

Supplemental Digital Content is Available in the Text.
BACKGROUND
PrabotulinumtoxinA is a 900-kDa botulinum toxin Type A produced by Clostridium botulinum.
OBJECTIVE
To investigate the efficacy and safety of prabotulinumtoxinA for the treatment of glabellar lines.
MATERIALS AND METHODS
Adult subjects (n = 330 in EV-001; n = 324 in EV-002) with moderate to severe glabellar lines at maximum frown on the 4-point Glabellar Line Scale (GLS; 0 = no lines, 1 = mild, 2 = moderate, and 3 = severe) were enrolled in 1 of 2 identical 150-day, double-blind, placebo-controlled, single-dose, Phase III studies. Subjects were randomized 3:1 to receive 20-U prabotulinumtoxinA or placebo. The primary efficacy end point was the proportion of responders on Day 30 where the investigator and subject independently agreed that a ≥2-point improvement had occurred on the GLS at maximum frown from Day 0. Adverse events (AEs) were evaluated throughout the study.
RESULTS
Responder rates in the prabotulinumtoxinA and placebo groups were 67.5% and 1.2% in EV-001 and 70.4% and 1.3% in EV-002; absolute differences between groups were 66.3% and 69.1% in EV-001 and EV-002, respectively (both p < .001). No serious AE in either study was assessed as study drug related.
CONCLUSION
In these studies, a single dose of 20-U prabotulinumtoxinA was safe and effective for the treatment of glabellar lines.
Treatment with botulinum toxin Type A has become the leading nonsurgical cosmetic procedure performed in the United States and internationally.1,2 The 900-kDa toxin onabotulinumtoxinA (Botox Cosmetic; Allergan, Irvine, CA) was the first botulinum toxin approved by the FDA for use in facial aesthetic procedures in the United States. It is widely recognized as the market leader in this field; the temporary improvement of glabellar lines continues to be the most common aesthetic application for this type of product.3
PrabotulinumtoxinA is a new 900-kDa botulinum toxin Type A preparation produced by Clostridium botulinum that was originally developed by Daewoong Pharmaceutical Co., Ltd., of Seoul, South Korea. It was licensed to Evolus, Inc., of Newport Beach, CA, for clinical development and distribution in several countries including the United States, Europe, Canada, and Australia. Results from a 268-subject, randomized, double-blind, Phase III comparator study conducted in South Korea using the original formulation provided the first evidence that 20-U prabotulinumtoxinA were both safe and effective for the treatment of moderate to severe glabellar lines in adult subjects and was noninferior to 20 U of onabotulinumtoxinA.4 The final formulation of prabotulinumtoxinA is vacuum-dried rather than freeze-dried, and uses a different source for the excipient human serum albumin (HSA); excipients include 0.5-mg HSA and 0.9-mg NaCl per 100-U vial.
Two identical Phase III clinical trials (EV-001 and EV-002) were undertaken in the United States to investigate the efficacy and safety of the final formulation of prabotulinumtoxinA for the treatment of glabellar lines in adult subjects. These studies were conducted in adherence with guidance published by the FDA in August 2014 that set new standards for developing botulinum toxin products for the treatment of upper facial lines.5 Two of the key FDA recommendations were (1) to make use of coprimary efficacy end points that are based on well-defined and reliable clinician-reported and patient-reported assessments and (2) to ensure that the degree of improvement associated with treatment is clinically meaningful—that is, success for treatment of glabellar lines should be defined by achievement of a score at maximum frown of 0 or 1, and a 2-grade improvement from baseline, on both investigator and subject assessment scales.
Methods
Study Design and Conduct
EV-001 and EV-002 were prospectively designed, multicenter, randomized, double-blind, placebo-controlled, single-dose studies conducted between January and September 2015 at 20 unique US study centers (10 centers each). Study protocols were approved by a centralized institutional review board (IRB) review process, and both studies were conducted in accordance with the ethical principles that have their origin in the 1975 Declaration of Helsinki. ClinicalTrials.gov identifiers: NCT02334423 for EV-001; NCT02334436 for EV-002.
