

Abstract
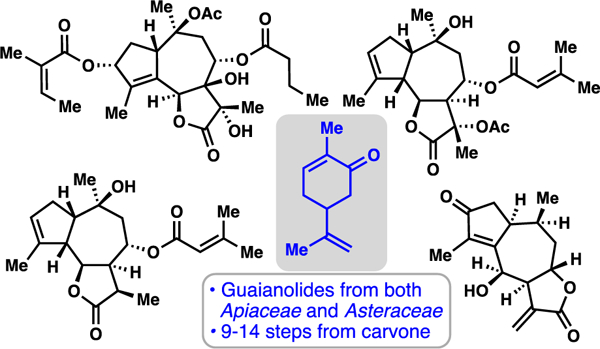
With hundreds of unique members isolated to date, guaianolide lactones represent a particularly prolific class of terpene natural products. Given their extensive documented therapeutic properties and fascinating chemical structures, these metabolites have captivated the synthetic chemistry community for many decades. As a result of divergent biosynthetic pathways, which produce a wide array of stereochemical and oxidative permutations, a unifying synthetic pathway to this broad family of natural products is challenging. Herein we document the evolution of a chiral pool-based synthetic program aimed at accessing an assortment of guaianolides, particularly those from the plant family Apiaceae as well as Asteraceae, members of which possess distinct chemical substructures and necessitate deviating synthetic platforms. An initial route employing the linear monoterpene linalool generated a lower oxidation state guaianolide, but was not compatible with the majority of family members. A double allylation disconnection using a carvone-derived fragment was then developed to access first an Asteraceae-type guaianolide and then various Apiaceae congeners. Finally, using these findings in conjunction with a tandem polyoxygenation cascade, we developed a pathway to highly-oxygenated nortrilobolide. A variety of interesting observations in metal-mediated aldehyde allylation and alkene polyoxygenation are reported and discussed.
Graphical Abstract
Introduction
Humans have experimented with plant-derived sesquiterpenes as medicine, poison, flavoring, and fragrance for several millenia.1 Amongst such natural products, the large family of guaianolide lactones represents a particularly diverse and important source of bioactive metabolites.2 Notably, such compounds have been heavily investigated for the treatment of inflammation and cancer among other ailments; certain derivatives have even advanced into human clinical trials.1–3
Although detailed biosynthetic blueprints are missing for most guaianolide members, it is generally assumed that an oxidized germacrene-type macrocycle (see 1) serves as the precursor to guaianolides (Figure 1).4 Of critical importance is the stereochemistry surrounding the lactone ring, particularly the configuration of the proton labelled Ha in orange. In the plant family Asteraceae, guaianolides produced (see 2) typically have a β-configured proton at this center (guaianolide C6 position) with the 5,7-fused lactone being trans configured and thus costunolide (1 where R = H and Ha is β) appears to be a biosynthetic precursor. Notably terpene cyclases in this family forge products with cis stereochemistry at the 5,7-fused carbocycle ring junction with α-configurations of the protons at the newly-formed C1 and C5 positions. Also, present in many members is a Δ11,13 alkene, thus making many Asteraceae guaianolides potentially protein reactive via hetero-Michael reactions with reactive cysteine residues,5,6 a theme that presumably underpins much of their significant documented bioactivity.1–2
Figure 1.

General overview of guaianolide lactone biosynthesis in Asteraceae and Apiaceae.
Guaianolides obtained from plants of the Apiaceae family (aka slovanolides, see 3) generally feature the opposite configuration at C6 (see Ha) and possess cis stereochemistry at the 5,7-fused lactone ring junction. Upon enzymatic cyclization of 1, a cis-fused 5,7 ring system is also made, but with the opposite configurations at C1 and C5 relative to Asteraceae guaianolides. These differences can have important ramifications when using chiral pool starting materials available in only one enantiomeric series (vide infra). Apiaceae guaianolides typically do not possess Δ11,13 unsaturation and thus are not likely covalent modifiers of cysteine residues in proteins in most cases. Finally, 2 and 3 are both 6,12-guaianolides owing to the position of the lactone, but when a free hydroxyl group is present at C8, an isomeric 8,12-guaianolide (see 4) can arise; this process has been noted in a variety of saponification studies of guaianolides and is presumably more favorable for the Apiaceae-derived systems.
The stereochemical divergence of these pathways combined with extensive late-stage C-H oxidation leads to a truly incredible array of highly complex sesquiterpenes (Figures 2 and 3). Figure 2 highlights a small sampling of Asteraceae members (5–11) showcasing various oxidation patterns. Notable is the 8,12-guaianolide mikanokryptin (8) which features unusual, inverted C6 stereochemistry. Absinthin (10) and ainsliatrimer (11) highlight the occurrence of higher order Asteraceae members which are often formed via pericyclic reactions amenable to biomimetic synthesis.6c,7
Figure 2.

Select guaianolide natural products from the plant family Asteraceae.
Figure 3.

Select, complex guaianolide lactones from Apiaceae (slovanolides).
The complexity found in slovanolides can exceed that of the Asteraceae guaianolides from a stereochemical and oxidative perspective (Figure 3).2b In analogy to Asteraceae members, the simplest congeners (see 12–17) do not possess oxidation at the C8 position, but can range from lowly oxidized sinodielide A (12) to the more heavily oxidized ferulide (17). The majority of Apiaceae members, however, do possess oxidation at C8 although many are unoxidized at the C11 position (see 18–22). Of note, ferulidin (22) is an example of an 8,12-guaianolide from Apiaceae. The most highly oxidized guaianolides possess hydroxyl groups at C8, C11, and often elsewhere (23–30). From an oxidation state perspective, the ferupennin (see 25–27) and trilobolide/thapsigargin (see 28–30) members represent the pinnacle of complexity in this series. Unlike most guaianolides, these structures can be found with oxidation at the C5, C7, C14, and C15 positions.
Given the structural complexity and medicinal properties of guaianolide lactones it is not surprising that such natural products have served as popular synthetic targets for decades resulting in over 50 syntheses.8,9 The stereochemical and oxidative diversity found in the guaianolides makes a unifying synthetic strategy challenging. Most synthetic work has focused on the syntheses of a small subset of the Asteraceae guaianolides, while the trilobolide/thapsigargin subtypes represent the only Apiaceae members chemically synthesized. Herein we detail the evolution of a synthetic program aimed at synthesizing multiple, diverse guaianolide members, work that has resulted in syntheses of mikanokryptin (8), sinodielide A (12), slovanolide 18, montanolide (23), and nortrilobolide (29).
Results and Discussion
Synthetic Planning.
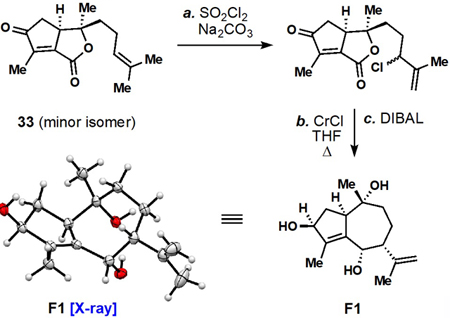
The chiral pool of terpenes has been featured prominently in past syntheses of guaianolides,9 and given our laboratories interest in this area,10 we also began our investigations there. In analyzing past chiral pool approaches to guaianolide skeletons (see 31) two major strategies have been employed (Figure 4). In the first, the abundant sesquiterpene lactone (–)-α-santonin has been utilized,11 often employing classic dienone photochemical rearrangements.12 Remarkably, over 30 unique guaianolides have been assembled from this chiral pool building block.11 This work is best applied to Asteraceae members owing to the absolute configuration of natural (–)-α-santonin. The second major disconnection involves the use of a ring-contracted cyclic monoterpene as a surrogate for the guaianolide cyclopentane ring system.9 The availability of many cyclic monoterpenes in either enantiomeric form makes this route potentially applicable to a wide array of members. In this work, we further investigated this structural disconnection as well as a third route which traces the guaianolide skeleton back to a linear monoterpene building block.
Figure 4.

Chiral pool-based disconnections of the guaianolide skeleton.
Initial Linalool-Based Design: Total Synthesis of Sinodielide A.
The linear monoterpene linalool, which possesses an asymmetric tertiary alcohol stereocenter evocative of the C10 position of many guaianolides, was chosen as a starting point (Scheme 1). We envisioned that the cyclopentane ring system could be constructed rapidly via a Pauson-Khand reaction and the 7-membered ring closed by exploiting the nucleophilic terminal prenyl group.13,14 Thus (–)-linalool was converted into ester 32 via deprotonation and reaction with the mixed anhydride of 2-butynoic acid. This material underwent smooth Pauson-Khand (PK) reaction using dicobalt octacarbonyl resulting in strained bicyclic lactone 33 (65% yield, 5:2 dr) which was rapidly reduced with DIBAL to afford triol 34.15 The major isomer formed in the PK reaction was found to have a cis relationship between the C1 proton and C10 methyl group which is unusual for most guaianolides. Nevertheless, this stereochemical pattern is found in absinthin (10) and thus we viewed this outcome at the time as potentially amenable to the preparation of ent-10 via Diels-Alder dimerization of 35 (See inset, Scheme 1).16 Notably, a past route to 10, which relies on the α-santonin photorearrangement, has to invert this center via a multi-step procedure.7a
Scheme 1.

