

The 52-week open-label extension of the RECONNECT studies demonstrates bremelanotide's favorable safety profile, with sustained efficacy in treating hypoactive sexual desire disorder in premenopausal women.
OBJECTIVE:
To evaluate the long-term safety and efficacy of bremelanotide as treatment for hypoactive sexual desire disorder in premenopausal women.
METHODS:
Women who completed the 24-week double-blind core phase of RECONNECT, composed of two parallel phase 3 trials (301 and 302) examining the safety and efficacy of bremelanotide compared with placebo in premenopausal women with hypoactive sexual desire disorder, could enroll in the 52-week open-label extension, provided they had not experienced serious adverse events during the core phase. Efficacy was assessed using the coprimary endpoints from the core phase, and all adverse events were collected during the open-label extension. All statistical analyses were descriptive.
RESULTS:
The study 301 open-label extension began on July 17, 2015, and concluded on July 13, 2017; the study 302 open-label extension began on October 5, 2015, and concluded on June 29, 2017. Of the 856 eligible patients who completed the core phase, 684 elected to participate in the open-label extension, and 272 completed it. The most common treatment-emergent adverse events considered related to study drug were nausea (40.4%), flushing (20.6%), and headache (12.0%), and the only severe treatment-emergent adverse event experienced by more than one participant in both studies was nausea during the open-label extension. The change in Female Sexual Function Index–desire domain score and Female Sexual Distress Scale–Desire/Arousal/Orgasm item 13 from baseline to end of the open-label extension ranged from 1.25 to 1.30 and −1.4 to −1.7, respectively, for patients who received bremelanotide during the core phase, and 0.70–0.77 and −0.9, respectively, for patients who received placebo during the core phase.
CONCLUSION:
During the 52-week open-label extension of RECONNECT, no new safety signals were observed, and premenopausal women treated with bremelanotide exhibited sustained improvements in hypoactive sexual desire disorder symptoms.
CLINICAL TRIAL REGISTRATION:
ClinicalTrials.gov, NCT02333071 (study 301) and NCT02338960 (study 302).
FUNDING SOURCE:
Palatin Technologies, Inc., and AMAG Pharmaceuticals, Inc.
Female sexual dysfunction comprises a group of sexual disorders related to desire, arousal, and interest, as well as orgasm.1 Hypoactive sexual desire disorder,2 defined as the persistent or recurrent deficiency or absence of sexual fantasies or thoughts and desire for—or receptivity to—sexual activity, is the most prevalent female sexual dysfunction associated with personal distress and is not explained by another mental disorder, medical condition, or medications.3–5 According to a U.S.-based survey study, hypoactive sexual desire disorder occurs in 8.9% of women aged 18–44 years, 12.3% of women aged 45–64 years, and 7.4% of women aged 65 years or older.2
Current models for female sexuality are nonlinear, as women may participate in sexual activity without the need for desire.6 Multiple factors contribute to the pathology of hypoactive sexual desire disorder, including biologic, psychologic, and social components.7 Functional neuroimaging studies of patients with hypoactive sexual desire disorder have demonstrated decreased blood flow to areas of the brain associated with sexual desire.8–10 Hypoactive sexual desire disorder is thought to be caused by an imbalance in the excitatory and inhibitory neural pathways in the prefrontal cortex and limbic system implicated in human sexual response. This imbalance leads to increased inhibition, decreased excitation, and ultimately a diminished responsiveness to sexual cues. In recent years, there has been increased interest in developing evidence-based pharmacologic treatments to address the unmet need for effective hypoactive sexual desire disorder interventions.11–16 Flibanserin, a 5-HT1A agonist and 5-HT2A antagonist, is the first U.S. Food and Drug Administration–approved drug available for the condition,17 and bremelanotide was recently approved for the treatment of acquired, generalized hypoactive sexual desire disorder in premenopausal women.18 Bremelanotide is a melanocortin receptor agonist and a novel cyclic heptapeptide analog of the endogenous neuropeptide α-melanocyte-stimulating hormone.19–22 Based on findings from preclinical studies, bremelanotide acts on physiologic and neurobiologic components of female sexual function by modulating neurotransmitter pathways involved in sexual desire and arousal in women with hypoactive sexual desire disorder.20
The efficacy and safety of bremelanotide, taken as needed over 24 weeks, demonstrated statistically significant and clinically meaningful improvements in low sexual desire and related distress in the core phase of the RECONNECT studies.23 Here we report on the safety and efficacy of bremelanotide taken for an additional 52 weeks in the open-label extension of the RECONNECT studies.
