

Abstract
Advancing therapeutics for traumatic brain injury (TBI) remains a challenge, necessitating testable targets with interventions appropriately timed to intercede on evolving secondary insults. Neuroproteomics provides a global molecular approach to deduce the complex post-translational processes that underlie secondary events after TBI. Yet method advancement has outpaced approaches to interrogate neuroproteomic complexity, in particular when addressing the well-recognized temporal evolution of TBI pathobiology. Presented is a detailed account of the temporal neuroproteomic response to mild-moderate rat controlled cortical impact within perilesioned somatosensory neocortex across the first two weeks after injury. Further, this investigation assessed use of artificial neural network and functional enrichment analyses to discretize the temporal response across some 2047 significantly impacted proteins. Results were efficiently narrowed onto ion transporters with phenotypic relevance to abnormal GABAergic transmission and a delayed decline amenable to intervention under managed care conditions. The prototypical target potassium/chloride co-transporter 2 (KCC2 or SLC12A5) was investigated further with the KCC2-selective modulator CLP290. Guided by post-translational processing revealed one-day after insult to precede KCC2 protein loss a day after, CLP290 was highly effective at restoring up to 70% of lost KCC2 localization, which was significantly correlated with recovery of sham-level function in assessed somatosensory behavioral tasks. The timing of administration was important, with no significant improvement observed if given earlier, one-hour after insult, or later when KCC2 protein decline begins. Results portend importance for a detailed post-translational characterization when devising TBI treatments, and support the therapeutic promise of KCC2-targeted CLP290 intervention for positive functional recovery after brain injury.
Keywords: CLP290, KCC2, proteomics, therapy, traumatic brain injury
Introduction
Traumatic brain injury (TBI) continues as a significant cause of morbidity in the United States and abroad, yet therapeutic advancement remains elusive.1–4 More precise therapeutic approaches are regarded as necessary to address the lack of effective outcomes from randomized trials.5,6 Interventions devised around testable mechanisms that can be assessed across time are needed for selective clinical application to whom and when appropriate.7
Yet, therapeutic target selection is challenged by TBI's multi-faceted pathobiology, augmented by a dynamic combination of secondary insults and injuries.8,9 Hypoxic and hypercarbic events, for example, disrupt ionic homeostasis and worsen secondary cellular injury and excitotoxic outcomes.10,11 A TBI further promotes a variety of secondary neurological complications, such as a 29-fold greater prevalence of seizure after severe TBI.12,13 Thus, more global, temporal molecular assessments stand to improve therapeutic target identification and address the inherent biochemical complexity underlying TBI neurobiology.
Mass spectrometry has evolved into a powerful interrogational tool with which to study the complex biochemical response to TBI.14 Recent advancements in data-independent methods for label-free quantification have broadened the utility of mass spectrometry for unbiased characterization of translational and post-translational shifts within the proteome.15,16 Moreover, these newer methods are more cost-effective for large-scale study designs, enabling temporal analysis of the TBI neuroproteome with sufficient statistical power.
Decoding the temporal processes that follow TBI is vital to the effective selection and optimization of new therapies.17,18 While the onset of pathology is defined clearly by the insult, the timing of secondary events is varied, overlapping, and responsive to external and internal factors that influence ensuing damage and the evolution of disease.1,2 Hence, therapeutics need to be properly timed, which is less of a concern for events of immediate gestation; however, the onset of secondary events can be significantly delayed from the primary injury, rendering them less amenable to therapy without a temporal guide to their initiation. Yet, treating delay-onset events offers a practical advantage for patient care, permitting intervention under controlled intensive care conditions.
In addressing this opportunity, proteomic analysis can provide a useful temporal scaffold with which to identify novel targets and assess therapeutic effectiveness. Many pharmaceuticals impact enzymatic actions, such as kinase or phosphatase regulation of biochemical pathways.19–23 Again, proteomics affords broad interrogation of such post-translational processes, providing guidance on secondary event initiation and a potential therapeutic window. To achieve such goals, reductionist approaches are essential to address the complexity of the TBI neuroproteomic response, moving what otherwise would just be another large dataset onto a practical application in therapeutic development.
In this study, we assessed the use of dimensionality reduction with biochemical interrogation informatic techniques to draw out new therapeutic targets from the temporal TBI neuroproteome. Potassium/chloride co-transporter 2 (KCC2, SLC12a5 gene product) was resolved as a promising delayed-onset target—an ion-transporter essential to chloride homeostasis and maintenance of inhibitory gamma-aminobutyric acid (GABA)ergic neurotransmission.24–27 Moreover, we assessed how post-translational processing of this prototypical target may help define an effective treatment window. Properly timed, the selective agent CLP29028 provided effective KCC2 preservation and enhanced functional recovery after focal TBI.
Methods
Controlled cortical impact (CCI) animal model
Adult male Sprague-Dawley rats (∼275 g, Harlan Laboratories) were used after a week of acclimation under approval from the Virginia Commonwealth University Institutional Laboratory Animal Care and Use Committee in accordance with National Institutes of Health guidelines and the Guide for the Care and Use of Laboratory Animals (U.S. Department of Health and Human Services). Animal facilities were accredited fully by the Association for Assessment and Accreditation of Laboratory Animal Care International. Food and water were provided ad libitum in ventilated cages in a temperature and humidity-controlled environment on 12 h light/dark cycle.
The CCI was performed using a Leica Impact One Stereotaxic Impactor as described previously (Supplementary Fig. S1A).29 Briefly, rats were anesthetized using 4% isoflurane and maintained at 2% throughout the surgical procedure. Rectal temperature was monitored to maintain 37°C via heating pad. Further, heart rate, respiratory rate, and arterial blood oxygenation were monitored via pulse oximetry (MouseOx, Starr Life Sciences) using a rat foot sensor to affirm physiological homeostasis (Supplementary Fig. S2A–E). A 5 mm craniotomy was centered 1.6 mm caudal from bregma and 3.4 mm lateral (Supplementary Fig. 1B). Focal injury was administered using a 3 mm flat-tip stainless steel impactor translating perpendicular to the brain surface at 4.0 m/sec to a depth of 2 mm with a dwell time of 0.5 sec. The scalp was sutured closed with Neosporin and 2% lidocaine applied topically. Anesthesia was discontinued and righting reflex was monitored as a correlate measure of injury severity, with TBI animals falling within the mild to moderate range (4.1 ± 0.94 min), which was significantly greater than sham (2.8 ± 0.56 min; p = 0.001).30
Brains were collected at 1, 2, 4, 7, and 14 days post-injury by decapitation under isoflurane anesthesia. Brains were removed from the skull, rinsed with ice-cold phosphate buffered saline (PBS) and flash-frozen in liquid nitrogen. A 2 mm thick coronal frozen tissue block centered on the lesion was mounted on a ThermoFisher HM 525 cryostat. The block was sectioned interleaving two adjacent 150 μm sections for protein lysate with five 10 μm sections for imaging. Each 150 μm cut was dissected further by scalpel to extract perilesioned tissue between 0.5 mm and 1.5 mm lateral from the core boundary (Supplementary Fig. 1C), with pieces combined for protein analysis.
