this trial, mice with KPC32908 tumours were euthanized at 9 days, those with KPC32047 at 13 days, and KPC32043 at 14 days (N = 4 or 5 per group). Tumour size at euthanasia was not different across groups cell lines (one‐way analysis of variance, 0.576). Compared with the normal pancreas from sham‐operated controls, the KPC32043, KPC32047, and KPC32908 tumours expressed high levels of Inhba, with 201‐fold, 85‐fold, and 931‐fold increase, respectively, although due to large variance, only KPC32047 met the statistical definition of significance (Figure 1C, left). These tumours also expressed Inhbb, with the highest KPC32908 being ~10‐fold greater than normal pancreas (Figure 1C, right). This differential expression pattern generally agreed with that displayed by the original KPC cell lines, namely, high levels of Inhba and low/no levels of Inhbb. To confirm the presence of activin proteins in the PDAC tumour tissue, we performed IHC staining with antibodies against activin subunits. Both Inhibin βA and βB subunits were detected in the tumour epithelial cells and in the stromal cells of orthotopic PDAC tumours (Figure 1D) as well as in tumour epithelial and stromal cells present in tumours from the genetically engineered KPC model (Figure 1E). These results indicate that PDAC tumour cells express activin A but that they also elicit expression of activins from surrounding host cells. Moreover, tumour growth and systemic effects scaled with level of activin expression by tumours.
There was a systemic functional decline with orthotopic pancreatic ductal adenocarcinoma tumours
We assessed the organ response to factors released by activinLow‐KPC32043 tumours over time and at the experimental endpoint. Mouse body weights were monitored after intra‐pancreatic implantation of 50 000 KPC cells, up until the time of euthanasia at the specified humane endpoint. Tumour‐bearing mice ceased gaining body weight by 12 days and lost body weight over time. On the other hand, the sham‐operated mice steadily gained weight, leading to a final difference of approximately 9% between KPC mice and sham controls (P < 0.001) (Figure 2A). Evaluation of the final activity and body condition of the tumour‐bearing mice demonstrated significant reductions, P < 0.001 (Figure 2B) and P < 0.0001 (Figure 2C), respectively, compared with the sham controls. Furthermore, KPC mouse organ weights were significantly reduced (Figure 2D). Because the KPC tumour‐derived cell lines release activin A into the culture medium as shown in Figure 1B, the tumour would be expected to release activin A into the blood system. Indeed, activin A was increased by an 11.5‐fold in KPC32043 mouse plasma (P < 0.0001) (Figure 2E).
Figure 2.
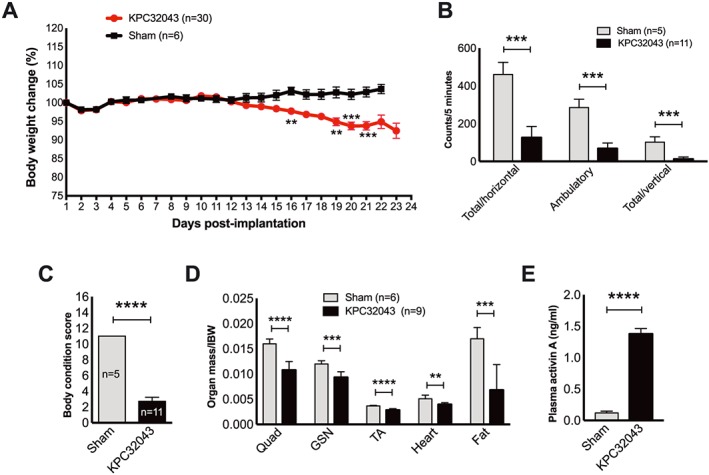
There is a systemic functional decline and organ wasting due to PDAC. (A–D) Systemic changes in body weight, activity, body condition, and organ weight in response to PDAC tumour developed by orthotopic injection of KPC32043 cancer cell line, compared with the sham‐operated mice. (E) Elevated activin A level in plasma from the tumour‐bearing mice as determined by ELISA. Data are presented as mean ± SEM. ** P < 0.01, *** P < 0.001, **** P < 0.0001.
There was a systemic activin response to orthotopic pancreatic ductal adenocarcinoma tumours
Multiple organs responded to the presence of the PDAC tumour by increasing activin expression. Inhba mRNA was significantly increased up to a few‐fold in most of the organs including the spleen, heart, fat, and kidney but down‐regulated in the liver and quadriceps skeletal muscle (Figure 3A). However, all organs demonstrated increased Inhbb expression (Figure 3B), and the magnitude of the increase was generally much greater than that of Inhba in the same tissues. The increased mRNA levels led to increased activin proteins in these organs as detected by immunohistochemistry in the orthotopic Activinlow‐KPC32043 (Figure 3C) and the orthotopic activinhigh‐KPC32908 models (Figure 3D). Consistent with this systemic, multi‐organ activin response being a general feature in pancreatic cancer, the muscle, kidney, liver, and heart from genetically modified KPC mice with autochthonic tumours also showed strong staining for activins (Figure 3E), while low to no staining was observed in normal genotype control tissues.
Figure 3.
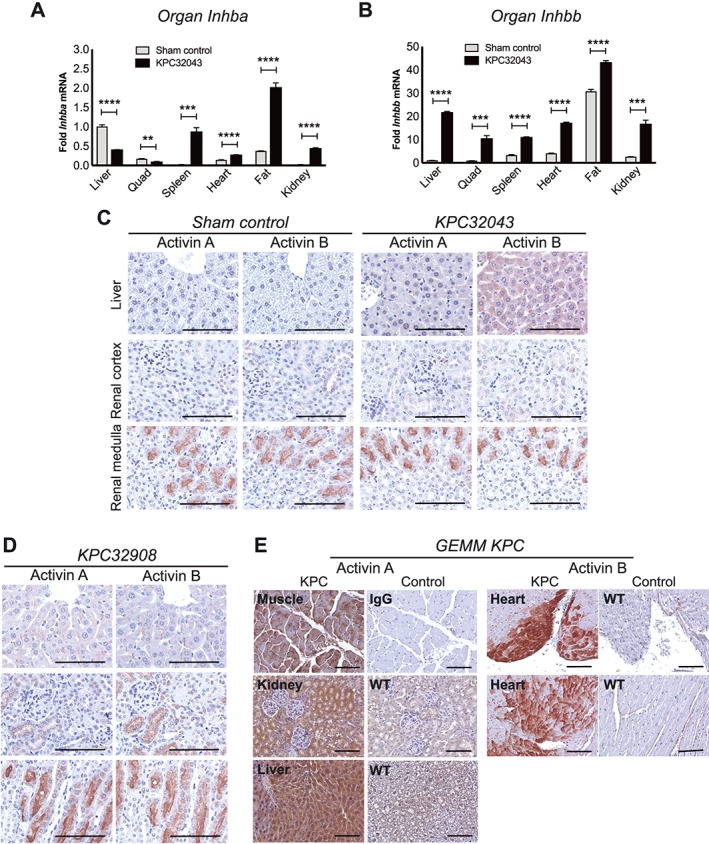
There is a systemic activin response to PDAC. (A, B) Inhba or Inhbb mRNA expression by quantitative RT–PCR in organs harvested at the study endpoint from sham control and orthotopic KPC32043 tumour‐bearing mice. Expression values in all organs for each gene were normalized to the value from the control liver. IHC for activin A and activin B in organs from mice with (C) orthotopic KPC32043 tumours, (D) orthotopic KPC32908 tumours, or (E) genetic PDAC (KPC mice). Data are presented as mean ± SEM. Scale bar: 100 μm.