Subjects
Study subjects were selected from a population of healthy adults, at least 18 years of age, who had moderate to severe glabellar lines at maximum frown, as independently agreed by investigator and subject assessment using the same validated 4-point photonumeric Glabellar Line Scale (GLS, Figure 1). Key exclusion criteria included previous treatment with botulinum toxin in any body region within the last 6 months or any planned treatment during the study period; previous treatment with any facial aesthetic procedure in the glabellar area within the last 12 months; previous insertion of permanent material in the glabellar area; any surgery in the glabellar area or any other planned facial aesthetic procedure during the study; marked facial asymmetry; and ptosis of eyelid and/or eyebrow, or history of eyelid and/or eyebrow ptosis. Women of childbearing potential had to have a negative pregnancy test result and be willing to use an acceptable form of contraception. All subjects provided written informed consent before entering the study.
Figure 1.
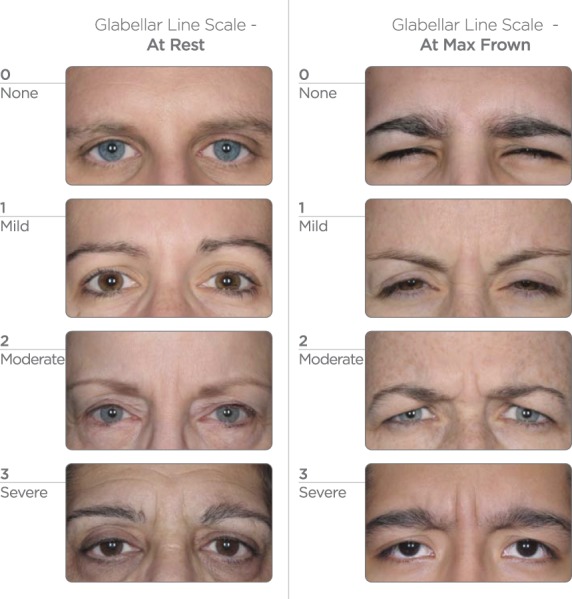
Glabellar Line Scale—at rest and at maximum frown. The instruction given to subjects to elicit a maximum frown was to “frown as much as possible, as if concentrating hard.” Note: this figure has been replaced since the article was originally published online ahead of print to correct the numerical scale.
Treatments and Follow-up
On Day 0, eligible subjects were randomly assigned in a 3:1 ratio to receive a single treatment (0.1 mL injected into each of 5 sites) of either prabotulinumtoxinA (total of 20 U, administered as 4 U/0.1 mL) or placebo (0.9% saline). Random numbers had been generated using SAS PROC PLAN (SAS Institute, Inc., Cary NC); a block randomization scheme with no stratification was used, with each block containing assignments for 3 prabotulinumtoxinA subjects and 1 placebo subject. At each study site, designated protocol-trained study personnel selected a study vial, which contained either 100 U of prabotulinumtoxinA or placebo according to the randomization schedule, reconstituted the vial with 2.5 mL of 0.9% sterile saline, filled the injection syringe, and provided it to the investigator in a blinded manner. The investigator administered the study treatment by intramuscular injection (Figure 2). Subjects were then followed for 150 days with site visits on Days 2, 7, 14, 30, 90, 120, and 150.
Figure 2.
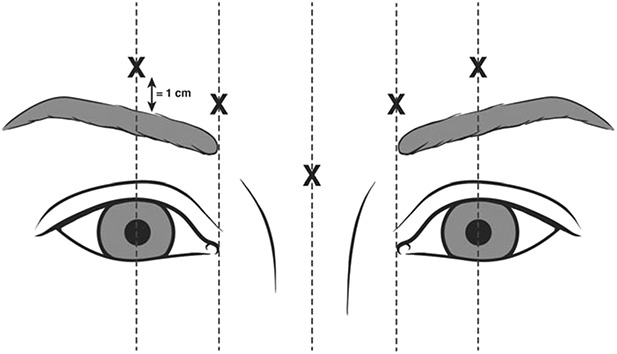
Target injection sites. The 5 target injection sites were the midline of the procerus, the inferomedial aspect of each corrugator muscle, and the superior middle aspect of each corrugator, at least 1 cm above the bony orbital rim.