Proteomic mass spectrometry
Tissue lysis (n = 6/group) and mass spectrometric analysis were performed as described previously.29 In summary, we sequentially homogenized brain tissue to resolve the neuroproteome into matrix-associated and membrane-associated compartments. Sample protein concentration was assessed and balanced to a nominal 1 μg/μL concentration. A 50-μg aliquot of each sample was reduced, alkylated, trypsin-digested, and concentrated into 20 μL of 100-mM ammonium formate buffer (pH 10). We injected protein digests (4 μL each) in a treatment-interspersed order for five-fraction two-dimensional nanoACQUITY ultra performance liquid chromatography.
An XBridge BEH C18 1 mm x 50 mm fractionation column was used ahead of a Symmetry C18 180 μm × 20 mm trap column and an HSS T3 75 μm × 150 mm analytical column (Waters). Eluting peptides were electrosprayed via a 10 μm i.d. PicoTip emitter (New Objective) online with a Synapt G2 mass spectrometer (Waters) operated in a data-independent acquisition mode with ion mobility enabled. For quality control, instrument tuning, mass calibration, and a system performance evaluation for chromatographic separation (mean 0.16 min peak width), resolution (nominally 26,000 at 785 m/z), accuracy (0.5 ppm, RMS), and sensitivity (nominally 1e5 intensity, a.u., at 750 pg/μL) were performed daily using a [Glu1]-Fibrinopeptide B external mass standard (Waters) and a Pierce HeLa Protein Digest Standard (ThermoFisher).
Proteomic informatic analysis
Ion-mobility enabled data-independent analysis (HDMSe™) results were processed using Waters PLGS software (v.3.0.2). Peak picked ion tables were generated using precursor and product intensity cutoffs of 325 and 30 counts for matrix fractions and 200 and 30 counts for membranous fractions, respectively. Processed results for the five chromatography fractions per sample were combined into a single ion table, merging duplicate ions within ±1 min and ±10 ppm difference in elution time and mass for precursor ions and ±1 min and ±30 ppm difference for product ions. Ion tables were then searched against a Uniprot KB Rattus Database (rel. 2017_01) with parameters: trypsin specificity; one missed cleavage; a minimum of two peptides per protein ID each with a minimum of three fragment ions; fixed carbamidomethylation; variable oxidation, phosphorylation, acetylation, and methylation; neutral loss of ammonia or water; ±10 ppm precursor ion tolerance, ±20 ppm product ion tolerance. Peptide annotations were filtered to a 1% false discovery rate (FDR) using the reversed-decoy method.
Processed data were imported into IsoQuant (v1.7b) software for peptide clustering across biological replicates using the tolerance bands: ±12 ppm for mass; ±2 min for elution time; ±4 bins for drift time.31 Peptide intensity data were merged when variably split by methionine oxidation, missed cleavage, or the neutral loss of water or ammonia, as can occur during experimentation. Data were then filtered to preserve replicating ions, measured in ≥four of six biological replicates per group. We chose this conservative level of replication to balance the need for sufficient statistical power and comprehensive assessment of the TBI-responsive neuroproteome. Data were imputed for non-random missing values that consistently fell below the limit of detection (across all replicates per group) as described previously.29 Intensity values were then median-centered within each treatment group and log2 transformed. Analysis of variance (ANOVA) testing was performed at the peptide-level, adjusted to a 5% false discovery rate (FDR) using the Q-value method. The TBI responsive peptide features were then dimensionally reduced in an unsupervised fashion by generating a self-organizing feature map (SOM), from which concerted temporal trends were identified (Multiple Experiment Viewer, v4.9).32 Proteins matched to each distinct temporal node from the feature map were assessed by enrichment analysis, using a Fisher inverse chi-squared method corrected to a 5% FDR (ToppGene).33
Immunoblot analysis
Protein-balanced samples (2 μg) were resolved on a NuPAGE 4–12% Bis-Tris gel system with MOPS buffer (ThermoFisher). Protein was transferred via the semi-dry method to 0.22 μm PVDF membrane (Millipore). Membranes were blocked in 0.5% (w/v) powdered milk for 1 h at room temperature. Blots were then probed with primary antibodies overnight (∼16 h) at 4°C: anti-mouse KCC2 (Antibodies Inc., 75-013, 1:300); anti-rabbit KCC2 (Cell Signaling 94725, 1:2500); anti-mouse β-actin (load control, Sigma A5441, 1:5000). Horseradish peroxidase-conjugated secondary antibodies and the SuperSignal West Femto chemiluninescence reagent were used for detection (ThermoFisher).
Blots were imaged on an Image Station 4000MM Pro 16-bit CCD imager (Carestream). Blot measure were normalized for load and standardized to control. The KCC2 relative abundance was assessed across post-injury time using the average control-standardized measure for two independent KCC2 antibodies for a robust assessment. An effect of time post-injury was assessed using JMP software (v13) by one-way ANOVA, with Tukey's honest significant difference post hoc analysis for paired comparisons against control.
Immunofluorescence microscopy
Fresh-frozen brains were cut into 2 mm thick coronal blocks centered about the lesion and thin-sectioned (10 μm) on a Microm HM 525 cryostat. Sections were thaw-mounted on charged glass slides, air dried, and fixed in 3% (w/v) paraformaldehyde. Slides were blocked for 1 h at room temperature with a buffer containing: 5% goat serum (v/v); 2.5% bovine serum albumin (w/v); 0.1% Triton in 1x PBS (v/v). The sections were then probed overnight at 4°C with a mixture of anti-mouse lgG2a KCC2 (Antibodies Inc., 75-013, 1:40), and the excitatory neuronal marker anti-mouse IgG1 NeuN (Millipore MAB377, 1:500). Washed slides were then probed with Alexa Fluor 488 and 568 conjugated secondary antibodies (ThermoFisher) for 1 h at room temperature. Prolong Diamond Antifade Mountant and a 0.17 mm thick coverslip were applied (ThermoFisher).
Images were collected at 400x magnification on a multi-channel fluorescence Axioimager.M2 motorized microscope (Zeiss) equipped with a constant-illumination solid-state light source, structured illumination, and a model 506 m high-sensitive CCD camera. Image capture and analysis were performed in Zen v2.0 with consistent exposure settings across sample groups (five animals per group, four sections per animal). Slides were stained in cohorts that contained an equal representative number of animals per sample group. Each slide cohort was imaged in one session to further minimize within-cohort variance.
Nine multi-channel fluorescence 150 μm2 perilesional images per section were collected in a sequential-random stereological fashion within layers 4/5 of barrel cortex, between 500 μm and 1500 μm lateral from the lesion periphery. Integrated mean intensity gray-level measures (14-bit) on the target fluorescence channel were averaged across images per animal and background subtracted using the mean immunofluorescence imaged over the adjacent corpus callosum (exhibited negative staining for target antibodies). Measures from each staining cohort (with equal sample group representation) were mean-centered to correct for between-cohort variance. Measures were then standardized as a percentage of sham control staining. Statistical testing in JMP was performed across animals between sample groups with an effect of CLP290 on post-TBI KCC2 recovery assessed at each administration time relative to vehicle control using the Student t test with a Šidák correction for multiple comparisons.
