

Bremelanotide significantly improves sexual desire and related distress in premenopausal women with hypoactive sexual distress disorder and has a favorable safety profile.
OBJECTIVE:
To evaluate the safety and efficacy of bremelanotide for the treatment of premenopausal women with hypoactive sexual desire disorder.
METHODS:
Two identical phase 3, randomized, double-blind, placebo-controlled, multicenter clinical trials (RECONNECT) evaluated the safety and efficacy of bremelanotide 1.75 mg administered subcutaneously as needed in premenopausal women with hypoactive sexual desire disorder. Patients were randomized 1:1 to 24 weeks of treatment with bremelanotide or placebo. Sample size was estimated based on simulations from key endpoints in patients with hypoactive sexual desire disorder from a prior trial. Coprimary efficacy endpoints were change from baseline to end-of-study in the Female Sexual Function Index–desire domain score and Female Sexual Distress Scale–Desire/Arousal/Orgasm item 13.
RESULTS:
Study 301 began on January 7, 2015, and concluded on July 26, 2016. Study 302 began on January 28, 2015, and concluded on August 4, 2016. Of the 1,267 women randomized, 1,247 and 1,202 were in the safety and efficacy (modified intent-to-treat) populations, respectively. Most participants were white (85.6%), from U.S. sites (96.6%), and had a mean age of 39 years. From baseline to end-of-study, women taking bremelanotide had statistically significant increases in sexual desire (study 301: 0.30, P<.001; study 302: 0.42, P<.001; integrated studies 0.35, P<.001) and statistically significant reductions in distress related to low sexual desire (study 301: −0.37, P<.001; study 302: −0.29, P=.005; integrated studies −0.33, P<.001) compared with placebo. Patients taking bremelanotide experienced more nausea, flushing, and headache (10% or more in both studies) compared with placebo.
CONCLUSIONS:
Both studies demonstrated that bremelanotide significantly improved sexual desire and related distress in premenopausal women with hypoactive sexual desire disorder. The safety profile was favorable. Most treatment-emergent adverse events were related to tolerability and the majority were mild or moderate in intensity.
CLINICAL TRIAL REGISTRATION:
ClinicalTrials.gov, NCT02333071 (study 301) and NCT02338960 (study 302).
FUNDING SOURCE:
Palatin Technologies, Inc., and AMAG Pharmaceuticals, Inc.
Female sexual dysfunction refers to a heterogenous group of sexual disorders, including difficulties with desire, interest, arousal, orgasm, or pain (dyspareunia) that may result in personal and interpersonal distress, and impair a woman's overall health and quality of life.1 The most prevalent sexual dysfunction among women is hypoactive sexual desire disorder, defined as persistent (or recurrent) diminished or lack of desire for sexual activity, accompanied by distress (not better accounted for by a medical condition, substance use, or relationship conflict).1–3 Although hypoactive sexual desire disorder affects ∼10% of women aged 18–44 years in the United States,4,5 there is only one U.S. Food and Drug Administration (FDA)–approved therapy for hypoactive sexual desire disorder in premenopausal women at the time of this study, and this option is administered once daily.6
Hypoactive sexual desire disorder may be caused by an imbalance in the excitatory and inhibitory neural pathways in the prefrontal cortex and limbic system implicated in human sexual response, leading to increased inhibition, decreased excitation, and diminished responsiveness to sexual cues. Melanocortins have been associated with excitatory pathways and linked to appetitive sexual behaviors.6–9 Bremelanotide is a novel cyclic heptapeptide analog of the endogenous neuropeptide α-melanocyte-stimulating hormone with high affinity for the melanocortin-4 receptor.10 Preclinical studies suggest that bremelanotide acts on physiologic and neurobiologic components of female sexual function by modulating neurotransmitter pathways involved in sexual desire and arousal in women with hypoactive sexual desire disorder.8
A phase 2b, randomized, double-blind, placebo-controlled, dose-finding trial (NCT01382719) was conducted with 397 premenopausal women with hypoactive sexual desire disorder, female sexual arousal disorder, or both. Self-administered subcutaneous bremelanotide, taken as needed for up to 12 weeks, showed dose-responsive improvements in desire, arousal, and associated distress, as well as increases in the number of satisfying sexual events compared with placebo. The 1.75 mg dose of bremelanotide was selected for phase 3 studies based on optimal efficacy and an acceptable safety profile.11
After discussion with the FDA, two separate, identically designed phase 3 clinical trials were conducted as per their recommendation. The current report details the individual study results and the integrated data from the core phase 3 RECONNECT pivotal studies, designed to evaluate the safety and efficacy of bremelanotide in premenopausal women with acquired, generalized hypoactive sexual desire disorder.
