

Summary
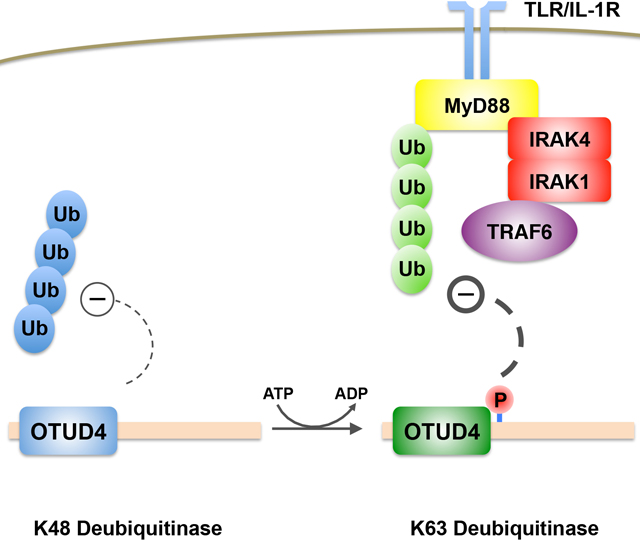
Ubiquitination is a major mechanism that regulates numerous cellular processes, including autophagy, DNA damage signaling, and inflammation. While hundreds of ubiquitin ligases exist to conjugate ubiquitin onto substrates, approximately one hundred deubiquitinases are encoded by the human genome. Thus, deubiquitinases are likely regulated by unidentified mechanisms to target distinct substrates and cellular functions. Here, we demonstrate that the deubiquitinase OTUD4, which nominally encodes a K48-specific deubiquitinase, is phosphorylated near its catalytic domain, activating a latent K63-specific deubiquitinase. Besides phosphorylation, this latter activity requires an adjacent ubiquitin-interacting motif, which increases the affinity of OTUD4 for K63-linked chains. We reveal the Toll-like receptor (TLR) associated factor MyD88 as a target of this K63 deubiquitinase activity. Consequently, TLR-mediated activation of NF-κB is negatively regulated by OTUD4, and macrophages from Otud4−/− mice exhibit increased inflammatory signaling upon TLR stimulation. Our results reveal insights into how a deubiquitinase may modulate diverse processes through post-translational modification.
eTOC Blurb
OTUD4 nominally encodes a K48-specific deubiquitinase previously shown to counteract proteasomal degradation of DNA repair proteins. Zhao et al. demonstrate that OTUD4 is phosphorylated near its catalytic domain, activating a dormant K63-specific deubiquitinase activity. This enzymatic activity is shown to modulate TLR-dependent signaling by targeting the scaffolding protein MyD88.
Graphical Abstract
Introduction
As implied by its name, ubiquitin is an abundant small polypeptide found in all eukaryotes. The covalent modification of proteins by ubiquitin, typically on lysine residues, is promoted by ubiquitin ligase complexes, and was originally found to target the modified protein to the proteasome for degradation (Komander and Rape, 2012). However, ubiquitination has emerged as a versatile modification that regulates numerous cellular processes independent of the proteasome. The process of polyubiquitination, where multiple ubiquitins are linked together head-to-tail, or via any of seven lysines present on ubiquitin, creates chains with different structures, each of which execute distinct functions (Komander and Rape, 2012; Swatek and Komander, 2016). Of the different ubiquitin linkage types, the three that are best understood include K48-linked and K11-linked polyubiquitin, which are recognized by the proteasome as degradation signals, and K63-linked polyubiquitin, which serves as a platform triggering many signal transduction pathways. While the functions of other ubiquitin linkage types are still being uncovered, the likely overarching paradigm appears to be that distinct chains types are recognized by linkage-specific ‘readers’ with specialized downstream functions (Yau and Rape, 2016).
How specific substrates are targeted for ubiquitination, and which ubiquitin linkage is used for a given substrate, are critical questions that are only partially answered. The vast number of E3 ubiquitin ligases (>300 in the human genome), which function by direct interaction with individual targets, likely provides much of the specificity for substrate selection (Zheng and Shabek, 2017). Certain E2 ligases are known to have linkage type specificity, each of which may function with numerous E3s, providing a substantial combinatorial potential for target protein and linkage type selection (Stewart et al., 2016; Zheng and Shabek, 2017). Yet this type of diversity may not exist for deubiquitinases (DUBs), as only ~100 are believed to be encoded within the human genome (Mevissen and Komander, 2017; Wolberger, 2014). It is possible that interacting factors provide functional diversity for individual DUBs in vivo, which may not be apparent from biochemical studies alone. For example, the deubiquitinase OTUD7B/Cezanne targets K11-linked ubiquitin in vitro, but appears to promote K48-linked deubiquitination of its substrate TRAF3 in vivo (Bremm et al., 2010; Hu et al., 2013). Certain DUBs, such as USP7, associate with specific scaffolding factors that bridge the DUB and the substrate, thereby promoting DUB function (Schwertman et al., 2012; Zhang et al., 2012). Deubiquitinases may act as scaffolds themselves, recruiting additional DUB activities to their substrates (Zhao et al., 2015). Therefore, DUBs likely have additional unrecognized mechanisms that regulate differential substrate selection and linkage specificity.
Regulation of DUBs by accessory domains or additional post-translational modifications has emerged as an important mechanism by which ubiquitin signaling is controlled. Several OTU DUB family members, such as OTUD1 and TRABID, containing ubiquitin-binding domains (UBDs) that promote selectivity towards K63 and K29/K33 linked ubiquitin chains, respectively (Kristariyanto et al., 2015; Licchesi et al., 2012; Mevissen et al., 2013; Michel et al., 2015). On the other hand, OTUD2 is a relatively non-selective DUB, but this broad activity requires its zinc-finger domain (Mevissen et al., 2013). Certain DUBs have also been shown to become activated by post-translational modifications, including OTUD5/DUBA, A20, and OTUB1 (Herhaus et al., 2015; Huang et al., 2012; Wertz et al., 2015). How these modifications alter the linkage specificity and function of these DUBs in vivo, or how they may cooperate with UBDs, however, are only starting to be understood.
In this study, we show that OTUD4, a K48-specific deubiquitinase which we previously showed to be important for maintaining the stability of alkylation repair enzymes (Zhao et al., 2015), has a completely distinct function in modulating innate immune signaling via NF-κB. We find that OTUD4 is phosphorylated in vivo, activating an unrecognized K63-linked deubiquitinase activity. This activity relies not only on its phosphorylation but also a UBD adjacent to its OTU catalytic domain. OTUD4 directly recognizes and deubiquitinates MyD88 to inhibit NF-κB signaling. This mechanism provides functional flexibility to OTUD4 and establishes MyD88 K63-linked deubiquitination as an additional node for canonical NF-κB regulation.
Results
Characterization of the deubiquitinase activity of OTUD4
To gain greater insight into the function and activity of OTUD4, we initially purified Flag-His tagged OTUD4 (residues 1–300) from E. coli and tested its DUB activity. As previously described, bacterially purified OTUD41−300 had DUB activity with preference against K48-linked ubiquitin chains (Ub2–7), but no apparent activity with K63-linked Ub2–7 (Figure 1A) (Mevissen et al., 2013; Zhao et al., 2015). Mutation of the catalytic cysteine residue to alanine (C45A) completely abrogated DUB activity against K48-linked Ub2–7. In striking contrast to bacterially purified OTUD41−300, Flag-tagged OTUD41−300 purified from HEK293 cells preferentially hydrolyzed K63-linked Ub2–7 (Figure 1B). Again, the C45A OTUD41−300 mutant purified from HEK293 cells had no detectable catalytic activity (Fig 1B). To determine whether OTUD4 is a bona fide K63-linked deubiquitinase, we purified Flag-tagged full-length OTUD4 (OTUD4FL) from HEK293 cells (Supplemental Figure S1A). We also expressed and purified two catalytically inactive mutants (C45A and C45S) from these cells. Indeed, wildtype OTUD4FL from HEK293 cells had DUB activity preferentially against K63-linked Ub2–7 (Figure 1C). The C45A and C45S OTUD4 mutants had minimal DUB activity against K63-linked Ub2–7. This residual activity was likely due to the previously shown association between OTUD4 and USP-family DUBs (Zhao et al., 2015).
Figure 1. OTUD4 deubiquitinase chain specificity.