CLP290 pharmacological TBI treatment
A follow-up cohort of CCI animals then were treated by oral gavage (p.o.) with the KCC2-selective enhancer compound CLP290 (5-fluoro-2-[(Z)-(2-hexahydrophyridazin-1-yl-4-oxo-thiazol-5-ylidene)methyl]phenyl]-pyrrolidine-1-carboxylate, Supplementary Fig. S3A, PharmaBlock USA, Sunnyvale, CA) as demonstrated previously.28,34 CLP290 is a carbamate prodrug designed to enhance the pharmacokinetics of the active compound CLP257, which itself was effective in modulating KCC2 at a 100 mg/kg intraperitoneal (i.p.) dose, but was limited pharmaceutically by a short <15 min half-life. The kinetic profile was substantially improved with the metabolism of the orally administered prodrug CLP290 at the same 100 mg/kg dose, extending the active compound's circulating half-life out to 5 h along with a greater overall plasma concentration in the rat.28
To examine CLP290's pharacodynamics after TBI, we conducted a dose-response analysis starting with the previously published 100 mg/kg dose. A minimum effective dose of 50 mg/kg (p.o.) was identified in demonstrating improved somatosensory functional recovery by four days after TBI using the whisker adhesive removal and Rotarod behavioral tasks described below (Supplementary Fig. 3B,C). Animals were weighed daily with an appropriate mass of CLP290 freshly suspended in 2 mL of 1% methylcellulose vehicle (w/v). The CLP290 (2 mL) was delivered via gavage, with different administrations tested, initiated at 1 h, 1 day, and 2 days after injury, or vehicle alone. Following behavioral testing four days after CCI, animals were sacrificed and brain tissue collected as described earlier. Animals showed no abnormal behavior or significant weight changes in response to the drug (Supplementary Fig. 2F), which previously was found to show no overt toxicological effects in a seven-day repeat-dose toxicity study up to a maximum 2000 mg/kg dose.28
Whisker adhesive removal task
The whisker adhesive removal task (w-ART)35 was performed to assess whisker sensory perception after injury adjacent to barrel somatosensory neocortex. Briefly, rats were trained before injury to remove a 113 mm2 adhesive sticker within 5 sec of being placed on their whiskers. The sticker removal task was performed again with video recording four days after CCI or sham procedures, with CLP290 drug or vehicle administration as indicated. Latency to initiate sticker removal was then assessed. In JMP, an effect of CLP290 on post-TBI w-ART performance was tested at each administration time relative to vehicle control using the Student t test with a Šidák correction for multiple comparisons. Within-animal correlation between latency outcomes and perilesional KCC2 abundance were assessed at day 4 using a Spearman rank-order test.
Rotarod coordination performance task
The Rotarod task36 was performed to assess recovery of sensory integration for limb placement after CCI to limb-associated areas of somatosensory neocortex using the previously described method.37 Briefly, animals were trained to walk on the rotating device before TBI or sham injury, which was accelerated from 0 revolutions per minute (rpm) to a maximum of 30 rpm over a 100 sec time interval. Animals were then tested one and four days after CCI or sham procedures, with CLP290 drug or vehicle administration as indicated. Latency to fall from the Rotarod on day 4 was determined as the average time on the device over three trials, normalized within each animal to day 1 performance, with a maximum cutoff time of 120 sec for any given trial. In JMP, an effect of CLP290 on post-TBI Rotarod performance was tested at each administration time relative to vehicle control using the Student t test with a Šidák correction for multiple comparisons. Within-animal correlation between Rotarod performance and perilesional KCC2 abundance were assessed at day 4 using a Spearman rank-order test.
Results
Temporal proteomics reveals a concerted delay in perilesional transporter loss after TBI as amenable therapeutic targets
The neuroproteome of surviving perilesional tissue at risk of secondary insult was evaluated across the first two weeks post-TBI for delayed-onset therapeutic targets. In all, 35,625 unique peptides were identified reproducibly from 3494 protein isoforms of 2378 protein families at a 1% false positive peptide identification rate. Peptide measures were tested for a main effect of time post-TBI, corrected to a 5% FDR. Significant measures comprised the TBI-responsive neuroproteome, which contained 11,745 (33.0% of total) unique peptides, including post-translational modifications, derived from 2047 protein isoforms, implicating nearly 60% of the neuroproteome within the perilesioned tissue as having some post-translational effect of TBI (Supplementary Table S1). An unsupervised, SOM approach reduced the TBI neuroproteomic response to concerted temporal trends.
The three most populated nodes, illustrated in Fig. 1, were assessed informatically for biomolecular and functional relevance. The densest node, with 264 (14.7% of total) protein families, was associated most significantly with altered transcription/translation, showing an elevated profile between days 2 and 7 post-injury (Fig. 1A). The second-most populated node, with 125 (7.0%) protein families, was enriched in association with vesicular transport (e.g., synaptic vesicles) and had a profile showing early suppression within the first 24 h, recovery at one week, and a second sizeable decline by two weeks post-TBI (Fig. 1B).
FIG. 1.

Functional relevance of the three most prominent temporal profiles within the traumatic brain injury responsive neuroproteome as revealed by Self-Organizing Map (SOM) data reduction. (A) SOM node 1 profile of 264 temporally tracking proteins with an enriched functional association with transcription and translation processes. (B) SOM node 2 profile of 125 temporally tracking proteins with an enriched functional association with vesicular transport processes. (C) SOM node 3 profile of 123 temporally tracking proteins with an enriched functional association with transmembrane transport processes. Results reported as the median log-2 fold-change per time point, per SOM node.
The third-most dense node, with 123 (6.9%) protein families, was enriched in transmembrane transport proteins (e.g., ion channels). With a delayed onset beyond 24 h after injury and a protracted period of decline (Fig. 1C), this node's temporal profile suggested an amenable window for managed intervention. Relative to the first node's involvement in complex transcriptional mechanisms, proteins of the third node were of greater focus given their more discrete nature as therapeutic targets.
An in-depth phenotypic classification of the third node is provided in Table 1. From 2047 TBI-responsive proteins, temporally and functionally guided data reduction narrowed focus here to eight membrane transporters with an enriched association to GABAergic transmission and phenotypic relevance to seizure and epilepsy. The neuron-specific ion channel KCC2 was of interest, having received recent attention as a therapeutic target in other neurodegenerative conditions.38–41 KCC2 is an essential transporter critical to maintaining chloride homeostasis as needed for effective hyperpolarizing GABAergic neurotransmission. Abnormal GABAergic signaling is linked to the onset and progression of epilepsy, to include post-traumatic epilepsy (PTE), a significant TBI comorbidity that remains poorly managed and understood. Thus, KCC2 was particularly favorable as a prototypical therapeutic, with it addressing GABAergic dysfunction after TBI, having an established selective drug modulator, and a delayed onset response after TBI that is advantageous for managed care.
Table 1.