ROLE OF THE FUNDING SOURCE
The two studies, NCT02333071 (study 301) and NCT02338960 (study 302), were sponsored by Palatin Technologies, Inc., the innovator of bremelanotide. The sponsor had a role in the design, execution, analysis, reporting, and funding of the studies. The authors had access to relevant individual and aggregated study data and other information (such as study protocol, analytic plan and report, validated data tables, and clinical study report) required to understand and report research findings. The authors take responsibility for the presentation and publication of the research findings, have been fully involved at all stages of publication and presentation development, and are willing to take public responsibility for all aspects of the work. All individuals included as authors and contributors who made substantial intellectual contributions to the research, data analysis, and publication or presentation development are listed appropriately. The role of the sponsor in the design, execution, analysis, reporting, and funding is fully disclosed. The authors' personal interests, financial or nonfinancial, relating to this research and its publication have been disclosed.
METHODS
The RECONNECT studies were two identically designed, randomized, double-blind, placebo-controlled, multicenter trials evaluating the safety and efficacy of bremelanotide administered subcutaneously as needed compared with placebo in premenopausal women with hypoactive sexual desire disorder (Appendix 1, available online at http://links.lww.com/AOG/B585). “RECONNECT” refers to a common response from patient interviews, during which women expressed their wish to reconnect to the desire they had lost with hypoactive sexual desire disorder. Investigators obtained approval from institutional review boards (U.S.) or independent ethics committees (Canada) from all related study sites. The studies were conducted in accordance with Good Clinical Practice requirements, and all patients provided written informed consent.
The studies included a core study phase consisting of a 4-week screening period, a 4-week baseline period (single-blind, placebo-only treatment period), and a 24-week randomized, double-blind, placebo-controlled treatment period. A 52-week open-label extension study phase was optional for patients who completed the core study phase. A total of 1,500 premenopausal female patients were planned to be screened in each study to obtain approximately 550 patients for randomization and to provide 2 or more months of data from the double-blind treatment period in 450 patients or more. Sample sizes were based on simulations from the observed distributions of the coprimary and key secondary endpoints in patients with hypoactive sexual desire disorder (with and without arousal) in a phase 2 study.11 Simulations predicted the estimated mean treatment difference (placebo compared with bremelanotide 1.75 mg) of 1.0 for change from baseline for the Female Sexual Function Index-desire domain (FSFI-D) and −0.43 for the Female Sexual Distress Scale-Desire/Arousal/Orgasm (FSDS-DAO) item 13. Based on these simulations, it was calculated that 450 evaluable patients would provide 100% power to detect a difference between the two treatment groups for both FSFI-D and FSDS-DAO item 13. As there were two primary endpoints, the inference threshold was 0.025. Similar calculations established that 480 evaluable patients would provide 93.8% power to detect a difference between the two groups for the key secondary endpoint. Patients were randomized in a 1:1 ratio to receive bremelanotide 1.75 mg or placebo using an Interactive Response System. The randomization scheme was generated using permuted blocks by country.
After completing the 4-week screening period (visit 1), all patients were included in the 4-week, single-blind, placebo-only treatment period (visit 2) and were expected to engage in sexual activity at least once. This single-blind placebo period was used to establish the baseline to which end-of-treatment changes were compared and to minimize the placebo effect typically associated with subjective-endpoint trials. All patients who completed the placebo run-in period were randomized at visit 3 to a 24-week, double-blind treatment period of either bremelanotide or placebo administered subcutaneously. In a prior dose-finding trial, the bremelanotide 1.75 mg dose showed optimal efficacy and an acceptable safety profile.11 Patients were observed while self-administering an in-clinic dose, and blood pressure (BP) measurements were taken at up to 1 hour predose and 1.0, 1.5, and 2 hours postdose. They could self-administer a maximum of 12 doses as needed (approximately 45 minutes before anticipated sexual activity) in each 4-week period as outpatients, with no more than one dose per 24-hour period. Safety evaluations were performed at screening, then monthly to the end of the core study phase (visit 9 at 8 months or at premature discontinuation). Patients had a physical examination, including BP assessments and other vital sign data collection, measurement of electrocardiograms, clinical laboratory testing, adverse event monitoring, and administration of the Beck Scale for Suicidal Ideation.12
The studies recruited healthy, premenopausal women 18 years of age or older in stable monogamous relationships with a male or female partner. Patients were recruited from clinical sites such as private practices, academic medical centers, and research clinics. Diagnosis of the patient with hypoactive sexual desire disorder (with or without symptoms of decreased arousal) was performed using the Diagnostic Screening Guide for hypoactive sexual desire disorder and was completed by the investigator or an appropriately licensed health care provider. All patients had a diagnosis of acquired, generalized hypoactive sexual desire disorder, with or without decreased arousal, for 6 months or more; had prior experience of normal sexual function (self-defined) for 2 years or more; and were willing to engage in sexual activities one or more times per month during the study. Patients were excluded if they were pregnant or nursing; had any other female sexual dysfunction; or were diagnosed with or being treated for depression, psychosis, bipolar disorder, or substance abuse within 6 months before screening. Patients taking neuroleptics, lithium, antidepressants, mood stabilizers, benzodiazepines, cognitive enhancers, or γ-aminobutyric acid antagonists within 3 months of screening were also excluded.