Assessments
Efficacy was evaluated at site visits by investigator and subject assessment of:
(1) Glabellar lines at maximum frown and at rest on the 4-point GLS (0 = no lines, 1 = mild, 2 = moderate, and 3 = severe);
(2) Aesthetic outcomes on the 5-point Global Aesthetic Improvement Scale (GAIS; 2 = much improved, 1 = improved, 0 = no change, −1 = worse, and −2 = much worse).
In addition, subjects assessed their level of overall satisfaction on the 5-point Subject Satisfaction Scale (SSS, 2 = very satisfied, 1 = satisfied, 0 = indifferent, −1 = unsatisfied, and −2 = very unsatisfied). To ensure competency, investigators performing GLS evaluations were trained and certified on use of the GLS; in addition, all subjects received on-site training on the GLS.
Safety was evaluated by assessing adverse events (AEs), medical histories, physical examinations, vital signs, laboratory tests (including anti–botulinum toxin antibodies), electrocardiograms (ECGs), and concomitant medications. A directed questionnaire and directed review of body systems were performed as part of the medical history taken at each clinic visit. Findings based on these assessments were used to help guide the physical examination and ensure that the reporting of AEs—particularly those of special interest—was comprehensive. Adverse events of special interest (AESIs) were those 50 events listed in the FDA guidance document5—for example, blurred vision, dysphonia, eyelid ptosis, and speech disorder.
Outcomes and Statistical Analysis
The primary efficacy end point was defined as the proportion of subjects classified as responders on Day 30. This was a composite end point in which a subject was a responder only if both the investigator and subject independently agreed that a ≥2-point improvement had occurred on the GLS at maximum frown from Days 0 to 30. Secondary efficacy end points included the proportion of subjects, as independently agreed by both investigator and subject assessment, with a ≥2-point improvement on the GLS at maximum frown from Day 0 on each of Days 90, 120, and 150. Glabellar Line Scale, GAIS, and SSS scores by visit, and the proportion of subjects with a ≥1-point improvement on the GLS at maximum frown by visit, were considered exploratory efficacy end points.
The primary analysis of the primary efficacy end point was performed using the intent-to-treat (ITT) population, defined as all subjects randomized to treatment; subjects without a Day 0 or 30 primary outcome measure were excluded. The primary efficacy end point assessed the effectiveness of prabotulinumtoxinA against placebo on Day 30 in a superiority design. The primary null hypothesis (H0) was that the proportion of subjects classified as responders in the prabotulinumtoxinA group at Day 30 was equal to the proportion of subjects classified as responders in the placebo group. The null hypothesis was tested using the exact unconditional test; the corresponding 95% exact confidence intervals (CIs) were calculated by inversion of 2 one-sided intervals. The overall studywide Type I error (alpha) was 0.05. Secondary end points were tested sequentially using gatekeeper methods to maintain the Type I error. Calculations were performed using the unconditional procedure in StatExact 10 (Cytel software 2013) called “CI for difference of proportions.” Exploratory end points were evaluated using descriptive statistics.
All AEs were coded according to the Medical Dictionary for Regulatory Activities (MedDRA Version 17.0) and grouped by system organ class and preferred term. The incidences of AEs were summarized for each treatment group as frequencies and proportions. A 2-sided 95% CI was calculated for the differences between groups for the proportion of subjects with any AE and for the most common AE.
Sample Size
In each study, 324 subjects were to be enrolled. Assuming a 10% dropout rate, about 292 subjects were expected to complete the study: 219 treated with prabotulinumtoxinA and 73 treated with placebo. If no AE was observed among a sample size of 219, the upper bound of the 2-sided 95% CI would equal 0.016, and it was therefore unlikely that the true incidence of an event would exceed the upper bound of the 95% CI (i.e., <1.6%).
Results
Subject Disposition and Demographics
A total of 330 subjects in EV-001 and 324 in EV-002 were randomized to treatment, received the treatment as allocated, and thus by definition (Figure 3) formed the ITT and safety populations. These included 246 prabotulinumtoxinA and 84 placebo subjects in EV-001 and 246 prabotulinumtoxinA and 78 placebo subjects in EV-002. Most subjects (96.1% in EV-001; 96.9% in EV-002) attended the Day 150 visit and completed the study.