Functional Enrichment Analysis of Proteins from Self-Organizing Map Cluster C
| Category | Term | Description | Count | FDR | Symbols |
|---|---|---|---|---|---|
| Molecular function | GO:0005215 | Transporter activity | 29 | 2.43E-10 | KCC2, GABRB2, ATP5F1, TTYH1, CACNB4, GC2, TOMM20, TTYH3, SFXN1, CACNG2, PTP, VGAT, GAT3, OSBP, SFXN5, NCX1, MCT1, FATP1, SCN2A, SFXN3, LRP1, LAT1, ATP1A4, ATP1B3, FXYD1, HBZ, NNT, ANT4, GABRA1 |
| Biological process | GO:0055085 | Transmembrane support | 35 | 2.23E-14 | KCC2, SHANK1, GABRB2, ATP5F1, TTYH1, DPP6, CACNB4, GC2, TSC22D3, RAB8B, TOMM20, PAM16, TTYH3, PPIF, SFXN1, CACNG2, PTP, VGAT, GAT3, CHCHD4, SFXN5, NCX1, MCT1, SCN2A, AFG3L2, SFXN3, LAT1, NLGN2, SAMM50, ATP1A4, ATP1B3, FXYD1, NNT, ANT4, GABRA1 |
| Cellular component | GO:0097458 | Neuron part | 26 | 6.08E-07 | KCC2, SHANK1, TTYH1, DPP6, SNPH, TPRN, NTRK2, GNG13, CNTNAP1, VTI1A, GABBR2, GRM3, CPNE5, ADAM22, TULP1, VGAT, GAT3, HEPACAM, NCX1, NRXN3, SCN2A, SYNGR3, SYNGR1, LRP1, NLGN2, PCDH9 |
| Mouse phenotype | MP:0002064 | Seizures | 11 | 6.02E-05 | KCC2, CACNB4, NTRK2, GABBR2, ADAM22, CACNG2, GAT3, AIFM1, SYNGR3, SYNGR1, GABRA1 |
| Disease | C0014544 | Epilepsy | 10 | 1.01E-03 | KCC2, GABRB2, CACNB4, GRM3, ADAM22, CACNG2, VGAT, GAT3, SCN2A, GABRA1 |
| Pathway | 377263 | GABAergic synapse | 8 | 6.38E-08 | KCC2, GABRB2, GNG13, GNG2, GABBR2, VGAT, GAT3, GABRA1 |
FDR, false discovery rate; GABA, gamma-aminobutyric acid.
Post-translational plasmalemmal KCC2 events portend a therapeutic window
A targeted look at the KCC2 proteomic results indicated a significant decline in total concentration when applying a peptide-to-protein rollup function to average across the peptides. An initial drop was observed starting at day 1, although it leveled off into day 2 before a sharp decline thereafter through day 7, followed by a partial recovery by day 14 after injury (Fig. 2A). The escalated loss of KCC2 through the first week was validated via immunoblotting, except that the decline in total protein was delayed until day 2, affirmed across two independent antibodies (Fig. 2B).
FIG. 2.

Loss of perilesional KCC2 within the first two weeks after traumatic brain injury (TBI) involves dynamic post-translational processing that precedes total protein decline. (A) Peptide-to-protein KCC2 rollup results reflecting post-translational processing within the first two weeks after TBI. Fold change data reported as mean ± standard error of the mean (SEM), n = 6, log2 transformed and normalized to time-zero control. *Q-value corrected to a 5% false discovery rate (FDR). (B) KCC2 immunoblotting reflecting perilesional total protein decline beginning two days after injury. Inset images show KCC2 densitometric results after background subtraction and adjusting for load and transfer variability using matched β-actin staining. Values reported as mean ± SEM across two independent KCC2 antibodies, n = 6/group, Tukey HSD: *p < 0.05. (C) Representative micrographs from barrel cortex layers 4/5 of KCC2 Immunofluorescence staining morphologically depicting perilesional KCC2 neuronal loss (NeuN-stained soma) over the first two weeks after TBI. (D) Post-translational KCC2 dynamics involves n-terminal acetylation and multimodal phosphorylation, shown here at threonine 34 (T34) and serine 649 (S649) across the first two weeks after injury. Fold change data reported as mean ± SEM, n = 6, log2 transformed and normalized to time-zero control. *Q-value corrected to a 5% FDR. CCI, controlled cortical impact.
Morphological assessment within layers 4/5 of barrel cortex revealed that on day 1, there was a notable increase in somal localization to suggest a biphasic KCC2 response that in total did not amount to a decline in overall KCC2 until two days after injury, escalating through the first week and recovering somewhat by day 14 (Fig. 2C). Thus, while the KCC2 findings are overall quite similar, the proteomic platform's greater sensitivity and specificity to post-translational modifications allowed it to pick up on earlier dynamics beginning on day 1 after injury, before a bulk decline in protein as reflected in the immunoblot and immunofluorescent results. Regulatory KCC2 phosphorylation at threonine 34 (T34), for example, was 70% more decreased at day 1 than the decline in total KCC2 (Fig. 2D, purple), while another phosphor-motif on serine 649 (S649) showed no effect at day 1, but a sizeable 110% increase in phosphorylation indicating further regulatory modification coincident with total protein decline (Fig. 2D, brown).
Separately, the proteomic results picked up a significant decline in n-terminal KCC2 acetylation, which decreased 75% more at day 1 than total KCC2 (Fig. 2D, red), a modification demarking an initial destabilization event.42 Thus, while the findings together denote the pronounced loss of KCC2 total protein from day 2 after TBI, the proteomics results provide greater insight into preceding post-translational processing of KCC2, demarking a putative window within which to target therapeutic intervention.
Correlated recovery of KCC2 and somatosensory function after CLP290 TBI intervention
The novel compound CLP290, a carbamate prodrug of CLP257, has been described recently as a highly selective enhancer of KCC2 membrane localization,28 which effectively restores chloride homeostasis to correct GABAergic dysfunction.28,34,43 Thus, here we evaluated CLP290's KCC2 enhancing abilities after TBI and the concomitant potential for functional improvement. Based on temporal proteomic findings, we selected three intervention start times within the first 48 h post-TBI: 1 h post CCI (Tx0d) to assess an immediate effect within the often-targeted “golden-hour”; one day post-CCI (Tx1d) to coincide with early KCC2 post-translational modification; two days post-CCI (Tx2d) to coincide with the initial onset of total KCC2 decline.
First, restoration of KCC2 by four days after TBI was assessed, with recovery evident within layers 4/5 of barrel cortex (Fig. 3). Yet, the most pronounced and significant effect was observed with the Tx1d administration, recovering 70% of lost KCC2, to within 84% of sham-vehicle control animals. Administering the drug earlier (Tx0d) or later (Txd2) was approximately half as effective, restoring only 41% or 39% of lost KCC2, respectively, neither of which met significance. Thus, these results imply a time-sensitive nature to restoring KCC2 post-TBI.
FIG. 3.

CLP290-enhanced recovery of KCC2 with day 1 administration after traumatic brain injury (TBI). Animals were treated with CLP290 initiating 1 h (Tx0d), one day (Tx1d), two days (Tx2d) after injury or with vehicle alone. Representative micrographs from barrel cortex layers 4/5 depicting recovery of perilesional KCC2 immunofluorescence neuronal staining (NeuN-stained soma). Immunodensitometry results with background subtraction reported as mean ± standard error of the mean, n = 4/group, normalized to sham-vehicle control. Student t test: *p < 0.05 (CNT/Veh vs. TBI/Veh). #p < 0.017 (TBI/Veh vs. TBI/Tx1d), with Šidák multiple comparisons correction.