(a) Flag-His-tagged wildtype (WT) or catalytically inactive mutant (C45A) OTUD41−300 were purified from E.coli, incubated with K48- or K63-linked Ub2–7 chains, and analyzed by Western blot (WB). Results are representative of three independent experiments. (b) Flag-tagged WT or C45A OTUD41−300 were purified from HEK293 cells and tested for DUB activity using the same chain substrates as in (a). Results are representative of two independent experiments. (c) Full-length Flag-OTUD4 wildtype (WT), or two catalytically inactive mutants (C45A and C45S) were purified from HEK293 cells and tested for DUB activity using K63-linked Ub2–7 chains and analyzed by Western blot. Silver staining of the full-length Flag-OTUD4 proteins from the DUB assay is shown below. Results are representative of three independent experiments. (d) Schematic of the OTUD4 protein, along with the OTUD4 fragments (residue 1–300 and 1–180) used in the DUB assays. (e) Full-length and OTUD4 fragments as shown in (d) were purified from HEK293 cells or E.coli, and incubated with Ub2 of all linkage types for the indicated times, resolved on SDS-PAGE and Western blotted. Enzyme concentration is as indicated for each reaction. Results shown (for each panel of deubiquitinase assays) are representative of at least two independent experiments.
We next wished to determine whether OTUD4 truly preferred K63-linked ubiquitin chains, or any other ubiquitin linkage. We then tested all ubiquitin linkage types and found that both OTUD4FL and OTUD41−300 purified from HEK293 cells specifically hydrolyzed K63-Ub2 and showed little to no apparent activity for other chain types (Figure 1D–E). We then tested the DUB activity of the OTU domain alone (OTUD41−180). When purified from HEK293 cells, its DUB activity was preferentially against K48-Ub2 (Figure 1E). This K48 DUB activity was noticeably weaker than the K63 DUB activity of the OTUD4FL and OTUD41−300, as significantly greater time was required to observe the K48 activity of the shorter construct. We also observed a preference for K48 linked chains for OTUD41−180 and OTUD41−300 purified from E. coli (Figure 1E). Therefore, these results demonstrate that a region adjacent to the OTU domain (residues 181–300) is responsible for OTUD4 DUB specificity against K63-linked chains in mammalian cells.
Phosphorylation of OTUD4 controls its K63 deubiquitinase activity
A common modification to alter enzyme activity is protein phosphorylation. To test whether phosphorylation affects OTUD4 DUB specificity, Flag-OTUD4FL purified from HEK293 cells was pre-incubated with calf intestinal alkaline phosphatase (CIP) and tested its DUB activity against K63-Ub2–7 chains. As shown in Figure 2A, CIP-treated, but not mock treated OTUD4, completely abrogated its K63-linked DUB activity. These results suggested that phosphorylation controls OTUD4 K63-linked DUB activity. To identify the phosphorylated sites of OTUD4, we purified Flag-OTUD41−300 from HEK293 cells and analyzed it by phosphoproteomic mass spectrometry (Supplemental Figure S1B and S1C). The most commonly phosphorylated peptide within 181–300 was one that contained two phospho-serines out of three potential sites (Figure 2B; Ser199, Ser202 and Ser204). We mutated these three serines (S) to alanine (A), alone or in double combinations (as indicated in Figure 2C). These OTUD41−300 mutants, along with WT OTUD41−300 were purified from HEK293 cells and tested for DUB activity. Of the three single mutants, S202A had the greatest reduction in DUB activity, and an antibody specific for pS202-OTUD4 confirmed loss of phosphorylation at this site (Figure 2C). Notably, in combination with S204A, the double mutant (S202/204A) completely abrogated its K63-linked DUB activity. However, the less potent K48 DUB activity was still apparent in the S202/S04A mutant (Supplemental Figure S1D), suggesting that the phosphorylation of these sites specifically affects the K63-linked DUB activity.
Figure 2. Phosphorylation of OTUD4 controls its K63 deubiquitinase activity.

(a) Flag- OTUD41−300 protein purified from HEK293 cells was pre-incubated with calf intestinal alkaline phosphatase (CIP) or buffer as control. The proteins were then tested for DUB activity with K63-linked Ub2–7 chains and analyzed by Western blot. Results are representative of two independent experiments. (b) Schematic and sequence conservation of phosphorylation sites on OTUD4 identified by phosphoproteomic mass spectrometry analysis (shown in red). (c) HEK293 cells purified Flag-OTUD41−300 wildtype or indicated phospho-mutants were tested for DUB activity with K63-linked Ub2–7 chains. Results are representative of three independent experiments. (d) HEK293 cells were infected with control shRNA, or shRNAs targeting CK2α and/or CK2α as indicated. Full-length Flag-OTUD4 was purified from these cells and DUB activity assay was performed as in (c). Results are representative of three independent experiments. (e) E. coli purified His-Flag-tagged phospho-mimic OTUD41−300 mutant proteins were tested for DUB activity as indicated. Coomassie blue staining of the proteins from the DUB assay is shown below. Results are representative of two independent experiments.
Amino acid sequence analysis suggested that both of these phosphorylation sites in OTUD4, S202DSE and S204EDD have the same general recognition motif (SXXE/D) and may, at least under steady state conditions, be potentially phosphorylated by Casein Kinase II (CK2) (Olsten and Litchfield, 2004). Knockdown of the kinase CK2α or CK2α prime, or both in HEK293 cells decreased phosphorylation of purified OTUD4, and similarly impaired OTUD4 K63 DUB activity (Supplemental Figure S1E and Figure 2D). Substitution of aspartate for serine at positions 202 and 204 (S202D/S204D) had little effect on the K63-linked DUB activity of E. coli purified OTUD41–300. However, replacement of these residues with glutamate (S202E/S204E) appeared to increase its K63-linked DUB activity (Figure 2E). Taken together, these results strongly suggested that phosphorylation of S202/S204 is critical for OTUD4 K63-linked DUB activity.
A ubiquitin-interacting motif (UIM) in OTUD4 is necessary for K63 deubiquitinase activity
To gain greater sight into how OTUD4 activity is regulated, we constructed a number of additional deletions between residues 1–180 and 1–300, which were expressed and purified from HEK293 cells. Surprisingly any additional deletion, including OTUD41−270, OTUD41−254 and OTUD41−227, abolished the K63-DUB activity, even though the phosphorylation at S202 was not affected in these deletions (Figure 3A). This suggested that an additional domain exists between residues 271–300 critical for this activity. This region is highly conserved (Supplemental Figure S2A) and alignment of this domain suggested the presence of a ubiquitin-interacting motif (UIM) with homology to the UIM domain of OTUD1 (Figure 3B). Mutation of the conserved KRD motif in OTUD4 (KRD273–5AAA), or substitution of arginine for a conserved alanine in this domain (A279R), abrogated its K63 DUB activity without affecting S202 phosphorylation (Figure 3B). Importantly, these mutants had similar DUB activities against K48-linked chains (Supplemental Figure S2B). To test whether this domain functions to promote substrate binding, we purified the OTUD4KRD and OTUD4A279R mutant proteins in the context of a catalytically inactive (C45S) OTUD41−300 and tested its binding to K63 Ub4. In comparison to WT OTUD41−300 C45S, both of these mutants pulled down significantly lower amounts of K63 Ub4 (Figure 3C). This was in contrast to the binding of these proteins to K48 Ub4, which was apparently unchanged (Figure 3D). Taken together, these results suggested that the OTUD4 UIM promotes its K63 DUB activity by increasing its ability to interact with its substrate.
Figure 3. An ubiquitin-interacting motif (UIM) in OTUD4 is needed for K63 deubiquitinase activity.

(a) Flag-tagged OTUD4 deletions were purified from HEK293 cells, and tested for DUB activity assay as indicated. The samples were Western blotted with the antibodies as shown. Results are representative of three independent experiments. (b) Full-length Flag-OTUD4 WT or the indicated mutant were purified from HEK293 cells and tested for DUB activity. Results are representative of three independent experiments. Alignment with the UIM of OTUD1 is shown on top. (c) and (d) Flag-tagged proteins as indicated were purified from HEK293 cells by using anti-Flag (M2) beads, then used for pull-down assay with (c) K63-linked Ub4 chains or (d) K48-linked Ub4 chains. Results are representative of two independent experiments.