Coprimary efficacy endpoints were change from baseline to end-of-study (defined as the patients' last visit) in the FSFI-D score, comprised of questions 1 and 2, and change from baseline to end-of-study in the score for feeling bothered by low sexual desire as measured by the FSDS-DAO item 13 (Appendix 2, available online at http://links.lww.com/AOG/B585). The key secondary endpoint was the change from baseline to end-of-study in satisfying sexual events that occurred within 16 hours of study drug dosing and reported within 72 hours.
The primary efficacy analysis used the last-observation-carried-forward approach for primary and secondary endpoints in the modified intent-to-treat population, which was defined as all patients in the safety population who had one or more double-blind follow-up visits. The safety population was defined as all patients who were randomized and received one or more doses of double-blind medication (in or outside the clinic). An Independent Anchor Assessment Committee evaluated anchors used to define responders and to determine clinically meaningful responder thresholds for the coprimary and secondary endpoints before unblinding. As the conduct of both studies was identical (inclusion and exclusion criteria, and all efficacy and safety assessments), data from the individual studies are presented and were integrated for this publication. P-values were reported to no more than three decimal places per journal requirements.
RESULTS
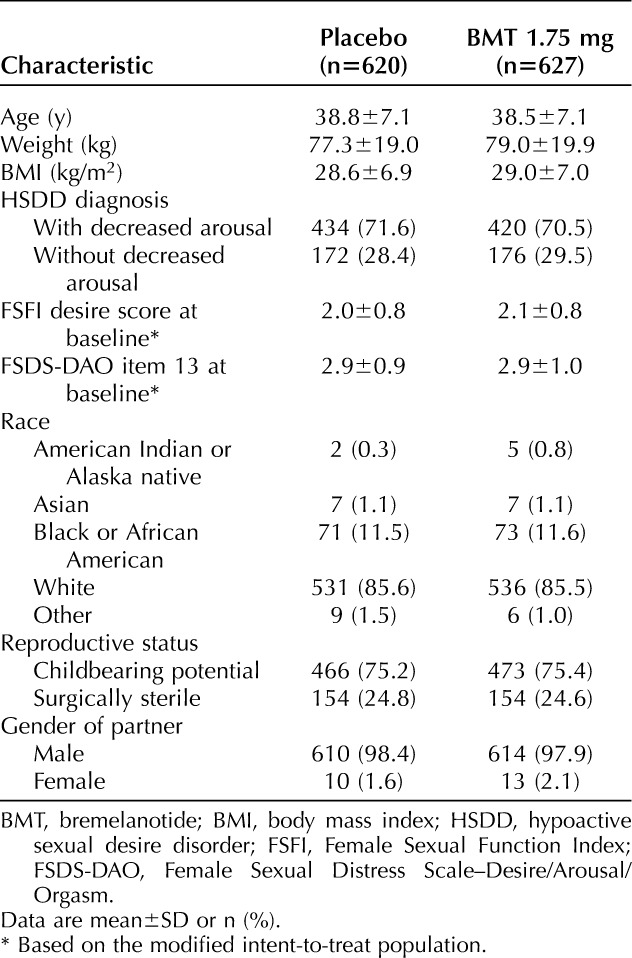
A total of 1,267 women were randomized in the two trials; 1,247 comprised the safety population, and 1,202 comprised the primary efficacy, modified intent-to-treat population. In studies 301 and 302, the modified intent-to-treat populations consisted of 630 and 572 patients, respectively, and the safety populations comprised 643 and 604 patients, respectively. Patient disposition in each study is presented in Appendix 3, available online at http://links.lww.com/AOG/B585. Study 301 began on January 7, 2015, and concluded on July 26, 2016; study 302 began on January 28, 2015, and concluded on August 4, 2016. Participants were mostly white (85.6%) and non-Hispanic or Latina (91.5%) (Table 1). Demographics were well balanced between the groups in both studies. Almost all randomized patients were from U.S. sites (96.6%).
Table 1.