Figure 3.
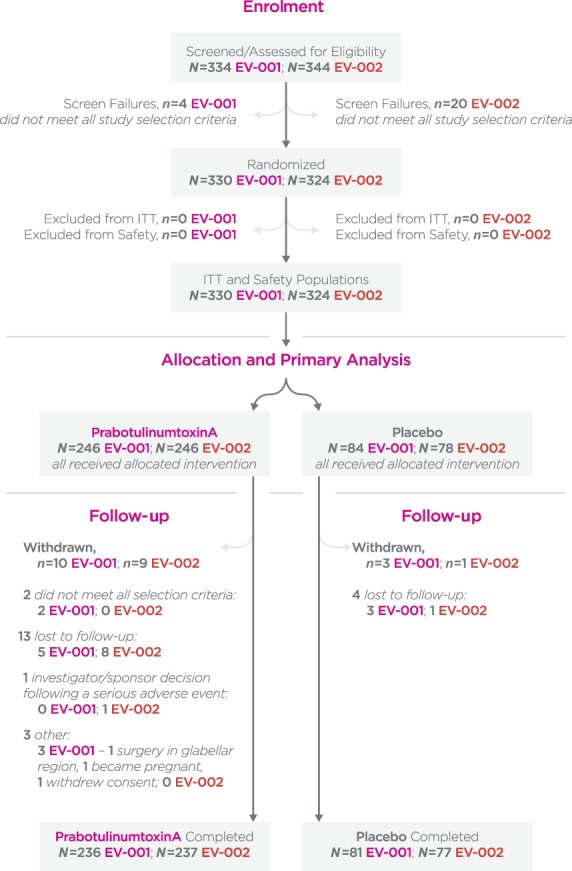
CONSORT flow diagram. ITT population, all subjects who were randomly assigned to treatment; Safety population, all subjects who were randomized and received at least 1 injection of prabotulinumtoxinA or placebo. ITT, intent-to-treat.
Within each of the studies and across studies, prabotulinumtoxinA and placebo groups were mostly similar in their demographic and other baseline characteristics (Table 1). Subjects had a mean age between 50 to 51 years; most (89.4% in EV-001; 89.8% in EV-002) were less than 65 years of age. Most subjects (92.7% in EV-001; 89.5% in EV-002) were female; most (81.2% in EV-001; 87.7% in EV-002) were racially identified as white. Many subjects (40.6% in EV-001; 36.1% in EV-002) had a history of botulinum toxin treatment. In EV-001 and EV-002, respectively, 67.9% and 83.3% of subjects had severe glabellar lines on the GLS at maximum frown by investigator assessment; 77.6% and 81.8% of subjects did by subject assessment.
TABLE 1.
Demographic and Glabellar Line Characteristics at Baseline—ITT/Safety Populations

Efficacy
For the primary efficacy end point (Table 2), the percentages of responders in each of the prabotulinumtoxinA and placebo groups in the ITT population were 67.5% and 1.2% in EV-001 and 70.4% and 1.3% in EV-002, respectively; the absolute difference between groups was 66.3% in EV-001 and 69.1% in EV-002 (both p < .001). There was no evidence of statistically significant differences between investigative sites in either study. Compared with the composite scores, absolute differences between prabotulinumtoxinA and placebo groups were more pronounced by each of investigator and subject assessment but overall were otherwise similar to each other: 76.3% and 73.1%, respectively (both p < .001), in EV-001; 79.8% and 72.3%, respectively (both p < .001), in EV-002.
TABLE 2.