ROLE OF THE FUNDING SOURCE
The two studies, NCT02333071 (study 301) and NCT02338960 (study 302), were sponsored by Palatin Technologies, Inc., who pioneered the development of bremelanotide for the treatment of hypoactive sexual desire disorder and was responsible for the conduct of the phase 3 clinical trials. AMAG Pharmaceuticals, Inc. contributed to the further development of bremelanotide and, together with Palatin Technologies, submitted the new drug application to the U.S. Food and Drug Administration. The sponsor had a role in the design, execution, analysis, reporting, and funding of the studies. The authors had access to relevant individual and aggregated study data and other information (such as study protocol, analytic plan and report, validated data tables, and clinical study report) required to understand and report research findings. The authors take responsibility for the presentation and publication of the research findings, have been fully involved at all stages of publication and presentation development, and are willing to take public responsibility for all aspects of the work. All individuals included as authors and contributors who made substantial intellectual contributions to the research, data analysis, and publication or presentation development are listed appropriately. The role of the sponsor in the design, execution, analysis, reporting, and funding is fully disclosed. The authors' personal interests, financial or nonfinancial, relating to this research and its publication have been disclosed.
METHODS
The RECONNECT core phase comprised two identically designed, phase 3, randomized, double-blind, placebo-controlled, multicenter trials of bremelanotide 1.75 mg administered subcutaneously and as needed using an autoinjector pen for treatment of hypoactive sexual desire disorder in premenopausal women. The study protocol and informed consent materials were approved by the institutional review boards or independent ethics committees (Canada) from all related study sites in accordance with the International Conference on Harmonisation, Good Clinical Practices, and local requirements. All patients provided informed written consent before performance of any study procedures.
The core phase of the trials included a 4-week screening period, a 4-week single-blind placebo treatment period to establish baseline, and a 24-week randomized, double-blind treatment period (Appendix 1, available online at http://links.lww.com/AOG/B587). The RECONNECT study design has been reported in detail.23 At the last visit of the core phase, patients completed the following questionnaires to assess efficacy: the Female Sexual Distress Scale–Desire/Arousal/Orgasm (FSDS-DAO) item 13, the Female Sexual Function Index–desire domain (FSFI-D), and the General Assessment Questionnaire. Safety assessments, including physical examination, standardized blood pressure (BP) and other vital signs, measurement of electrocardiograms, clinical laboratory testing, adverse event monitoring, and administration of the Beck Scale for Suicide Ideation were also performed at the last visit. All participants in the open-label extension received bremelanotide 1.75 mg administered subcutaneously on an as-needed basis (restricted to no more than one dose within a 24-hour period). During the open-label extension, patients were allowed to return to the clinic for a resupply of the study drug at any time and could be supplied with up to 30 doses per month.
Eligibility criteria for the core phase have been reported previously.23 Participants in the open-label extension came from both arms (bremelanotide and placebo) of the core phase. Although all patients received bremelanotide during the open-label extension, patients fell into two groups: women randomized to bremelanotide in the core phase and continuing on bremelanotide in the open-label extension study phase (bremelanotide→bremelanotide), and women switching from placebo in the core phase to bremelanotide in the open-label extension (placebo→bremelanotide). Women continuing in the open-label extension must have successfully completed the core phase, experienced no treatment-emergent serious adverse events during the core phase, and agreed to continue in the open-label extension. Patients who became pregnant during the study were withdrawn and followed to the end of their pregnancy. Patients were enrolled using the interactive web (internet) response system.
Study evaluations for safety and key efficacy outcomes occurred at 4-week intervals through week 12 of the open-label extension and every 8 weeks thereafter. Safety was assessed during the study by recording and monitoring adverse events, concomitant medication use, and clinically significant changes in physical examinations, electrocardiograms, vital signs, and laboratory assessments.
The open-label extension did not have formal efficacy endpoints, but measures used during the core phase23 were continued during the open-label extension, and are summarized in Appendix 2, available online at http://links.lww.com/AOG/B587. Minimal clinically important differences were determined in a responder analysis of the phase 2b study as +0.6 for FSFI-D and −1.0 for FSDS-DAO item 13.24
Safety and efficacy results for the open-label extension were summarized based on descriptive analyses, as there were no comparator arms. Data for patients who received bremelanotide or placebo in the double-blind portion of the core phase are displayed separately, although all patients received the same fixed dose (as needed) of bremelanotide in the open-label extension. For some assessments, data from the core phase are shown for context.
Baseline values for event data were derived from the core phase and defined as the last 28 days in the single-blind placebo period for patients randomized to bremelanotide, and the last 28 days before the final visit of the core phase for those randomized to placebo. All patients who used at least one dose of bremelanotide in the open-label extension were included in the open-label extension study population.