Identification of MyD88 as a specific target of OTUD4
To determine the functional significance of the K63 DUB activity of OTUD4, we took an unbiased approach and analyzed its interacting partners through proteomics, as previously described (Zhao et al., 2015). Our mass spectrometry results suggested that OTUD4 is associated with the Toll-like receptor (TLR) associated factor MyD88 in cytosol extracts (Supplementary Table S1). We confirmed the interaction of OTUD4 with MyD88 by immunoprecipitation (IP) of Flag-tagged MyD88, along with HA-tagged OTUD4 co-expressed in HEK293 cells (Figure 4A). Consistently, reciprocal IP of HA-tagged OTUD4 confirmed its association with Flag-MyD88 (Figure 4B). Immunoprecipitation of OTUD4 from mouse embryonic fibroblast (MEF) cell extracts demonstrated the interaction between OTUD4 and MyD88, suggesting that this interaction exists at the endogenous level (Figure 4C). We also found that recombinant MBP-tagged OTUD4 binds to His-Myd88 when purified from bacteria (Figure 4D). Taken together, these results demonstrated that OTUD4 associates directly with MyD88.
Figure 4. Identification of MyD88 as a specific target of OTUD4.

(a) Flag immunoprecipitation was performed from HEK293 cells expressing the indicated vectors and Western blotted as shown. Results are representative of three independent experiments. (b) HA immunoprecipitation was performed after expression of the indicated vectors and analyzed as shown. Results are representative of two independent experiments. (c) Whole cell extracts from mouse embryonic fibroblasts (MEF WCE) was immunoprecipitated using control IgG or OTUD4 antibody and Western blotted as shown. Results are representative of three independent experiments. (d) MBP and MBP-OTUD4 were co-expressed in E. coli with His-Flag-MyD88. After MBP pulldown, the bound material was analyzed by Western blot using the indicated antibodies. Results are representative of three independent experiments. (e) Flag-MyD88 and HA-Ub WT or mutants were expressed in HEK293 cells, Flag-immunoprecipitated after SDS denaturation, and blotted as shown. Results are representative of two independent experiments. (f) Flag immunoprecipitation was performed after SDS denaturation from HEK293 cells expressing Flag-MyD88 and HA-Ub (WT), along with OTUD4WT, OTUD4C45A, or empty vector as indicated. (g) K63-linked (K63 only) ubiquitination status of MyD88 was assessed as in (f). Results for (f) and (g) are representative of two independent experiments for each. Flag immunoprecipitation was performed from HEK293 cells expressing HA-tagged WT ubiquitin (h) or K63-linked ubiquitin (i), along with vectors as indicated. Results for (h) and (i) are representative of two independent experiments for each.
MyD88 has been previously shown to be ubiquitinated by K48 as well as K63-linked ubiquitin chains (Emmerich et al., 2013; Lee et al., 2011). Using K48-only and K63-only ubiquitin mutants, we confirmed that both linkage types of ubiquitin are associated covalently with MyD88, as determined by Flag-IP under denaturing conditions (Figure 4E). To determine whether OTUD4 may contribute to the ubiquitination status of MyD88, we co-expressed HA-Ub with either WT or C45A mutant OTUD4. Denatured immunoprecipitation of Flag-MyD88 revealed a reduction of its ubiquitination with WT OTUD4 but not with the C45A catalytic mutant (Figure 4F). Consistent with our in vitro evidence that OTUD4 is a K63-specific deubiquitinase, WT but not C45A OTUD4 significantly reduced MyD88 K63-linked ubiquitination (Figure 4G). Furthermore, neither the UIM mutant (OTUD4KRD) nor the phospho-mutant (OTUD4S202/204A) were able to deubiquitinate MyD88 (Figure 4H and 4I). Thus, all the molecular determinants that activate the K63-deubiquitinase function of OTUD4 appear to be critical for counteracting MyD88 K63 ubiquitination.
OTUD4 regulates MyD88-dependent NF-κB signalling
While the above experiments suggest that MyD88 may be a substrate for OTUD4, they do not demonstrate a role for OTUD4 in MyD88 during signaling. Since MyD88 is an important protein involved in NF-κB signaling via IL-1β and certain TLRs, we investigated whether OTUD4 can regulate this pathway. We created Otud4 knockout (KO) MEFs using CRISPR/Cas9 technology (Supplemental Figure S3A and S3B). These Otud4−/− MEFs and their parental Otud4+/+ counterparts were stimulated with IL-1β for 15 or 30 minutes, and immunofluorescence was performed using for the NF-κB p65. As expected, IL-1β induced the nuclear translocation of p65 in both cell types (Figure 5A). However, the apparent rate of p65 nuclear import was significantly greater in the Otud4−/− MEFs (Figure 5A and 5B). We next assessed NF-κB transcriptional activity in Otud4+/+ and Otud4−/− MEFs cells using a luciferase reporter. The Otud4−/− MEFs demonstrated significantly higher NF-κB reporter activity relative to Otud4+/+ cells during IL-1β stimulation (Figure 5C). Consistently, upon IL-1β treatment, Otud4−/− MEFs demonstrated significantly higher levels of IκBα phosphorylation relative to Otud4+/+ MEFs, as well as a more rapid rate of IκBα degradation (Figure 5D). We also found that OTUD4 may play a role in regulating p38/MAPK signaling in response to IL-1β S3D). A lower dose of IL-1β was required to induce degradation of IκBα in Otud4−/− MEFs (Figure 5E), strongly suggesting that OTUD4 suppresses IL-1β-dependent NF-κB signaling. However, these differences in NF-κB signaling between Otud4+/+ and Otud4−/− MEFs were significantly less apparent upon stimulation with TNFα (Supplemental Figure S3C), consistent with the notion that TNFα signals independently of MyD88 (Parameswaran and Patial, 2010).
Figure 5. OTUD4 negatively regulates NF-κB signalling.

(a) WT (Otud4+/+) or OTUD4 knockout (Otud4−/−) MEFs were stimulated with IL-1β for 15 or 30 minutes, and immunofluorescence was performed using an anti-p65 antibody, with Hoechst as nuclear counter stain. Scale bar, 10 μm. (b) Quantification of p65 nuclear translocation from (a). N=3 biological replicates for each cell line; * = p < 0.05 and error bars indicate +/− S.D. of the mean. (c) Otud4+/+ or Otud4−/− MEFs expressing NF-κB luciferase reporter were stimulated with IL-1β for luciferase activity assay. N=3 biological replicates for each cell line; * = p < 0.05 and error bars indicate +/− S.D. of the mean. (d) and (e) Cells were stimulated with IL-1β (0.1 ng/ml) and lysed at the indicated time (d) or increasing IL-1β dose (0.004, 0.02, 0.1, or 0.5 ng/ml) (e). Lysates were Western blotted with the antibodies as shown. Results for (d) and (e) are representative of three and two independent experiments, respectively. (f) Otud4+/+ or Otud4−/− MEFs were stimulated with IL-1β for the indicated time, and cell lysates were collected and incubated with Halo-NZF2 beads to isolate proteins modified by K63-linked Ub chains. Bound and input material were analyzed by Western blot with the antibodies as shown. Results are representative of two independent experiments.
MyD88 is a scaffold critical for the assembly of downstream effector proteins during canonical NF-κB signaling (West et al., 2006). Ubiquitination of certain downstream effectors, such as IRAK proteins and TRAF6, may promote this signaling pathway by recruiting factors that bind to K63-linked ubiquitin chains (Lork et al., 2017). Thus, we reasoned that in Otud4−/− cells, there may be a relative increase in the degree of K63-linked ubiquitination of MyD88, IRAKs, and TRAF6. To assess this potential role for OTUD4, we utilized TUBE (tandem ubiquitin binding entity) technology to isolate endogenously ubiquitinated proteins (Hjerpe et al., 2009; Strickson et al., 2017). Lystes from Otud4+/+ and Otud4−/− MEFs stimulated with IL-1β were prepared, and proteins modified by K63-linked ubiquitin chains were isolated using immobilized Halo-NZF2 (Figure 5F). Strikingly, the degree of K63-ubiquitinated MyD88, IRAK1, and TRAF6, were significantly increased in Otud4−/− MEFs relative to Otud4+/+ cells during IL-1β stimulation. These data suggest that MyD88, as well as downstream factors involved in IL-1β signaling, likely serve as endogenous substrates of OTUD4.