Pancreatic ductal adenocarcinoma cells express factors that induced expression of activins and atrophy of muscle cells
Because muscle wasting is the major feature of cancer cachexia and activins are induced in distant organs by PDAC, we examined whether factors released from tumour cells can directly induce activin expression. Murine C2C12 cells were used to model muscle. Myoblasts were exposed to 75% of activinhigh‐KPC32908 CM for 24 or 48 h, while 4‐day‐old differentiated myotubes were exposed to 25%, 50%, or 75% of KPC32908 CM for 48 h (Figure 4A). Inhba and Inhbb expressions were assessed with RT–qPCR. CM significantly increased Inhba and Inhbb in myoblasts and myotubes, compared with their respective controls (Figure 4B), at levels that induced myotube atrophy after 48 h of CM treatment (Figure 4C, left). The average reductions in myotube diameter were statistically significant at all the three CM dilutions (25%, 50%, or 75%) in a dose‐dependent manner; the higher the CM concentration, the more severe the atrophy (Figure 4C, right). While Inhbb induction was consistent with skeletal muscle expression in vivo, the Inhba results are discrepant, suggesting differences between the systems.
Figure 4.
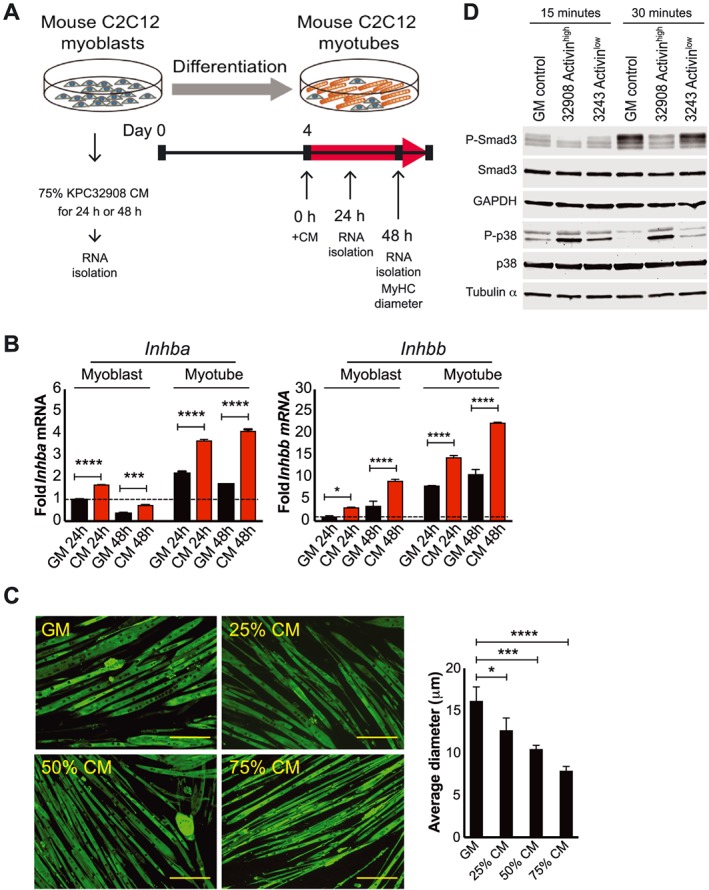
PDAC tumour cells express factors that induce muscle cell activin expression and atrophy. (A) Experimental scheme. (B) Quantitative RT–PCR demonstrating induction of Inhba and Inhbb by 75% KPC32908 CM. (C) Immunofluorescence staining for myosin heavy chain in C2C12 myotube cultures and quantification of myotube size (>200/condition) in growth medium GM, 25%, 50%, or 75% of KPC32908 CM. * P < 0.05, *** P < 0.001, **** P < 0.0001. Scale bar: 200 μm. (D) Western blot demonstrating activation of p38 MAPK by 75% CM from KPC32908 activinhigh and KPC32043 activinlow at 15 and 30 min after CM treatment.
To understand whether the KPC cells might also activate signalling downstream of activin receptors, we probed the phosphorylation/activation states of Smad2, Smad3, and p38 MAPK by western blotting analysis in myotubes treated with 75% CM from activinhigh‐KPC32908 or activinlow‐KPC32043 cells. No pSmad2 was detected in any samples (not shown). Neither was Smad3 activated by CM relative to growth media at any time points. However, p38 MAPK activation was prominent at 15 and 30 min (Figure 4D), consistent with the described signalling of activins on C2C12 myotubes.42 The activation state persisted through 1 and 5 h and disappeared by 24 h after CM treatment (data not shown). Notably, there was a much stronger p38 MAPK activation signal in cells treated with activinhigh‐KPC32908 CM than activinlow‐KPC32043 CM.
Activin trap in mice with activinlow pancreatic ductal adenocarcinoma blocked weight loss and prolonged survival
To determine whether blocking circulation activins would alleviate cachexia, we treated mice with ACVR2B/Fc in the lower Inhba‐expressing, activinlow‐KPC32043 orthotopic mouse model. Administration of ACVR2B/Fc (10 mg/kg i.p.) reduced body weight loss starting at approximately two weeks after the first injection (Figure 5A, left). By the end of the experiment, weight loss had been completely prevented in the ACVR2B/Fc treatment group. ACVR2B/Fc treatment also preserved body condition scoring (Figure 5A, right), hanging grid exploration activity (Figure 5B, left), and open‐field activity (Figure 5B, right). Survival was prolonged by treatment; median survival was 29.5 days for the ACVR2B/Fc treated mice vs. 25 days for the untreated controls (P < 0.05, log–rank test) (Figure 5C). Skeletal muscle, heart, and fat loss were attenuated, although the effect on fat did not reach significance (Figure 5D). Tumour growth was also attenuated ~30% (1763 ± 137.4 mg, n = 9 untreated vs. 1218 ± 114.5 mg, n = 10, treated, P =0.07). Muscle from ACVR2B/Fc‐treated mice showed reduction in two muscle‐specific E3 ligase genes, Atrogin‐1/MAFbx/Fbxo32 and Murf‐1/Trim63 (Figure 5E, left) and in the muscle growth inhibitor Mstn (Figure 5E, right) vs. vehicle‐only controls. Expression of Inhba and Inhbb was not reduced by activin blockade (Figure 5E, right), suggesting this is not an activin feed‐forward loop but rather that other factors not targeted by ACVR2B/Fc must elicit activin expression.
Activin trap in mice with activinhigh pancreatic ductal adenocarcinoma blocked weight loss without improving survival
We also administered ACVR2B/Fc in the higher Inhba‐expressing, activinhigh‐KPC32908 orthotopic mouse model to explore whether activin blockade could exert a therapeutic role at higher levels of activin production. As in the activinlow model, ACVR2B/Fc treatment prevented body weight loss (Figure 6A), but unlike the prior experiment, ACVR2B/Fc did not prolong survival (Figure 6B).