Total synthesis of sinodielide A (12) from (–)-linalool.
aReagents and conditions: (a) (–)-linalool (1.0 equiv), NaHMDS (2.0 equiv), but-2-ynoic pivalic anhydride (2.5 equiv), THF, −45 °C to 23 °C, 11 h, 65%; (b) Co2(CO)8 (1.1 equiv), NMO (9.5 equiv), DCM, 2 h, 65% (dr = 5:2); (c) DIBAL (4.0 equiv), DCM, −78 ºC to rt, 16 h, 81%; (d) 2,4,6-Colldine (10.0 equiv), TESOTf (5.0 eq.), SO2Cl2 (1.2 eq.), DCM, −78 ºC to 23 °C, 74%; (e) CrO3•2pyr (3.0 equiv), DCM, 0 ºC, 54%; (f) CrCl2 (5.0 equiv), NiCl2 (0.1 equiv), DMF, 60 ºC, 1 h, 57% (dr = 9:1); (g) BH3• THF (1.8 equiv), THF, 0 ºC, then add NaOH/H2O2, 1.5 h, 80% (dr = 5:4); (h) TEMPO (1 equiv), PIDA (10 equiv), DCM, 8 h, 76%; (i) LDA (2 equiv), 1 h, −78 ºC to −40 ºC, then AcOH (5 equiv), TBAF (6 equiv), −78 ºC to 23 °C, 8 h, 98%; (j) IPNBSH (3 equiv), DIAD (3 equiv), PPh3 (3 equiv), THF, 0 ºC, 1 h, then 23 °C, 4 h, then add TFE:H2O (v:v = 1:1), 0 ºC, 14 h, 64%; (k) SOCl2 (5 equiv), pyridine (8 equiv), DCM, −40 ºC to 23 °C, 95%; NaHMDS = sodium 1,1,1-trimethyl-N-(trimethylsilyl)silanaminide, NMO = N-methylmorpholine N-oxide, TESOTf = Triethylsilyl trifluoromethanesulfonate, PIDA = (Diacetoxyiodo)benzene, TEMPO = (2,2,6,6-Tetramethylpiperidin-1-yl)oxyl, LDA = lithium diisopropylamide, TBAF = tetrabutylammonium fluoride, DIAD = Diisopropyl azodicarboxylate, IPNBSH = N-Isopropylidene-N′−2-nitrobenzenesulfonyl hydrazine.
We envisioned closing the seven-membered ring, via Barbier-type coupling of an allylic chloride in analogy to our work on chatancin.17 Thus 34 was converted into aldehyde 37 via one-pot silylation (TESOTf, collidine) and allylic chlorination (SO2Cl2) to give 36 followed by a chemoselective chromium-mediated oxidation. Allylic chloride 37 smoothly participated in an intramolecular NHK-type coupling generating 38 in 57% yield and with good diastereoselectivity. We note that we were unable to directly forge the same corresponding C–C bond from a similar allylic chloride formed from ester 33 despite the apparent strain in the lactone ring.18 Unfortunately, the relative stereochemistry in 38 does not correspond to that found in Asteraceae-type guaianolides, such as 10, but instead has the correct relative configuration for Apiaceae members. We decided therefore to advance this material to lower oxidation state slovanolides featuring Δ1,10 unsaturation (Scheme 1). The hallmark cis-fused 6,12-lactone was assembled through straightforward means involving hydroboration and oxidation giving 39 which could be converted to isomerically pure 40 via deprotonation and kinetic proton quench. Notably the combination of TBAF and AcOH served as a both a proton source as well as desilylating reagent in this reaction. Following one-pot transpositive allylic reduction (DIAD, PPh3, IPNBSH)19,20 tertiary alcohol 41 was generated which could be cleanly dehydrated to give the simple, apoptosis-inducing guaianolide sinodielide A (12).21
While this initial route was successful in accessing a lower oxidation state guaianolide, the scope of natural products obtainable is lacking. Although the formation of a cis-lactone is desirable in many contexts, and not accessible by many guaianolide synthetic strategies, the need to install C-8 oxygenation–which is present in the majority of slovanolides–facilitated a new approach.
A Double Allylation Disconnection: Total Synthesis of Mikanokryptin, an Asteraceae Guaianolide.
Our second-generation retrosynthesis of guaianolides targeted structural motif 42 via a double allylation disconnection of a ten-carbon fragment (see 43) with that of a 5-carbon piece (see 44) (Figure 5). This strategy was expected to offer considerable flexibility as: i) the C-1 stereocenter is available in either enantiomeric form from the chiral pool of cyclic monoterpenes, ii) the Δ10,14 alkene could be reduced or hydrated to afford motifs common to many guaianolides, and iii) the C6–8 stereocenters could potentially be controlled by various allylation conditions.
Figure 5.

Revised double allylation retrosynthesis
We began our investigations by preparing aldehyde 47, the requisite ten-carbon fragment (Scheme 2).9n A three-step route to this piece was developed from carvone involving: i) one-pot allylic chlorination/Luche reduction, ii) hydroxyl silylation, and iii) one-pot chemoselective ozonolysis/aldol condensation. This chemistry was robust and allowed for multi-gram procurement of aldehyde 47. We then merged 47 with allylic bromide 48 under indium-mediated allylation conditions generating 49 (67%, 2:1 dr at C-6) on multigram scale. Notably, the inclusion of one equivalent of water both aided in the diastereoselectivity of this transformation as well as reduced formation of the corresponding 6,12-lactone framework. The stereochemical relationship between the C6 and C7 centers in 49 meant that we would inevitably again (as in the case of 38) form a cis-fused 6,12-lactone system upon ring closure, however, the relationship relative to C1 is incorrect for Apiaceae members. This finding though presented a unique opportunity to access mikanokryptin (8), which, while a member of the Asteraceae-type guaianolides, possesses inverted C6 stereochemistry. Notably, such members cannot be prepared in a straightforward manner from α-santonin.
Scheme 2.

Nine-step total synthesis of the unusual Asteraceae member mikanokryptin (8).a
aReagents and conditions: (a) SO2Cl2 (1.2 equiv), Na2CO3 (3.0 equiv), DCM, 0 ºC, 2 h, then add CeCl3•7H2O (1.1 equiv), NaBH4 (3.0 equiv), MeOH, 0 ºC, 1 h, 78%; (b) TBDPSCl (1.2 equiv), imidazole (3.0 equiv), DMAP (0.05 equiv), DMF, 23 °C, 8 h, 91%; (c) O3, pyridine (0.3 equiv), DCM, −78 ºC, 20–40 min, then add DMS (2.0 equiv), 25 °C, 8 h, then add piperidine (0.15 equiv), AcOH (0.2 equiv), 40 ºC, 16 h, 42%; (d) In0 (1.5 equiv), 48 (1.2 equiv), H2O (1 equiv), DMF, 23 °C, 1 h, 67% (dr = 2:1); (e) TESOTf (4 equiv), 2,4,6-collidine (6 equiv), DCM, 0 ºC, 24 h, 78%; (f) SnCl2 (4.5 equiv), NaI (9 equiv), DMF, 60 ºC, 12 h, 90%; (g) NaOMe (0.1 equiv), MeOH, 16 h, then add AcOH (0.1 equiv), PtO2 (0.1 equiv), H2 (1 atm), 6 h, 96%; (h) TBAF (3.0 equiv), THF, 23 °C, 60%; (i) MnO2 (30.0 equiv), DCM, 23 °C, 16 h, 97%; TBDPSCl = tert-butyldiphenylsilyl chloride, DMS = dimethylsulfide, DBU = 1,8-Diazabicyclo[5.4.0]undec-7-ene.
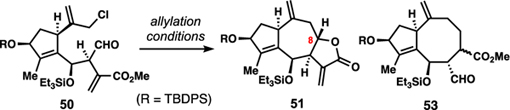
With large quantities of 49 we proceeded toward mikanokryptin (8) (Scheme 2). Mild deacetalization with concomitant silylation according to Fujioka and Kita’s protocol (TESOTf, collidine)22 generated 50 and set the stage for the second key intramolecular allylation. Treating 50 with SnCl2 in the presence of NaI lead to a remarkably clean and diastereoselective allylation reaction presumably via an in-situ generated allylic iodide. This reaction provided multi-gram batches of 51 which additionally possesses the desired 8,12-guaianolide lactone system.
The efficient conversion of 50 to 51 was the result of extensive optimization efforts involving a variety of known aldehyde/allyl halide coupling procedures (Table 1). Initial investigations involving the Nozaki–Hiyama–Kishi (NHK) coupling afforded the desired product (51), but with diminished yield (10%) and diastereoselectivity (2:1 dr at C8) (Entry 1). Interestingly, the major product produced under these conditions was cyclooctane 53, formed as a mixture of diastereomers. Competition between the formation of 7- and 8-membered ring–the latter of which we attribute to a radical process–was also observed in samarium iodide-mediated (entry 2) and zinc-mediated (entry 3) conditions.23 Indium-mediated allylation conditions of an in-situ generated allylic iodide were selective for formation of 51, but inefficient overall (entry 4). Finally, tin(II) chloride proved to be a superior reductant in this system, giving high yields of 51 even on multi-gram scales.
Table 1.