RESULTS
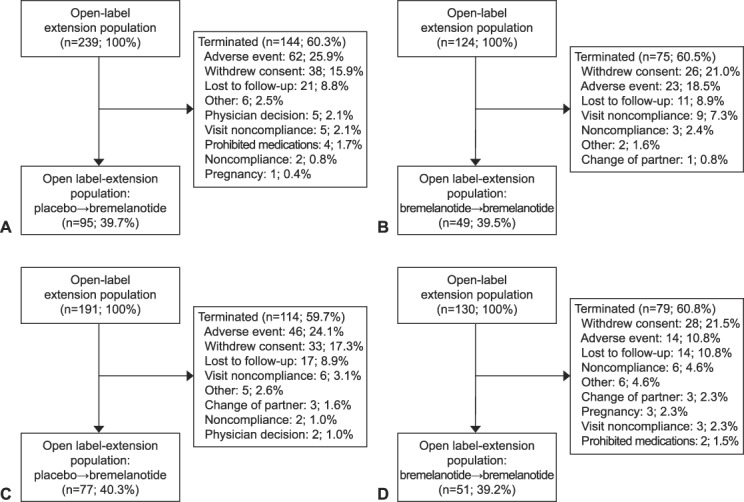
A total of 1,267 women were randomized in the RECONNECT core phase; of those, 32.4% (411 patients) did not complete the core phase. During the core phase, 1.1% (seven) of patients who received bremelanotide and 0.5% (three) of patients who received placebo reported serious adverse events. Among the 856 patients who completed the core phase, approximately 80% (684) elected to participate in the open-label extension: 363 participants in study 301 (124 bremelanotide→bremelanotide and 239 placebo→bremelanotide); 321 participants in study 302 (130 bremelanotide→bremelanotide and 191 placebo→bremelanotide). Approximately 40% of patients in both groups (272 patients; 39.7% in study 301 [Figs. 1A and B] and 39.9% in study 302 [Figs. 1C and D]) completed the 52-week open-label extension, with the most common reasons for the study discontinuation attributed to adverse events (study 301: 23.4%; study 302: 18.7%) and withdrawal of consent (17.6% and 19.0%) (Fig. 1). In study 301, patients who did not complete the open-label extension discontinued during the first 20 weeks (150/363 patients [41.3%]). In study 302, a similar number of patients discontinued the open-label extension throughout the 52-week period, except for week 4 (visit E2) and week 28 (visit E6) when higher numbers of patients discontinued (43 [13.4%] patients and 36 [11.2%] patients, respectively). Baseline characteristics of the open-label extension study population are shown in Table 1; this population was similar in distribution to the originally randomized population. All patients were premenopausal, the majority were white (more than 84%), and mean age for both studies was ∼39 years old (range 19–55 years).
Fig. 1. Patient disposition for the open-label extension of RECONNECT studies 301 (A and B) and 302 (C and D). Placebo to bremelanotide (A), bremelanotide to bremelanotide (B), placebo to bremelanotide (C), and bremelanotide to bremelanotide (D).
Simon. Long-term Safety and Efficacy of Bremelanotide. Obstet Gynecol 2019.
Table 1.
Baseline Demographics of the Open-Label Extension Study Population*

During the core phase, patients randomized to bremelanotide received a median of 14 injections in study 301 and 13.5 injections in study 302 (Appendix 3, available online at http://links.lww.com/AOG/B587). These patients received additional median 11 and 10 injections during the open-label extension, for a total median of 27 and 25 injections over a mean of 409 and 407 days in the two respective studies. Patients initially randomized to placebo in the core phase of studies 301 and 302 received a median of 12 and 13 injections during the open-label extension.
The most common treatment-emergent adverse events during the open-label extension were nausea, flushing, sunburn, and headache (Table 2). Nausea (which occurred in 42.7% of patients in study 301, 37.7% in study 302, and 40.4% across both studies), flushing (24.5%, 16.2%, and 20.6%, respectively), and headache (12.1%, 11.8%, and 12.0%, respectively) were the most common adverse events considered to be related to study drug. Patients switching from placebo experienced a higher incidence of adverse events than those continuing on bremelanotide during the open-label extension (78.8% vs 63.4%, respectively). This incidence of adverse events reported in the placebo→bremelanotide groups was consistent with that seen in patients randomized to bremelanotide in the core phase.23 Adverse events were considered the cause of treatment discontinuation in 25.9% and 24.1% of patients who switched from placebo to bremelanotide during the open-label extension of studies 301 and 302, respectively. Adverse events were considered the cause of treatment discontinuation in 18.5% and 10.8% of patients in the bremelanotide→bremelanotide group in studies 301 and 302, respectively. In study 301, the only severe treatment-emergent adverse events experienced by more than one patient were nausea (3/124 bremelanotide→bremelanotide vs 6/239 placebo→bremelanotide), fatigue (1/124 vs 2/239), flushing (0/124 vs 3/239), upper abdominal pain (2/124 vs 0/239), endometriosis (1/124 vs 1/239), and headache (1/124 vs 1/239). In study 302, no severe treatment-emergent adverse events occurred in more than one participant in the bremelanotide→bremelanotide group; nausea was the only severe treatment-emergent adverse event that occurred in more than one patient in the placebo→bremelanotide groups (4/191).