Next, CLP290-enhanced functional recovery was assessed using the w-ART task of sensorimotor coordination and the Rotarod test involving sensorimotor integration. In the w-ART task, TBI insult resulted in a 73% longer latency to sense and remove a sticker placed on the whiskers, as measured four days after injury (Fig. 4A). Administering CLP290 reversed this deficit; however, again maximal and significant recovery was achieved with one day (Tx1d) administration while 1 h and two day treatment paradigms were notably less effective. Indeed the one day therapy restored w-ART performance entirely to sham-vehicle levels. Further, there was a strong correlation between an animal's behavioral performance and recovery of perilesional KCC2 in the somatosensory neocortex, Spearman ρ = -0.61, p = 0.0078.
FIG. 4.
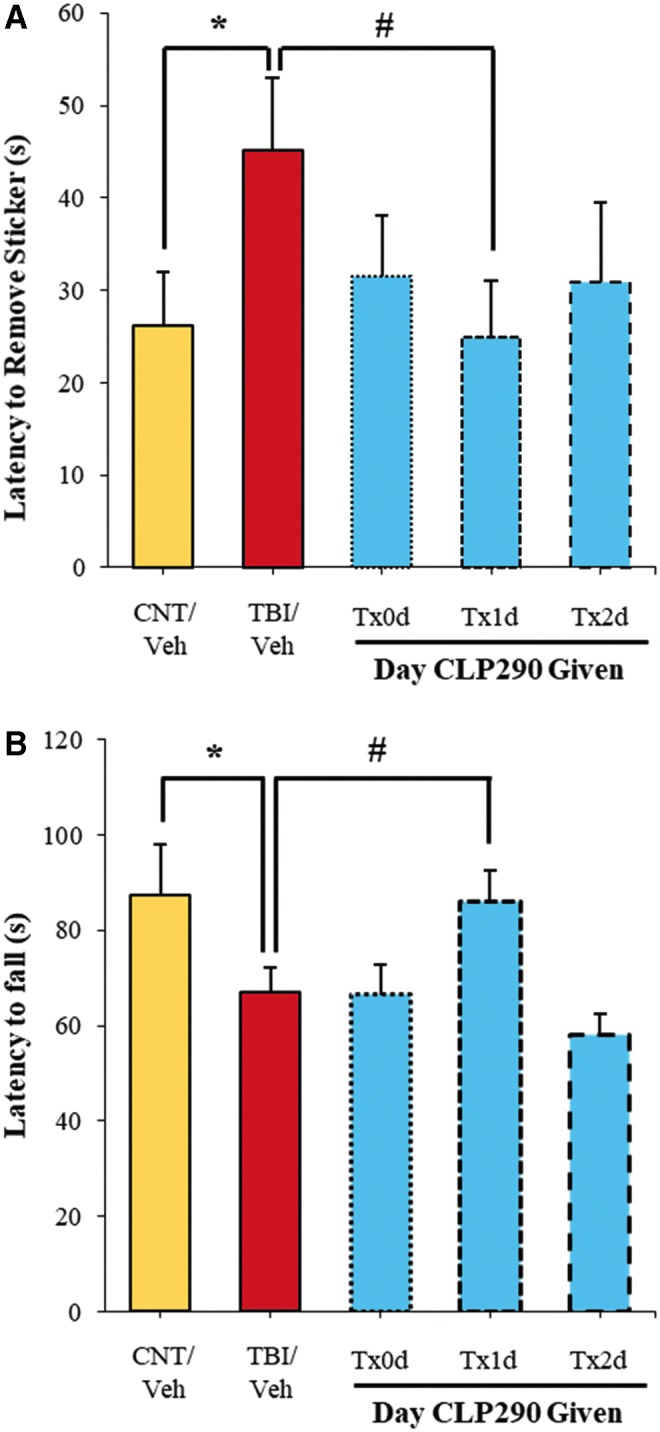
CLP290-ehanced recovery of function with day 1 administration after traumatic brain injury (TBI). Animals were treated with CLP290 initiating 1 h (Tx0d), one day (Tx1d), two days (Tx2d) after injury or with vehicle alone. (A) w-ART performance was assessed four days after injury as measured by the latency to remove a sticker placed on the whiskers. Data reported as mean ± standard error of the mean (SEM), n = 7/group. Student t test: *p < 0.05 (CNT/Veh vs. TBI/Veh). #p < 0.017 (TBI/Veh vs. TBI/Tx1d), with Šidák multiple comparisons correction. (B) Rotarod performance was assessed four days after injury as measured by the latency to fall from the device. Data reported as mean ± SEM, n = 7/group. Student t test: *p < 0.05 (CNT/Veh vs. TBI/Veh). #p < 0.017 (TBI/Veh vs. TBI/Tx1d), with Šidák multiple comparisons correction.
A similar trend was observed with Rotarod performance. As expected, with the CCI impact principally to hindlimb and forelimb somatosensory cortex, TBI-vehicle animals exhibited significantly worse sensorimotor integration on the Rotarod task, falling on average 20 sec (23%) sooner than sham-vehicle animals when assessed four days after injury (Fig. 4B). Impressively, animals administered CLP290 at one day (Tx1d) after TBI recovered near completely to within 98% of sham-vehicle animals. In contrast, CLP290 administered 1 h (Tx0d) or two days (Tx2d) produced no trend toward recovery. Thus, with properly timed CLP290 administration, somatosensory function four days after TBI could be restored to that of sham-vehicle animals.
Discussion
Findings from this study establish a reductionist approach to interrogate the highly complex temporal neuroproteomic response to TBI with its sensitivity to post-translational events portending clinically relevant intervention targets. Using unsupervised SOM analysis, the complex TBI response of some 2047 proteins across the first two weeks after injury was resolved into a discrete set of concerted temporal profiles. Enrichment analysis informed on the functional relevance of each temporal node.
One such node containing 123 proteins was enriched in association with dysregulated ionic homeostasis related to abnormal GABAergic transmission, and featured a delayed-response advantageous for intervention under managed intensive care. A prototypical target, KCC2, was selected given its direct role in secondary inhibitory neuronal dysfunction and the recent availability of a selective pharmacological modulator. Proteomic results here informed on regulatory post-translational processing of KCC2 that preceded its subsequent total protein decline, portending a testable intervention window. Molecular recovery of KCC2 and correlated functional improvement on treatment with the selective modulator CLP290 was attained distinctly one day after TBI, underscoring the promise of KCC2 as a TBI therapeutic target.
TBI is a highly dynamic and biologically complex disease, calling for global, temporally discriminant molecular assessment, such as at the proteomic level, to inform on ensuing pathobiology. This is especially instrumental in resolving novel interventions to secondary insults that may not respond effectively to generically applied therapy immediately after insult. The temporal evolution of TBI pathobiology is well recognized by researchers; yet, few large-scale proteomic studies have incorporated time in their design, hampering our understanding of the data.44–46
To that end, we report on the neuroproteomic response to mild-moderate rat CCI within non-lesioned somatosensory neocortex, excluding the focal core or developing glial scar. Enabling this work was the use of data-independent analysis, a recent mass spectrometric method that is touted for a high degree of measurement reproducibility advantageous for large-scale, label-free quantitative analysis, as needed for complex temporal study designs.47,48 Combined with orthogonal chromatographic and ion mobility characterization, the resultant spectral dataset provided a detailed assessment of some 3494 unique proteins. With known perilesional dysfunction in this model,49 it was anticipated that a majority of the proteome would be impacted, with 2047 proteins significantly responsive at some point within the first two weeks after injury when accounting for common post-translational modifications (Supplementary Table 1). Yet the complexity of these results is both advantageous in terms of the biochemical detail contained as well as a hindrance to extracting beneficial insight.