Intramolecular allylation of aldehyde 50: Key findings.
 | |||
|---|---|---|---|
| Entry | Conditions | yield of 51 | yield of 53 |
| 1 | CrCl2, cat. NiCl2, DMF, 60 °C | 10% (2:1 dr) | 17% |
| 2 | Nal, Sml2 THF/HMPA, −78 °C | 27% (2:1 dr) | 17% |
| 3 | Nal, Zn0, aq. NH4Cl, THF, rt | – | 51% |
| 4 | In0, Nal, DMF, 60 °C | 13% | – |
| 5 | Nal, SnCl2, DMF, 60 °Cb | 90% | – |
reactions performed on a 30 mg scale, isolated yields are shown.
yield on 7-gram scale
To complete the synthesis of mikanokryptin (8), a chemoselective reduction of the Δ10,14 alkene was required. While we were unable to perform such a reaction directly,9n the exo-methylene lactone was easily protected as its methanol adduct (cat. NaOMe, MeOH) allowing for clean olefin hydrogenation (cat. PtO2/H2) in the same vessel generating 52. Upon deprotecting 52, we gratifyingly found that TBAF was sufficiently basic to promote the retro conjugate addition reaction of methanol giving a 5:2 mixture of exo-methylene lactone:methanol adduct, both with concomitant deprotection of the secondary hydroxyl group. On larger scales, we elected to utilize DBU as base for the E1cB reaction, which was somewhat higher yielding. Finally, a near quantitative yielding allylic oxidation with manganese dioxide generated mikanokryptin. The robustness of this route allowed for the preparation of substantial quantities of material (>1g) which in turn helped fuel target identification studies of this unexplored cytotoxic natural product.24
Reversing Allylation Stereochemistry for Entry into Complex Slovanolides.
Given our success in forging stereochemical motifs needed for cis-fused 6,12-lactones, the complex slovanolides produced by the Apiaceae plant family (see Figure 3) were logically the next targets using the double allylation strategy. Many of these natural products, particularly those with cis-fused lactones, have remained inaccessible by synthesis, and the most complex members (i.e. trilobolide/thapsigargin-type) have served as grand challenges to the field of total synthesis for nearly two decades.9b,9c,9d,9m,9o,25 In order to gain access to slovanolides such as 18 and montanolide (23), a disparate stereochemical outcome in the initial allylation of aldehyde 47 (or more precisely ent-47 for these targets) was needed (Figure 6). Specifically, the configurations at C6 and C7 (relative to C1) requires inversion compared to the mikanokryptin allylation outcome (i.e ent-47 needs to produce diast-49 and not ent-49). Nucleophilic additions to aldehydes that are quite similar to 47 have been studied,26 and a variety of stereochemical outcomes at C6 have been reported depending on the nucleophile and conditions. We were therefore hopeful that alternative allylation conditions and/or reagents could provide us with the desired configuation.
Figure 6.
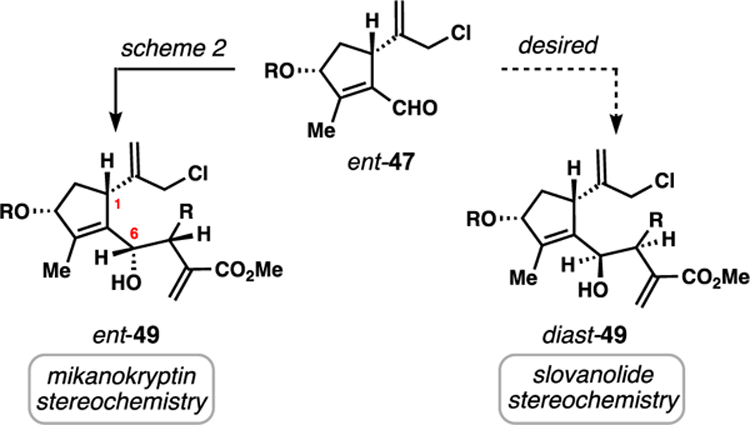
Stereochemical considerations for an allylative synthesis of slovanolides.
It was discovered that ent-47 could be merged with allylic bromide 54 under Zn0-mediated allylation conditions to give lactone 55 in good yield and with the correct C6 configuration in the major diastereomer (Scheme 3A).27 While the diastereoselectivity of this transformation was not high, this reaction was robust and enabled the procurement of gram-quantities of material. Worth noting, indium-mediated allylations employing 54 slightly favored the undesired diastereomer in analogy to the mikanokryptin work.9n The presence of catalytic Zn(II) chloride was necessary to promote cyclization to the requisite lactone. Sodium borohydride in methanol cleanly reduced the reactive exocyclic methylene lactone in nearly quantitative yield giving alcohol 56 after removal of the para-methoxybenzyl group. A Dess-Martin periodinane oxidation of 56 then generated the somewhat sensitive aldehyde 57 thus setting up another key 7-membered ring forming allylation.
Scheme 3.

Total synthesis of slovanolide 18 and montanolide (23).a
aReagents and conditions: (a) ent-47 (1.0 equiv), Zn0 (1.5 equiv), ZnCl2 (0.06 equiv), 54 (1.5 equiv), NMF, 23 °C, 65% (dr = 2:1); (b) NaBH4 (1.5 equiv), MeOH, 23 °C; (c) DDQ (3.0 equiv), pH = 7.5 buffer, DCM, 23 °C, 1 h, 75% (two steps); (d) DMP (1.5 equiv), NaHCO3 (1.5 equiv) DCM, 23 °C, 1 h, 75%; (e) TiCp2Cl2 (1.0 equiv), Zn0 (3.0 equiv), THF, 55 ºC, 2 h, 75% (dr = 10:1) (two steps); (f) 3,3-dimethylacrylic acid (2.0 equiv), DMAP (2.0 equiv), DCC (2.0 equiv) DCM, 23 °C, 84%; (g) 60 (0.2 equiv), PhSiH3 (2.5 equiv), EtOH, 23 °C, 2 h, then add PPh3 (2.0 eq.), 30 min, 50% (dr = 1.9:1); (h) TBAF (16.0 equiv), AcOH (10.0 equiv), 23 °C, 48 h, 79%; (i) IPNBSH (3 equiv), DIAD (3 equiv), PPh3 (3 equiv), THF, 0 ºC, 1 h, then add TFE/H2O (v:v = 1:1), 23 °C, 14 h, 39%; (j) NaHMDS (1.1 equiv), Davis’ oxaziridine (1.2 equiv), −78 ºC, 0.5 h, 92%; (k) Ac2O (1.5 equiv), DMAP (0.1 equiv), pyridine (3.0 equiv), 23 °C, 3 h, 99%; (l) 60 (0.2 equiv), PhSiH3 (2.5 equiv), EtOH, 23 °C, 2 h, then add PPh3 (2.0 equiv), 30 mins, 45%; (m) TBAF (16.0 equiv), AcOH (10.0 equiv), 48 h, 23 °C, 77%; (n) IPNBSH (3 equiv), DIAD (3 equiv), PPh3 (3 equiv), THF, 0 ºC, 8 h, then add p-TsOH (0.5 equiv), 55 ºC, 1 h, 42%; PMB = 4-methoxybenzyl, DDQ = 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone, DMP = Dess-Martin periodinane, DCC = N,N’-Dicyclohexylcarbodiimide, DMAP = 4-Dimethylaminopyridine, KHMDS = potassium 1,1,1-trimethyl-N-(trimethylsilyl)silanaminide, DIAD = Diisopropyl azodicarboxylate, IPNBSH = N-Isopropylidene-N′−2-nitrobenzenesulfonyl hydrazine.
A reductive, titanocene-mediated allylation (TiCp2Cl2, Zn0) generated tricycle 58,28 largely as a single isomer and with the correct configuration at the C-8 stereocenter as found in slovanolides. Significant experimentation went into achieving this stereochemical outcome which was crucial as we were unable to invert this center in one-step via Mitsunobu reaction of 8-epi-58 (i.e. 64) with a carboxylic acid side-chain found in slovanolides (Scheme 3B). Furthermore, our previously employed SnII-mediated conditions give exclusively the wrong stereochemical outcome at C8 both with and without exogenous transition metals (see entries 1–4). NHK coupling conditions afforded a small amount of desired 58, but were still dominated by formation of 64 (entry 5). We found that samarium (II) iodide, both with and without additives, was selective in the formation of 58, but ultimately low yielding (entries 6 and 7). Under these conditions chloride 66 was also formed, presumably via a 6-exo-trig ketyl radical cyclization. It is noteworthy that with only simple changes to the reductant employed, a near complete reversal in selectivity could be achieved (entry 1 vs. 8). With the 5,7,5-fused ring system in place, the senecioyl ester was attached via straightforward DCC-coupling giving 59 (Scheme 3A). This material was then subjected to Mukaiyama-type hydration employing cobalt catalyst 60.29,30 In accordance with Evans’ findings on a related scaffold containing a β-configured hydroxyl group at C-7,9o the C10 stereocenter was set correctly during this process, albeit with diminished diastereoselectivity in this system; the topological differences imparted by the configuration of the C-7 stereocenter likely influence the radical coupling event. Finally, ester 61 could be converted into slovanolide 18 via a two-step sequence involving removal of the tert-Butyldiphenylsilyl protecting group (TBAF/HOAc) and one-pot reductive allylic transposition (DIAD, IPNBSH, PPh3).31
Using a slightly modified route, key intermediate 59 could also be advanced to the more heavily oxidized slovanolide montanolide (23).32 Enolate oxidation of 59 with Davis’ oxaziridine followed by acetylation generated 62 in a stereoselective manner and with high yield. A similar endgame of formal radical hydration, deprotection, and reductive allylic transposition, resulted in the synthesis of 23. The routes to 18 and 23 proceeded in only 12- and 14-steps respectively from carvone. Moreover, it is easy to envision expanding this chemistry to include allylic oxidation products 24-27 (Figure 3).