Skeletal muscle‐specific activin blockade reduced body weight loss without prolonging survival in pancreatic ductal adenocarcinoma
Because ACVR2B/Fc administration inhibited cachexia in the KPC orthotopic model, we queried whether ACVR2B activity in skeletal muscle contributes to PDAC muscle wasting. We used transgenic (Tg) MLC‐dnACVR2B mice that express a skeletal muscle‐specific, dominant negative ACVR2B that lacks the kinase domain and thus blocks ACVR2B signalling43 and compared them with WT littermates. Activinlow‐KPC32043 cells were implanted in the pancreas. Body weights were monitored daily and normalized to starting weight to account for differences in size. There were no differences in relative body weight between WT sham and Tg sham over the whole experiment (Figure 7A). As previously, in WT mice activinlow‐KPC32043 tumours induced steady weight loss over 23 days (Figure 7A), reaching 11.5% reduction from starting weight (P < 0.01). However, Tg mice showed no body weight loss compared with tumour‐bearing WT mice and Tg sham controls. We also evaluated activinhigh‐KPC32908 tumours in Tg and WT mice. As for activinlow tumours, there was no body weight loss in activinhigh‐KPC32908 tumour‐bearing Tg mice. Regardless, survival was not prolonged in Tg vs. WT mice with activinhigh‐KPC32908 tumours (Figure 7C). These results indicate that muscle‐specific inhibition of activin signalling was protective against PDAC‐induced muscle wasting but did not promote survival.
Figure 7.
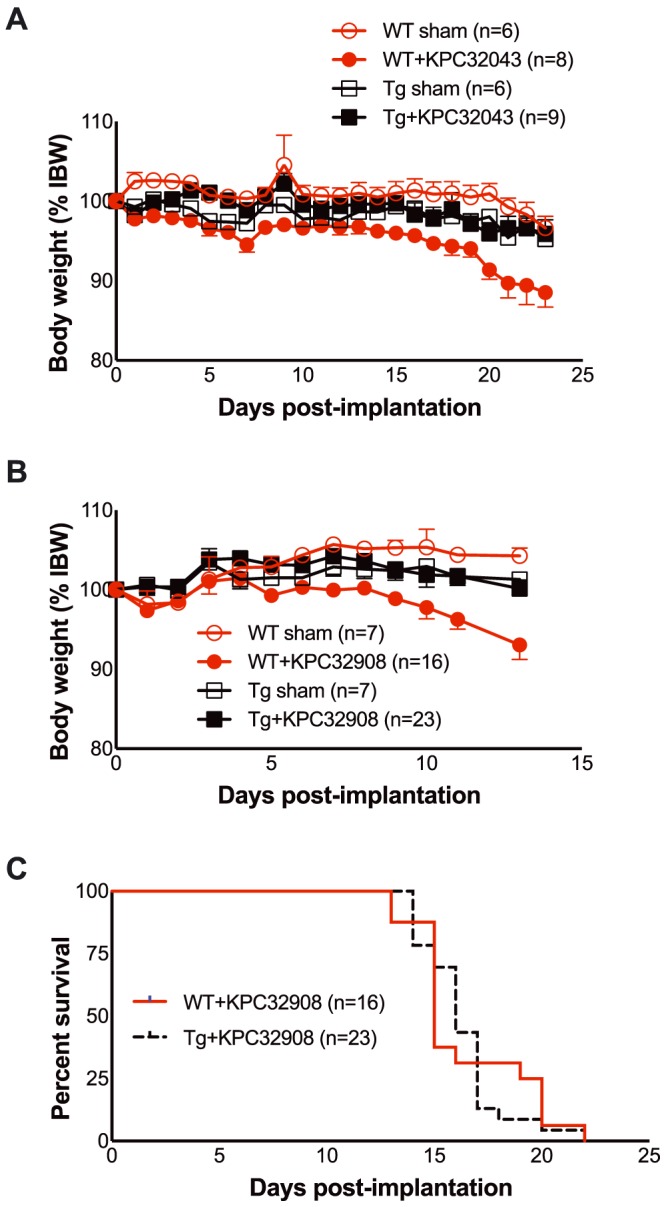
Skeletal muscle‐specific blockade of activin signalling via expression of dnACVR2B in MLC‐dnACVR2B mice prevents PDAC‐induced weight loss but does not improve survival. (A) Body weights of KPC32043 pancreatic tumour‐bearing wild‐type (WT) C57BL/6 mice (WT + KPC32043) or transgenic (Tg) MLC‐dnACVR2B mice (Tg + KPC32043), normalized to each mouse's own initial body weight. (B) Body weights of KPC32908 pancreatic tumour‐bearing wild‐type (WT) C57BL/6 mice (WT + KPC32908) or transgenic (Tg) MLC‐dnACVR2B mice (Tg + KPC32908), normalized to each mouse's own initial body weight. (C) Survival of WT vs. Tg mice with KPC32908 tumour cells is not different (log–rank test). Mean ± SEM. * P < 0.05, ** P < 0.01.
Activin proteins are expressed by human pancreatic ductal adenocarcinoma tumours
To determine the relevance to human PDAC, we evaluated activin expression in human tumours from patients treated at our institution. Antibodies to activin βA and βB both detected strong expression in human PDAC tumour sections (shown are representative images), compared with normal adjacent pancreas (Figure 8A, top panel). Staining was not significantly different between tumour and stroma, although both showed wide ranges of staining intensities. We also stained a commercially available pancreatic tissue microarray with the same antibodies and staining protocol, although with hand scoring by three pathologists. Both activin A and activin B expressions were significantly increased in PDAC tumour compared with the normal pancreas (P < 0.0001) (Figure 8B). Consistent with these findings, an Oncomine query of studies in pancreatic cancer demonstrates three‐fold to 33‐fold increases in INHBA gene expression in tumours vs. controls (Table 1), while INHBB showed no significant results.
Figure 8.
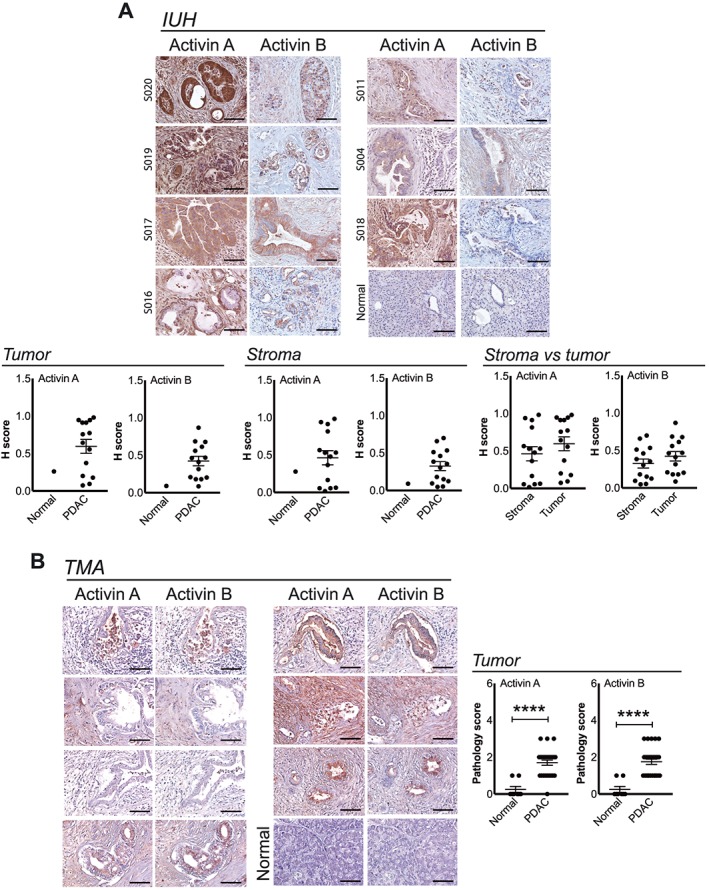
Activin proteins are expressed by human PDAC tumours. (A) Representative IHC for activin A and activin B in tumour tissues and quantification from PDAC patients from Indiana University (IUH). (B) Representative images and quantification of IHC for activin A and B in a PDAC cancer tissue microarray (TMA). Normal pancreas (n = 8) and PDAC tumour (n = 27). Mean ± SEM. Scale bar: 100 μm. **** P < 0.0001.