Temporal proteomic datasets (log transformed) can be assessed via ANOVA and post hoc methods for an effect of time after injury, with appropriate correction for multiple comparisons to control the FDR. Yet this approach only marginally simplified the number of individual measures to be assessed, from 35,625 to a still large 11,745, which hardly can be appreciated on its own. Thus, an artificial neural network procedure was applied, SOM, an unsupervised method to produce a feature map that in the case of temporal proteomic results in a discretized representation of time profiles. Thereby the data could be reduced to a handful of nodes, each representing correlative proteomic responses in time after injury.
We then focused on those nodes that suggested a delayed progression, which would facilitate more effective intervention of secondary insults with time to stabilize and assess the individual. Nodes were then effectively summarized by their enriched association with cellular processes (Fig. 1) and further examined for phenotypic importance to secondary insults such as abnormal GABAergic transmission and seizure in the case of node 3 (Table 1). Thus, what started as a list of some 2047 affected proteins was effectively reduced to a short list of eight plausible targets amenable to delayed treatment with potential to impact inhibitory dysfunction and post-traumatic functional consequences.
To further test the utility of this approach, we selected a prototypical target from Table 1, KCC2. First, in an earlier preliminary consideration of KCC2's declining proteomic profile, we reported that its decrease after CCI preceded perilesional declines in inhibitory synapse markers neuroligin-2 and gephyrin, which suggested that KCC2 dynamics were among early impacts to the GABAergic system.17 Second, KCC2 separately had been reported decreased after modeled diffuse mouse fluid percussion injury, which though that study was limited to one-week observations, it substantiated KCC2's applicability as a multi-modal, multi-species target.50 Further, our initial reporting on the temporal decline of rat KCC218 was affirmed independently out to one week as had been reported in the mouse.50,51
Last, KCC2 was considered an ideal prototypical target given the recent reporting on a specific KCC2 modulator, CLP290, which was well characterized as having minimal toxicological burden and efficacy in rescuing chloride homeostasis and GABAergic neurotransmission in neurodegenerative models other than TBI.28,34,43 Importantly, KCC2 is neuron specific, expressed broadly across all neuronal subtypes receiving hyperpolarizing inhibitory input, apart from vasopressin neurons, to extrude chloride in response to GABAergic receptor-mediated chloride influx.52 KCC2 is also exclusively expressed in the nervous system, in contrast to other chloride symporters, providing for broad-based, neuron-selective therapeutic targeting in addressing inhibitory network dysfunction after TBI.28,53
The selective modulator CLP290 was then used to assess the criticality in timing administration, as informed by the post-translational data on KCC2. Of particular interest was whether treatment would be best if timed to co-occur with the total protein decline two days after TBI, or beforehand when post-translational changes such as decreased n-terminal acetylation denoted diminished stabilization at one day after injury. We further wanted to compare intervention at 1 h after TBI, approximating the ideal golden-hour treatment often sought in trauma care.54 CLP290 was effective in recovering perilesional KCC2. Results showed an improved trend across all administrations; however, the one day treatment significantly restored as much as 70% of lost KCC2, which was nearly twice as effective as with 1 h or two day therapy (Fig. 3).
Next, it was assessed as to whether CLP290 intervention would enhance recovery of perilesional somatosensory function. As seen with KCC2, one day treatment was maximally effective, restoring performance on w-ART and Rotarod tasks back to sham-vehicle control levels. Indeed, w-ART performance significantly correlated with perilesional KCC2 in barrel neocortex. Yet, administering CLP290 either earlier or later had no significant effect on functional recovery.
Findings here thus demonstrate that there is a time dependency to CLP290's acute efficacy, requiring the drug to be onboard before total protein decline at two days, yet not too early so as to miss the appropriate post-translational target. These results support the need for greater detail on the acute timing of post-translational KCC2 processing, because this initial temporal proteomic assessment was focused more broadly across a two week time frame. While studies here focused on establishing proof-of-principle in using post-translational proteomic assessment for developing new therapeutic opportunities, the promising CLP290 results justify further interrogation of the drug mechanism of action related to KCC2 restoration after TBI and evaluation of additional cellular and functional benefits consequent recovered inhibitory signaling, which may reduce the burden of excitotoxic cell death or epileptogenesis following trauma.
Given additional relevance of the two-day time point to the resolved SOM nodes, future studies would also be warranted to address this period in greater detail. It is expected that therapeutic development of other putative targets with a similar delayed-onset to KCC2 are as likely to depend on a detailed appreciation of their post-translational dynamics to ensure the most effective therapeutic window and an understanding of the underlying mechanism of action.
Conclusion
The temporal TBI neuroproteomic dataset reported in this investigation offers a wealth of post-translational detail on the biochemical response two weeks after focal TBI. Yet to provide a tangible output, a dimensionality reduction approach was devised to resolve temporally concerted proteomic events that are clinically relevant to managed intervention. Focus was narrowed effectively onto a subset of ion transporters that presented a delayed decline within two days of injury. An exemplary target KCC2 was then assessed for therapeutic relevance using the selective modulator CLP290.
Timing of administration was critical in CLP290-mediated restoration of KCC2 localization and enhancement of functional recovery. The optimal day 1 intervention point coincided with extensive post-translational regulation of KCC2 that preceded its eventual degradation. These results ultimately support the utility of temporal neuroproteomics and indeed call for greater detail in interrogating post-translational dynamics within the first two days after injury, in light of the need for more refined intervention strategies for secondary insults. Mechanistic insight into the acute processing of KCC2, as well as other membrane transporters following the same temporal profile, may further translate to other neurodegenerative disorders that involve dysregulated ionic homeostasis.
Supplementary Material
Acknowledgments
A monoclonal KCC2 antibody was provided through the UC Davis/NIH NeuroMAB Facility, supported by NIH grant NS050606 and maintained by the Department of Neurobiology, Physiology and Behavior, College of Biological Sciences, University of California, Davis. This research was supported by the National Institute of Neurological Disorders and Stroke grants NS055012 and NS100322.