OTUD4 catalytic activity, phosphorylation and ubiquitin-interacting motif (UIM) regulate NF-κB signaling
Since OTUD4 appears to regulate MyD88-dependent signaling, we wished to determine whether phosphorylation of OTUD4 is altered during pathway activation. Immunoprecipitation of endogenous OTUD4 with or without prior stimulation with IL-1β revealed that a significantly greater amount of OTUD4 is phosphorylated during such signaling (Supplemental Figure S3E). On the other hand, the amount of MyD88 associated with OTUD4 appeared minimally changed under the same conditions (Supplemental Figure S3F). We then asked whether the catalytic activity of OTUD4, as well as its phosphorylation and UIM domain, were critical for regulation of IL-1β signaling. Otud4−/− MEFs were transduced with vectors to re-express WT OTUD4 or the catalytically inactive C45A. Expression of OTUD4WT, but not OTUD4C45A, reduced the relative accumulation of polyubiquitinated TRAF6 during IL-1β signaling, as well as the more rapid IκBα degradation apparent in Otud4−/− cells (Figure 6A). Similarly, re-expression of OTUD4WT in Otud4−/− cells, but not the phospho-mutant (OTUD4S202/204A) or the UIM mutant (OTUD4KRD), reduced IκBα phosphorylation and rescued IκBα degradation (Figure 6B). We also rescued p65 nuclear translocation in Otud4−/− cells during IL-1β signaling (Figure 6C–6E). Significantly, all three mutants defective for K63-DUB activity (OTUD4C45A, OTUD4S202/204A, and OTUD4KRD) mimicked the Otud4−/− phenotype in p65 nuclear translocation. Thus, the catalytic activity of OTUD4, as well as its phosphorylation and its UIM domain, are all important for its role in the regulation of canonical NF-κB signaling.
Figure 6. OTUD4 catalytic activity, phosphorylation and ubiquitin-interacting motif are critical in NF-κB signaling.

(a) Otud4−/− MEFs were transduced with HA-OTUD4 (WT or C45A) or control vector and stimulated with IL-1β. After incubation for the indicated time, lysates were collected and incubated with Halo-NZF2 beads. Bound and input material were analyzed by Western blot with the antibodies as shown. Results are representative of two independent experiments. (b) Otud4−/− MEFs were transduced with the indicated Flag vector, stimulated with IL-1β (1ng/ml) and lysed at indicated time for Western blot analysis as shown. Results are representative of two independent experiments. (c) Otud4−/− MEFs were transduced with Flag-OTUD4 (WT, C45A, KRD or S202A/S204A) or Flag-vector. Expression levels of the exogenous OTUD4 was determined by Western blot as shown. Results are representative of two independent experiments. (d) Cells from (c) were stimulated with IL-1β as indicated. Immunofluorescence was performed using an anti-p65 antibody, with Hoechst as nuclear counter stain. Scale bar, 10 μm. (e) Quantification of p65 nuclear translocation from (d). N=3 independent experiments for each cell line; * = p < 0.05 and error bars indicate +/− S.D. of the mean.
OTUD4 modulates innate immune signaling
To better understand the role of OTUD4 in vivo, we created Otud4 knockout mice using CRISPR/Cas9 technology (Figure 7A and Supplemental Figure S4A–C). While these mice were viable and did not have any gross phenotype, they were born at sub-Mendelian ratios (Supplemental Figure S4D–E). Because of the importance of MyD88 in IL-1 and TLR-dependent signaling, we focused on the role of OTUD4 in immune cells. Under basal conditions, analysis of splenocytes from Otud4−/− mice revealed no apparent difference in the various immune cell populations compared to WT littermate controls (Supplemental Figure S5A–B). We then focused on bone marrow-derived GM-CSF cell cultures, as OTUD4 is expressed in these cells (Figure 7B), and MyD88-dependent signaling is important for their activation (West et al., 2006). At baseline, IκBα levels were not significantly different in these cells from Otud4+/+ compared to Otud4−/− animals (Figure 7C). However, upon IL-1β stimulation, these cells exhibited significantly greater degradation of IκBα, consistent with a role of OTUD4 in modulating NF-κB signaling. We then analyzed a prototypical NF-κB target gene, Mip1a, and found that upon LPS Mip1a transcripts were significantly more sustained in the Otud4−/− bone marrow-derived cells (Figure 7D). Consistent with these data, TNFα and IL-6 production were both increased significantly in the Otud4−/− cells in response to LPS, a potent TLR ligand (Figure 7E–F) (West et al., 2006). Macrophages produce increased levels of nitric oxide (NO) upon activation, and loss of OTUD4 increased NO production as measured by nitrite assay (Figure 7G), further suggesting that OTUD4 may play a role in modulating the innate immune system. In response to heat-killed Listeria monocytogenes (HKLM), another TLR ligand, TNFα and IL-6 production were also increased in Otud4−/− cells. Taken together, these results provide a mechanistic basis for a non-proteasomal function of OTUD4 in vivo. In conjunction with our previous work on the role of OTUD4 in stabilizing alkylation repair enzymes (Zhao et al., 2015), our data suggests that OTUD4 can play multiple roles through very distinct mechanisms.
Figure 7. Otud4 suppresses inflammatory TLR signaling in vivo.

(a) CRISPR/Cas9 strategy for targeting mouse Otud4−/− using dual gRNAs (red arrows). One resulting allele deleted 113 nucleotides in exon 7 and was used for further studies. (b) Lysates of GM-CSF-derived bone marrow cultured cells from Otud4+/+ or Otud4−/− mice were Western blotted as shown. Results are representative of two independent experiments. (c) Cells from (b) were stimulated with IL-1β (0.1 pg/ml) for 10–30 minutes, and lysates were analyzed by Western blot using the indicated antibodies. Results are representative of three independent experiments. (d) Expression of the NF-κB target gene Mip1a was quantified by qRT-PCR from LPS-stimulated GM-CSF-derived bone marrow cultured cells. Values were normalized to β-actin. N=4 biological replicates from each group, and error bars indicate +/−S.D. of the mean. (e) and (f) TNFα and IL-6 production from GM-CSF-derived bone marrow cultured cells incubated with IFN-γ and stimulated with LPS was determined by ELISA. N=4 biological replicates from each group, and error bars indicate +/−S.D. of the mean. (g) Nitric oxide production was determined from the same cells by nitrite assay. N=4 mice from each group, and error bars indicate +/−S.D. of the mean. (h) and (i) TNFα and IL-6 production was assessed by ELISA from GM-CSF-derived bone marrow cultured cells incubated with IFN-γ and stimulated with heat-killed Listeria monocytogenes (HKLM). N=4 biological replicates from each group, and error bars indicate +/−S.D. of the mean. * = p < 0.05 by Student’s t-test.
Discussion
We had previously shown that OTUD4 regulates the stability of alkylation repair enzymes (ALKBH2 and ALKBH3) by counteracting K48-linked ubiquitination in a manner that was independent of its catalytic activity (Zhao et al., 2015). This was due to the ability of OTUD4 to associate with two other deubiquitinases of the USP family, thus functioning as a scaffold to bring together substrate and additional deubiquitinase activities. However, the catalytic domain of OTUD4 is highly conserved in many species, thus suggesting that it has another unidentified function. In this study we reveal a surprising and distinct role for its catalytic activity, by showing that OTUD4 specifically deubiquitinates K63-linked chains (Figure 1). This activity depends on the phosphorylation of OTUD4 near its catalytic domain (Figure 2) as well as a putative UIM motif adjacent to the OTU domain (Figure 3). At least one function of this catalytic activity is to bind and counteract the K63-linked ubiquitination of MyD88 and other factors associated with MyD88-dependent signaling (Figures 4–5). Consistent with this, loss of OTUD4 hypersensitizes cells to canonical NF-κB signaling (Figure 5).
Unlike its role in the stabilization of alkylation repair factors, the catalytic activity of OTUD4 is critical for this function in countering MyD88 ubiquitination (Figure 4). The phosphorylation of OTUD4, as well its putative UIM motif, are also important for MyD88 deubiquitination (Figure 4), and consistently, these features are also critical for regulating IL-1β signaling (Figure 6). Similar to OTUD1, which also deubiquitinates K63-linked chains but without phosphorylation (Mevissen et al., 2013), the UIM motif appears to increase affinity for this substrate (Figure 3). While the precise molecular mechanism by which phosphorylation activates the K63 DUB activity of OTUD4 will require structural studies, it is possible that its phosphorylation cooperates with the UIM motif to increase its affinity for this substrate. This is somewhat reminiscent of the activation mechanism of DUBA/OTUD5 by phosphorylation, which also depends on a UIM, yet in the case of DUBA phosphorylation activates a relatively less chain-specific DUB (Huang et al., 2012). We surmise that this phosphorylation-dependent regulation found in OTUD4 may be a more general feature found in other OTU family members. For example, A20 phosphorylation by IKK has also been shown to activate a relatively latent K63 DUB activity (Wertz et al., 2015). In this regard, it is interesting to note that at least two other OTU DUB family members, OTUD3 and Cezanne/OTUD7B, have putative phosphorylation sites in an acidic amino acid context near their catalytic domains, similar to OTUD4. Thus their substrate specificities may be regulated in a similar fashion.