Table 2.
Treatment-Emergent Adverse Events in the Open-Label Extension (5% of Patients or More in at Least One Study)*

No deaths occurred during the entire duration of the RECONNECT studies. A total of 10 patients became pregnant during the core phase (three placebo, three bremelanotide) and open-label extension (one placebo→bremelanotide, three bremelanotide→bremelanotide). Three patients (two placebo→bremelanotide and one bremelanotide→bremelanotide) experienced one treatment-emergent serious adverse event each (cerebrovascular accident, endometriosis, cholecystitis), but none were considered related to study drug and did not result in study discontinuation. In study 301, an additional patient developed acute hepatitis. The patient discontinued study drug after her 20th injection owing to postinjection rhinorrhea, at which point she was noted to have liver-related laboratory abnormalities (more than 20 times elevation in transaminases and more than two times bilirubin) and mild scleral icterus. The patient denied any relevant risk factors for hepatitis, including relevant exposures, medication, or notable travel. Despite a comprehensive workup, no alternative etiology could be determined. A re-review of preclinical toxicology data, including laboratory results, histopathology, and a full evaluation of all liver function tests across the entire bremelanotide development program did not demonstrate evidence of an association of bremelanotide with hepatotoxicity. In addition, an in-depth consultation with an independent panel of hepatologists with special expertise in drug-induced liver injury was also convened. Based on the totality of data, the independent expert panel concluded that this case of hepatitis was unlikely due to bremelanotide, but, in the absence of an alternative etiology, its role could not be excluded. At the 6-month follow-up, the patient's alanine aminotransferase and aspartate aminotransferase levels had decreased to two times the upper limit of normal (with normal bilirubin); they had returned to normal by the next evaluation 6 months later.
In both studies, placebo-adjusted changes in BP for patients taking bremelanotide were consistent with ambulatory BP readings from the phase 2 studies, with a mean increase of approximately 3 mm Hg for systolic BP and 2 mm Hg for diastolic BP. These mild transient changes were noted approximately 0–2 hours postbremelanotide dosing, returned to baseline within 8–10 hours, and were accompanied by similarly mild decreases in pulse rates, such that there were no mean increases in overall myocardial workload. No clinically significant effects on electrocardiograms, weight, or effect of alcohol consumption were observed.
Efficacy
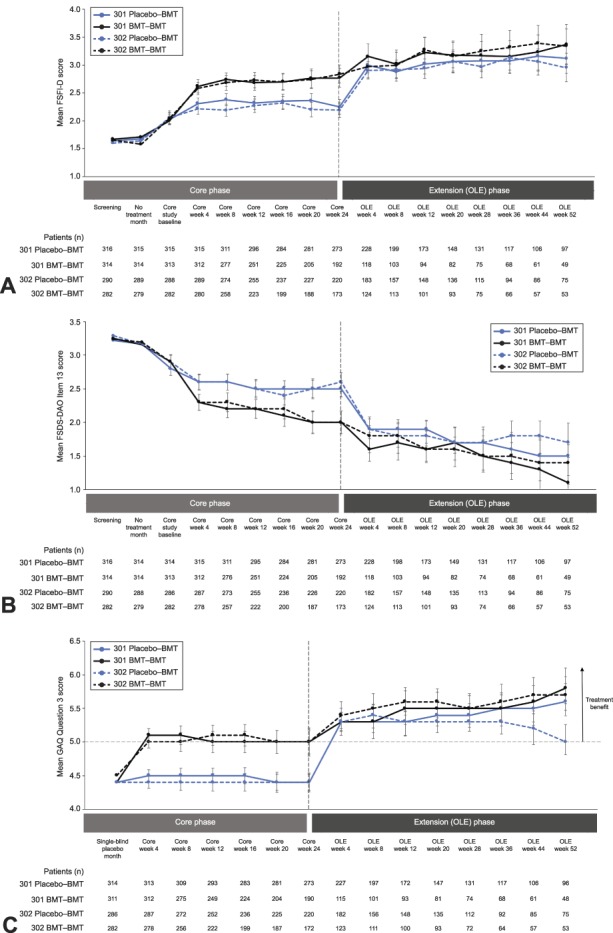
Women taking bremelanotide had statistically significant increases in sexual desire and reductions in distress related to low sexual desire compared with placebo from baseline to the end of the core phase (both P<.001 for integrated studies).23 Bremelanotide treatment was associated with sustained improvements in low sexual desire (FSFI–D scores25) throughout the 52-week open-label extension (Fig. 2A). In study 301, the change in FSFI-D from baseline to the end of the open-label extension was 0.77 (95% CI 0.54, 1.00) for the placebo→bremelanotide group and 1.30 (95% CI 0.98, 1.62) for the bremelanotide→bremelanotide group. In study 302, the change in FSFI-D from baseline to the end of the open-label extension was 0.70 (95% CI 0.42, 0.98) for the placebo→bremelanotide group and 1.25 (95% CI 0.93, 1.56) for the bremelanotide→bremelanotide group. All changes were greater than the minimal clinically important difference of +0.6. Despite slightly greater increases in desire in the bremelanotide→bremelanotide groups compared with the placebo→bremelanotide groups, after 1 month of bremelanotide treatment, patients in the placebo→bremelanotide groups demonstrated sustained increases in FSFI-D scores that were similar to changes seen in the bremelanotide groups during the core phase.