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
References
- 1. Alexander D.G., Shuttleworth-Edwards A.B., Kidd M., and Malcolm C.M. (2015). Mild traumatic brain injuries in early adolescent rugby players: long-term neurocognitive and academic outcomes. Brain Inj. 29, 1113–1125 [DOI] [PubMed] [Google Scholar]
- 2. Najafi M.R., Tabesh H., Hosseini H., Akbari M., and Najafi M.A. (2015). Early and late posttraumatic seizures following traumatic brain injury: a five-year follow-up survival study. Adv. Biomed. Res. 4, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chase A. (2015). Structural changes can progress for months after brain injury. Nat. Rev. Neurol. 11, 309. [DOI] [PubMed] [Google Scholar]
- 4. Kochanek P.M., and Clark R.S. (2016). Traumatic brain injury research highlights in 2015. Lancet Neurol. 15, 13–15 [DOI] [PubMed] [Google Scholar]
- 5. Narayan R.K., Michel M.E., Ansell B., Baethmann A., Biegon A., Bracken M.B., Bullock M.R., Choi S.C., Clifton G.L., Contant C.F., Coplin W.M., Dietrich W.D., Ghajar J., Grady S.M., Grossman R.G., Hall E.D., Heetderks W., Hovda D.A., Jallo J., Katz R.L., Knoller N., Kochanek P.M., Maas A.I., Majde J., Marion D.W., Marmarou A., Marshall L.F., McIntosh T.K., Miller E., Mohberg N., Muizelaar J.P., Pitts L.H., Quinn P., Riesenfeld G., Robertson C.S., Strauss K.I., Teasdale G., Temkin N., Tuma R., Wade C., Walker M.D., Weinrich M., Whyte J., Wilberger J., Young A.B., and Yurkewicz L. (2002). Clinical trials in head injury. J. Neurotrauma 19, 503–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kochanek P.M., Jackson T.C., Ferguson N.M., Carlson S.W., Simon D.W., Brockman E.C., Ji J., Bayir H., Poloyac S.M., Wagner A.K., Kline A.E., Empey P.E., Clark R.S., Jackson E.K., and Dixon C.E. (2015). Emerging therapies in traumatic brain injury. Semin. Neurol. 35, 83–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. DeWitt D.S., Hawkins B.E., Dixon C.E., Kochanek P.M., Armstead W., Bass C.R., Bramlett H.M., Buki A., Dietrich W.D., Ferguson A.R., Hall E.D., Hayes R.L., Hinds S.R., LaPlaca M.C., Long J.B., Meaney D.F., Mondello S., Noble-Haeusslein L.J., Poloyac S.M., Prough D.S., Robertson C.S., Saatman K.E., Shultz S.R., Shear D.A., Smith D.H., Valadka A.B., VandeVord P., and Zhang L. (2018). Pre-clinical testing of therapies for traumatic brain injury. J. Neurotrauma 35, 2737–2754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Levin H.S., and Diaz-Arrastia R.R. (2015). Diagnosis, prognosis, and clinical management of mild traumatic brain injury. Lancet Neurol. 14, 506–517 [DOI] [PubMed] [Google Scholar]
- 9. McGinn M.J., and Povlishock J.T. (2015). Cellular and molecular mechanisms of injury and spontaneous recovery., in: Handbook of Clinical Neurology. Department of Anatomy and Neurobiology, Medical College of Virginia Campus of Virginia Commonwealth University, Richmond, VA, USA.; Department of Anatomy and Neurobiology, Medical College of Virginia Campus of Virginia Commonwealth University. Elsevier B.V, Richmond, VA, pps. 67–87 [DOI] [PubMed] [Google Scholar]
- 10. Mazzeo A.T., and Gupta D. (2018). Monitoring the injured brain. J. Neurosurg. Sci. 62, 549–562 [DOI] [PubMed] [Google Scholar]
- 11. Wells A.J., and Hutchinson P.J. (2018). The management of traumatic brain injury. Surgery 36, 613–620 [Google Scholar]
- 12. Herman S.T. (2002). Epilepsy after brain insult: targeting epileptogenesis. Neurology 59, Suppl 5, S21–S26 [DOI] [PubMed] [Google Scholar]
- 13. Piccenna L., Shears G., and O'Brien T.J. (2017). Management of post-traumatic epilepsy: an evidence review over the last 5 years and future directions. Epilepsia Open 2, 123–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li X., Wang W., and Chen J. (2017). Recent progress in mass spectrometry proteomics for biomedical resarch. Sci. China Life Sci. 60, 1093–1113 [DOI] [PubMed] [Google Scholar]
- 15. Heap R.E., Gant M.S., Lamoliatte F., Peltier J., and Trost M. (2017). Mass spectrometry techniques for studying the ubiquitin system. Biochem. Soc. Trans. 45, 1137–1148 [DOI] [PubMed] [Google Scholar]
- 16. Leitner A. (2018). A review of the role of chemical modification methods in contemporary mass spectrometry-based proteomics research. Anal. Chim. Acta 1000, 2–19 [DOI] [PubMed] [Google Scholar]
- 17. Lizhnyak P.N. and Ottens A.K. (2015). Proteomics: in pursuit of effective traumatic brain injury therapeutics. Expert Rev. Proteomics 12, 75–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Elkind J.A., Lim M.M., Johnson B.N., Palmer C.P., Putnam B.J., Kirschen M.P., and Cohen A.S. (2015). Efficacy, dosage, and duration of action of branched chain amino acid therapy for traumatic brain injury. Front. Neurol. 6, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nikolian V.C., Georgoff P.E., Pai M.P., Dennahy I.S., Chtraklin K., Eidy H., Ghandour M.H., Han Y., Srinivasan A., Li Y., and Alam H.B. (2017). Valproic acid decreases brain lesion size and improves neurologic recovery in swine subjected to traumatic brain injury, hemorrhagic shock, and polytrauma. J. Trauma Acute Care Surg. 83, 1066–1073 [DOI] [PubMed] [Google Scholar]
- 20. Shen M., Wang S., Wen X., Han X.R., Wang Y.J., Zhou X.M., Zhang M.H., Wu D.M., Lu J., and Zheng Y.L. (2017). Dexmedetomidine exerts neuroprotective effect via the activation of the PI3K/Akt/mTOR signaling pathway in rats with traumatic brain injury. Biomed. Pharmacother. 95, 885–893 [DOI] [PubMed] [Google Scholar]
- 21. Wang G., Shi Y., Jiang X., Leak R.K., Hu X., Wu Y., Pu H., Li W.W., Tang B., Wang Y., Gao Y., Zheng P., Bennett M.V., and Chen J. (2015). HDAC inhibition prevents white matter injury by modulating microglia/macrophage polarization through the GSK3β/PTEN/Akt axis. Proc. Natl. Acad. Sci. U. S. A. 112, 2853–2858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen X., Wang H., Zhou M., Li X., Fang Z., Gao H., Li Y., and Hu W. (2018). Valproic acid attenuates traumatic brain injury-induced inflammation in vivo: involvement of autophagy and the Nrf2/ARE signaling pathway. Front. Mol. Neurosci. 11, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Plantier D., and Luauté J. (2016). Drugs for behavior disorders after traumatic brain injury: systematic review and expert consensus leading to French recommendations for good practice. Ann. Phys. Rehabil. Med. 59, 42–57 [DOI] [PubMed] [Google Scholar]
- 24. Blaesse P., and Schmidt T. (2015). K-Cl cotransporter KCC2—a moonlighting protein in excitatory and inhibitory synapse development and function. Pflugers Arch. 467, 615–624 [DOI] [PubMed] [Google Scholar]
- 25. Payne J.A., Rivera C., Voipio J., and Kaila K. (2003). Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci. 26, 199–206 [DOI] [PubMed] [Google Scholar]
- 26. Ben-Ari Y. (2014). The GABA excitatory/inhibitory developmental sequence: a personal journey. Neuroscience 279, 187–219 [DOI] [PubMed] [Google Scholar]