Baseline Characteristics (Safety Population)
Relative to placebo, women in the bremelanotide group had statistically significant increases in sexual desire from baseline to end-of-study, as measured by FSFI-D scores (study 301: 0.30, P<.001; study 302: 0.42, P<.001; integrated studies 0.35, P<.001), and had statistically significant reductions in distress related to low sexual desire from baseline to end-of-study, as measured by FSDS-DAO item 13 scores (study 301: −0.37, P<.001; study 302: −0.29, P=.005; integrated studies −0.33, P<.001). In the integrated dataset, the effect size of bremelanotide (relative to placebo) for improving sexual desire was 0.39, and the effect size associated with reducing related distress was 0.27. For both coprimary endpoints, changes were observed at week 4 (earliest evaluated time point) and sustained over the course of the study (Fig. 1A and B). By comparing the empirical distribution for the coprimary endpoints between the two treatment groups, the cumulative distribution showed highly statistical significant differences (FSFI-D: P<.001; FSDS-DAO: P<.001) for both studies (data not shown). These statistically significant differences between treatment groups in favor of bremelanotide in both studies were supported by an anchored responder analysis, with prespecified definitions to support clinical meaningfulness.1 The General Assessment Questionnaire question 3 primary dynamic anchor assessment of the coprimary endpoints (patients' perceived benefit of study drug) yielded clinically meaningful and statistically significant (P<.001) differences between the treatment groups in each study, with 58.3% and 58.2% responder rates for bremelanotide and 36.1% and 35.4% for placebo in studies 301 and 302, respectively (Fig. 1C). In a sensitivity analysis, three additional static anchor assessments, Elements of Desire Questionnaire question 9, FSDS-DAO item 1, and FSFI question 16, yielded similar responder rates for both studies. Importantly, additional sensitivity analyses demonstrated treatment benefits favoring bremelanotide even when all premature discontinuations were assumed to be nonresponders (data not shown).
Fig. 1. Co-primary endpoints and key dynamic anchor over the course of the core phase of the phase 3 studies (modified intent-to-treat population). A. Female Sexual Function Index–desire domain (FSFI-D). B. Female Sexual Distress Scale–Desire/Arousal/Orgasm (FSDS-DAO). C. General Assessment Questionnaire (GAQ) question 3 responders (≥5, dynamic anchor). A and B. Estimated treatment difference and P value from mixed-model repeated measures using change from baseline of all double-blind period data (baseline through core week 24), with uncorrelated covariance structure. C. P value from unadjusted Wilcoxon rank-sum test. Scores range from 1 (very much worse) to 7 (very much better). Summary statistics are based on the number of nonmissing values. Error bars for integrated populations represent 95% CIs. BMT, bremelanotide.

Kingsberg. Bremelanotide for Hypoactive Sexual Desire Disorder. Obstet Gynecol 2019.
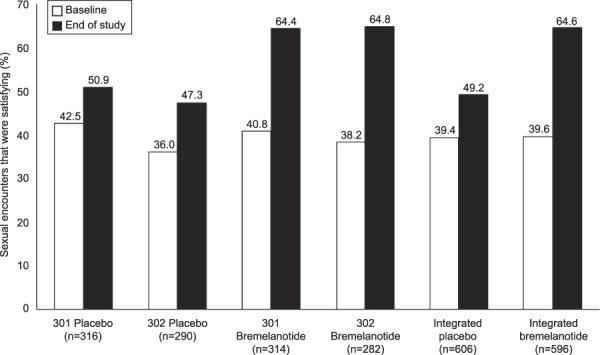
Because the difference in the change from baseline to end-of-study for the number of satisfying sexual events reported did not reach statistical significance between treatment groups (study 301: bremelanotide 0.0, placebo −0.1, P=.764; study 302: bremelanotide 0.0, placebo 0.0, P=.702; integrated studies: bremelanotide 0.0, placebo −0.1, P=.630), the remaining ranked secondary endpoints became supportive and exploratory per the statistical analysis plan. A post hoc exploratory analysis showed that the difference in the percentages of sexual events that were considered satisfying sexual events (number of satisfying sexual events per total number of sexual encounters) increased substantially in the bremelanotide group compared with the placebo group. Across both studies, the difference in the percentages of satisfying sexual events increased greater than two-fold in the bremelanotide group compared with the placebo group (25.0% vs 9.8%, P<.001) (Fig. 2).
Fig. 2. Percentage of sexual encounters that were satisfying sexual events (modified intent-to-treat populations, post hoc analysis). No significant difference between treatment groups was observed for satisfying sexual events associated with study drug and reported within 72 hours.
Kingsberg. Bremelanotide for Hypoactive Sexual Desire Disorder. Obstet Gynecol 2019.
Overall sexual function and associated distress were assessed according to mean changes from baseline to end-of-study for FSFI and FSDS-DAO total scores, respectively. The bremelanotide to placebo odds ratios for the proportion of responders (defined as 4.2 or more and –10 or below for FSFI and FSDS-DAO total scores, respectively) significantly favored bremelanotide for those endpoints (Fig. 3). Overall, the bremelanotide groups in both studies demonstrated a trend of having a greater number of responders compared with placebo for secondary endpoints measuring changes in the mean levels of desire and arousal per FSFI, as well as satisfaction with those aspects of sexual function.