Fig. 2. Female Sexual Function Index–desire domain (FSFI–D) (A) and Female Sexual Distress Scale–Desire/Arousal/Orgasm (FSDS-DAO) item 13 scores (B) throughout RECONNECT studies for the open-label extension population and General Assessment Questionnaire (GAQ) question 3 mean score by visit (C). A. FSFI-D scores range from 1.2 to 6.0, with higher scores indicating greater sexual desire. Reference 25 explains the scoring system for the FSFI-D (see Appendix B in that article). B. Item 13 asks, “How often do you feel bothered by low sexual desire?” Scores range from 0 to 4, where 0=never; 1=rarely; 2=occasionally; 3=frequently; 4=always. C. GAQ question 3 asks, “Compared with the start of the study [before taking the study drug], to what degree do you think you benefited from taking the study drug?” Scores range from 1 (very much worse) to 7 (very much better); a score of 5 or higher indicates benefit. BMT, bremelanotide; OLE, open-label extension.
Simon. Long-term Safety and Efficacy of Bremelanotide. Obstet Gynecol 2019.
Bremelanotide was also associated with continued reductions in related distress (FSDS–DAO item 13 scores) throughout the 52-week open-label extension (Fig. 2B). Among the placebo→bremelanotide groups, change in FSDS-DAO item 13 from baseline to end of the open-label extension was −0.9 (95% CI −1.15, −0.71) and −0.9 (95% CI −1.10, −0.61) for studies 301 and 302, respectively. The bremelanotide→bremelanotide groups reported changes in FSDS-DAO item 13 from baseline to end of the open-label extension of −1.7 (95% CI −2.02, −1.32) and −1.4 (95% CI −1.77, −1.10) for studies 301 and 302, respectively. Although there were slightly greater reductions in related distress in the bremelanotide→bremelanotide groups compared with the placebo→bremelanotide groups, patients in the placebo→bremelanotide groups demonstrated a reduction in the item 13 score after 1 month that was similar to that reported in the bremelanotide groups during the core phase. Of note, continued improvement in low sexual desire during the open-label extension was coupled with even greater reductions in related distress over time.
According to the responses to General Assessment Questionnaire question 3, which served as a dynamic anchor for the coprimary endpoints of the core phase, participants felt that they had benefited from use of the study drug (Fig. 2C).
DISCUSSION
The safety and efficacy of bremelanotide, an approved treatment self-administered subcutaneously using an autoinjector on an as-needed basis, was demonstrated previously in a large 12-week, phase 2b trial in premenopausal women with hypoactive sexual desire disorder, female sexual arousal disorder, or both disorders, and more recently in two large, identically designed, phase 3, randomized, double-blind, placebo-controlled, 24-week trials in premenopausal women with hypoactive sexual desire disorder.16,23 Both phase 3 studies met their prespecified coprimary endpoints, demonstrating statistically significant and clinically meaningful increases in sexual desire and decreases in related distress for participants treated with bremelanotide compared with placebo during the core phase.23 In the phase 3 RECONNECT trials, the effect size of as-needed bremelanotide alone for improving sexual desire ranged from 0.49 to 0.61, and from 0.60 to 0.62 in reducing related distress. Relative to placebo, these values were 0.29–0.43 and 0.26–0.32, respectively. To provide context for the effect sizes observed with bremelanotide, the effect size of once-daily flibanserin relative to placebo was reported to range from 0.29 to 0.44 in improving sexual desire, and from 0.24 to 0.44 in reducing related distress across three phase 3 studies.26,27
Participants who received bremelanotide in the core phase showed continued improvements in desire and related distress during the open-label extension. Likewise, those who received placebo in the core phase had treatment benefits that mirrored bremelanotide-treated patients once they began treatment with bremelanotide during the open-label extension. Interestingly, the continued improvement in low sexual desire during the open-label extension was coupled with even greater reductions in related distress over time, suggesting that reductions in psychological distress, not surprisingly, lag behind improvements in desire.
