

Abstract
Background
Nonobstructive azoospermia (NOA) is one of the most severe forms of male infertility because of impaired spermatogenesis with the absence of spermatozoa in the ejaculate. The causes of this disease can be partly attributed to genetic factors. Some common structural variants and single nucleotide polymorphisms (SNPs) were reported to be associated with NOA. However, the underlying etiology and genetic mechanism(s) remain largely unclear. The aim of this study was to investigate the associated mutations of spermatogenic genes in Chinese infertile men with NOA.
Methods
The entire coding region of 25 genes associated with spermatogenesis was sequenced from 200 infertile men with NOA. Screening was carried out using the targeted exome sequencing to identify genetic variations and SNPs of the entire coding region of these genes.
Results
After the targeted exome sequencing data were filtered through several currently existing variation databases, a series of variations were found. In this paper, we report one novel stopgain variation c.G992A (p.W331X) in the exon 4 of TACR3 gene. The variant was heterozygous and categorized as pathogenic.
Conclusion
In conclusion, our study revealed a novel stopgain mutation c.G992A (p.W331X) in TACR3 which expanded the mutation spectrum of TACR3 in Chinese NOA infertile men and advanced our understanding of the genetic susceptibility to NOA.
Keywords: male infertility, mutation, next‐generation sequencing, NOA, TACR3
1. INTRODUCTION
Infertility affects approximately 15% of couples at childbearing age worldwide with equivalent male and female factor.1 Male infertility accounts for almost half of all infertility cases.2 Nonobstructive azoospermia (NOA) is one of the most severe forms of male infertility because of impaired spermatogenesis with the absence of spermatozoa in the ejaculate.1 It occurs in approximately 1% of all adult men.3 The causes of this disease can be partly attribute to genetic factors.4 A series of studies have reported that genetic factors including karyotype abnormalities, Y‐chromosome microdeletions, single‐gene or polygenic defects including structural variants or SNPs in multiple biological pathways were involved in the development of NOA.4, 5 However, the underlying etiology and genetic mechanism(s) remain largely unclear.
In this study, we aimed to discover and investigate genetic mutations of a few spermatogenic genes in a population of infertile men with NOA. Then we presented a novel stopgain mutation of TACR3 gene in a patient with NOA which next‐generation sequencing (NGS) indicated.
TACR3 gene encodes the receptor for the tachykinin neurokinin 3(NKR or known as NK3R), which is the endogenous receptor for neurokinin B (NKB), which is encoded by TAC3.11 The receptors belonging to this family are characterized by interactions with G proteins and seven hydrophobic transmembrane regions. TACR3 is expressed in uterus, placenta, skeletal muscle, liver, lung, intestinal tract, and widely expressed in the central nervous system, including hypothalamic nuclei involved in regulating GnRH release.12, 13
In the recent years, missense loss‐of‐function mutations inTAC3 and TACR3 have been reported in cases with nonsyndromic normosmic congenital hypogonadotropic hypogonadism (nCHH),14 the disease which result in azoospermia at the pre‐testicular (NOA) level.15 TACR3 was considered to be one of the first two genes to be screened in a clinical setting for equivocal cases such as constitutional delay in puberty vs idiopathic HH.16 NKB action via the NK3R plays a fundamental role in the physiology of human gonadotrope axis and this pathway is necessary for the central neuroendocrine control of human reproduction.14, 17, 18
2. MATERIALS AND METHODS
This study was approved by the Ethics Committee of the First Hospital of Jilin University. All the participants were Han Chinese. Written informed consent was obtained after the nature of the procedures had been fully explained prior to the study. We recruited patients who consulted for sterility and diagnosed as nonobstructive azoospermia. By performing the comprehensive examinations including a detailed medical history, physical examination, chromosome analysis, Y‐chromosome microdeletions, semen analysis, and hormone profiles, these patients were diagnosed as having nonobstructive azoospermia. If patients were detected with Y‐chromosomal microdeletion, they were excluded by the study. Finally a total of 200 patients were recruited. They were subjected to sequencing analysis of the spermatogenesis associated genes.