Fig. 3. Forest plot of supportive secondary efficacy endpoints in the phase 3 studies (modified intent-to-treat population). Q, question; FSEP-R, Female Sexual Encounter Profile–Revised; BMT, bremelanotide; FSDS-DAO, Female Sexual Distress Scale–Desire/Arousal/Orgasm; FSFI, Female Sexual Function Index; PBO, placebo.

Kingsberg. Bremelanotide for Hypoactive Sexual Desire Disorder. Obstet Gynecol 2019.
In both studies, the most common treatment-emergent adverse events that occurred at a higher rate in the bremelanotide group (10% or more in both studies) than the placebo group were nausea, flushing, and headache (Table 2). The majority of treatment-emergent adverse events in both studies were mild or moderate in severity. In both studies, severe treatment-emergent adverse events that occurred in more than one patient who received bremelanotide included nausea (13 [2.1%] patients), headache (5 [0.8%]), vomiting (3 [0.5%]), and myalgia (2 [0.3%]). No other severe treatment-emergent adverse events were reported by more than one patient in the bremelanotide group, and there were no severe treatment-emergent adverse events that occurred in more than one patient in the placebo group.
Table 2.
Treatment-Emergent Adverse Events Reported More Frequently in the Bremelanotide Treatment Group (1% or More in Bremelanotide Group With Incidence 1% or Higher Than for Placebo Patients) by Preferred Term (Safety Population)

The incidence of nausea with bremelanotide was 40.0% compared with 1.3% with placebo across both studies. Nausea had a median onset of 30 minutes after dosing, with a median duration of 2.4 hours, tended to occur sporadically with dosing, and was mild to moderate in severity (approximately 98% of cases in both studies) with most cases resolving spontaneously (Appendix 4, available online at http://links.lww.com/AOG/B585). Among the subset with one or more nausea events who chose to use an antiemetic, the subsequent nausea rate was lower (∼5%) compared with those who did not use an antiemetic (∼15%). Although 40% of patients taking bremelanotide experienced nausea, only 8.1% discontinued study drug during the two studies due to nausea, and only 3.5% simultaneously reported vomiting.
Across both studies, seven patients who received bremelanotide reported 10 treatment-emergent serious adverse events, and three patients who received placebo reported four treatment-emergent serious adverse events. No specific treatment-emergent serious adverse event occurred more than once (Appendix 5, available online at http://links.lww.com/AOG/B585). There was one treatment-emergent serious adverse event in study 301 that was considered related to the study drug in a patient who had received bremelanotide and subsequently experienced vomiting and headache. Both symptoms were resolved after overnight hospitalization with hydromorphone hydrochloride, ketorolac tromethamine, morphine sulfate, and intravenous sodium chloride for the headache, and promethazine and ondansetron for nausea and vomiting. No treatment-emergent serious adverse events in study 302 were considered related to study drug, and no deaths occurred during either study.
In both studies, placebo-adjusted changes in BP for patients taking bremelanotide were consistent with ambulatory BP readings from the phase 2 studies, with a mean increase of approximately 3 mm Hg for systolic BP and 2 mm Hg for diastolic BP. These mild transient changes were noted approximately 0–2 hours postbremelanotide dosing, returned to baseline within 8–10 hours, and were accompanied by similarly mild decreases in pulse rates, such that there were no mean increases in overall myocardial workload. No clinically significant effects on clinical laboratory tests, electrocardiograms, weight, depression, or suicidal ideation were observed in either study. There were no differences in the adverse event profiles among patients based on the amount of alcohol they consumed during the study.
Among the 856 patients who completed the double-blind core study phase, approximately 80% (684 patients) elected to participate in the open-label extension. This included 87% (430/493) of patients from the placebo groups and 70% (254/363) of patients from the bremelanotide groups.