An Oxygen Stitching Strategy for the Synthesis of Heavily Oxidized Apiaceae Members.
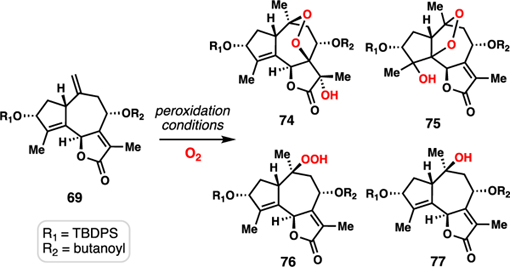
While our studies so far had realized syntheses of a variety of guaianolides, our strategy did not easily lend itself to the preparation of the most heavily oxidized subtypes, namely the trilobolide (28) and thapsigargin (30) members. In particular, the presence of hydroxylation at C7 is troublesome synthetically given the chemistry developed so far (vide supra). Taking note of the relative spatial orientations of the oxygen atoms at C10, C7, and C11, however, we wondered if a polyoxygenation cascade using molecular oxygen could construct this motif in analogy to our work on the antimalarial cardamom peroxide (67) (Figure 7).33 In this scenario, the complexity of trilobolide (29) can be reduced to that of butenolide 69 by way of a metal-catalyzed radical hydroperoxidation cascade.34 If successful, much of the stereochemical complexity of the target could be installed in a single operation.
Figure 7.

Oxygen stitching retrosynthesis of nortrilobolide (29).
Our efforts began by retooling our prior slovanolide synthesis to access butenolide 69 (Scheme 4). Starting from lactone 55, the PMB group was removed with DDQ and the exocyclic olefin isomerized into the lactone ring using catalytic quantities of RuHCl(CO)(PPh3)3 in refluxing DCE.35 Under optimized conditions this step could be telescoped with a mild, buffered TEMPO/bleach oxidation delivering sensitive aldehyde 70. With this material in hand, yet another seven-membered ring-forming allylation was investigated. We found that our previously utilized SnCl2/NaI-mediated conditions were quite effective in this setting, yet unlike in the cases of substrates 50 (Scheme 2) and 57 (Scheme 3), this reaction afforded a near equimolar mixture of alcohol diastereomers at C8 (Scheme 4). The lack of stereochemistry at C7 and C11 may contribute to this outcome. Inspired by reports on the use of exogenous transition-metals in combination with SnCl2 as catalysts for allylation chemistry,36 we found that addition of catalytic quantities of PdCl2 afforded 71 as essentially a single diastereomer.37 Moreover, this chemistry was easily scaled without a depression in yield. While the C8 hydroxyl group was set with an incorrect β-configuration during this process, it underwent facile Mitsunobu inversion with butyric acid to give 69, a result in stark contrast to the reactivity of 64 (vide supra).
Scheme 4.

12-Step formal synthesis of nortrilobolide (29).a
aReagents and conditions: (a) 55 (1.0 equiv), DDQ (3.0 equiv), pH = 7.5 buffer, DCM, 23 °C, 1 h, 94%; (b) RuHCl(CO)(PPh3)3 (0.1 equiv), DCE, 60 ºC, 16 h, then add TEMPO (0.1 equiv), KBr (1.0 equiv), aq. NaOCl, pH = 8.6 buffer, 23°C, 1 h, 86%; (c) SnCl2 (5.0 equiv), PdCl2(PhCN)2 (0.15 equiv), DMF, 60 ºC, 8 h, 95%; (d) butyric acid (3.0 equiv) DIAD (3.0 equiv), 0 ºC to 23 °C, 8 h, 70%. (e) Co(acac)2 (0.2 equiv), Et3SiH (5.0 equiv), O2 (1 atm), EtOH 24 h, 23 °C, then add Zn (3.0 equiv), aq. NH4Cl (5 equiv), 15%; (f) pTSA (1.2 equiv), isopropenyl acetate, 23 °C, 80%; (g) HF/MeCN (1:5 = v/v), 23 °C, 56%; DDQ = 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone [RuH] = RuHCl(CO)(PPh3)3, acac = acetylacetone.
With 69 now secure, we were poised to examine the key polyoxygenation cascade. Subjecting 69 to classic Mukaiyama hydroperoxidation conditions (Co(acac)2, Et3SiH, O2) lead to desired triol 72 after reductive workup in 15% yield and as a single diastereomer. While the yield of this transformation is low, three tertiary alcohols are formed in a single step and the stereochemical outcome at C7 and C11 was remotely controlled by the initial hydroperoxidation at C10 via the strategic peroxide handle. The successful reactivity window for this transformation was exceedingly small – nearly any variation to these conditions led to no desired product (Table 2). Only Co(acac)2-based systems formed any product (entries 1 and 2). The major competing pathways were premature reduction of the peroxy radical intermediate forming alcohol 77, and formation of an unstable peroxide tentatively assigned as 75.38 The prolonged exposure of these reactions to oxygen led to significant oxidative degradation resulting in mass balances of only ~50%. Notably, while manganese and iron-based catalysts afforded none of these products (entries 5 and 6), cobalt catalyst 60 was reasonably efficient in producing peroxide 76, again the result of premature reduction of the radical intermediate (entry 4).39 Finally, acetylation of 72 followed by desilylation with hydrofluoric acid intercepted triol 73, thus marking the completion of a 12-step formal synthesis of 29.9m,9o,40
Table 2.
Catalyst evaluation in the key polyoxygenation Step.
 | |||||
|---|---|---|---|---|---|
| Entry | Conditions | 74 | 75 | 76 | 77 |
| 1 | 25 mol% Co(acac)2, Et3SiH, EtOH | 16% | 10% | – | 19% |
| 2 | 25 mol% Co(acac)2, PhSiH3, i-PrOH/DCM | 13% | 18% | – | 15% |
| 3 | 30 mol% Co(modp)2, Et3SiH,DCE | – | – | – | – |
| 4 | 30 mol% 60, Et3SiH, EtOH | – | – | 63% | – |
| 5 | 30 mol% Mn(dpm)3, PhSiH3, i-PrOH/DCM | – | – | – | |
| 6 | 30 mol% Fe(acac)3, PhSiH3 or Et3SiH, EtOH | – | – | – | |
A major challenge in the polyoxygenation cascade is presumably the addition of a peroxy radical to an electron-deficient π-system in combination with the ring flip in the 7-membered carbocycle necessary to accommodate this 6-exo cyclization geometry.41 These requirements presumably compete with simple reduction of the reactive intermediates. We wondered, however, if the oxygen stitching cascade could be a useful tool in other terpene settings, particularly those in which the cyclization requirements are more accommodating.
To this end, we targeted the eudesmane sesquiterpene boariol (80) as a way to examine the efficiency of this strategy in an alternative environment (Scheme 5).42,43 Carvone Robinson annulation product 78, prepared in one-step, was chosen as the starting material. Subjecting this enone to the tandem peroxidation conditions formed triol 79 after reductive work-up, presumably via the bis-peroxide intermediate shown. This transformation, which like 69, also involves peroxy radical addition to an electron deficient π-system and 6-exo cyclization, proceeded in a respectable 50% isolated yield. Finally, lithium aluminum hydride induced stereoselective reduction of 79, producing a tetraol which cyclized to boariol in 73% yield overall yield after treatment with protic acid (HCl). Overall, these results suggest the oxygen stitching strategy has utility in the rapid synthesis of polyol natural products. Moreover, in terms of efficiency, this 3-step route compares quite favorably to terpene synthetic strategies targeting similar molecules, yet assembling them via the oxidation of C-H bonds rather than the polyoxygenation of alkenes.44
Scheme 5.

Three-step synthesis of the eudesmane sesquiterpene boariol (80).
aReagents and conditions: (a) 78 (1 equiv), Co(acac)2 (0.2 equiv), Et3SiH (2.4 equiv.), O2 (1 atm), EtOH, then add Zn0 (2.0 equiv), NH4Cl EtOH, 50%; (b) LiAlH4 (1.5 equiv), Et2O, 0 ºC, 45 mins, then add aq. 1N HCl, 0 ºC to 23 °C, 73%. acac = acetylacetone.
Conclusion
In this report, we have detailed the evolution of an allylative approach to five complex guaianolide natural products from both the Apiaceae and Asteraceae plant families in 9–14 synthetic steps. Seeking to leverage abundant chiral pool building blocks, several strategies were explored. First, we examined the annulation and cyclization of the linear monoterpene linalool, ultimately finding a pathway toward the simple, stereochemical complexity-deficient Apiaceae members sinodielide A (12). Several oxidative and stereochemical challenges, however, limited the utility of this route in broader guaianolide contexts. A double allylation strategy employing a carvone-derived fragment proved more general and led to syntheses of several more complex guaianolides (see 8, 18, 23), not accessible by previous strategies. Key to the success of this overarching blueprint were five distinct metal-mediated allylation events, each of which presented their own unique reactivity and stereochemical challenges. It is noteworthy that many of the stereocenters forged during these processes could be influenced by very simple changes to reagents and conditions utilized–in no instance were chiral reagents employed. Finally, we extended a previous polyoxygenation cascade strategy for endoperoxide synthesis to the formation of complex hydroxylated terpenes, namely nortrilobolide (29) and the eudesmane sesquiterpene boariol (80). In cases where peroxyradical addition is geometrically favorable, this reaction can quite efficiently and rapidly increase hydroxyl complexity. While significant interest has recently been placed on C-H activation routes to complex natural products, particularly terpenes, these results suggest polyoxygenation-based cascades could serve as alternative tools in the retrosynthetic planning stages. Further work in this area is underway and will be reported in due course.