The ability of OTUD4 to target K63-linked MyD88 establishes another major node of regulation in the IL-1/TLR-mediated NF-κB pathway. Very recent work has demonstrated that TRAF6 as well as Pellino E3 ubiquitin ligases are responsible for the induction of this post-translational modification on MyD88 (Strickson et al., 2017). Our results strongly suggest that OTUD4 reverses K63-linked ubiquitination on MyD88 and associated proteins, thus playing a key role in modulating MyD88 dependent signaling (Figure 5). While a number of other deubiquitinases, in particular A20 and CYLD, have been established to regulate this pathway, to our knowledge neither have been shown to directly target MyD88. More importantly, their substrate specificity may be relatively broader, such that they also govern TNFα and NOD signaling by targeting downstream effectors common to these pathways, including TRAF proteins, RIPK, and likely others (Brummelkamp et al., 2003; Kovalenko et al., 2003; Trompouki et al., 2003; Wertz et al., 2004). Our proteomics data does not identify factors belonging to these pathways in association with OTUD4, consistent with additional data that loss of OTUD4 has minimal effect on TNFα-mediated activation of NF-κB (Supplemental Figure S3). Thus OTUD4 is uniquely positioned as a deubiquitinase that targets MyD88-dependent signaling, potentially without having an effect on other innate immune pathways.
What could be connection between the regulation of innate immune signaling by OTUD4 and its role in the stabilization of alkylation repair enzymes? Inflamed tissues are known to have increased levels of alkylated nucleotides, indirectly as a result of increased reactive oxygen and nitrogen species (RONS), which in turn react with polyunsaturated lipids to produce peroxides that produce alkylated bases on DNA and RNA (Chung et al., 1996). Strikingly, alkylation damage repair is essential for survival in mice after induction of chemically induced colitis, and loss of the dealkylating enzyme ALKBH3 induces an inflammatory gene signature (Calvo et al., 2012; Liefke et al., 2015), suggesting a strong connection between removal of alkylated lesions and preserving an anti-inflammatory state. Thus the overall function of OTUD4 may be to maintain such a state by stabilizing alkylation repair enzymes while counteracting MyD88-dependent inflammatory signaling. Future work will be needed to further understand connection between these two apparently disparate processes.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Nima Mosammaparast (nima@wustl.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
MEFs and HEK293 cells were cultured in Dulbecco’s modified eagle medium (Invitrogen), supplemented with 10% fetal bovine serum (Atlanta Biologicals), 100 U/ml of penicillin-streptomycin (Gibco) at 37°C and 5% CO2.
Mouse bone marrow culture
Bone marrow was harvested from femur and tibias by flushing to generate bone marrow-derived GM-CSF cell cultures. Cells were cultured for 7 days in RPMI 1640 media containing 10% fetal calf serum (FCS), 100 U/ml of penicillin-streptomycin, L-glutamine, 2-mercaptoethanol (2-ME) and GM-CSF (20 ng/ml; PeproTech) in 6-well plates at 37°C and 8% CO2.
Mice
C57BL/6 mice were bred and maintained in our animal facility according to institutional guidelines and with protocols approved by the Animal Studies Committee of Washington University in St. Louis.
METHOD DETAILS
Plasmids
Full-length, human OTUD4 cDNA was used as the template for the creation of subsequent mutants (Zhao et al, 2015). All OTUD4 mutations were produced by PCR-mediated mutagenesis and cloned into pENTR-3C or pENTR-4 (Invitrogen), then subcloned into pHAGE-CMV-Flag or pHAGE-CMV-HA by Gateway recombination. MyD88 cDNA was obtained from Addgene (clone #12287), cloned by PCR into pENTR-3C and subcloned into the expression vectors as above. For recombinant protein expression, the relevant portion of OTUD4 was cloned into pET28a-Flag (Zhao et al, 2015). All constructs derived by PCR, including deletions and point mutations (as shown in the Key Resources table), were confirmed by Sanger sequencing.
KEY RESOURCES TABLE.
| Reagent or Resource: | Source: | Identifier: |
|---|---|---|
| Antibodies (for Western blotting and immunofluorescent microscopy) | ||
| 6x-His (Mouse) | Abcam | ab18184 |
| Flag (Mouse) | Sigma | F3165 |
| HA (Mouse) (Rabbit) | BioLegend | 901501 |
| IκBα(Mouse) | Cell Signaling | 9242 |
| p-IκBα (Mouse) | Cell Signaling | 9246 |
| IRAK4 (Rabbit) | Cell Signaling | 4363 |
| MBP (Rabbit) | NEB | E8030 |
| OTUD4 (Rabbit) | Sigma | HPA036623 |
| P65 (Rabbit) | Cell Signaling | 8242 |
| pCK2 substrate (pS/pT-DXE; Rabbit) | Cell Signaling | 8738 |
| pS202 OTUD4 (Rabbit) | This paper (Primm Biotech) | N/A |
| TRAF6 (Rabbit) | Abcam | ab33915 |
| Ubiquitin (Mouse) | Santa Cruz | SC-8017 |
| β-actin HRP (Mouse) | Sigma | A3854 |
| Antibodies (for Western blotting and immunofluorescent microscopy) | ||
| Alexa Fluor 488 B220 | BioLegend | RA3–6B2 |
| Biotin DX5 (CD49b) | BioLegend | DX5 |
| Streptavidin PE | Tonbo | |
| PE/Cy7 CD4 | BioLegend | RM4–5 |
| APC TCRγδ | BioLegend | GL3 |
| APC/Cy7 CD19 | BioLegend | 6D5 |
| PerCP/Cy5.5 CD8α | Tonbo | 53–6.7 |
| Pacific Blue TCRβ | BioLegend | H57–597 |
| PE F4/80 | Tonbo | BM8.1 |
| Alexa Fluor 647 CD11c | BioLegend | N418 |
| APC/Cy7 CD11b | Tonbo | M1/70 |
| Pacific Blue Ly6C | BioLegend | HK1.4 |
| Chemicals, Peptides, and Purified Proteins | ||
| Protein A/G Beads | Santa Cruz | SC-2003 |
| Anti-Flag (M2) Beads | Sigma | A2220 |
| Anti-HA Beads | Santa Cruz | SC-7392AC |
| Ni-NTA Beads | Thermo | 88221 |
| Amylose Resin | NEB | E8021 |
| Flag-His-OTUD4 (1–300; bacterial) | This paper | N/A |
| Flag-His-OTUD4 (1–300; C45A; | This paper | N/A |
| bacterial) | ||
| Flag-OTUD4 (FL, 1–300; 1–270, 1–254, 1–227, 1–180, and point mutants; HEK293) | This paper | N/A |
| Flag-OTUD4 (FL C45A, C45S; HEK293) |
This paper | N/A |
| Flag Pepide | Sigma | F3290 |
| Critical Commercial Assays and Reagents | ||
| Luciferase Assay System | Promega | E1500 |
| TNFα ELISA | BioLegend | 430901 |
| IL-6 ELISA | BioLegend | 431301 |
| IL-1β | Peprotech | 211–11B |
| TNFα | Peprotech | 300–01A |
| LPS | Sigma | L2880 |
| Recombinant DNA | ||
| pHAGE-Flag-OTUD4 | Zhao et al., 2015 | N/A |
| pHAGE-Flag-OTUD4 (C45A) | Zhao et al., 2015 | N/A |
| pHAGE-Flag-OTUD4 (C45S) | Zhao et al., 2015 | N/A |
| pHAGE-Flag-OTUD4 (1–300; 1270; 1–254; 1–227) | Zhao et al., 2015 and this paper | N/A |
| pHAGE-Flag-OTUD4 (1–300 C45A) | This paper | N/A |
| pHAGE-Flag-OTUD4 (1–180) | Zhao et al., 2015 | N/A |
| pET-Flag-OTUD4 (1–300) | This paper | N/A |
| pET-Flag-OTUD4 (1–300 C45A) | This paper | N/A |
| pET-Flag-OTUD4 (1–180) | Zhao et al., 2015 | N/A |
| pHAGE-Flag-OTUD4 (1–300 phosphomutants) | This paper | N/A |
| pHAGE-Flag-OTUD4 (KRD273–5AAA) | This paper | N/A |
| pHAGE-Flag-OTUD4 (1–300, C45S) | This paper | N/A |
| pHAGE-Flag-OTUD4 (1–300, C45S, KRD273–5AAA) | This paper | N/A |
| pHAGE-HA-OTUD4 | This paper | N/A |
| pHAGE-Flag-MyD88 | This paper | N/A |
| pMAL-OTUD4 | Zhao et al., 2015 | N/A |
| pET-Flag-MyD88 | This paper | N/A |
| pcDNA-HA-Ub | Laboratory of Yang Shi | N/A |
| pRK5-HA-Ub (K48 only) | Addgene | 17605 |
| pRK5-HA-Ub (K63 only) | Addgene | 17606 |
| Luciferase reporter construct | Addgene | 49343 |
| shRNA Vectors | ||
| CK2α | Thermo Scientific | TRCN0000000607 |
| CK2α | Thermo Scientific | TRCN0000000611 |
| Oligonucleotides – mouse genotyping | ||
| Otud4 (forward) | IDT | 5’-AAAACATGCCTCTAACGTTG-3’ |
| Otud4 (reverse) | IDT | 5’-CCCTGGATGACACTAGAAAGAC-3’ |
| Oligonucleotides – qRT-PCR | ||
| Mip1a (forward) | IDT | 5’-CCCAGCCAGGTGTCATTTTCC-3’ |
| Mip1a (reverse) | IDT | 5’-GCATTCAGTTCCAGGTCAGTG-3’ |
| Actb (forward) | IDT | 5’-TCATCACTATTGGCAACGAGCGGTTC-3’ |
| Actb (reverse) | IDT | 5’-TACCACCAGACAGCACTGTGTTGGCA-3’ |
| Deposited Data Link: https://data.mendeley.com/datasets/d576pn2h5s/draft?a=e43e7821-9769-4bda-b6ef-f8270f238313 | ||
Transfection, virus production, and transduction
HEK293T cells were transfected using TransIT-293 reagent (Mirus Bio). All virus production was performed by transfection of HEK293T cells with helper vectors (pVSV-G and pGag-Pol for pMSCV vectors; MISSION lentiviral packaging mix for pLKO.1 and pHAGE vectors). Supernatant was collected 48–72 hours after transfection, and added to cells to be transduced in the presence of polybrene (4 μg/ml). For OTUD4 knockout cell rescue experiments, cells were transduced with the Flag-OTUD4 lentiviral vectors.