DISCUSSION
These two large, identically designed, randomized, phase 3, double-blind, placebo-controlled registration trials evaluating the safety and efficacy of bremelanotide compared with placebo successfully met their coprimary efficacy endpoints of improving sexual desire and related distress in premenopausal women with hypoactive sexual desire disorder. Additionally, the bremelanotide group showed significantly greater numbers of responders compared with placebo, thus demonstrating clinically meaningful benefits from bremelanotide treatment in alignment with FDA guidances.1 The coprimary endpoints used components of the FSFI and FSDS-DAO, namely, the desire domain and item 13, respectively, which are robust and validated instruments for the assessment of hypoactive sexual desire disorder.13–17 Moreover, the independently defined thresholds for the dynamic anchor assessment of the coprimary endpoints confirmed both clinically meaningful and statistically significant differences between treatment groups in both studies. This was achieved despite the high placebo response rates of approximately 35%; as there was a 25% increase in the percentage of satisfying sexual events among patients who received placebo, 40% of the relative increase among patients treated with bremelanotide could be attributed to the placebo effect. A high placebo response rate is typical of subjective endpoints where expectations, anxiety, and reward are likely involved.18
Effect size is important to consider as it indicates the magnitude of the treatment's effect: an effect size of 0.2 is considered small, 0.5 medium, and 0.8 large.19 In the RECONNECT trials, the effect size of as-needed bremelanotide alone for improving sexual desire ranged from 0.49 to 0.61, and from 0.60 to 0.62 in reducing related distress. Relative to placebo, these values were 0.29–0.43 and 0.26–0.32, respectively. Across three phase 3 studies, the effect size of once-daily flibanserin relative to placebo was reported to range from 0.29 to 0.44 in improving sexual desire, and from 0.24 to 0.44 in reducing related distress.20,21 For additional perspective, an analysis of pharmacologic treatments for generalized anxiety disorder reported effect sizes relative to placebo of 0.36 and 0.38 for SSRIs and benzodiazepines, respectively.22
Prespecified exploratory subgroup analyses of the integrated phase 3 studies have demonstrated that bremelanotide is efficacious across all age and weight quartiles and all baseline FSFI total, FSFI-D, and FSDS-DAO total scores in premenopausal women with HSSD, supporting its use across a wide range of patients (unpublished data). The totality of improvement in the supportive secondary efficacy endpoints that evaluated overall sexual function and distress related to level of sexual function (total scores for FSFI and FSDS-DAO), as well as specific aspects of sexual distress and arousal, all provide robust and consistent data that align with the statistically significant results for the coprimary endpoints. The FSFI total score, although not specific to low sexual desire, reflects other important components of sexual function (eg, arousal, orgasm, satisfaction) that are affected by low sexual desire and provides additional insight to patients and prescribers around overall sexual health. The FSFI total score is often improved by treating low desire, as evidenced by the RECONNECT studies. Similarly, the FSDS-DAO total score highlights reduction in overall distress and parallels the overall improvement in the FSFI-D score.
Treatment effects were noted early after treatment initiation, with statistically significant treatment benefits in both studies in favor of bremelanotide, and were maintained throughout the 24-week double-blind treatment period. Early onset of treatment effect and as-needed dosing will allow patients to quickly decide whether they are benefiting from the drug, thereby minimizing unnecessary exposure and unwanted tolerability issues.
As with any investigational drug, patients may speculate whether they are on active drug by experiencing adverse events or the perception of efficacy. Although 40.0% of patients in the bremelanotide arm (vs 1.3% in the placebo arm) reported nausea, the overall incidence of treatment-emergent adverse events was more similar between the two study arms (76.6% vs 58.2%). Moreover, the high placebo effect rate in sexual dysfunction studies suggests that those taking placebo could also have falsely speculated that they were on active drug. Thus, the randomized, placebo-controlled design of the study shields speculation that efficacy was driven by patient speculation to any significant extent.
The effects of bremelanotide on BP have been well characterized in several clinical studies, which consistently revealed small, transient BP increases that peak within 4 hours postdose and return toward baseline by approximately 8–10 hours. These small changes in BP were accompanied by corresponding reductions in heart rate, resulting in lower or neutral heart rate-BP product values. There have been no cumulative or sustained effects on BP related to bremelanotide and, given the limited intermittent use of the product, no untoward cardiovascular effects would be anticipated.
The most common treatment-emergent adverse events occurring in more than 10% of bremelanotide patients compared with placebo were nausea, flushing, and headache. The majority of treatment-emergent adverse events in the bremelanotide group were transient and mild to moderate in severity. Furthermore, the most common adverse events were related to tolerability issues that patients may be willing to accept and manage in exchange for improvements in sexual desire and reductions in associated distress.
Clinical research questions remain as the safety and efficacy of bremelanotide has yet to be established in postmenopausal women and in women whose comorbidities include psychiatric disorders requiring medication. The majority of the study population was white and non-Hispanic, and further clinical data are needed to establish the efficacy and safety of bremelanotide in other populations. Finally, a differential study withdrawal rate was observed in both studies (study 301: 41.9% bremelanotide vs 16.0% placebo; study 302: 43.8% bremelanotide vs 28.4% placebo).
The data obtained from these two identically designed, phase 3, randomized, double-blind, placebo-controlled trials demonstrate that bremelanotide can improve sexual desire and decrease distress related to sexual desire with a manageable side-effect profile as an as-needed potential treatment for premenopausal women with hypoactive sexual desire disorder independent of decreased arousal.
Authors' Data Sharing Statement
Will individual participant data be available (including data dictionaries)? No.
What data in particular will be shared? Not available.
What other documents will be available? Not available.
When will data be available (start and end dates)? Not applicable.
By what access criteria will data be shared (including with whom, for what types of analyses, and by what mechanism)? Not applicable.