Supplementary Material
ACKNOWLEDGMENT
Financial support is acknowledged from the NIH (R01GM116952). Early work was supported by the ACS PRF fund (53703-DNI1). X.H. thanks Bristol-Myers Squibb for a graduate fellowship. A.J.M. acknowledges the NIH for a post-doctoral fellowship (F32GM126655). Y.T. thanks the UC-Berkeley College of Chemistry for a summer undergraduate research award. We are grateful to Dr. Hasan Celik and Dr. Jeffrey Pelton for NMR spectroscopic assistance and NIH grants GM68933 and S10OD024998. Dr. Nicholas Settineri and Dr. Antonio DiPasquale are acknowledged for X-ray crystallographic analysis wherein support from NIH Shared Instrument Grant (S10-RR027172) is also acknowledged.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b08001.
X-ray crystallographic data for F1 (CIF)
X-ray crystallographic data for 80 (CIF)
Experimental procedures and spectroscopic data (PDF)
The authors declare no competing financial interests.
REFERENCES
- (1).Chadwick M; Trewin H; Gawthrop F; Wagstaff C Sesquiterpenoids Lactones: Benefits to Plants and People. Int. J. Mol. Sci 2013, 14, 12780; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Breitmaier E Terpenes: Flavors, Fragrance, Pharmaca, Pheromones, 2006. Wiley-VCH. [Google Scholar]
- (2).(a) For reviews on guaianolides and their chemistry, see: Schaal A; Reiser O Synthesis of Biologically Active Guaianolides with a trans-Annulated Lactone Moiety. Eur. J. Org. Chem 2008, 2353;; (b) Drew DP; Krichau N; Reichwald K; Simonsen HT Guaianolides in apiaceae: perspectives on pharmacology and biosynthesis. Phytochem Rev 2009, 8, 581; [Google Scholar]; (c) Simonsen HT; Weitzel C; Christensen SB Guaianolide sesquiterpenoids: Pharmacology and biosynthesis In Natural Products; Ramawat KG, Merillon JM, Eds.; Springer-Verlag: Berlin, 2013; Vol. 5, pp. 3069; [Google Scholar]; (d) Santana A; Molinillo JMG; Macías FA Trends in the Synthesis and Functionalization of Guaianolides. Eur. J. Org. Chem 2015, 2093.
- (3).(a) Andersen TB; López CQ; Manczak T; Martinez K; Simonsen HT Thapsigargin—From Thapsia L. to Mipsagargin. Molecules, 2015, 20, 6113; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ghantous A; Gali-Muhtasib H; Vuorela H; Saliba NA; Darwiche N What made sesquiterpene lactones reach cancer clinical trials? Drug. Discov. Today 2010, 15, 668–678. [DOI] [PubMed] [Google Scholar]; (c) Li J; Li S; Guo J; Li Q; Long J; Ma C; Ding Y; Yan C; Li L; Wu Z; Zhu H; Li KK; Wen L; Zhang Q; Xue Q; Zhao C; Liu N; Ivanov I; Luo M; Xi R; Long H; Wang PG; Chen Y Natural Product Micheliolide (MCL) Irreversibly Activates Pyruvate Kinase M2 and Suppresses Leukemia. J. Med. Chem 2018, 61, 4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) For recent biosynthetic investigations into these steps, see: Andersen TB; Martinez-Swatson KA; Rasmussen SA; Boughton BA; Jørgensen K; Andersen-Ranberg J; Nyberg N; Christensen SB; Simonsen HT Localization and in-Vivo Characterization of Thapsia garganica CYP76AE2 Indicates a Role in Thapsigargin Biosynthesis. Plant Physiol 2017, 174, 56; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ramirez AM; Saillard N; Yang T; Franssen MCR; Bouwmeester HJ; Jongsma MA Biosynthesis of Sesquiterpene Lactones in Pyrethrum (Tanacetum cinerariifolium). PLoS ONE, 2013, 8(5): e65030 10.1371/journal.pone.0065030. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Liu Q; Kashkooli AB; Manzano D; Pateraki I; Richard L; Kolkman P; Lucas MF; Guallar V; de Vos RCH; Franssen MCR; van der Krol A; Bouwmeester H Kauniolide synthase is a P450 with unusual hydroxylation and cyclization-elimination activity. Nat. Commun 2018, 9, 4657; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) de Kraker J-W; Franssen MCR; Joerink M; de Groot A; Bouwmeester HJ Biosynthesis of Costunolide, Dihydrocostunolide, and Leucodin. Demonstration of Cytochrome P450-Catalyzed Formation of the Lactone Ring Present in Sesquiterpene Lactones of Chicory. Plant Physiol 2002, 129, 257; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Pickel B; Drew DP; Manczak T; Weitzel C; Simonsen HT; Ro DK Identification and characterization of a kunzeaol synthase from Thapsia garganica: implications for the biosynthesis of the pharmaceutical thapsigargin. Biochem. J 2012, 448, 261. [DOI] [PubMed] [Google Scholar]
- (5).Jackson PA; Widen JC; Harki DA; Brummond KM Covalent Modifiers: A Chemical Perspective on the Reactivity of α,β-Unsaturated Carbonyls with Thiols via Hetero-Michael Addition Reactions, J. Med. Chem 2017, 60, 839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Schmidt TJ Structure-Activity Relationships of Sesquiterpene Lactones In Studies in Natural Products Chemistry; Attaur-Rahman, Ed.; Elsevier, 2006; Vol. 33, p. 309; [Google Scholar]; (b) Gersch M; Kreuzer J; Sieber SA Electrophilic natural products and their biological targets. Nat. Prod. Rep 2012, 29, 659; [DOI] [PubMed] [Google Scholar]; (c) Li C; Jones AX; Lei X Synthesis and mode of action of oligomeric sesquiterpene lactones, Nat. Prod. Rep 2016, 33, 602; [DOI] [PubMed] [Google Scholar]; (d) Lagoutte R; Winssinger N Following the Lead from Nature with Covalent Inhibitors. Chimia 2017, 71, 703. [DOI] [PubMed] [Google Scholar]
- (7).(a) For examples, see: Zhang W; Luo S; Chen Q; Hu H; Jia X; Zhai H Total Synthesis of Absinthin. J. Am. Chem. Soc 2005, 127, 18; [DOI] [PubMed] [Google Scholar]; (b) Li C; Yu X; Lei X A Biomimetic Total Synthesis of (+)-Ainsliadimer A. Org. Lett 2010, 12, 4284; [DOI] [PubMed] [Google Scholar]; (c) Li C; Dian L; Zhang W; Lei X Biomimetic Syntheses of (–)-Gochnatiolides A-C and (–)-Ainsliadimer B. J. Am. Chem. Soc 2012, 134, 12414; [DOI] [PubMed] [Google Scholar]; (d) Li C; Dong T; Dian L; Zhang W; Lei X Biomimetic syntheses and structural elucidation of the apoptosis-inducing sesquiterpenoid trimers: (–)-ainsliatrimers A and B. Chem. Sci 2013, 4, 1163; [Google Scholar]; (e) Li C; Lei X Strategies toward the Biomimetic Syntheses of Oligomeric Sesquiterpenoids, J. Org. Chem 2014, 79, 3289. [DOI] [PubMed] [Google Scholar]
- (8).(a) For syntheses of guaianolide lactones, see: Devreese AA; De Clerq PJ; Vandewalle M A general entry to guaianolides. An illustrative synthesis of (±)-compressanolide. Tett. Lett 1980, 21, 4767; [Google Scholar]; (b) Demuynck M; Devreese AA; De Clercq PJ; Vandewalle M GUAIANOLIDES: THE TOTAL SYNTHESIS OF (±)-ESTAFIATIN. Tett. Lett 1982, 23, 2501; [Google Scholar]; (c) Rigby JH; Wilson JZ Total synthesis of guaianolides: (±)-dehydrocostus lactone and (±)-estafiatin. J. Am. Chem. Soc 1984, 106, 8217; [Google Scholar]; (d) Rigby JH; Senanayake C Total synthesis of (±)-grosshemin C., J. Am. Chem. Soc 1987, 109, 3147; [Google Scholar]; (e) Carret S; Depres JP Access to Guaianolides: Highly Efficient Stereocontrolled Total Synthesis of (±)-Geigerin. Angew. Chem. Int. Ed 2007, 46, 6870; [DOI] [PubMed] [Google Scholar]; (f) Kalidindi S; Jeong WB; Schall A; Bandichhor R; Nosse B; Reiser O Enantioselective synthesis of arglabin. Angew. Chem. Int. Ed 2007, 46, 6361; [DOI] [PubMed] [Google Scholar]; (g) Hirose T; Miyakoshi N; Mukai C Total Synthesis of (+)-Achalensolide Based on the Rh(I)-Catalyzed Allenic Pauson−Khand-Type Reaction. J. Org. Chem 2008, 73, 1061; [DOI] [PubMed] [Google Scholar]; (h) Banchelin TSL; Carret S; Giannini A; Depres JP Short and Stereoselective Total Synthesis of Δ-11,13-Didehydroguaianes and -guaianolides: Synthesis of (±)-Achalensolide and (±)-Pechueloic Acid; Revision of the Structure of (+)-Rupestonic Acid. Eur. J. Org. Chem 2009, 3678;; (i) Valot G; Garcia J; Duplan V; Serba C; Barluenga S; Winssinger N Diversity-oriented synthesis of diverse polycyclic scaffolds inspired by the logic of sesquiterpene lactones biosynthesis. Angew. Chem. Int. Ed 2012, 51, 5391; [DOI] [PubMed] [Google Scholar]; (j) Shimomaki K; Kusama H; Iwasawa N Total Synthesis of (+)-Integrifolin. Chem. Eur. J 2016, 22, 9953–9957. [DOI] [PubMed] [Google Scholar]; (k) Sun W-B; Wang X; Sun B-F; Zou J-P; Lin G-Q Catalytic Asymmetric Total Synthesis of Hedyosumins A, B, and C. Org. Lett 2016, 18, 1219; [DOI] [PubMed] [Google Scholar]; (l) Wang X; Sun W-B; Zou J-P; Lin G-Q; Sun B-F Asymmetric total synthesis of hedyosumin E aglycon, 7,10-epoxyhedyosminolide and ent-zedolactone A. Org. Biomol. Chem, 2016, 14, 10581. [DOI] [PubMed] [Google Scholar]