Mutation screening of genes was performed by targeted exome sequencing. Genomic DNA was isolated from blood lymphocyte samples and subjected to exome capture using the in house Targeted genes Panel (Peking Medriv Academy of Genetics and Reproduction, Peking) followed by next‐generation sequencing on the Illumina MiSeq sequencing platform including 25 spermatogenesis associated genes. Capture probes were prepared based on these spermatogenesis associated genes. Sequence reads were mapped to the human reference genome assembly (NCBI build 37/hg19) using the Burrow‐Wheeler Aligner (BWA version 0.7.12). Duplicated reads from library and PCR preparation were removed with Picard tools. After alignment to the human reference genome (GRCh37/hg19), most likely nondeleterious variants were filtered as previously described.19 Variants with minor allele frequencies greater than 1% in the databases including dbSNP, 1000 Genomes Project, Exome Aggregation Consortium, Exome Variant Server, were excluded because they were unlikely to be deleterious. Intronic variants and synonymous variants that were located within intronic regions, regulatory regions and nonregulatory intergenic regions were excluded. Nonsynonymous variants and splice site variants were remained. Pathogenicity of nonsynonymous variants was assessed using SIFT (https://sift.jcvi.org/) and PolyPhen2 (https://genetics.bwh.harvard.edu/pph2/). Pathogenicity of a splicing site mutation was assessed using Mutation Taster (https://www.mutationtaster.org/) and Human Splicing Finder (https://umd.be/HSF3/). The variant classification was determined followed the American College of Medical Genetics and Genomics (ACMG) standards and guidelines.20
The suspected pathogenic variant was confirmed by conventional PCR and Sanger sequencing (ABI 3730XL, BGI Genomics, Beijing Genomics Institute‐Shenzhen, Shenzhen). Primers for the detected potential disease‐causing mutation p.W331X in TACR3 were 5′‐ TTTGTCCGTGTTTGAGT‐3′ (forward) and 5′‐ TTGGAGGAAGAAGTTGG‐3′ (reverse).
Multiple protein alignments were performed with BLASTP 2.8.0+ (blast.ncbi.nlm.nih.gov/Blast.cgi). The amino acid sequences of the TACR3 in humans, Orycteropus afer afer, Dasypus novemcinctus, Nannospalax galili, Acinonyx jubatus, Mus musculus, Bos Taurus, Myotis davidii, Propithecus coquereli, Felis catus, and Camelus ferus was determined. The identification numbers of TACR3 protein were as follows: Human, NP_001050.1; Orycteropus afer afer, XP_007957420.1; Dasypus novemcinctus, XP_012378583.1; Nannospalax galili, XP_008824373.1; Acinonyx jubatus, XP_014938373.1; Mus musculus, BAC32723.1; Bos Taurus, CAD67556.1; Myotis davidii, ELK33228.1; Propithecus coquereli, XP_012510544.1; Felis catus, XP_003985169.2; Camelus ferus, XP_014419730.1.
3. RESULTS
After targeted exome sequencing was performed in 200 individuals with nonobstructive azoospermia and evaluated for mutations in the 25 spermatogenesis associated genes, we examined whether TACR3 genetic defects were associated with nonobstructive azoospermia. As a result, the present study discovered a novel stopgain mutation c.G992A (p.W331X) in a patient, in whom no variations were found in other 24 spermatogenesis associated genes. The novel mutation, which was located in the exon 4 of TACR3, was heterozygous and categorized as pathogenic. We performed PCR and Sanger sequencing on this patient and confirmed the heterozygous stopgain mutation in the patient but not in the control (Figure 1A). Alignment of the amino acid sequence of TACR3 to its orthologs in 10 different species demonstrated that the c.G992A (p.W331X) variant affected a highly conserved amino acid, as shown in Figure 1B. Scrotal color Doppler ultrasonography of this patient revealed that his testicular volume was 10.9 mL left and 11 mL right, with a homogeneous echotexture and wide hypoechogenicity. No solid or cystic lesions were observed. The clinical and hormone data of this patient were summarized in Table 1.
Figure 1.