The open-label extension demonstrates the long-term safety of bremelanotide in women with hypoactive sexual desire disorder who received the drug for up to 76 weeks (when including those treated with bremelanotide during the core study phase and open-label extension). Most adverse events were mild or moderate in severity and transient in nature. Nausea, flushing, and headache were the most common treatment-emergent adverse events considered to be related to study drug in both studies, and the only severe treatment-emergent adverse event considered study drug-related and experienced by more than one participant in both studies was nausea. One patient was diagnosed with acute hepatitis that resolved without further complications. Although this was regarded as possibly related to study drug, no indications of hepatitis related to bremelanotide have been reported previously.16 There were no other new safety signals or findings in patients who received treatment throughout the study for up to 76 weeks compared with those who only received bremelanotide during the 24-week double-blind period of the core phase.
The effects of bremelanotide on BP have been characterized in several clinical studies, which consistently revealed small, transient BP increases that peak within 4 hours postdose and return toward baseline by approximately 8–10 hours. These small changes in BP were accompanied by corresponding reductions in heart rate, resulting in lower or neutral heart rate-BP product values. Based on these studies, there is no evidence of any cumulative or sustained effects on BP related to bremelanotide. Because the product is used on a limited, as-needed basis, no long-term adverse cardiovascular effects would be anticipated.16
The most notable limitation of this study is that patients had to complete the double-blind portion of the trial before entry to the open-label extension. This introduced selection bias as those who discontinued bremelanotide or declined to enroll in the open-label portion of the study were not included. Fewer bremelanotide-treated patients completed the core phase and elected to continue in the open-label extension compared with placebo-treated patients. Women who had an initial satisfactory clinical response might have more readily elected participation in the open-label extension. Conversely, nonresponders are likely underrepresented in the open-label extension, which may result in an overestimation of the robust efficacy results. Additionally, only about 40% of patients who enrolled in the open-label extension completed the 52-week treatment period. As the major reason for study discontinuation was adverse events (21.2%), it is likely that the incidence of adverse events is higher in the “real world” compared with the population who participated in the open-label extension. The second most common reason for discontinuing the study was withdrawal of consent (18.3%), which is consistent with study discontinuation rates seen in other studies of similar duration. Overall, the results herein may be biased toward participation by women who had fewer adverse reactions and better clinical responses compared with those of the initial cohort. Finally, as hypoactive sexual desire disorder is a disorder of biopsychosocial etiology, treatment modalities such as psychotherapy may also be beneficial for patients in addition to pharmacologic interventions.
The results from the open-label extension confirm and highlight the robust data from the double-blind core phase and provide additional support for use of bremelanotide in the treatment of hypoactive sexual desire disorder in premenopausal women. Treatment benefits were maintained for participants who received bremelanotide during the core phase and were rapidly observed for participants who received placebo during the core phase once they began bremelanotide in the open-label extension. There were no new safety signals in patients who received long-term treatment with bremelanotide for a period of up to 76 weeks in the core and open-label extension phases of the study.23 Although potential selection biases may have affected the results of this study, based on the totality of data from this open-label extension of the RECONNECT studies, as-needed treatment with bremelanotide for up to 76 weeks demonstrated an acceptable safety profile accompanied by treatment benefits that support long-term use.
Authors' Data Sharing Statement
Will individual participant data be available (including data dictionaries)? No.
What data in particular will be shared? Not available.
What other documents will be available? Not available.
When will data be available (start and end dates)? Not applicable.
By what access criteria will data be shared (including with whom, for what types of analyses, and by what mechanism)? Not applicable.
Footnotes
The RECONNECT studies were sponsored and funded by Palatin Technologies, Inc., the innovator of bremelanotide. Editorial support in the preparation of this manuscript was provided by Phase Five Communications, supported by AMAG Pharmaceuticals, Inc., the licensee of bremelanotide.