- (9).(a) For syntheses of guaianolide lactones using chiral terpenes, see: Lee E; Lim JW; Yoon CH; Sung YS; Kim YK; Yun M; Kim S Total Synthesis of (+)-Cladantholide and (−)-Estafiatin: 5-Exo,7-Endo Radical Cyclization Strategy for the Construction of Guaianolide Skeleton. J. Am. Chem. Soc 1997, 119, 8391; [Google Scholar]; (b) Oliver SF; Högenauer K; Simic O; Antonello A; Smith MD; Ley SV A route to the thapsigargins from (S)-carvone providing a substrate-controlled total synthesis of trilobolide, nortrilobolide, and thapsivillosin F. Angew. Chem. Int. Ed 2003, 42, 5996; [DOI] [PubMed] [Google Scholar]; (c) Ley SV; Antonello A; Balskus EP; Booth DT; Christensen SB; Cleator E; Gold H; Högenauer K; Hünger U; Myers RM; Oliver SF; Simic O; Smith MD; Søhoel H; Woolford AJ, Synthesis of the thapsigargins. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 12073; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ball M; Andrews SP; Wierschem F; Cleator E; Smith MD; Ley SV Total Synthesis of Thapsigargin, a Potent SERCA Pump Inhibitor. Org. Lett 2007, 9, 663; [DOI] [PubMed] [Google Scholar]; (e) Andrews SP; Ball M; Wierschem F; Cleator E; Oliver S; Hogenauer K; Simic O; Antonello A; Hunger U; Smith MD; Ley SV Total Synthesis of Five Thapsigargins: Guaianolide Natural Products Exhibiting Sub-Nanomolar SERCA Inhibition. Chem. Eur. J 2007, 13, 5688; [DOI] [PubMed] [Google Scholar]; (f) Yang H; Qiao X; Li F; Ma H; Xie L; Xu X Diastereoselective total synthesis of 8-epigrosheimin, Tett. Lett 2009, 50, 1110; [Google Scholar]; (g) Elford TG; Hall DG Total synthesis of (+)-chinensiolide B via tandem allylboration/lactonization. J. Am. Chem. Soc 2010, 132, 1488; [DOI] [PubMed] [Google Scholar]; (h) Yang H; Gao Y; Qiao X; Xie L; Xu X Concise total synthesis of (–)-8-epigrosheimin. Org. Lett 2011, 13, 3670; [DOI] [PubMed] [Google Scholar]; (i) Zhai JD; Li D; Long J; Zhang HL; Lin JP; Qiu CJ; Zhang Q; Chen Y Biomimetic semisynthesis of arglabin from parthenolide. J. Org. Chem 2012, 77, 7103; [DOI] [PubMed] [Google Scholar]; (j) Anagnostaki EE; Demertzidou VP; Zografos AL Divergent pathways to furosesquiterpenes: first total syntheses of (+)-zedoarol and (Rac)-gweicurculactone. Chem. Commun 2015, 51, 2364; [DOI] [PubMed] [Google Scholar]; (k) Johnson TC; Chin MR; Han T; Shen JP; Rana T; Siegel D Synthesis of Eupalinilide E, a Promoter of Human Hematopoietic Stem and Progenitor Cell Expansion. J. Am. Chem. Soc 2016, 138, 6068; [DOI] [PubMed] [Google Scholar]; (l) Barthel A; Kaden F; Jäger A; Metz P Enantioselective Synthesis of Guaianolides in the Osmitopsin Family by Domino Metathesis. Org. Lett 2016, 18, 3298. [DOI] [PubMed] [Google Scholar]; (m) Chu H; Smith JM; Felding J; Baran PS Scalable Synthesis of (–)-Thapsigargin. ACS. Cent. Sci 2017, 3, 47; [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Hu X; Xu S; Maimone TJ A Double Allylation Strategy for Gram-Scale Guaianolide Production: Total Synthesis of (+)-Mikanokryptin. Angew. Chem. Int. Ed 2017, 56, 1624; [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Chen D; Evans PA A Concise, Efficient and Scalable Total Synthesis of Thapsigargin and Nortrilobolide from (R)-(–)-Carvone. J. Am. Chem. Soc 2017, 139, 6046; [DOI] [PubMed] [Google Scholar]; (p) Hajra S; Acharyya S; Mandal A; Maity R Unified total synthesis of (+)-chinensiolide B and (+)-8-epigrosheimin. Org. Biomol. Chem 2017, 15, 6401. [DOI] [PubMed] [Google Scholar]
- (10).(a) Brill ZG; Condakes ML; Ting CP; Maimone TJ Navigating the Chiral Pool in the Total Synthesis of Complex Terpene Natural Products. Chem. Rev 2017, 117, 11753. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Brill ZG; Grover H; Maimone TJ Enantioselective Synthesis of an Ophiobolin Sesterterpene via a Programmed Radical Cascade. Science, 2016, 352, 1078. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hung K; Condakes M; Morimoto T; Maimone TJ Oxidative Entry into the Illicium Sesquiterpenes: Enantiospecific Syntheses of (+)-Pseudoanisatin. J. Am. Chem. Soc 2016, 138, 16616. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Condakes M; Hung K; Harwood SL; Maimone TJ Total Syntheses of (–)-Majucin and (–)-Jiadifenoxolane A, Complex Majucin-type Illicium Sesquiterpenes. J. Am. Chem. Soc 2017, 139, 17783. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Condakes ML; Rosen R; Harwood SJ; Maimone TJ A Copper-catalyzed Doubling Coupling Enables 3-Step Entry into the Quassinoid Core Architecture. Chem. Sci 2019, 10, 768 [Google Scholar]; (f) Condakes ML; Hung K; Novaes LFT; Morikawa T; Harwood S Yang, Z.; Maimone, T. J. Development of a Terpene Feedstock-Based Oxidative Approach to the Illicium Sesquiterpenes. J. Am. Chem. Soc 2019, 141, 3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) For guaianolide semisyntheses employing santonin, see: Barton DHR; Levisalles JED Sesquiterpenoids. Part XI. The constitution of geigerin. J. Chem. Soc 1958, 4518;; (b) Barton DHR; Pinhey JT; Wells RJ Photochemical transformations. Part XV. Synthetic studies on geigerin and its derivatives J. Chem. Soc 1964, 2518;; (c) Barton DHR; Pinhey JT The Stereochemical Correlation of Artemisin and Geigerin. Proc. Chem. Soc 1960, 279;; (d) White EH; Marx JN The synthesis and stereochemistry of deacetoxymatricarin and achillin. J. Am. Chem. Soc 1967, 89, 5511; [Google Scholar]; (e) Marx JN; White EH The stereochemistry and synthesis of Achillin. Tetrahedron 1969, 25, 2117; [Google Scholar]; (f) White EH; Eguchi S; Marx JN The synthesis and stereochemistry of desacetoxymatricarin and the stereochemistry of matricarin. Tetrahedron 1969, 25, 2099; [Google Scholar]; (g) Winter REK; Lindauer RF The photoisomerization of dihydrocostunolide. Tetrahedron, 1976, 32, 955 [Google Scholar]; (h) Edgar MT; Greene AE; Crabbe P Stereoselective Synthesis of (–)-Estafiatin. J. Org. Chem 1978, 44, 159. [Google Scholar]; (i) Greene AE; Edgar MT Synthesis of Oxoisodehydroleucodin: A Novel Guaianolide from Montanoa imbricate. J. Org. Chem 1989, 54, 1468. [Google Scholar]; (j) Ando M; Yoshimura H Syntheses of Four Possible Diastereoisomers of Bohlmann’s Structure of Isoepoxyestafiatin. The Stereochemical Assignment of Isoepoxyestafiatin. J. Org. Chem 1993, 58, 4127; [Google Scholar]; (k) Ando M; Ibayashi K; Minami N; Nakamura T; Isogai K; Yoshimura H Studies on the Synthesis of Sesquiterpene Lactones, 16. The Syntheses of 11β,13-Dihydrokauniolide, Estafiatin, Isodehydrocostuslactone, 2-Oxodesoxyligustrin, Arborescin, 1,10-Epiarborescin, 11β,13-Dihydroludartin, 8-Deoxy-11β,13-dihydrorupicolin B, 8-Deoxyrupicolin B, 3,4-Epiludartin, Ludartin, Kauniolide, Dehydroleucodin, and Leucodin. J. Nat. Prod 1994, 57, 433; [Google Scholar]; (l) Delair P; Kann N; Greene AE Synthesis (in ent-form) of a Novel Jalcaguaianolide from Ferula arrigonii Bocchieri. J. Chem. Soc. Perkin Trans 1 1994, 1651; [Google Scholar]; (m) Bargues