Protein purification
Recombinant His-tagged proteins were purified from Rosetta (DE3) using an ÄKTA-pure FPLC (GE Healthcare). Cells were resuspended in His-lysis buffer (50 mM Tris-HCl pH 7.9, 150 mM NaCl, 1% Triton X-100, 2 mM DTT, 20 mM imidazole, and protease inhibitors) and lysed by sonication. After centrifugation and filtration, the extract was loaded onto a HisTrap HP column (GE Healthcare). After extensive washing with lysis buffer, the protein was eluted using lysis buffer containing 400 mM imidazole. The eluted proteins were dialyzed into TBS buffer (50 mM Tris-HCl pH 7.9, 150 mM NaCl, 5% glycerol and 3 mM β−ME). For purification of OTUD4 from HEK293 cells, Flag-OTUD4 was expressed by transient transfection or by stable lentiviral expression. Cells were harvested in 1X PBS, resuspended in lysis buffer (50 mM Tris-HCl pH 7.9, 150 mM NaCl, 1% Triton X-100, 2 mM DTT, protease inhibitors, and phosphatase inhibitor cocktail), and the lysate was incubated with anti-Flag (M2) agarose (Sigma) after clearing the lysate by centrifugation. The bound material was eluted using lysis buffer containing 0.5 mg/ml Flag peptide.
Phosphorylation site identification by mass spectrometry
The excised gel band containing Flag-OTUD41−300 was cut into approximately 1 mm pieces. Gel pieces were then subjected to a modified in-gel trypsin digestion procedure (Shevchenko et al., 1996). Gel pieces were washed and dehydrated with acetonitrile for 10 minutes. Acetonitrile was removed and gel pieces were completely dried in a speed-vac. The gel was rehydrated with 50 mM ammonium bicarbonate solution containing 12.5 ng/μl modified sequencing-grade trypsin (Promega) at 4°C. After 45 minutes, the excess trypsin solution was removed and replaced with 50 mM ammonium bicarbonate solution to just cover the gel pieces. Samples were incubated at 37°C overnight. Peptides were extracted by removing the ammonium bicarbonate solution, followed by one wash with a solution containing 50% acetonitrile and 1% formic acid. The extracts were dried in a speed-vac. For LC-MS/MS, peptides were resuspended in 6μl 1% formic acid and analyzed on an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific) equipped with a Proxeon Easy nLC 1000 for online sample handling and peptide separations. A portion of the peptides was loaded onto a 100 μm inner diameter fused-silica micro capillary with a needle tip pulled to an internal diameter less than 5 μm. The column was packed in-house to a length of 35 cm with a C18 reverse phase resin (GP118 resin 1.8 μm, 120 Å, Sepax Technologies). The peptides were separated using a 120 minute linear gradient from 3% to 25% buffer B (100% ACN + 0.125% formic acid) equilibrated with buffer A (3% ACN + 0.125% formic acid) at a flow rate of 600 nL/min across the column. The scan sequence for the Fusion Orbitrap began with an MS1 spectrum (Orbitrap analysis, resolution 120,000, 400−1400 m/z scan range, AGC target 2 × 105, maximum injection time 100 ms, dynamic exclusion of 30 seconds). The most intense precursor from each MS1 scan was selected for MS2 analysis. Peptides were isolated in the quadrupole using an isolation window of 0.5 Da, fragmented by HCD with a collision energy of 35%, and analyzed in the orbitrap (resolution 60,000, 350–1400 m/z scan range, AGC target 2 × 104) with a maximum injection time of 150 ms. The remaining peptides were analyzed over an 85 minute linear gradient using a “most intense” method in which MS1 collection was the same as above, but MS2 analysis after quadrapole isolation (isolation window 0.7 Da) was performed using CID activation with a collision energy of 30% followed by fragment ion detection in the ion trap (350–1400 m/z scan range, 35 ms maximum injection time, AGC target 1 × 104). Data analysis was performed using a suite of in-house software tools for .RAW file processing and controlling peptide and protein level false discovery rates, assembling proteins from peptides, and phosphopeptide identification (Huttlin et al., 2010).
CRISPR/Cas9 mediated knockout of OTUD4 in MEFs
MEF knockout cells were created using CRISPR/Cas9 genome editing at the Genome Engineering and iPSC Center (GEiC) at Washington University School of Medicine (St. Louis). Clones were initially assessed by deep sequencing and confirmed by Western blotting. The gRNA sequence used to generate the OTUD4 knockout cell line was: 5’-TGAAGAATATTTAAAGCGTT-3’.
Immunofluorescence microscopy
After stimulation with IL-1β, MEFs were fixed with 3.2% paraformaldehyde, then washed extensively with IF Wash Buffer (1X PBS, 0.5% NP-40, and 0.02% NaN3). The cells were blocked with IF Blocking Buffer (IF Wash Buffer plus 10% FBS) for at least 30 minutes. Primary antibodies were diluted in IF Blocking Buffer and incubated overnight at 4°C. After staining with secondary antibodies (conjugated with Alexa Fluor 488 or 594; Millipore) and Hoechst 33342 (Sigma-Aldrich), where indicated, samples were mounted using Prolong Gold mounting medium (Invitrogen). Epifluorescent microscopy was performed on an Olympus fluorescent microscope (BX-53) using an ApoN 60X/1.49 NA oil immersion lens or an UPlanS-Apo 100X/1.4 oil immersion lens and cellSens Dimension software (Zhao et al., 2015).
In vitro deubiquitinase assays
All ubiquitin substrates were from Boston Biochem. Deubiquitination (DUB) assays were performed in enzyme buffer (50 mM Tris-HCl pH 7.9, 150 mM NaCl, 5% glycerol, 2 mM DTT) at 37°C containing the indicated amount of enzyme in a volume of 25 μl. The reactions were incubated for the times indicated in the figures. Reactions were stopped by the addition of Laemmli buffer and analyzed by Western blot.