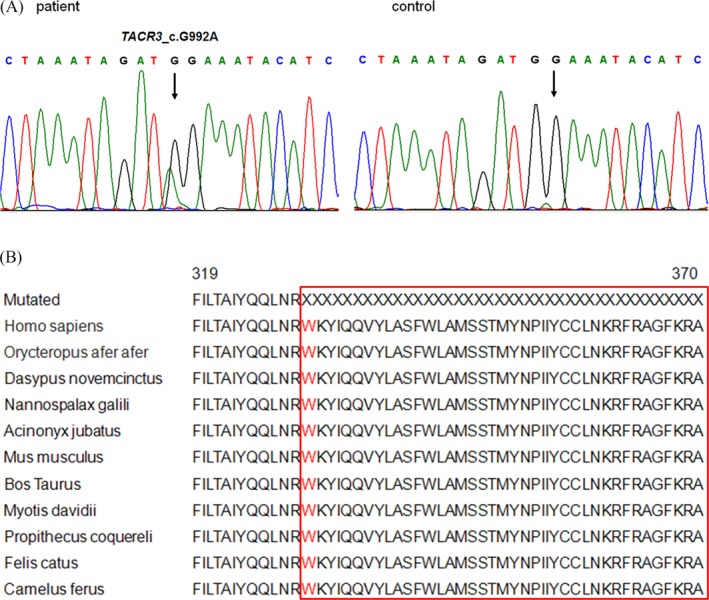
A, The TACR3 C.G992A mutation in patient compared to the control was confirmed by Sanger sequencing. The position is indicated by an arrow. B, Multiple‐sequence alignment in TACR3 from different species reveals that codon 331, where the mutation(P.W167X) occurred, is highly conserved (highlighted in red box)
Table 1.
Clinical and hormone profile of patient with nonobstructive azoospermia with novel TACR3 stopgain mutation
| Sample ID | Age, years | FSH, mIU/mla | LH, mIU/mlb | Estradiol, pg/mlc | PRL, uIU/mld | Testosterone, nmol/le | Inhibin B, pg/mlf |
|---|---|---|---|---|---|---|---|
| 2017A114 | 42 | 21.9 | 7.3 | 23.6 | 565 | 9.4 | 16.13 |
FSH, follicle stimulating hormone; LH, luteinizing hormone; PRL, prolactin.
Normal ranges are.
1.5‐12.4 mIU/mL.
1.7‐8.6 mIU/mL.
25.8‐60.7 pg/mL.
86.0‐324.0 uIU/mL.
9.90‐27.80 pg/mL.
≥80 pg/mL.
4. DISCUSSION
Congenital hypogonadotropic hypogonadism (CHH), which result in azoospermia at the pre‐testicular (NOA) level,15 has classically been categorized as a monogenic disorder in the past few years.21 Lots of genes have been reported to be related with CHH, such as GNRHR, KISS1, KISS1R, FSHB, DAX1, HESX‐1, LHX3, PROP‐1, SOX, POLR3A, POLR3B, PNPLA6.16 Missense loss‐of‐function mutations in TAC3 and TACR3 have also been reported in cases with nonsyndromic normosmic congenital hypogonadotropic hypogonadism (nCHH) from different families and population.14 Although mutations in CHH gene identified so far account for <10% of cases,21 TACR3 was considered to be one of the first two genes to be screened in a clinical setting for equivocal cases such as constitutional delay in puberty versus CHH.16
Hypogonadotropic hypogonadism (HH) is characterized by low sex steroid and low gonadotropin levels resulting from a defect in the normal pulsatile secretion pattern of GnRH from the hypothalamus.22 TACR3 gene encodes the receptor for the tachykinin neurokinin 3(NKR or known as NK3R), which is a member of the rhodopsin family of G‐protein‐coupled receptors.11 In rodents the receptor is expressed in GnRH neurons, which suggests it has a possible role in the regulation of GnRH secretion.11, 18 Topaloglu et al first confirmed the key role of loss‐of‐function mutations in TACR3 (encoding NK3R) and TAC3 (encoding NKB) in the pathogenesis of GnRH deficiency.18 Other investigators have also reported additional loss‐of‐function mutations in the genes in the cases with GnRH deficiency subsequently, which confirmed the two genes’ critical role in normal reproductive development and function.13, 14, 17, 23, 24 These facts highlight the importance of TAC3 and TACR3 in reproduction. However, the HH disease phenotype does not occur in the patient with the novel TACR3 stopgain mutation c.G992A (p.W331X) in this study. We hypothesized that the less severe affected phenotype was due to the fact that the stopgain mutation was heterozygous. The patients with heterozygous nonsense mutations in TACR3 might be have less severe phenotypes than those bearing homozygous mutations. This hypothesis was also suggested by Gianetti et al.23However, it was also reported that both dominant and recessive forms of CHH transmission were present in patients with TACR3 mutations and patients with heterozygous mutations were clinically similar to those with homozygous mutations.12 But most researches suggested that TACR3 was autosomal recessive inheritance, offsprings with heterozygous mutations were not affected.13, 17, 18, 21, 25
The pathogenicity of the variant may be due to the dominant negative effect. Noel et al reported the mechanisms of another heterozygous mutation in TACR3 disrupting NK3R function in GnRH‐deficient patients. This work found that R295S NK3R heterozygous mutation could interfere with dissociation of G proteins and IP signaling from wild‐type NK3R, indicative of dominant negative effects.24 This locus is R295S, which is close to W331X.