V; Blay G; Cardona L; García B; Pedro JR Regio- and stereoselective oxyfunctionalization at C-1 and C-5 in sesquiterpene guaianolides. Tetrahedron 1998, 54, 1845; [Google Scholar]; (n) Yuuya S; Hagiwara H; Suzuki T; Ando M; Yamada A; Suda K; Kataoka T; Nagai K Guaianolides as Immunomodulators. Synthesis and Biological Activities of Dehydrocostus Lactone, Mokko Lactone, Eremanthin, and Their Derivatives. J. Nat. Prod 1999, 62, 22; [DOI] [PubMed] [Google Scholar]; (o) Blay G; Bargues V; Cardona L; García B; Pedro JR Stereoselective synthesis of 7,11-guaien-8,12-olides from santonin. Synthesis of podoandin and (+)-zedolactone A. J. Org. Chem 2000, 65, 6703; [DOI] [PubMed] [Google Scholar]; (p) Blay G; Bargues VV; Cardona L; Collado AM; Garcia B; Munoz MC; Pedro JR Stereoselective synthesis of 4 alpha-hydroxy-8,12-guaianolides from santonin. J. Org. Chem 2000, 65, 2138; [DOI] [PubMed] [Google Scholar]; (q) Bargues V; Blay G; Cardona L; García B; Pedro JR Stereoselective synthesis of (+)-11βH,13-dihydroestafiatin, (+)-11βH,13-dihydroludartin, (−)-compressanolide, and (−)-11βH,13-dihydromicheliolide from santonin. J. Nat. Prod 2002, 65, 1703; [DOI] [PubMed] [Google Scholar]; (r) Macías FA; Santana A; Yamahata A; Varela RM; Fronczek FR; Molinillo JMG Facile Preparation of Bioactive seco-Guaianolides and Guaianolides from Artemisia gorgonum and Evaluation of Their Phytotoxicity. J. Nat. Prod 2012, 75, 1967; [DOI] [PubMed] [Google Scholar]; (s) Zhang L; Dai X; Tao L; Xie C; Wang M Total Synthesis of (+)-Chinensiolide B from α-Santonin. Chin. J. Chem 2017, 35, 1284. [Google Scholar]
- (12).For early studies, see: Barton DHR; de Mayo P; Shafiq M Photochemical transformations. Part I. Some preliminary investigations. J. Chem. Soc 1957, 929–935 and references therein.
- (13).(a) For Pauson-Khand reactions toward guaianolide carbocycles, see: Wells SM; Brummond KM Conditions for a Rh(I)-catalyzed [2+2+1] cycloaddition reaction with methyl substituted allenes and alkynes. Tetrahedron Lett 2015, 56, 3546; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Grillet F; Huang C; Brummond KM An Allenic Pauson–Khand Approach to 6,12-Guaianolides. Org. Lett 2011, 13, 6304; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wen B; Hexum JK; Widen JC; Harki DA; Brummond KM A Redox Economical Synthesis of Bioactive 6,12-Guaianolides. Org. Lett 2013, 15, 2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).For the exploration of a related C–C bond disconnection, see: Hudlicky T; Govindan SV; Frazier JO Heteroatom Cyclopentene Annulation. Synthesis of Guaiane Ring System. J. Org. Chem 1985, 50, 4166. [Google Scholar]
- (15).For a Pauson-Khand reaction on a linalool derivative, see: Olagnier D; Costes P; Berry A; Linas M-D; Urrutigoity M; Dechy-Cabaret O; Benoit-Vical F Modifications of the chemical structure of terpenes in antiplasmodial and antifungal drug research. Bioorg. Med. Chem. Lett 2007, 17, 6075. [DOI] [PubMed] [Google Scholar]
- (16).Although (–)-linalool is most easily obtained, either enantiomer can be easily prepared from geraniol, see: Wu Y-K; Liu H-J; Zhu J-L An Efficient Procedure for the 1,3-Transposition of Allylic Alcohols Based on Lithium Naphthalenide Induced Reductive Elimination of Epoxy Mesylates. Synlett 2008, 621.
- (17).Zhao Y-M; Maimone TJ Short, Enantioselective Total Synthesis of Chatancin, Angew. Chem. Int. Ed 2015, 54, 1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
(18).Of note, treatment of the minor diastereomer of 33 with SO2Cl2 afforded an allylic chloride which could open the strained lactone under Barbier-type conditions as shown below. Following double reduction with DIBAL, a product suitable for X-ray crystallographic analysis (F1) was obtained.
- (19).Movassaghi M; Ahmad OK N-Isopropylidene-N’-2-nitrobenzenesulfonyl Hydrazine, a Reagent for Reduction of Alcohols via the Corresponding Monoalkyl Diazenes. J. Org. Chem 2007, 72, 1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).For the use of similar reductive allylic transpositions in related 5,7-fused ring systems, see refs 8l, 11h, 11k.
- (21).(a) Wang N-H; Taniguchi M; Tsuji D; Doi M; Ohishi H; Yoza K; Baba K Four Guaianolides from Sinodielsia yunnanensis. Chem. Pharm Bull 2003, 51, 68; [DOI] [PubMed] [Google Scholar]; (b) Hatashita M; Taniguchi M; Baba K; Koshiba K; Sato T; Jujo Y; Suzuki R; Hayashi S Sinodielide A exerts thermosensitizing effects and induces apoptosis and G2/M cell cycle arrest in DU145 human prostate cancer cells via the Ras/Raf/MAPK and PI3K/Akt signaling pathways. Int. J. Mol. Med 2014, 33, 406. [DOI] [PubMed] [Google Scholar]
- (22).(a) Fujioka H; Sawama Y; Murata N; Okitsu T; Kubo O; Matsuda S; Kita Y Unexpected highly chemoselective deprotection of the acetals from aldehydes and not ketones: TESOTf-2,6-lutidine combination. J. Am. Chem. Soc 2004, 126, 11800; [DOI] [PubMed] [Google Scholar]; (b) Fujioka H; Okitsu T; Sawama Y; Murata N; Li R; Kita Y Reaction of the acetals with TESOTf-base combination; speculation of the intermediates and efficient mixed acetal formation. J. Am. Chem. Soc 2006, 128, 5930. [DOI] [PubMed] [Google Scholar]
- (23).Hung K; Hu X; Maimone TJ Total Synthesis of Complex Terpenes Employing Radical Cascade Processes, Nat. Prod. Rep 2018, 35, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Berdan CA; Ho R; Lehtola HS; To M; Hu X; Huffman TR; Petri Y; Altobelli CR; Demeulenaere SG; Olzmann JA; Maimone TJ; Nomura DK Parthenolide Covalently Targets and Inhibits Focal Adhesion Kinase in Breast Cancer Cells. Cell Chem. Biol 2019, 26, 1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).(a) For the synthesis of advanced precursors towards 28–30, see: Manzano FL; Guerra FM; Moreno-Dorado FJ; Jorge ZD; Massanet GM Toward the Synthesis of Thapsigargin: Enantioselective Synthesis of 7,11-Dihydroxyguaianolides. Org. Lett 2006, 8, 2879; [DOI] [PubMed] [Google Scholar]; (b) Marín-Barrios R; García-Cabeza AL; Moreno-Dorado FJ; Guerra FM; Massanet GM Acyloxylation of Cyclic Enones: Synthesis of Densely Oxygenated Guaianolides. J. Org. Chem 2014, 79, 6501. [DOI] [PubMed] [Google Scholar]
- (26).(a) For recent examples, see: Liu P; Cui Y; Chen K; Zhou X; Pan W; Ren J; Wang Z Total Syntheses of (−)-Englerins A/B, (+)-Orientalols E/F, and (−)-Oxyphyllol. Org. Lett 2018, 20, 2517; [DOI] [PubMed] [Google Scholar]; (b) Srikrishna A; Pardeshi VH; Satyanarayana G Enantioselective formal total syntheses of Clavukerin A and isoclavukerin A via a ring-closing metathesis reaction. Tett. Asymm 2010, 21, 746; [Google Scholar]; (c) Jouanneau M; Bonepally KR; Jeuken A; Tap A; Guillot R; Ardisson J; Férézou J-P; Prunet J Diastereoselective Synthesis of an Advanced Intermediate of Thapsigargin and Other 6,12-Guaianolides Using a RCEYM Strategy. Org. Lett 2018, 20, 2176. [DOI] [PubMed] [Google Scholar]
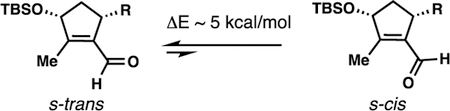
-
(27).In a closely related system, Prunet and co-workers have reported that the s-trans conformer shown below is ~5 kcal/mol more stable than the s-cis one, but suggested that the transition state for nucleophilic carbonyl addition to s-cis is more favorable thus introducing the possibility of a Curtin-Hammett scenario based on the nucleophile employed (ref. 26c). This rationale is consistent with the findings in refs. 26a,b. For reactivity comparisons between Zn0 and In0 in an aldehyde allylation using allylic halides, see: Kong W; Fu C; Ma S
Indium and zinc-mediated Barbier-type addition reaction of 2,3-allenals with allyl bromide: an efficient synthesis of 1,5,6-alkatrien-4-ols. Org. Biomol. Chem
2008, 6, 4587.).