MBP or Flag-tagged protein pulldown assays
All in vitro binding assays were performed with amylose agarose resin (NEB) or Flag (M2) agarose (Sigma). MBP-tagged protein pulldown assays were performed by resuspension of E. coli co-expressing MBP and His-Flag-tagged proteins in lysis buffer. After sonication and centrifugation, proteins were isolated on amylose resin, washed extensively with lysis buffer, and eluted using Laemmli buffer. For ubiquitin binding assays, Flag-tagged catalytically inactive mutant (C45S) Flag-OTUD41−300 or mutant (C45S/KRD) was added to each reaction, along with 500ng of the indicated type of ubiquitin chain (Boston Biochem). The indicated proteins were added to 10μl of beads in a total volume of 100μl with lysis buffer. Reactions were incubated at 4°C with rotation for 1 hour, then washed extensively with lysis buffer. The bound material was eluted with Laemmli buffer or using Flag peptide, analyzed by SDS-PAGE, and then Western blotted as indicated.
Immunoprecipitation and Western blotting
HEK293 cells were transfected with plasmids as indicated using TransIT-293 (Mirus Bio) for 2–3 days. Cells were harvested, resuspended in lysis buffer (50 mM Tris–HCl pH 7.9, 150 mM NaCl, 10% glycerol, 1% Triton X-100, 2 mM DTT, phosphatase inhibitors and protease inhibitors), cleared by centrifugation, and incubated at 4°C with M2-agarose beads (Sigma) or HA-agarose beads (Santa Cruz). The beads were then washed in the same buffer five times, and bound material was eluted using Laemmli buffer and analyzed by Western blotting. Endogenous immunoprecipitation was performed from MEF cell extracts. Cells were harvested as above, resuspended in lysis buffer, and incubated overnight at 4°C with 3 μg of antibody and 2.5 mg of BSA. The extract was then incubated with Protein A/G- agarose (Santa Cruz) for 1 h at 4°C, centrifuged, and washed extensively in lysis buffer. For immunoprecipitation after denaturation, the extracts were resuspended in TBS with 1% SDS, boiled, and then diluted with lysis buffer to 0.1% SDS before immunoprecipitation. Bound material was eluted using Laemmli sample buffer and analyzed by Western blotting. All Western blotting was carried out using PVDF membrane and the indicated antibodies. Protein bands were visualized by enhanced chemiluminescence (Bio-Rad).
TUBE pulldowns with Halo-NZF2
K63-ubiquitin chains and ubiquitinated proteins were captured from cell extracts using Halo-NZF2 beads (Strickson et al., 2017). After stimulation with IL-1β, cells were resuspended in lysis buffer (50 mM Tris-HCl pH 7.9, 150 mM NaCl, 1% Triton X-100, 2 mM DTT, plus protease inhibitors). Extracts were incubated with 30 μl of Halo-Tag beads (Promega) bound to Halo-NZF2 (20 μg/pulldown). After overnight incubation at 4°C, the beads were washed twice with 1.0 mL of 50 mM Tris-HCl pH 7.9, 500 mM NaCl, and 1% Triton X-100, and three times with lysis buffer. The bound material was eluted with Laemmli buffer, analyzed by SDS-PAGE, and then Western blotted with the indicated antibodies.
CRISPR/Cas9 knockout mouse
Otud4−/− mice were created using CRISPR/Cas9 technology at the Washington University Genome Engineering and iPSC Center. Female C57BL/6 mice were super-ovulated using 5 IU of Pregnant Mares Serum Gonadotropin followed by 5 IU of Human Chorionic Gonadotropin 48 hours later. The females were then mated to C57BL/6 male mice and day 0.5 embryos were isolated the morning after mating. The fertile single cell embryos underwent pronuclear micro-injection delivering the CRISPR gRNA mixed with RNA encoding Cas9. The concentration of the injection mix was 5ng/μl gRNA with 10 ng/μl Cas9 RNA. Tail samples were taken from pups and deep sequencing was performed to identify animals carrying indels, and the exact modification that occurred. The founder male mouse with the Δ113 allele was selected and mated to a C57BL/6 female to isolate this allele, and heterozygous progeny were mated to generate the homozygous mutant mice.
FACS analysis of mouse splenocytes
Spleens were harvested and digested with collagenase B (250 μg/ml; Roche) and DNase I (30 U/ml; EMD) for 40–60 min at 37°C with stirring in Iscove’s modified Dulbecco’s media containing 10% FCS, L-glutamine, sodium pyruvate, non-essential amino acids, penicillin/streptomycin and β-ME. The reaction was stopped by addition of EDTA to a final concentration of 5 mM, and cells were incubated on ice for 5 min. Red blood cells were lysed with ACK lysis buffer. Cells were passed through a 70-μm strainer before cell counting with acetic acid. Cell surface staining was performed in FACS buffer (0.5% BSA, 2 mM EDTA, 0.02% NaN3 in 1X PBS) in the presence of Fc receptor blocking antibody (Clone 93, BioLegend) for 20 min at 4 °C followed by streptavidin staining if necessary. Cells were fixed in 4% paraformaldehyde (Electron Microscopy Sciences) for 20 min at room temperature. Flow cytometry was performed on a FACSCanto II (BD Bioscience) and data was analyzed with FlowJo (Tree Star Inc.).
ELISA and NO assay
Responses of bone marrow-derived GM-CSF cell cultures to heat-killed Listeria monocytogenes strain EGD (HKLM) and LPS macrophages were assessed after seven days of culture. Cells were incubated in media with or without 50 ng/ml murine IFN-γ (as indicated in the figure legends) in 96-well plates at 5 X 104 cells/well. Supernatants were collected after 48 h and assessed for TNFα and IL-6 by ELISA per manufacturer’s instructions. Nitrite was measured using Greiss reagent (Edelson and Unanue, 2002).
RNA extraction and real-time quantitative PCR (RT-qPCR) analysis
Total RNA was isolated from 1 X 106 BMDMs, with or without LPS stimulation for three or six hours, using the RNeasy Mini Kit (Qiagen) following the manufacturer’s instructions. 400 ng of RNA was used for reverse transcription using poly(dT) oligonucleotide and M-MuLV Reverse Transcriptase (NEB) in a total volume of 20 μL. Each reaction was then diluted with 80 μl of water, and 2.5 μl of the diluted cDNA was used for real-time PCR. Real-time PCR was performed with SYBR Green Jumpstart Taq ReadyMix (Sigma) on a Stratagene Mx3000 real-time PCR machine. Relative quantification was calculated using the ΔΔCt method with β-actin as the internal control.
QUANTIFICATION AND STATISTICAL ANALYSIS
Where relevant, data is represented as the mean of the indicated number of independent replicates. Error bars indicate +/− standard deviation of the mean. All statistical significance was determined by Student’s T-test.
DATA AND SOFTWARE AVAILABILITY
Primary data can be found on Mendeley (doi:10.17632/d576pn2h5s.1).
Supplementary Material
SUPPLEMENTAL TABLE S1. Related to Figure 4. Interactome analysis of cytosolic Flag-OTUD4. Total and unique peptides for each interacting protein is shown.
Highlights.
OTUD4 phosphorylation activates a K63-specific deubiquitinase activity
OTUD4 K63 deubiquitinase activity requires a ubiquitin-interacting motif
OTUD4 K63 deubiquitinase activity targets MyD88
Otud4−/− cells are hypersensitive to MyD88-dependent TLR signaling
Acknowledgments
We wish to thank Gene Oltz, Yang Shi, Alessandro Vindigni, and members of the Mosammaparast and Edelson labs for their advice on this manuscript. J.M.S. is supported by a Monsanto Graduate Program Fellowship. A.K.B. is supported by the NIH Cell and Molecular Biology Training Grant (5T32GM007067-40). We thank J. Michael White of the Department of Pathology and Immunology Micro-Injection core facility, as well as the Alvin J. Siteman Cancer Center at Washington University and Barnes-Jewish Hospital for the use of the GEiC Core. The Siteman Cancer Center is supported in part by an NCI Cancer Center Support Grant (P30 CA091842; Eberlein, PI). This work was supported by the NIH (R01 AI113118 to B.T.E., and K08 CA158133 and R01 CA193318 to N.M.), a Career Award for Medical Scientists from the Burroughs Wellcome Fund (to B.T.E), the American Cancer Society (IRG-58-010-56 to N.M.), the Alvin Siteman Cancer Research Fund (15-FY17-01 to N.M.), the Siteman Investment Program (to N.M.), and the Children’s Discovery Institute of St. Louis Children’s Hospital (MC-II-2015-453 to N.M.).