Table 1.
Tumour INHBA but not INHBB expression is increased in pancreatic cancer revealed by Oncomine database
| Study | Comparison | Gene rank | % tile | P value | t‐test | Fold change |
|---|---|---|---|---|---|---|
| Badea Pancrea | PDAC vs. normal | 4 | Top 1% | 1.68E‐20 | 13.27 | 15.87 |
| Bittner Multi‐cancer | PDAC vs. all cancer | 29 | Top 1% | 2.01E‐06 | 6.40 | 3.02 |
| Ramaswarny Multi‐cancer | PDAC vs. all cancer | 129 | Top 2% | 2.20E‐05 | 5.60 | 4.36 |
| London | PDAC vs. normal | 512 | Top 10% | 2.00E‐03 | 5.35 | 33.50 |
| Lacobuzio‐Donahue Pancras | PDAC vs. normal | 962 | Top 7% | 3.00E‐03 | 4.44 | 12.19 |
| Pei Pancreas | PC vs. normal | 1227 | Top 7% | 3.27E‐05 | 5.04 | 8.95 |
PDAC, pancreatic ductal adenocarcinoma.
Tumour INHBA but not INHBB correlates with reduced survival in patients with PDAC
Our studies indicate that tumour‐expressed activin A may play an important role in human PDAC disease. Indeed, analysis of TCGA pancreatic adenocarcinoma data set demonstrates that high INHBA expression associated with shorter overall survival vs. low/medium expression (Table 2, Figure 9). Expression by stage was not statistically informative, because 146 of the 162 samples comprising this data set represent Stage 2 disease, consistent with tissue collection from surgical resection of the tumour (Supporting Information, Figure S1). In contrast, no association with mortality was shown for expression of Inhibin‐α (INHA), activin B (INHBB), GDF‐15 (GDF15), activin binding proteins Follistatin (FST) and FLRG (FSTL3), or any of the receptors ACVR2A, ACVR2B, ACVR1B, or ACVR1C (not shown), either by analysis using dichotomous (high vs. low/medium) (Figure 9) or continuous variables (Table 2). Expression of two related TGF‐β family proteins that also share binding to ACVR2B, myostatin (MSTN), and GDF‐11 (GDF11) actually associated with improved survival (Table 2). While myostatin is generally considered to be muscle specific, the extra‐muscular expression of MSTN by pancreatic tumours is consistent with its expression in other tissues, in transformed fibroblasts, and in cancer cell lines (Supporting Information, Figure S2A–C). There was insufficient data to assess another ACVR2B‐binding family member, GDF‐2/BMP‐9 (not shown).
Table 2.
Relationship between PDAC tumour gene expression abundance vs. survival in the TCGA pancreatic adenocarcinoma database, adjusted for age, by the univariate Cox proportionate hazards (CoxPH) model
| Gene | Hazard ratio | P |
|---|---|---|
| INHBA | 1.218 | 0.003 |
| INHBB | 1.145 | 0.195 |
| INHA | 0.972 | 0.659 |
| FST | 0.966 | 0.554 |
| FSTL3 | 1.181 | 0.073 |
| GDF15 | 1.038 | 0.551 |
| GDF11 | 0.679 | 0.004 |
| MSTN | 0.809 | 0.028 |
Bold text indicate statistically significant results.
PDAC, pancreatic ductal adenocarcinoma; TCGS, The Cancer Genome Atlas.
Figure 9.
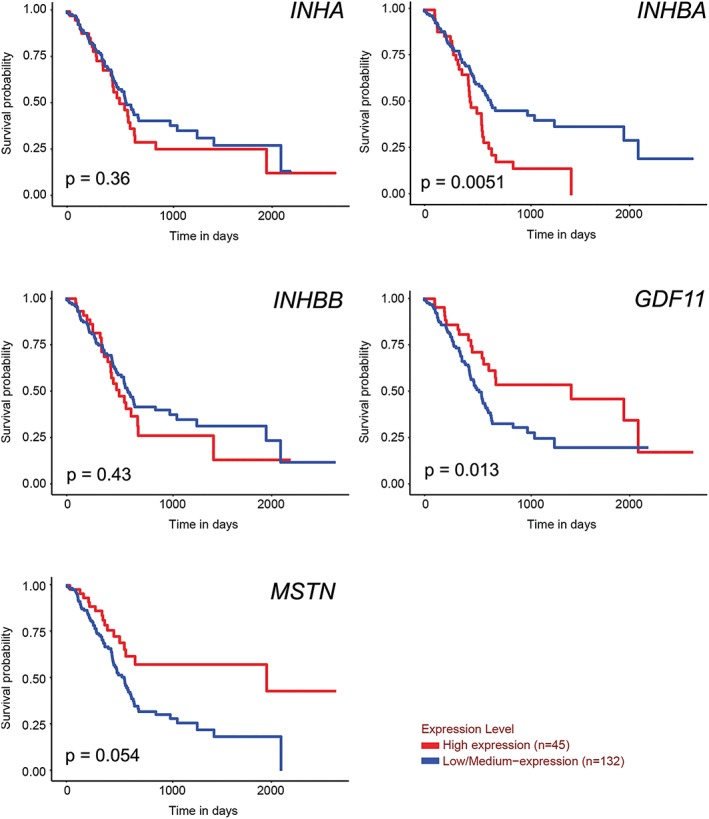
Survival of patients according to expression of activin family genes in pancreatic tumours. Gene expression vs. survival in the TCGA pancreatic adenocarcinoma data set. Ualcan.path.uab.edu accessed on 31 October 2018.
Discussion
The myriad functions of activin A in multiple biological processes such as reproduction and inflammation are well established although its roles in cancer and cancer cachexia are only more recently explored. Understanding its role in pancreatic cancer and associated wasting is vital because PDAC cachexia reduces treatment efficacy and quality and length of life.44 Substantial data document an association of activin and cachexia in pancreatic cancer and other cancers.23, 29, 30 As well, a large body of literature demonstrates a protective effect of activin receptor‐based inhibitors across a spectrum of experimental and pre‐clinical cachexia cancer models34, 35,45 isolated chemotherapy,46 or the combination of cancer and chemotherapy.47 A clinical trial (https://clinicaltrials.gov/ct2/show/NCT01433263) of an anti‐activin receptor antibody has also been carried out in 57 patients with late stage lung and pancreatic cancer, although the results have not been formally reported in the literature. Interpretation of those results and ultimately effective targeting of activin in PDAC will require an intimate understanding of the mechanisms of its expression and action in tumour cells, in the tumour microenviroment, and in the host body.