- (28).Estévez RE; Justicia J; Bazdi B; Fuentes N; Paradas M; Choquesillo-Lazarte D; García-Ruiz JM; Robles R; Gansäuer A; Cuerva JM; Oltra JE Ti-Catalyzed Barbier-Type Allylations and Related Reactions. Chem. Eur. J 2009, 15, 2774. [DOI] [PubMed] [Google Scholar]
- (29).(a) For lead references, see: Isayama S; Mukaiyama T A New Method for Preparation of Alcohols from Olefins with Molecular Oxygen and Phenylsilane by the Use of Bis(acetylacetonato)cobalt(II). Chem. Lett 1989, 18, 1071; [Google Scholar]; (b) Mukaiyama T; Isayama S; Inoki S; Kato K; Yamada T; Takai T Oxidation-Reduction Hydration of Olefins with Molecular Oxygen and 2-Propanol Catalyzed by Bis(acetylacetonato)cobalt(II). Chem. Lett 1989, 18, 449; [Google Scholar]; (c) Inoki S; Kato K; Takai T; Isayama S; Yamada T; Mukaiyama T Bis(trifluoroacetylacetonato)cobalt(II) Catalyzed Oxidation-Reduction Hydration of Olefins Selective Formation of Alcohols from Olefins Chem. Lett 1989, 18, 515. [Google Scholar]
- (30).For use of catalyst 60, see: Waser J; Gaspar B; Nambu H; Carreira EM Hydrazines and Azides via the Metal-Catalyzed Hydrohydrazination and Hydroazidation of Olefins. J. Am. Chem. Soc 2006, 128, 11693. [DOI] [PubMed] [Google Scholar]
- (31).Barrero AF; Herrador MM; Arteaga P Sesquiterpene Lactones and Other Constituents of Seseli Vayredanum. Phytochemistry 1994, 37, 1351. [Google Scholar]
- (32).(a) Smítalová Z; Buděšínský M; Šaman D; Vašíčková S; Holub M Components of the extract from the underground parts of Laserpitium siler L. of slovenian origin, mainly sesquiterpenic lactones. Collect. Czech Chem. Commun 1984, 49, 852; [Google Scholar]; (b) Popović V; Stojković D; Nikolić M; Heyerick A; Petrović S; Soković M; Niketić M Extracts of three Laserpitium L. species and their principal components laserpitine and sesquiterpene lactones inhibit microbial growth and biofilm formation by oral Candida isolates. Food Funct 2015, 6, 1205; [DOI] [PubMed] [Google Scholar]; (c) Holub M; Samek Z; Vašíčková S; Masojídková M 11-Hydroxy-1βH,5βH,6αH,7αH-guaian-6,12-olides: Relative and absolute configuration of the sesquiterpenic lactones montanolide, isomontanolide, acetylisomontanolide and related substances. Collect. Czech Chem. Commun 1978, 43, 2444. [Google Scholar]
- (33).(a) Hu X; Maimone TJ Four-step Synthesis of the Antimalarial Cardamom Peroxide via an Oxygen Stitching Strategy, J. Am. Chem. Soc 2014, 136, 5287; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hu X; Lim P; Fairhurst R; Maimone TJ Synthesis and Study of the Antimalarial Cardamom Peroxide, Tetrahedron, 2018, 74, 3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).For a review on radical hydrofunctionalization chemistry, see: Crossley SWM; Obradors C; Martinez RM; Shenvi RA Mn-, Fe-, and Co-Catalyzed Radical Hydrofunctionalizations of Olefins. Chem. Rev 2016, 116, 8912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wakamatsu H; Nishida M; Adachi N; Mori M Isomerization Reaction of Olefin Using RuClH(CO)(PPh3)3. J. Org. Chem 2000, 65, 3966. [DOI] [PubMed] [Google Scholar]
- (36).(a) For representative examples, see: Imai T; Nishida S A new type of catalysis by copper(I) salts in the Barbier-type aldehyde allylation with tin(II) chloride. Short syntheses of (±)-lavandulol and its γ, δ-dihydro derivative J. Chem. Soc., Chem. Commun 1994, 277;; (b) Tan XH; Shen B; Deng W; Zhao H; Liu L; Guo QX Novel Carbonyl Allylation Mediated by SnCl2/TiCl3 in Water. Org. Lett 2003, 5, 1833; [DOI] [PubMed] [Google Scholar]; (c) Chaudhuri MK; Dehury SK; Hussain S Barbier coupling in water: SnCl2-mediated and Co(acac)2-catalyzed allylation of carbonyls. Tetrahedron Lett 2005, 46, 6247; [Google Scholar]; (d) Masuyama Y; Kaneko Y; Kurusu Y Rhodium-catalyzed carbonyl allylations by allylic alcoholswith tin(II) chloride. Tetrahedron Lett 2004, 45, 8969. [Google Scholar]
- (37).(a) For Pd/Sn systems, see: Masuyama Y; Hayashi R; Otake K; Kurusu Y Charge reversal of electrophilic π-allylpalladium intermediates; carbonyl allylation by allylic acetates with PdCl2(PhCN)2–SnCl2. J. Chem. Soc., Chem. Commun 1988, 44;; (b) Masuyama Y; Otake K; Kurusu Y Diastereoselectivity in carbonyl allylation by allylic carbonates using PdCl2(PhCN)2-SnCl2 system. Tetrahedron Lett 1988, 29, 3563; [Google Scholar]; (c) Masuyama Y; Takahara JP; Kurusu Y Palladium-catalyzed carbonyl allylation by allylic alcohols with SnCl2. A solvation-controlled diastereoselection. Tetrahedron Lett 1989, 30, 3437; [Google Scholar]; (d) Masuyama Y; Hayakawa A; Kurusu Y Ultrasound-promoted carbonyl allylation by allylic alcohols with palladium–tin dichloride in nonpolar solvents: inversed regiocontrol of carbonyl allylation in polar solvents. J. Chem. Soc., Chem. Commun 1992, 1102;; (e) Masuyama Y; Ito A; Kurusu Y Either γ-syn- or γ-anti-selective palladium-catalysed carbonyl allylation by mixed (E)- and (Z)-1,3-dichloropropene with tin(II) halides. Chem. Commun 1998, 315;; (f) Marshall JA Synthesis and Reactions of Allylic, Allenic, Vinylic, and Arylmetal Reagents from Halides and Esters via Transient Organopalladium Intermediates. Chem. Rev 2000, 100, 3163. [DOI] [PubMed] [Google Scholar]; (g) Kashyap B; Phukan P Rapid access to homoallylic alcohols via Pd(OAc)2 catalyzed Barbier type allylation in presence of DMAP Tetrahedron Lett 2013, 54, 6324. [Google Scholar]
- (38).Both peroxides 74 and 75 underwent partial decomposition during chromatographic purification and characterization.
- (39).for a similar finding, see: Michaudel Q; Journot G; Regueiro-Ren A; Goswami A; Guo Z; Tully TP; Zou L; Ramabhadran RO; Houk KN; Baran PS Improving Physical Properties via C–H Oxidation: Chemical and Enzymatic Approaches, Angew. Chem. Int. Ed 2014, 53, 12091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Doan NTQ; Crestey F; Olsen CE; Christensen SB Chemo- and Regioselective Functionalization of Nortrilobolide: Application for Semisynthesis of the Natural Product 2-Acetoxytrilobolide. J. Nat. Prod 2015, 78, 1406. [DOI] [PubMed] [Google Scholar]
- (41).(a) For reviews on peroxy radical cyclizations, see: Korshin EE; Bachi MD Synthesis of Cyclic Peroxides, Patai’s Chemistry of Functional Groups, Online; Wiley: 2009; pp 1–117; [Google Scholar]; (b) Dussault P Synlett 1995, 997.; (c) Hu X Maimone TJ “Peroxy Radical Additions,” in Science of Synthesis, Applications of Domino Transformations in Organic Synthesis, Vol. 1, Snyder SA Ed.; Georg Thieme Verlag: Stuttgart, Germany, 2016; 157. [Google Scholar]
- (42).González AG; Muñoz OM; Ravelo AG; Crespo A; Bazzocchi IL; Jiménez IA; Solans X; Ruiz-Pérez C Rodríguez-Romero, V. A New Sesquiterpene from Maytenus boaria (Celastraceae); Crystal Structure and Absolute Configuration. Tetrahedron. Lett 1992, 33, 1921. [Google Scholar]
- (43).For a synthesis of 80, see: Nan F; Chen X; Xiong Z; Li T; Li Y First Total Synthesis of 3α,4α-Oxidoagarofuran and (–)-3β,4α-Dihydroxy-β-dihydroagarofuran, Synth. Commun 1994, 24, 2319. [Google Scholar]
- (44).Chen K; Baran PS Total synthesis of eudesmane terpenes by site-selective C–H oxidations. Nature, 2009, 459, 824. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.