Footnotes
The RECONNECT studies were sponsored by Palatin Technologies, Inc., the innovator of bremelanotide. Editorial support in the preparation of this manuscript was provided by Phase Five Communications, supported by AMAG Pharmaceuticals, Inc., the licensee of bremelanotide.
Financial Disclosure Dr. Kingsberg has served on advisory boards or has been a consultant for AMAG Pharmaceuticals, Inc., Daré Bioscience, Duchesnay, Emotional Brain, Valeant, Endoceutics, Ivix, Palatin Technologies, Inc., Pfizer, Shionogi, Materna, Nuelle, Mitsubishi Tanaka North America, TherapeuticsMD, SST, and Lupin/Symbiomix. She has stock options in Viveve Medical. Dr. Clayton has served on advisory boards or has been a consultant for Acadia, Alkermes, Allergan, AMAG Pharmaceuticals, Inc., Fabre Kramer, Lundbeck, Palatin Technologies, Inc., S1 Biopharma, Sage Therapeutics, Sprout, and Takeda. She has received grants from Endoceutics, Inc., Janssen, Sage Therapeutics, and Takeda. In addition, she has received royalties and/or owns the copyright to works published by Ballantine Books/Random House and Guilford Publications, and the Changes in Sexual Functioning Questionnaire. She has shares or restricted stock units in Euthymics and S1 Biopharma. Dr. Portman has served on advisory boards or has been a consultant to AMAG Pharmaceuticals, Inc., Palatin Technologies, Inc., Endoceutics, Valeant Pharmaceuticals, and Sprout, and is on the speaker's bureau for AMAG Pharmaceuticals, Inc.; he is an employee and stockholder of Sermonix Pharmaceuticals. Dr. Williams and Dr. Krop are employees and stockholders of AMAG Pharmaceuticals, Inc. (the licensee of bremelanotide). Mr. Jordan is Vice President, Clinical Operations and Project Management, and a stockholder of Palatin Technologies, Inc. (the sponsor of the trial). He has a BS in Biology and has worked in clinical development for over 20 years. He led the Palatin team in the development of bremelanotide (clinical, regulatory, etc.) since 2007, including having a very significant role in designing the phase 3 studies, conduct of the studies, and analysis of the data. Dr. Lucas was an employee of Palatin Technologies, Inc. at the time of the study. She has no remaining financial interests in Palatin Technologies. She was the medical lead for the entire trial. She was the physician who ran the trial and provided safety evaluation and analysis for all adverse events as they occurred during the trial. She was also responsible for aggregate assessment of the adverse event database. She was the physician who provided medical information to the DSMB during the trial. Dr. Simon has served on advisory boards or has been a consultant for AbbVie, Allergan, AMAG Pharmaceuticals, Inc., Amgen, Ascend Therapeutics, Azure Biotech, Bayer HealthCare Pharmaceuticals, Inc., CEEK Enterprises, Covance Inc., Daré Bioscience, Duchesnay, Hologic Inc., KaNDy/NeRRe Therapeutics Ltd., Millendo, Mitsubishi Tanabe Pharma, ObsEva, Radius Health, Sanofi, Sebela, Sermonix, Shionogi, Symbiotec Pharmalab, TherapeuticsMD, and Valeant. He has also served on the speaker's bureau for AbbVie, AMAG Pharmaceuticals, Inc., Duchesnay, Novo Nordisk, Shionogi, and Valeant. He has received grants/research funding from AbbVie, Allergan, Agile Therapeutics, Bayer Healthcare LLC, Dornier MedTech, Endoceutics Inc., GTx Inc., Hologic Inc., Myovant Sciences, New England Research Institutes, ObsEva, Palatin Technologies, Inc., Symbio Research, TherapeuticsMD, Tissue Genesis, and Viveve Medical. He owns stock in Sermonix.
Presented at the ISSWSH Annual Meeting in Atlanta, Georgia, February 23–26, 2017.
The authors thank all participants in these studies, their families, and personnel at all study sites.
Each author has confirmed compliance with the journal's requirements for authorship.
Peer reviews are available at http://links.lww.com/AOG/B586.
Figure.

No available caption
REFERENCES
- 1.U.S. Food and Drug Administration. Guidance for industry: low sexual interest, desire, and/or arousal in women: developing drugs for treatment. Available at: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM526362.pdf. Retrieved December 12, 2018.