In the present study, we have identified a systemic, multiple organ activin response to PDAC tumours both in pre‐clinical models and in patients. While PDAC tumour cells and tumours expressed a high level of activin A and activin A expression was associated with mortality, multiple distant organs expressed both activin A and activin B in response to a PDAC tumour in the pancreas. PDAC tumour‐derived factors are causally involved in eliciting activin expression and muscle wasting because direct exposure of PDAC cell CM on myotubes caused both activin expression and a reduction in myotube size. In vivo, activin blockade using the soluble receptor ACVR2B/Fc reduced tumour growth and associated PDAC cachexia in both models and prolonged survival in the less‐aggressive, activinlow mouse model. This protection was not mediated by reduced systemic activin production, apparently, because activin expression in muscle remained high, implicating non‐activin factors in the systemic response. Furthermore, skeletal muscle‐specific inhibition of activin signalling using the MLC‐dnACVR2B mouse model prevented PDAC‐induced weight loss, although it did not improve survival. These studies are relevant to human disease because human tumours clearly express activin A and B and levels of INHBA expression correlate with mortality. Overall, our results suggest that a systemic activin‐targeting therapeutic strategy in combination with anti‐cancer agents will be necessary to combat PDAC and its associated fatal wasting syndrome.
Our observation that the PDAC induced activin A and activin B in multiple host organs has implications for designing strategies to reduce activin signalling. First, both the circulating activin and endogenous activin should be targeted to achieve stronger inhibition. Although the soluble receptor ACVR2B/Fc can block the circulating activins and inhibit paracrine and endocrine signalling, it cannot prevent endogenous activin from initiating autocrine signalling due to its inability to bind cell surface heparin‐sulfated proteoglycans.48, 49 Such endogenous activin and autocrine signalling may indeed play an important part in muscle wasting.
Targeting upstream inducers of activin could also prevent activin‐mediated wasting. Activin A can induce INHBB (activin B) expression, through a feed‐forward, SMAD‐dependent mechanism.50 Thus, one might speculate that organ activins induced in response to PDAC tumour were the result of tumour‐derived activins. However, while ACVR2B/Fc administration alleviated cachexia in the orthotopic mouse model, it did not alter the expression levels of Inhba and Inhbb in the organs examined. This suggests that organ activin induction is not via feed‐forward endocrine activin loop; instead, other factors released by the tumour are responsible for the induction, as illustrated in our myoblast/myotube cultures with PDAC CM. The promoters of INHBA and INHBB demonstrate quite different activity and transcription factor binding sites, suggesting that each might be induced by different molecular stimuli and signalling pathways. Identifying and targeting those pathways could offer gene‐specific means to prevent endogenous activin A vs. activin B production.
ACVR2B/Fc has a broad spectrum of ligand binding beyond activins that includes myostatin, activin, GDF11, and other TGF‐β family ligands, raising concerns of off‐target effects. To overcome this issue, Chen et al. developed antagonists specific to activin A vs. activin A and B.51, 52 The use of the antagonist that targeted both activin A and activin B resulted in a greater increase in muscle mass than activin A ‐only targeting. In this context, our finding of a systemic induction of activin B in addition to activin A in PDAC cachexia is significant. In fact, while activin A was induced in some of the organs we examined, activin B was consistently induced in all (Figure 3A and B). However, like the ACVR2B/Fc, the antagonists that Chen et al. generated only block circulating activins and endocrine signalling but not the endogenous ones.52 Therefore, newer generations of antagonists targeting autocrine activin A and activin B signalling are needed to achieve more potent effects.48, 49, 52
Activin blockade was effective here in reducing weight loss but not always in increasing survival. In the present study, we used two murine PDAC models, orthotopic implantation of activinlow and activinhigh cell lines. ACVR2B/Fc was less effective in activinhigh than in activinlow in terms of survival benefit. This result suggests that with more activin, perhaps a higher dose of ACVR2B/Fc is necessary to be effective. Alternatively, higher levels of Inhba expression in tumour may either reflect or confer a more aggressive phenotype on the tumour itself, and this would in turn pose a more profound impact on the host. Such effects have been observed experimentally in other in vitro and murine studies.16, 21, 53 These distinctions and their relevance to the human condition require further investigation. Of course, the lack of survival benefit in this study could also be due to the lack of chemotherapy and other clinical treatment. Such pre‐clinical considerations must be a focus of future studies in PDAC, given that activin receptor inhibition prolongs survival dramatically in combination with chemotherapy.47 Alternatively, a limitation of this approach could be the lesser protection of cardiac muscle vs. skeletal muscle with activin blockade54 or the requirement for inhibition of autocrine signalling in other yet unknown organs.
The primary focus of most cancer cachexia research has been skeletal muscle. Muscle represents more than 40% of body weight, and thus, its wasting accounts for a large part of body weight loss in cachexia. Other organs also waste, however, including cardiac muscle and adipose tissue, two organs that clearly play an important role in cachexia and pancreatic cancer outcomes.55, 56, 57, 58, 59 Here, we observed wasting of skeletal muscle, heart, fat, and multiple other organs, as well as activin induction in most of those organs. Mice with skeletal muscle‐specific activin inhibition here showed preserved body weight with both activinlow and activinhigh tumours but did not show prolonged survival, unlike WT mice bearing the same activinlow tumours and treated with ACVR2B/Fc. Two possible explanations for this discrepancy include (i) a critical role for activin signalling in other tissues, including heart and fat, which might or might not be achieved with systemic activin blockade but clearly not by transgene‐produced muscle‐specific inhibition or (ii) a greater intrinsic propensity for cancer death in mice with the dominant negative ACVR2B transgene, perhaps due to lower adipose mass.43 Further studies would be required to distinguish these possibilities.
Finally, our analysis of human tumours and available data sets indicate specificity of activin isoforms and family members in the pathogenesis of PDAC and its associated cachexia. RNAseq data from TCGA showed a positive association of only tumour activin A expression on mortality and not for genes encoding Inhibin‐alpha, follistatin and related proteins, activin B, or receptors ACVR1B, ACVR1C, ACVR2A, and ACVR2B (not shown), despite empirical evidence of certain of these receptors influencing tumour phenotype.60, 61, 62, 63 Moreover, RNA expression of the closely related proteins, myostatin, and GDF‐11 in tumours was actually associated with reduced mortality, despite their ability to induce systemic wasting.64, 65 These results suggest highly context‐specific roles of these factors in PDAC disease progression and mortality. Finally, the use of targeting strategies that non‐specifically bind all of these family members (as does ACVR2B/Fc) or their shared receptors (e.g. antibodies to ACVR2B or ACVR2A) rather than binding activin A or activin B selectively might ablate both the positive and negative activities of this family in PDAC.