- 27. Kahle K.T., Deeb T.Z., Puskarjov M., Silayeva L., Liang B., Kaila K., and Moss S.J. (2013). Modulation of neuronal activity by phosphorylation of the K-Cl cotransporter KCC2. Trends Neurosci. 36, 726–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gagnon M., Bergeron M.J., Lavertu G., Castonguay A., Tripathy S., Bonin R.P., Perez-Sanchez J., Boudreau D., Wang B., Dumas L., Valade I., Bachand K., Jacob-Wagner M., Tardif C., Kianicka I., Isenring P., Attardo G., Coull J.A., and De Koninck Y. (2013). Chloride extrusion enhancers as novel therapeutics for neurological diseases. Nat. Med. 19, 1524–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cortes D.F., Landis M.K., and Ottens A.K. (2012). High-capacity peptide-centric platform to decode the proteomic response to brain injury. Electrophoresis 33, 3712–3719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Morehead M., Bartus R.T., Dean R.L., Miotke J.A., Murphy S., Sall J., and Goldman H. (1994). Histopathologic consequences of moderate concussion in an animal model: correlations with duration of unconsciousness. J. Neurotrauma 11, 657–667 [DOI] [PubMed] [Google Scholar]
- 31. Distler U., Kuharev J., Navarro P., Levin Y., Schild H., and Tenzer S. (2014). Drift time-specific collision energies enable deep-coverage data-independent acquisition proteomics. Nat. Methods 11, 167–170 [DOI] [PubMed] [Google Scholar]
- 32. Saeed A.I., Bhagabati N.K., Braisted J.C., Liang W., Sharov V., Howe E.A., Li J., Thiagarajan M., White J.A., and Quackenbush J. (2006). [9] TM4 microarray software suite. Methods Enzymol. 411, 134–193 [DOI] [PubMed] [Google Scholar]
- 33. Chen J., Bardes E.E., Aronow B.J., and Jegga A.G. (2009). ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 37, W305–W311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ferrini F., Lorenzo L.-E., Godin A.G., Quang M. Le, and De Koninck Y. (2017). Enhancing KCC2 function counteracts morphine-induced hyperalgesia. Sci. Rep. 7, 3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bouet V., and Freret T. (2012). A master key to assess stroke consequences across species: The Adhesive Removal Test., in: Balestrino M. (ed). Advances in the Preclinical Study of Ischemic Stroke. InTech: Rijeka, Croatia, pps. 47–64 [Google Scholar]
- 36. Hamm R.J. (2001). Neurobehavioral assessment of outcome following traumatic brain injury in rats: an evaluation of selected measures. J. Neurotrauma 18, 1207–1216 [DOI] [PubMed] [Google Scholar]
- 37. Hamm R.J., Pike B.R., O'Dell D.M., Lyeth B.G., and Jenkins L.W. (1994). The rotarod test: an evaluation of its effectiveness in assessing motor deficits following traumatic brain injury. J. Neurotrauma 11, 187–196 [DOI] [PubMed] [Google Scholar]
- 38. Cote M.P., Gandhi S., Zambrotta M., and Houle J.D. (2014). Exercise modulates chloride homeostasis after spinal cord injury. J. Neurosci. 34, 8976–8987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gackiere F., and Vinay L. (2014). Serotonergic modulation of post-synaptic inhibition and locomotor alternating pattern in the spinal cord. Front. Neural Circuits 8, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bos R., Sadlaoud K., Boulenguez P., Buttigieg D., Liabeuf S., Brocard C., Haase G., Bras H., and Vinay L. (2013). Activation of 5-HT2A receptors upregulates the function of the neuronal K-Cl cotransporter KCC2. Proc. Natl. Acad. Sci. U. S. A. 110, 348–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hubner C.A., Stein V., Hermans-Borgmeyer I., Meyer T., Ballanyi K., and Jentsch T.J. (2001). Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron 30, 515–524 [DOI] [PubMed] [Google Scholar]
- 42. Friedel P., Ludwig A., Pellegrino C., Agez M., Jawhari A., Rivera C., and Medina I. (2017). A novel view on the role of intracellular tails in surface delivery of the potassium-chloride cotransporter KCC2. eNeuro 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hamidi S., and Avoli M. (2015). KCC2 function modulates in vitro ictogenesis. Neurobiol. Dis. 79, 51–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Evans T.M., Van Remmen H., Purkar A., Mahesula S., Gelfond J.A.L., Sabia M., Qi W., Lin A.L., Jaramillo C.A., and Haskins W.E. (2014). Microwave and magnetic (M2) proteomics of a mouse model of mild traumatic brain injury. Transl. Proteomics 3, 10–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bales J.W., Ma X., Yan H.Q., Jenkins L.W., and Dixon C.E. (2010). Expression of protein phosphatase 2B (calcineurin) subunit A isoforms in rat hippocampus after traumatic brain injury. J. Neurotrauma 27, 109–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wu P., Zhao Y., Haidacher S.J., Wang E., Parsley M.O., Gao J., Sadygov R.G., Starkey J.M., Luxon B.A., Spratt H., Dewitt D.S., Prough D.S., and Denner L. (2013). Detection of structural and metabolic changes in traumatically injured hippocampus by quantitative differential proteomics. J. Neurotrauma 30, 775–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ottens A.K., Kobeissy F.H., Fuller B.F., Liu M.C., Oli M.W., Hayes R.L., and Wang K.K.W. (2007). Novel neuroproteomic approaches to studying traumatic brain injury. Prog. Brain Res. 161, 401–418 [DOI] [PubMed] [Google Scholar]
- 48. Souza G.H., Guest P.C., and Martins-de-Souza D. (2017). LC-MSE, multiplex MS/MS, ion mobility, and label-free quantitation in clinical proteomics. Methods Mol. Biol. 1546, 57–73 [DOI] [PubMed] [Google Scholar]
- 49. Lighthall J.W. (1988). Controlled cortical impact: a new experimental brain injury model. J. Neurotrauma 5, 1–15 [DOI] [PubMed] [Google Scholar]
- 50. Bonislawski D.P., Schwarzbach E.P., and Cohen A.S. (2007). Brain injury impairs dentate gyrus inhibitory efficacy. Neurobiol. Dis. 25, 163–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wu H., Shao A., Zhao M., Chen S., Yu J., Zhou J., Liang F., Shi L., Dixon B.J., Wang Z., Ling C., Hong Y., and Zhang J. (2016). Melatonin attenuates neuronal apoptosis through up-regulation of K + -Cl − cotransporter KCC2 expression following traumatic brain injury in rats. J. Pineal Res. 61, 241–250 [DOI] [PubMed] [Google Scholar]
- 52. Kanaka C., Ohno K., Okabe A., Kuriyama K., Itoh T., Fukuda A., and Sato K. (2001). The differential expression patterns of messenger RNAs encoding K-Cl cotransporters (KKC1,2) and Na-K-2C1cotransporter (NKCC1) in the rat nervous system. Neuroscience 104, 933–946 [DOI] [PubMed] [Google Scholar]
- 53. Mercado A., Mount D.B., and Gamba G. (2004). Electroneutral cation-chloride cotransporters in the central nervous system. Neurochem. Res. 29, 17–25 [DOI] [PubMed] [Google Scholar]
- 54. Newgard C.D., Meier E.N., Bulger E.M., Buick J., Sheehan K., Lin S., Minei J.P., Barnes-Mackey R.A., and Brasel K. (2015). Revisiting the “Golden Hour”: an evaluation of out-of-hospital time in shock and traumatic brain injury. Ann. Emerg. Med. 66, 30-41.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.