- 2.Parish SJ, Goldstein AT, Goldstein SW, Goldstein I, Pfaus J, Clayton AH, et al. Toward a more evidence-based nosology and nomenclature for female sexual dysfunctions—part II. J Sex Med 2016;13:1888–906. [DOI] [PubMed] [Google Scholar]
- 3.Goldstein I, Kim NN, Clayton AH, DeRogatis LR, Giraldi A, Parish SJ, et al. Hypoactive sexual desire disorder: International Society for the Study of Women's Sexual Health (ISSWSH) expert consensus panel review. Mayo Clin Proc 2017;92:114–28. [DOI] [PubMed] [Google Scholar]
- 4.Rosen RC, Shifren JL, Monz BU, Odom DM, Russo PA, Johannes CB. Correlates of sexually related personal distress in women with low sexual desire. J Sex Med 2009;6:1549–60. [DOI] [PubMed] [Google Scholar]
- 5.Shifren JL, Monz BU, Russo PA, Segreti A, Johannes CB. Sexual problems and distress in United States women: prevalence and correlates. Obstet Gynecol 2008;112:970–8. [DOI] [PubMed] [Google Scholar]
- 6.Addyi (flibanserin) prescribing information. Bridgewater, NJ: Valeant Pharmaceuticals North America LLC; 2018. [Google Scholar]
- 7.Kingsberg SA, Clayton AH, Pfaus JG. The female sexual response: current models, neurobiological underpinnings and agents currently approved or under investigation for the treatment of hypoactive sexual desire disorder. CNS Drugs 2015;29:915–33. [DOI] [PubMed] [Google Scholar]
- 8.Pfaus J, Giuliano F, Gelez H. Bremelanotide: an overview of preclinical CNS effects on female sexual function. J Sex Med 2007;4(suppl 4):269–79. [DOI] [PubMed] [Google Scholar]
- 9.Pfaus JG, Shadiack A, Van Soest T, Tse M, Molinoff P. Selective facilitation of sexual solicitation in the female rat by a melanocortin receptor agonist. Proc Natl Acad Sci U S A 2004;101:10201–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Molinoff PB, Shadiack AM, Earle D, Diamond LE, Quon CY. PT-141: a melanocortin agonist for the treatment of sexual dysfunction. Ann N Y Acad Sci 2003;994:96–102. [DOI] [PubMed] [Google Scholar]
- 11.Clayton AH, Althof SE, Kingsberg S, DeRogatis LR, Kroll R, Goldstein I, et al. Bremelanotide for female sexual dysfunctions in premenopausal women: a randomized, placebo-controlled dose-finding trial. Womens Health (Lond) 2016;12:325–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beck AT, Kovacs M, Weissman A. Assessment of suicidal intention: the scale for suicide ideation. J Consult Clin Psychol 1979;47:343–52. [DOI] [PubMed] [Google Scholar]
- 13.Meston CM. Validation of the Female Sexual Function Index (FSFI) in women with female orgasmic disorder and in women with hypoactive sexual desire disorder. J Sex Marital Ther 2003;29:39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rosen R, Brown C, Heiman J, Leiblum S, Meston C, Shabsigh R, et al. The Female Sexual Function Index (FSFI): a multidimensional self-report instrument for the assessment of female sexual function. J Sex Marital Ther 2000;26:191–208. [DOI] [PubMed] [Google Scholar]
- 15.DeRogatis L, Clayton A, Lewis-D'Agostino D, Wunderlich G, Fu Y. Validation of the Female Sexual Distress Scale-Revised for assessing distress in women with hypoactive sexual desire disorder. J Sex Med 2008;5:357–64. [DOI] [PubMed] [Google Scholar]
- 16.DeRogatis LR, Edelson J, Revicki DA. Reliability and validity of the Female Sexual Distress Scale-Desire/Arousal/Orgasm instrument in a phase 2B dose-ranging study of bremelanotide. Available at: https://www.palatin.com/assets/PAL-P4078-APA-DeRogatis-FSDS-DAO-Reliability-and-Validity-Poster-0421-2-1.pdf. Retrieved June 11, 2019. [Google Scholar]
- 17.DeRogatis LR, Rosen R, Leiblum S, Burnett A, Heiman J. The Female Sexual Distress Scale (FSDS): initial validation of a standardized scale for assessment of sexually related personal distress in women. J Sex Marital Ther 2002;28:317–30. [DOI] [PubMed] [Google Scholar]
- 18.Benedetti F, Carlino E, Pollo A. How placebos change the patient's brain. Neuropsychopharmacology 2011;36:339–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cohen J. Statistical power analysis for the behavioral sciences. 2nd ed Hillsdale, NJ: Lawrence Erlbaum Associates; 1988. [Google Scholar]
- 20.U.S. Food and Drug Administration. Summary review for regulatory action. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/022526Orig1s000SumRedt.pdf. Retrieved March 26, 2019. [Google Scholar]
- 21.Data on file. AMAG Pharmaceuticals, Inc. Waltham, MA. [Google Scholar]
- 22.Hidalgo RB, Tupler LA, Davidson JR. An effect-size analysis of pharmacologic treatments for generalized anxiety disorder. J Psychopharmacol 2007;21:864–72. [DOI] [PubMed] [Google Scholar]