Our report has some limitations. We were not able to perform any functional study in order to demonstrate the pathogenicity of the variant. We will demonstrate the pathogenicity of the variant by proving the dominant negative effect of the TACR3 mutation p.W331X according to the method described in Reference [24] in the future study.
Although we have not yet made a definite inference about the consequences of the stopgain mutation c.G992A (p.W331X) in TACR3, what is clear is that the mutation results in the loss of residues after 331st amino acids and the truncation of C terminal peptide. Therefore, the novel stopgain mutation was likely to affect the function of NK3R protein. In a word, our study revealed a novel stopgain mutation c.G992A (p.W331X) in TACR3 which expanded the mutation spectrum of TACR3 in Chinese NOA infertile men and advanced our understanding of the genetic susceptibility to NOA.
AUTHORS' CONTRIBUTIONS
Conceived and designed the experiments: Ruizhi Liu, Dongfeng Geng. Performed the experiments: Xiao Yang, Ruixue Wang, Shu Deng. Analyzed the data: Leilei Li, Xiaonan Hu, Yuting Jiang. Wrote the paper: Dongfeng Geng, Xiao Yang.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This study was approved by the Ethics Committee of the First Hospital of Jilin University. All the participants were Han Chinese. Written informed consent was obtained after the nature of the procedures had been fully explained prior to the study.
ACKNOWLEDGMENTS
The authors alone are responsible for the content and the writing of the paper. We are sincerely grateful to all subjects who participated in this study.
Geng D, Yang X, Wang R, et al. A novel stopgain mutation c.G992A (p.W331X) in TACR3 gene was identified in nonobstructive azoospermia by targeted next‐generation sequencing. J Clin Lab Anal. 2019;33:e22700 10.1002/jcla.22700
Funding information
This study was supported by the National Natural Science Foundation of China (Grant No. 81471515).
REFERENCES
- 1. Jin Q, Pan H, Wang B, et al. The PGAM4 gene in non‐obstructive azoospermia. Syst Biol Reprod Med. 2013;59(4):179‐183. [DOI] [PubMed] [Google Scholar]
- 2. Lu C, Xu M, Wang R, et al. Pathogenic variants screening in five non‐obstructive azoospermia‐associated genes. Mol Hum Reprod. 2014;20(2):178‐183. [DOI] [PubMed] [Google Scholar]
- 3. Maduro MR, Lamb DJ. Understanding new genetics of male infertility. J Urol. 2002;168(5):2197‐2205. [DOI] [PubMed] [Google Scholar]
- 4. Hu Z, Li Z, Yu J, et al. Association analysis identifies new risk loci for non‐obstructive azoospermia in Chinese men. Nat Commun. 2014;5:3857. [DOI] [PubMed] [Google Scholar]
- 5. Zhang W, Yang Y, Su D, Ma Y, Zhang S. Absence of the H2AX mutations in idiopathic infertile men with spermatogenic impairment. Syst Biol Reprod Med. 2008;54(2):93‐95. [DOI] [PubMed] [Google Scholar]
- 6. Ferlin A, Tessari A, Ganz F, et al. Association of partial AZFc region deletions with spermatogenic impairment and male infertility. J Med Genet. 2005;42(3):209‐213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dohle GR, Halley DJ, Van Hemel JO, et al. Genetic risk factors in infertile men with severe oligozoospermia and azoospermia. Hum Reprod. 2002;17(1):13‐16. [DOI] [PubMed] [Google Scholar]
- 8. Ruan J, He XJ, Du WD, et al. Genetic variants in TEX15 gene conferred susceptibility to spermatogenic failure in the Chinese Han population. Reprod Sci. 2012;19(11):1190‐1196. [DOI] [PubMed] [Google Scholar]