Financial Disclosure Dr. Simon has served on advisory boards or has been a consultant for AbbVie, Allergan, AMAG Pharmaceuticals, Inc., Amgen, Ascend Therapeutics, Azure Biotech, Bayer HealthCare Pharmaceuticals, Inc., CEEK Enterprises, Covance Inc., Daré Bioscience, Duchesnay, Hologic Inc., KaNDy/NeRRe Therapeutics Ltd., Millendo, Mitsubishi Tanabe Pharma, ObsEva, Radius Health, Sanofi, Sebela, Sermonix, Shionogi, Symbiotec Pharmalab, TherapeuticsMD, and Valeant. He has also served on the speaker's bureau for AbbVie, AMAG Pharmaceuticals, Inc., Duchesnay, Novo Nordisk, Shionogi, and Valeant. He has received grants/research funding from AbbVie, Allergan, Agile Therapeutics, Bayer Healthcare LLC, Dornier MedTech, Endoceutics Inc., GTx Inc., Hologic Inc., Myovant Sciences, New England Research Institutes, ObsEva, Palatin Technologies, Inc., Symbio Research, TherapeuticsMD, Tissue Genesis, and Viveve Medical. He owns stock in Sermonix. Dr. Kingsberg has served on advisory boards or has been a consultant for AMAG Pharmaceuticals, Inc., Daré Bioscience, Duchesnay, Emotional Brain, Valeant, Endoceutics, Ivix, Palatin Technologies, Inc., Pfizer, Shionogi, Materna, Nuelle, Mitsubishi Tanaka North America, TherapeuticsMD, SST, and Lupin/Symbiomix. She has stock options in Viveve Medical. Dr. Portman has served on advisory boards or has been a consultant to AMAG Pharmaceuticals, Inc., Palatin Technologies, Inc., Endoceutics, Valeant Pharmaceuticals and Sprout, and is on the speaker's bureau for AMAG Pharmaceuticals, Inc. He is an employee and stockholder of Sermonix Pharmaceuticals. Dr. Williams and Dr. Krop are employees and stockholders of AMAG Pharmaceuticals, Inc. (the licensee of bremelanotide). Mr. Jordan is Vice President, Clinical Operations and Project Management, and a stockholder of Palatin Technologies, Inc. (the sponsor of the trial). He has a BS in Biology and has worked in clinical development for over 20 years. He led the Palatin team in the development of bremelanotide (clinical, regulatory, etc.) since 2007, including having a very significant role in designing the phase 3 studies, conduct of the studies, and analysis of the data. Dr. Lucas was an employee of Palatin Technologies, Inc. at the time of the study. She has no remaining financial interests in Palatin Technologies. She was the medical lead for the entire trial. She was the physician who ran the trial and provided safety evaluation and analysis for all adverse events as they occurred during the trial. She was also responsible for aggregate assessment of the adverse event database. She was the physician who provided medical information to the DSMB during the trial. Dr. Clayton has served on advisory boards or has been a consultant for Acadia, Alkermes, Allergan, AMAG Pharmaceuticals, Inc., Fabre Kramer, Ivix, Lundbeck, Palatin Technologies, Inc., S1 Biopharma, Sage Therapeutics, Sprout Pharmaceuticals, and Takeda. She has received grants from Endoceutics, Inc., Janssen, Sage Therapeutics, and Takeda. In addition, she has received royalties and/or owns copyright to works published by Ballantine Books/Random House and Guilford Publications, and the Changes in Sexual Functioning Questionnaire. She has shares or restricted stock units in Euthymics and S1 Biopharma.
Presented at the Annual Clinical and Scientific Meeting of the American College of Obstetricians and Gynecologists, April 27–29, 2018, Austin, Texas.
The authors thank all participants in these studies, their families, and personnel at all study sites.
Each author has confirmed compliance with the journal's requirements for authorship.
Peer reviews are available at http://links.lww.com/AOG/B588.
REFERENCES
- 1.U.S. Food and Drug Administration. Guidance for industry: low sexual interest, desire, and/or arousal in women: developing drugs for treatment. Available at: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM526362.pdf. Retrieved December 12, 2019.
- 2.Shifren JL, Monz BU, Russo PA, Segreti A, Johannes CB. Sexual problems and distress in United States women: prevalence and correlates. Obstet Gynecol 2008;112:970–8. [DOI] [PubMed] [Google Scholar]
- 3.Parish SJ, Hahn SR. Hypoactive sexual desire disorder: a review of epidemiology, biopsychology, diagnosis, and treatment. Sex Med Rev 2016;4:103–20. [DOI] [PubMed] [Google Scholar]
- 4.Goldstein I, Kim NN, Clayton AH, DeRogatis LR, Giraldi A, Parish SJ, et al. Hypoactive sexual desire disorder: International Society for the Study of Women's Sexual Health (ISSWSH) expert consensus panel review. Mayo Clin Proc 2017;92:114–28. [DOI] [PubMed] [Google Scholar]
- 5.Parish SJ, Goldstein AT, Goldstein SW, Goldstein I, Pfaus J, Clayton AH, et al. Toward a more evidence-based nosology and nomenclature for female sexual dysfunctions—part II. J Sex Med 2016;13:1888–906. [DOI] [PubMed] [Google Scholar]