Footnotes
Declaration of Interests
The authors have no conflicts of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bremm A, Freund SM, and Komander D (2010). Lys11-linked ubiquitin chains adopt compact conformations and are preferentially hydrolyzed by the deubiquitinase Cezanne. Nature structural & molecular biology 17, 939–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummelkamp TR, Nijman SM, Dirac AM, and Bernards R (2003). Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature 424, 797–801. [DOI] [PubMed] [Google Scholar]
- Calvo JA, Meira LB, Lee CY, Moroski-Erkul CA, Abolhassani N, Taghizadeh K, Eichinger LW, Muthupalani S, Nordstrand LM, Klungland A, et al. (2012). DNA repair is indispensable for survival after acute inflammation. The Journal of clinical investigation 122, 2680–2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung FL, Chen HJ, and Nath RG (1996). Lipid peroxidation as a potential endogenous source for the formation of exocyclic DNA adducts. Carcinogenesis 17, 2105–2111. [DOI] [PubMed] [Google Scholar]
- Edelson BT, and Unanue ER (2002). MyD88-dependent but Toll-like receptor 2-independent innate immunity to Listeria: no role for either in macrophage listericidal activity. J Immunol 169, 3869–3875. [DOI] [PubMed] [Google Scholar]
- Emmerich CH, Ordureau A, Strickson S, Arthur JS, Pedrioli PG, Komander D, and Cohen P (2013). Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proceedings of the National Academy of Sciences of the United States of America 110, 15247–15252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herhaus L, Perez-Oliva AB, Cozza G, Gourlay R, Weidlich S, Campbell DG, Pinna LA, and Sapkota GP (2015). Casein kinase 2 (CK2) phosphorylates the deubiquitylase OTUB1 at Ser16 to trigger its nuclear localization. Science signaling 8, ra35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjerpe R, Aillet F, Lopitz-Otsoa F, Lang V, England P, and Rodriguez MS (2009). Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin-binding entities. EMBO reports 10, 1250–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Brittain GC, Chang JH, Puebla-Osorio N, Jin J, Zal A, Xiao Y, Cheng X, Chang M, Fu YX, et al. (2013). OTUD7B controls non-canonical NF-kappaB activation through deubiquitination of TRAF3. Nature 494, 371–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang OW, Ma X, Yin J, Flinders J, Maurer T, Kayagaki N, Phung Q, Bosanac I, Arnott D, Dixit VM, et al. (2012). Phosphorylation-dependent activity of the deubiquitinase DUBA. Nature structural & molecular biology 19, 171–175. [DOI] [PubMed] [Google Scholar]
- Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ME, and Gygi SP (2010). A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143, 1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komander D, and Rape M (2012). The ubiquitin code. Annual review of biochemistry 81, 203–229. [DOI] [PubMed] [Google Scholar]
- Kovalenko A, Chable-Bessia C, Cantarella G, Israel A, Wallach D, and Courtois G (2003). The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature 424, 801–805. [DOI] [PubMed] [Google Scholar]
- Kristariyanto YA, Abdul Rehman SA, Campbell DG, Morrice NA, Johnson C, Toth R, and Kulathu Y (2015). K29-selective ubiquitin binding domain reveals structural basis of specificity and heterotypic nature of k29 polyubiquitin. Mol Cell 58, 83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Park JS, Kim JH, Jung SM, Lee JY, Kim SJ, and Park SH (2011). Smad6-specific recruitment of Smurf E3 ligases mediates TGF-beta1-induced degradation of MyD88 in TLR4 signalling. Nature communications 2, 460. [DOI] [PubMed] [Google Scholar]
- Licchesi JD, Mieszczanek J, Mevissen TE, Rutherford TJ, Akutsu M, Virdee S, El Oualid F, Chin JW, Ovaa H, Bienz M, et al. (2012). An ankyrin-repeat ubiquitin-binding domain determines TRABID’s specificity for atypical ubiquitin chains. Nature structural & molecular biology 19, 62–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liefke R, Windhof-Jaidhauser IM, Gaedcke J, Salinas-Riester G, Wu F, Ghadimi M, and Dango S (2015). The oxidative demethylase ALKBH3 marks hyperactive gene promoters in human cancer cells. Genome medicine 7, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lork M, Verhelst K, and Beyaert R (2017). CYLD, A20 and OTULIN deubiquitinases in NF-kappaB signaling and cell death: so similar, yet so different. Cell death and differentiation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mevissen TE, Hospenthal MK, Geurink PP, Elliott PR, Akutsu M, Arnaudo N, Ekkebus R, Kulathu Y, Wauer T, El Oualid F, et al. (2013). OTU deubiquitinases reveal mechanisms of linkage specificity and enable ubiquitin chain restriction analysis. Cell 154, 169–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mevissen TET, and Komander D (2017). Mechanisms of Deubiquitinase Specificity and Regulation. Annual review of biochemistry 86, 159–192. [DOI] [PubMed] [Google Scholar]
- Michel MA, Elliott PR, Swatek KN, Simicek M, Pruneda JN, Wagstaff JL, Freund SM, and Komander D (2015). Assembly and specific recognition of k29- and k33-linked polyubiquitin. Mol Cell 58, 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsten ME, and Litchfield DW (2004). Order or chaos? An evaluation of the regulation of protein kinase CK2. Biochem Cell Biol 82, 681–693. [DOI] [PubMed] [Google Scholar]
- Parameswaran N, and Patial S (2010). Tumor necrosis factor-alpha signaling in macrophages. Critical reviews in eukaryotic gene expression 20, 87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwertman P, Lagarou A, Dekkers DH, Raams A, van der Hoek AC, Laffeber C, Hoeijmakers JH, Demmers JA, Fousteri M, Vermeulen W, et al. (2012). UV-sensitive syndrome protein UVSSA recruits USP7 to regulate transcription-coupled repair. Nature genetics 44, 598–602. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, and Mann M (1996). Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem 68, 850–858. [DOI] [PubMed] [Google Scholar]
- Stewart MD, Ritterhoff T, Klevit RE, and Brzovic PS (2016). E2 enzymes: more than just middle men. Cell Res 26, 423–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickson S, Emmerich CH, Goh ETH, Zhang J, Kelsall IR, Macartney T, Hastie CJ, Knebel A, Peggie M, Marchesi F, et al. (2017). Roles of the TRAF6 and Pellino E3 ligases in MyD88 and RANKL signaling. Proceedings of the National Academy of Sciences of the United States of America 114, E3481–E3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swatek KN, and Komander D (2016). Ubiquitin modifications. Cell Res 26, 399–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, and Mosialos G (2003). CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature 424, 793–796. [DOI] [PubMed] [Google Scholar]
- Wertz IE, Newton K, Seshasayee D, Kusam S, Lam C, Zhang J, Popovych N, Helgason E, Schoeffler A, Jeet S, et al. (2015). Phosphorylation and linear ubiquitin direct A20 inhibition of inflammation. Nature 528, 370–375. [DOI] [PubMed] [Google Scholar]
- Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, et al. (2004). De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 430, 694–699. [DOI] [PubMed] [Google Scholar]
- West AP, Koblansky AA, and Ghosh S (2006). Recognition and signaling by toll-like receptors. Annual review of cell and developmental biology 22, 409–437. [DOI] [PubMed] [Google Scholar]
- Wolberger C (2014). Mechanisms for regulating deubiquitinating enzymes. Protein science : a publication of the Protein Society 23, 344–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau R, and Rape M (2016). The increasing complexity of the ubiquitin code. Nature cell biology 18, 579–586. [DOI] [PubMed] [Google Scholar]
- Zhang X, Horibata K, Saijo M, Ishigami C, Ukai A, Kanno S, Tahara H, Neilan EG, Honma M, Nohmi T, et al. (2012). Mutations in UVSSA cause UV-sensitive syndrome and destabilize ERCC6 in transcription-coupled DNA repair. Nature genetics 44, 593–597. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Majid MC, Soll JM, Brickner JR, Dango S, and Mosammaparast N (2015). Noncanonical regulation of alkylation damage resistance by the OTUD4 deubiquitinase. The EMBO journal 34, 1687–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng N, and Shabek N (2017). Ubiquitin Ligases: Structure, Function, and Regulation. Annual review of biochemistry. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMENTAL TABLE S1. Related to Figure 4. Interactome analysis of cytosolic Flag-OTUD4. Total and unique peptides for each interacting protein is shown.
Data Availability Statement
Primary data can be found on Mendeley (doi:10.17632/d576pn2h5s.1).