In summary, our study demonstrates that PDAC induces cachexia involving multiple organs. This effect is mediated by tumour‐derived activins and other cachectic factors that are released systemically and in turn stimulate a systemic organ response that includes induction of endogenous activin A and activin B. While soluble activin receptor ACVR2B/Fc can block this circulating, endocrine activin, it has only a limited capability of blocking autocrine activin signalling and might non‐specifically ablate positive effects of certain other ligands (Figure 10). The tumour‐derived, activin‐inducing factors have yet to be identified; however, finding novel agents that block production of all activins or that inhibit activin selectively is necessary to provide more effective therapy for PDAC cachexia.
Figure 10.
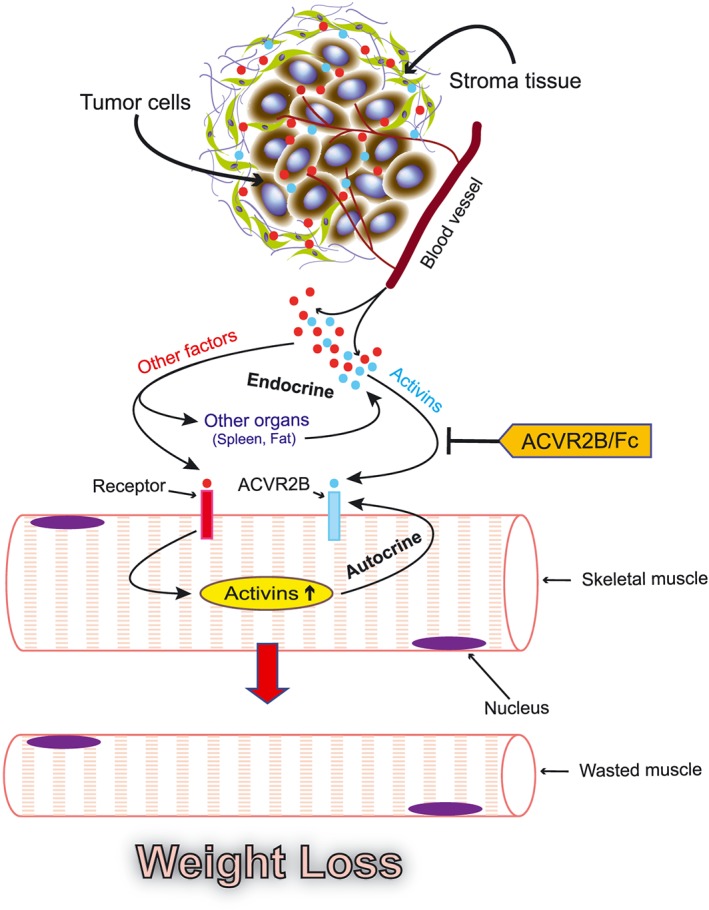
Proposed framework illustrating the systemic activin response to pancreatic cancer.
Author contributions
X. Z., M. P., Y. L., L. G. K., and T. A. Z. did the study conception and design. The acquisition of the data was performed by X. Z., M. P., C. P., Y. J., J. L., G. E. S., S. S., A. N., C. M. S., M. G. H., E. P. C., N. J. Z., G. J., T. A. Z. X. Z. drafted the manuscript. Critical revision of the manuscript was carried out by X. Z., M. C., L. G. K., T. A. Z.
Conflict of interest
The authors declare no conflicts of interest.
Supporting information
Figure S1: Expression of INHBA in pancreatic adenocarcinoma tumors by individual cancer stages from patients in the TCGA dataset as analyzed by UALCAN. (Ualcan.path.uab.edu accessed 04/03/2019)
Figure S2: Evidence for expression of MSTN/myostatin outside of skeletal muscle.
Acknowledgements
This work was funded in part by grants to T. A. Z. from the National Institutes of Health (NIH), including the National Cancer Institute (NCI) and National Institute of General Medical Sciences (grants R01CA122596, R01CA194593, and R01GM092758), the IU Simon Cancer Center (IUSCC), the Lustgarten Foundation, and the IUPUI Signature Center for Pancreatic Cancer Research and by grants to L. G. K. from NIH (R01DK096167) and the Lilly Endowment, Inc. We thank the IU Simon Cancer Center (NIH P30CA082709) at Indiana University School of Medicine support from the Clinical Trials Office, the Tissue Procurement & Distribution Core, and the Collaborative Core for Cancer Bioinformatics, resources supported by the IU Simon Cancer Center (NCI grant P30CA082709), the Purdue University Center for Cancer Research (grant P30CA023168), and the Walther Cancer Foundation. The Genotype‐Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health and by NCI, National Human Genome Research Institute, National Heart Lung and Blood Institute, National Institute on Drug Abuse, National Institute of Mental Health, and National Institute of Neurological Disorders and Stroke.
Zhong X., Pons M., Poirier C., Jiang Y., Liu J., Sandusky G. E., Shahda S., Nakeeb A., Schmidt C. M., House M. G., Ceppa E. P., Zyromski N. J., Liu Y., Jiang G., Couch M., Koniaris L. G., and Zimmers T. A. (2019) The systemic activin response to pancreatic cancer: implications for effective cancer cachexia therapy, Journal of Cachexia, Sarcopenia and Muscle, 10: 1083–1101. 10.1002/jcsm.12461.
References
- 1. Blum D, Stene GB, Solheim TS, Fayers P, Hjermstad MJ, Baracos VE, et al. Validation of the consensus‐definition for cancer cachexia and evaluation of a classification model—a study based on data from an international multicentre project (epcrc‐csa). Ann Oncol 2014;25:1635–1642. [DOI] [PubMed] [Google Scholar]
- 2. Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol 2011;12:489–495. [DOI] [PubMed] [Google Scholar]
- 3. Baracos VE, Martin L, Korc M, Guttridge DC, Fearon KCH. Cancer‐associated cachexia. Nat Rev Dis Primers 2018;4:17105. [DOI] [PubMed] [Google Scholar]
- 4. Werner S, Alzheimer C. Roles of activin in tissue repair, fibrosis, and inflammatory disease. Cytokine Growth Factor Rev 2006;17:157–171. [DOI] [PubMed] [Google Scholar]
- 5. Hedger MP, Winnall WR. Regulation of activin and inhibin in the adult testis and the evidence for functional roles in spermatogenesis and immunoregulation. Mol Cell Endocrinol 2012;359:30–42. [DOI] [PubMed] [Google Scholar]
- 6. Loomans HA, Andl CD. Intertwining of activin A and TGF‐β signaling: dual roles in cancer progression and cancer cell invasion. Cancers (Basel) 2014;7:70–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim SK, Hebrok M, Li E, Oh SP, Schrewe H, Harmon EB, et al. Activin receptor patterning of foregut organogenesis. Genes Dev 2000;14:1866–1871. [PMC free article] [PubMed] [Google Scholar]
- 8. Asashima M, Ariizumi T, Malacinski GM. In vitro control of organogenesis and body patterning by activin during early amphibian development. Comp Biochem Physiol B Biochem Mol Biol 2000;126:169–178. [DOI] [PubMed] [Google Scholar]
- 9. Ling N, Ying SY, Ueno N, Shimasaki S, Esch F, Hotta M, et al. A homodimer of the β‐subunits of inhibin a stimulates the secretion of pituitary follicle stimulating hormone. Biochem Biophys Res Commun 1986;138:1129–1137. [DOI] [PubMed] [Google Scholar]
- 10. Esquela AF, Zimmers TA, Koniaris LG, Sitzmann JV, Lee SJ. Transient down‐regulation of inhibin‐βc expression following partial hepatectomy. Biochem Biophys Res Commun 1997;235:553–556. [DOI] [PubMed] [Google Scholar]
- 11. Hotten G, Neidhardt H, Schneider C, Pohl J. Cloning of a new member of the TGF‐β family: a putative new activin βc chain. Biochem Biophys Res Commun 1995;206:608–613. [DOI] [PubMed] [Google Scholar]
- 12. Reader KL, Marino FE, Nicholson HD, Risbridger GP, Gold EJ. Role of activin C in normal ovaries and granulosa cell tumours of mice and humans. Reprod Fertil Dev 2017; 10.1071/RD17250. [DOI] [PubMed] [Google Scholar]
- 13. Hashimoto O, Tsuchida K, Ushiro Y, Hosoi Y, Hoshi N, Sugino H, et al. cDNA cloning and expression of human activin βe subunit. Mol Cell Endocrinol 2002;194:117–122. [DOI] [PubMed] [Google Scholar]
- 14. Gold E, Marino FE, Harrison C, Makanji Y, Risbridger G. Activin‐β(c) reduces reproductive tumour progression and abolishes cancer‐associated cachexia in inhibin‐deficient mice. J Pathol 2013;229:599–607. [DOI] [PubMed] [Google Scholar]