- 6.Basson R. Biopsychosocial models of women's sexual response: applications to management of “desire disorders.” Sex Relation Ther 2003;18:107–15. [Google Scholar]
- 7.Arnow BA, Millheiser L, Garrett A, Lake Polan M, Glover GH, Hill KR, et al. Women with hypoactive sexual desire disorder compared to normal females: a functional magnetic resonance imaging study. Neuroscience 2009;158:484–502. [DOI] [PubMed] [Google Scholar]
- 8.Bianchi-Demicheli F, Cojan Y, Waber L, Recordon N, Vuilleumier P, Ortigue S. Neural bases of hypoactive sexual desire disorder in women: an event-related FMRI study. J Sex Med 2011;8:2546–59. [DOI] [PubMed] [Google Scholar]
- 9.Bloemers J, Scholte HS, van Rooij K, Goldstein I, Gerritsen J, Olivier B, et al. Reduced gray matter volume and increased white matter fractional anisotropy in women with hypoactive sexual desire disorder. J Sex Med 2014;11:753–67. [DOI] [PubMed] [Google Scholar]
- 10.Woodard TL, Nowak NT, Balon R, Tancer M, Diamond MP. Brain activation patterns in women with acquired hypoactive sexual desire disorder and women with normal sexual function: a cross-sectional pilot study. Fertil Steril 2013;100:1068–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simon JA, Kingsberg SA, Shumel B, Hanes V, Garcia M, Jr, Sand M. Efficacy and safety of flibanserin in postmenopausal women with hypoactive sexual desire disorder: results of the SNOWDROP trial. Menopause 2014;21:633–40. [DOI] [PubMed] [Google Scholar]
- 12.Thorp J, Simon J, Dattani D, Taylor L, Kimura T, Garcia M, Jr, et al. Treatment of hypoactive sexual desire disorder in premenopausal women: efficacy of flibanserin in the DAISY study. J Sex Med 2012;9:793–804. [DOI] [PubMed] [Google Scholar]
- 13.Derogatis LR, Komer L, Katz M, Moreau M, Kimura T, Garcia M, Jr, et al. Treatment of hypoactive sexual desire disorder in premenopausal women: efficacy of flibanserin in the VIOLET Study. J Sex Med 2012;9:1074–85. [DOI] [PubMed] [Google Scholar]
- 14.Goldfischer ER, Breaux J, Katz M, Kaufman J, Smith WB, Kimura T, et al. Continued efficacy and safety of flibanserin in premenopausal women with hypoactive sexual desire disorder (HSDD): results from a randomized withdrawal trial. J Sex Med 2011;8:3160–72. [DOI] [PubMed] [Google Scholar]
- 15.Katz M, DeRogatis LR, Ackerman R, Hedges P, Lesko L, Garcia M, Jr, et al. Efficacy of flibanserin in women with hypoactive sexual desire disorder: results from the BEGONIA trial. J Sex Med 2013;10:1807–15. [DOI] [PubMed] [Google Scholar]
- 16.Clayton AH, Althof SE, Kingsberg S, DeRogatis LR, Kroll R, Goldstein I, et al. Bremelanotide for female sexual dysfunctions in premenopausal women: a randomized, placebo-controlled dose-finding trial. Womens Health (Lond) 2016;12:325–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Addyi (flibanserin) prescribing information. Bridgewater, NJ: Valeant Pharmaceuticals North America LLC; 2016. [Google Scholar]
- 18.Vyleesi (bremelanotide injection) prescribing information. Waltham, MA: AMAG Pharmaceuticals, Inc; 2019. [Google Scholar]
- 19.Molinoff PB, Shadiack AM, Earle D, Diamond LE, Quon CY. PT-141: a melanocortin agonist for the treatment of sexual dysfunction. Ann N Y Acad Sci 2003;994:96–102. [DOI] [PubMed] [Google Scholar]
- 20.Pfaus J, Giuliano F, Gelez H. Bremelanotide: an overview of preclinical CNS effects on female sexual function. J Sex Med 2007;4(suppl 4):269–79. [DOI] [PubMed] [Google Scholar]
- 21.Kingsberg SA, Clayton AH, Pfaus JG. The female sexual response: current models, neurobiological underpinnings and agents currently approved or under investigation for the treatment of hypoactive sexual desire disorder. CNS Drugs 2015;29:915–33. [DOI] [PubMed] [Google Scholar]
- 22.Pfaus JG, Shadiack A, Van Soest T, Tse M, Molinoff P. Selective facilitation of sexual solicitation in the female rat by a melanocortin receptor agonist. Proc Natl Acad Sci USA 2004;101:10201–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kingsberg SA, Clayton AH, Portman D, Williams LA, Krop J, Jordan R, et al. Bremelanotide for the treatment of hypoactive sexual desire disorder: two randomized phase 3 trials. Obstet Gynecol 2019;134:899–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Althof S, Derogatis LR, Greenberg S, Clayton AH, Jordan R, Lucas J, et al. Responder analyses from a phase 2b dose-ranging study of bremelanotide. J Sex Med 2019;16:1226–35. [DOI] [PubMed] [Google Scholar]
- 25.Rosen R, Brown C, Heiman J, Leiblum S, Meston C, Shabsigh R, et al. The Female Sexual Function Index (FSFI): a multidimensional self-report instrument for the assessment of female sexual function. J Sex Marital Ther 2000;26:191–208. [DOI] [PubMed] [Google Scholar]
- 26.U.S. Food and Drug Administration. Summary review for regulatory action. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/022526Orig1s000SumRedt.pdf. Retrieved March 26, 2019. [Google Scholar]
- 27.Data on file. AMAG Pharmaceuticals, Inc. Waltham, MA. [Google Scholar]