- 9. Massart A, Lissens W, Tournaye H, Stouffs K. Genetic causes of spermatogenic failure. Asian J Androl. 2012;14(1):40‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zc A, Yang Y, Zhang SZ, Li N, Zhang W. Single nucleotide polymorphism C677T in the methylenetetrahydrofolate reductase gene might be a genetic risk factor for infertility for Chinese men with azoospermia or severe oligozoospermia. Asian J Androl. 2007;9(1):57‐62. [DOI] [PubMed] [Google Scholar]
- 11. Bianco SD, Kaiser UB. The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism. Nat Rev Endocrinol. 2009;5(10):569‐576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Root AW. Reversible isolated hypogonadotropic hypogonadism due to mutations in the neurokinin B regulation of gonadotropin‐releasing hormone release. J Clin Endocrinol Metab. 2010;95(6):2625‐2629. [DOI] [PubMed] [Google Scholar]
- 13. Guran T, Tolhurst G, Bereket A, et al. Hypogonadotropic hypogonadism due to a novel missense mutation in the first extracellular loop of the neurokinin B receptor. J Clin Endocrinol Metab. 2009;94(10):3633‐3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Francou B, Bouligand J, Voican A, et al. Normosmic congenital hypogonadotropic hypogonadism due to TAC3/TACR3 mutations: characterization of neuroendocrine phenotypes and novel mutations. PLoS One. 2011;6(10):e25614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hamada AJ, Esteves SC, Agarwal A. A comprehensive review of genetics and genetic testing in azoospermia. Clinics (Sao Paulo). 2013;68(Suppl 1):39‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Topaloglu AK, Kotan LD. Genetics of Hypogonadotropic Hypogonadism. Endocr Dev. 2016;29:36‐49. [DOI] [PubMed] [Google Scholar]
- 17. Young J, Bouligand J, Francou B, et al. TAC3 and TACR3 defects cause hypothalamic congenital hypogonadotropic hypogonadism in humans. J Clin Endocrinol Metab. 2010;95(5):2287‐2295. [DOI] [PubMed] [Google Scholar]
- 18. Topaloglu AK, Reimann F, Guclu M, et al. TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for Neurokinin B in the central control of reproduction. Nat Genet. 2009;41(3):354‐358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim YM, Lee YJ, Park JH, et al. High diagnostic yield of clinically unidentifiable syndromic growth disorders by targeted exome sequencing. Clin Genet. 2017;92(6):594‐605. [DOI] [PubMed] [Google Scholar]
- 20. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Boehm U, Bouloux PM, Dattani MT, et al. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism–pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2015;11(9):547‐564. [DOI] [PubMed] [Google Scholar]
- 22. Tüttelmann F, Simoni M. Current recommendations for genetic testing in male infertility. Eur Urol Rev. 2008;3(2):88‐92. [Google Scholar]
- 23. Gianetti E, Tusset C, Noel SD, et al. TAC3/TACR3 mutations reveal preferential activation of gonadotropin‐releasing hormone release by neurokinin B in neonatal life followed by reversal in adulthood. J Clin Endocrinol Metab. 2010;95(6):2857‐2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Noel SD, Abreu AP, Xu S, et al. TACR3 mutations disrupt NK3R function through distinct mechanisms in GnRH‐deficient patients. FASEB J. 2014;28(4):1924‐1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aoyama K, Mizuno H, Tanaka T, et al. Molecular genetic and clinical delineation of 22 patients with congenital hypogonadotropic hypogonadism. J Pediatr Endocrinol Metab. 2017;30(10):1111‐1118. [DOI] [PubMed] [Google Scholar]