- 15. Marino FE, Risbridger G, Gold E. Activin‐βc modulates cachexia by repressing the ubiquitin‐proteasome and autophagic degradation pathways. J Cachexia Sarcopenia Muscle 2015;6:365–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kleeff J, Ishiwata T, Friess H, Buchler MW, Korc M. Concomitant over‐expression of activin/inhibin β subunits and their receptors in human pancreatic cancer. Int J Cancer 1998;77:860–868. [DOI] [PubMed] [Google Scholar]
- 17. Wildi S, Kleeff J, Maruyama H, Maurer CA, Buchler MW, Korc M. Overexpression of activin A in stage IV colorectal cancer. Gut 2001;49:409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hofland J, van Weerden WM, Steenbergen J, Dits NF, Jenster G, de Jong FH. Activin A stimulates akr1c3 expression and growth in human prostate cancer. Endocrinology 2012;153:5726–5734. [DOI] [PubMed] [Google Scholar]
- 19. Basu M, Bhattacharya R, Ray U, Mukhopadhyay S, Chatterjee U, Roy SS. Invasion of ovarian cancer cells is induced bypitx2‐mediated activation of TGF‐β and activin‐A. Mol Cancer 2015;14:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Leto G, Incorvaia L, Flandina C, Ancona C, Fulfaro F, Crescimanno M, et al. Clinical impact of cystatin C/cathepsin L and follistatin/activin A systems in breast cancer progression: a preliminary report. Cancer Invest 2016;34:415–423. [DOI] [PubMed] [Google Scholar]
- 21. Togashi Y, Kogita A, Sakamoto H, Hayashi H, Terashima M, de Velasco MA, et al. Activin signal promotes cancer progression and is involved in cachexia in a subset of pancreatic cancer. Cancer Lett 2015;356:819–827. [DOI] [PubMed] [Google Scholar]
- 22. Hoda MA, Dong Y, Rozsas A, Klikovits T, Laszlo V, Ghanim B, et al. Circulating activin A is a novel prognostic biomarker in malignant pleural mesothelioma—a multi‐institutional study. Eur J Cancer 2016;63:64–73. [DOI] [PubMed] [Google Scholar]
- 23. Loumaye A, de Barsy M, Nachit M, Lause P, Frateur L, van Maanen A, et al. Role of activin A and myostatin in human cancer cachexia. J Clin Endocrinol Metab 2015;100:2030–2038. [DOI] [PubMed] [Google Scholar]
- 24. Harada K, Shintani Y, Sakamoto Y, Wakatsuki M, Shitsukawa K, Saito S. Serum immunoreactive activin A levels in normal subjects and patients with various diseases. J Clin Endocrinol Metab 1996;81:2125–2130. [DOI] [PubMed] [Google Scholar]
- 25. Leto G, Incorvaia L, Badalamenti G, Tumminello FM, Gebbia N, Flandina C, et al. Activin A circulating levels in patients with bone metastasis from breast or prostate cancer. Clin Exp Metastasis 2006;23:117–122. [DOI] [PubMed] [Google Scholar]
- 26. Terpos E, Kastritis E, Christoulas D, Gkotzamanidou M, Eleutherakis‐Papaiakovou E, Kanellias N, et al. Circulating activin‐A is elevated in patients with advanced multiple myeloma and correlates with extensive bone involvement and inferior survival; no alterations post‐lenalidomide and dexamethasone therapy. Ann Oncol 2012;23:2681–2686. [DOI] [PubMed] [Google Scholar]
- 27. Chen JL, Walton KL, Qian H, Colgan TD, Hagg A, Watt MJ, et al. Differential effects of interleukin‐6 and activin A in the development of cancer‐associated cachexia. Cancer Res 2016;76:5372–5382. [DOI] [PubMed] [Google Scholar]
- 28. Chen JL, Walton KL, Winbanks CE, Murphy KT, Thomson RE, Makanji Y, et al. Elevated expression of activins promotes muscle wasting and cachexia. FASEB J 2014;28:1711–1723. [DOI] [PubMed] [Google Scholar]
- 29. Lerner L, Gyuris J, Nicoletti R, Gifford J, Krieger B, Jatoi A. Growth differentiating factor‐15 (gdf‐15): a potential biomarker and therapeutic target for cancer‐associated weight loss. Oncol Lett 2016;12:4219–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Loumaye A, de Barsy M, Nachit M, Lause P, van Maanen A, Trefois P, et al. Circulating activin A predicts survival in cancer patients. J Cachexia Sarcopenia Muscle 2017;8:768–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kita A, Kasamatsu A, Nakashima D, Endo‐Sakamoto Y, Ishida S, Shimizu T, et al. Activin B regulates adhesion, invasiveness, and migratory activities in oral cancer: a potential biomarker for metastasis. J Cancer 2017;8:2033–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xiong S, Klausen C, Cheng JC, Zhu H, Leung PC. Activin B induces human endometrial cancer cell adhesion, migration and invasion by up‐regulating integrin β3 via Smad2/3 signaling. Oncotarget 2015;6:31659–31673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xiong S, Klausen C, Cheng JC, Leung PC. Activin B promotes endometrial cancer cell migration by down‐regulating E‐cadherin via SMAD‐independent MEK‐ERK1/2‐SNAIL signaling. Oncotarget 2016;7:40060–40072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Benny Klimek ME, Aydogdu T, Link MJ, Pons M, Koniaris LG, Zimmers TA. Acute inhibition of myostatin‐family proteins preserves skeletal muscle in mouse models of cancer cachexia. Biochem Biophys Res Commun 2010;391:1548–1554, Epub 2009/12/29. [DOI] [PubMed] [Google Scholar]
- 35. Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010;142:531–543. [DOI] [PubMed] [Google Scholar]
- 36. von Haehling, S , Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53r172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005;7:469–483. [DOI] [PubMed] [Google Scholar]
- 38. Paster EV, Villines KA, Hickman DL. Endpoints for mouse abdominal tumor models: refinement of current criteria. Comp Med 2009;59:234–241, Epub 2009/07/22. [PMC free article] [PubMed] [Google Scholar]
- 39. Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, et al. Oncomine: a cancer microarray database and integrated data‐mining platform. Neoplasia 2004;6:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cancer Genome Atlas Research Network . Electronic address aadhe, Cancer Genome Atlas Research N. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 2017;32:185–203 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chandrashekar DS, Bashel B, Balasubramanya SAH, Creighton CJ, Ponce‐Rodriguez I, Chakravarthi B, et al. UALCAN: a portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 2017;19:649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ding H, Zhang G, Sin KW, Liu Z, Lin RK, Li M, et al. Activin A induces skeletal muscle catabolism via p38β mitogen‐activated protein kinase. J Cachexia Sarcopenia Muscle 2017;8:202–212, Epub 2016/11/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lee SJ, McPherron AC. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci U S A 2001;98:9306–9311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tan CR, Yaffee PM, Jamil LH, Lo SK, Nissen N, Pandol SJ, et al. Pancreatic cancer cachexia: a review of mechanisms and therapeutics. Front Physiol 2014;5:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Toledo M, Busquets S, Penna F, Zhou X, Marmonti E, Betancourt A, et al. Complete reversal of muscle wasting in experimental cancer cachexia: additive effects of activin type ii receptor inhibition and β‐2 agonist. Int J Cancer 2016;138:2021–2029. [DOI] [PubMed] [Google Scholar]
- 46. Barreto R, Kitase Y, Matsumoto T, Pin F, Colston KC, Couch KE, et al. ACVR2B/Fc counteracts chemotherapy‐induced loss of muscle and bone mass. Sci Rep 2017;7:14470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hatakeyama S, Summermatter S, Jourdain M, Melly S, Minetti GC, Lach‐Trifilieff E. ActRII blockade protects mice from cancer cachexia and prolongs survival in the presence of anti‐cancer treatments. Skelet Muscle 2016;6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nakamura T, Sugino K, Titani K, Sugino H. Follistatin, an activin‐binding protein, associates with heparan sulfate chains of proteoglycans on follicular granulosa cells. J Biol Chem 1991;266:19432–19437. [PubMed] [Google Scholar]
- 49. Sidis Y, Mukherjee A, Keutmann H, Delbaere A, Sadatsuki M, Schneyer A. Biological activity of follistatin isoforms and follistatin‐like‐3 is dependent on differential cell surface binding and specificity for activin, myostatin, and bone morphogenetic proteins. Endocrinology 2006;147:3586–3597. [DOI] [PubMed] [Google Scholar]
- 50. Eramaa M, Hilden K, Tuuri T, Ritvos O. Regulation of inhibin/activin subunit messenger ribonucleic acids (mRNAs) by activin A and expression of activin receptor mRNAs in cultured human granulosa‐luteal cells. Endocrinology 1995;136:4382–4389. [DOI] [PubMed] [Google Scholar]
- 51. Makanji Y, Walton KL, Chan KL, Gregorevic P, Robertson DM, Harrison CA. Generation of a specific activin antagonist by modification of the activin A propeptide. Endocrinology 2011;152:3758–3768. [DOI] [PubMed] [Google Scholar]
- 52. Chen JL, Walton KL, Al‐Musawi SL, Kelly EK, Qian H, La M, et al. Development of novel activin‐targeted therapeutics. Mol Ther 2015;23:434–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. De Waele E, Wauters E, Ling Z, Bouwens L. Conversion of human pancreatic acinar cells toward a ductal‐mesenchymal phenotype and the role of transforming growth factor β and activin signaling. Pancreas 2014;43:1083–1092. [DOI] [PubMed] [Google Scholar]
- 54. Hulmi JJ, Nissinen TA, Rasanen M, Degerman J, Lautaoja JH, Hemanthakumar KA, et al. Prevention of chemotherapy‐induced cachexia by ACVR2B ligand blocking has different effects on heart and skeletal muscle. J Cachexia Sarcopenia Muscle 2018;9:417–432, Epub 2017/12/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Haugen F, Labori KJ, Noreng HJ, Buanes T, Iversen PO, Drevon CA. Altered expression of genes in adipose tissues associated with reduced fat mass in patients with pancreatic cancer. Arch Physiol Biochem 2011;117:78–87. [DOI] [PubMed] [Google Scholar]
- 56. Michaelis KA, Zhu X, Burfeind KG, Krasnow SM, Levasseur PR, Morgan TK, et al. Establishment and characterization of a novel murine model of pancreatic cancer cachexia. J Cachexia Sarcopenia Muscle 2017;8:824–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kays JK, Shahda S, Stanley M, Bell TM, O'Neill BH, Kohli MD, et al. Three cachexia phenotypes and the impact of fat‐only loss on survival in FOLFIRINOX therapy for pancreatic cancer. J Cachexia Sarcopenia Muscle 2018;9:673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Argiles JM, Stemmler B, Lopez‐Soriano FJ, Busquets S. Nonmuscle tissues contribution to cancer cachexia. Mediators Inflamm 2015;2015:182872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. von Haehling S, Lainscak M, Kung T, Cramer L, Fulster S, Pelzer U, et al. Non‐invasive assessment of cardiac hemodynamics in patients with advanced cancer and with chronic heart failure: a pilot feasibility study. Arch Med Sci 2013;9, 2:261–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Su GH, Bansal R, Murphy KM, Montgomery E, Yeo CJ, Hruban RH, et al. ACVR1B (ALK4, activin receptor type 1B) gene mutations in pancreatic carcinoma. Proc Natl Acad Sci U S A 2001;98:3254–3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hempen PM, Zhang L, Bansal RK, Iacobuzio‐Donahue CA, Murphy KM, Maitra A, et al. Evidence of selection for clones having genetic inactivation of the activin A type II receptor (ACVR2) gene in gastrointestinal cancers. Cancer Res 2003;63:994–999. [PubMed] [Google Scholar]
- 62. Lonardo E, Hermann PC, Mueller MT, Huber S, Balic A, Miranda‐Lorenzo I, et al. Nodal/activin signaling drives self‐renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell 2011;9:433–446. [DOI] [PubMed] [Google Scholar]
- 63. Togashi Y, Sakamoto H, Hayashi H, Terashima M, de Velasco MA, Fujita Y, et al. Homozygous deletion of the activin A receptor, type IB gene is associated with an aggressive cancer phenotype in pancreatic cancer. Mol Cancer 2014;13:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zimmers TA, Davies MV, Koniaris LG, Haynes P, Esquela AF, Tomkinson KN, et al. Induction of cachexia in mice by systemically administered myostatin. Science 2002;296:1486–1488, Epub 2002/05/25. [DOI] [PubMed] [Google Scholar]
- 65. Zimmers TA, Jiang Y, Wang M, Liang TW, Rupert JE, Au ED, et al. Exogenous GDF11 induces cardiac and skeletal muscle dysfunction and wasting. Basic Res Cardiol 2017;112:48, Epub 2017/06/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Expression of INHBA in pancreatic adenocarcinoma tumors by individual cancer stages from patients in the TCGA dataset as analyzed by UALCAN. (Ualcan.path.uab.edu accessed 04/03/2019)
Figure S2: Evidence for expression of MSTN/myostatin outside of skeletal muscle.