Primary Efficacy End Point: Number and Percentage of Responders Based on a ≥2-Point Improvement on the GLS at Maximum Frown From Days 0 to 30—ITT Population

Analysis of the secondary end points allowed for investigation of the response at maximum frown beyond Day 30 (Table 3). For the composite end point, the absolute differences in the percentages of responders between groups at each of the Days 90, 120, and 150 or early termination visits were 25.2%, 7.0%, and 4.6%, respectively, for EV-001 (all p < .05), and 25.8%, 12.4%, and 4.6%, respectively, for EV-002 (all p < .05). Differences in responses between prabotulinumtoxinA and placebo groups were also clearly evident at each visit for exploratory end points based on the GLS—that is, a ≥1-point improvement on the GLS at maximum frown or a GLS score at maximum frown of 0 or 1 (See Supplemental Digital Content 1, Figure S1, http://links.lww.com/DSS/A142; Supplemental Digital Content 2, Figure S2, http://links.lww.com/DSS/A143; Supplemental Digital Content 3, Figure S3, http://links.lww.com/DSS/A144; and Supplemental Digital Content 4, Figure S4, http://links.lww.com/DSS/A145). Representative photographs of a subject's glabellar lines taken at baseline, and at 30 days, 90 days, 120 days, and 150 days after treatment with 20-U prabotulinumtoxinA are presented in Figures 4–8.
TABLE 3.
Secondary Efficacy End Points: Number and Percentage of Responders Based on a ≥2-Point Improvement on the GLS at Maximum Frown From Days 0 to 90, Day 120, and Day 150 or Early Termination, as Independently Agreed by Both Investigator and Subject Assessment—ITT Population

Figure 4.
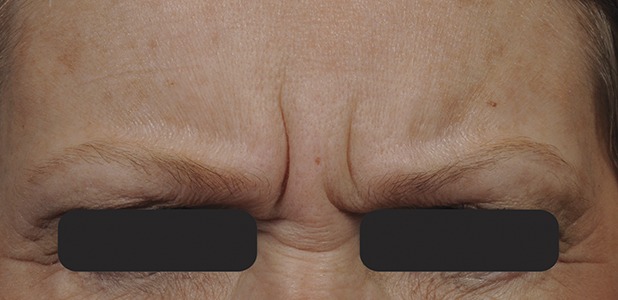
Glabellar lines at maximum frown at baseline before treatment with 20-U prabotulinumtoxinA.
Figure 8.
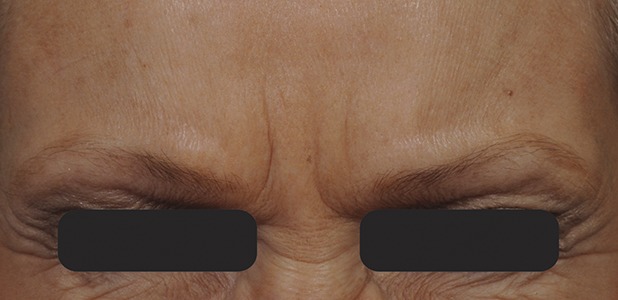
Glabellar lines at maximum frown at Day 150 after treatment with 20-U prabotulinumtoxinA.
Figure 5.
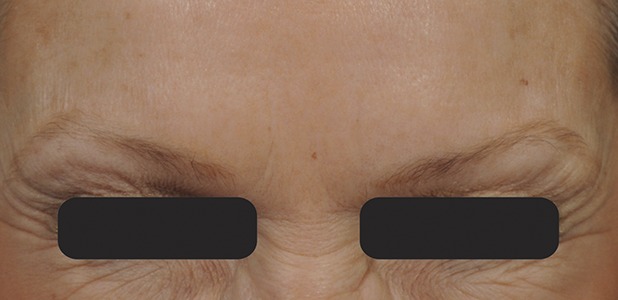
Glabellar lines at maximum frown at Day 30 after treatment with 20-U prabotulinumtoxinA.
Figure 6.
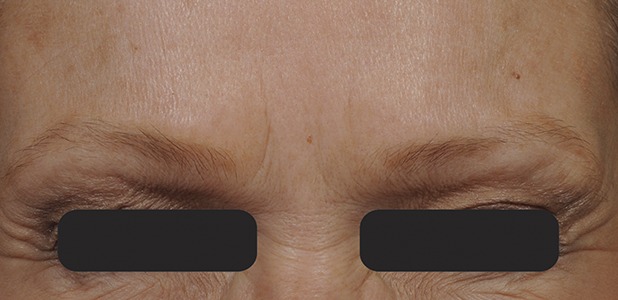
Glabellar lines at maximum frown at Day 90 after treatment with 20-U prabotulinumtoxinA.
Figure 7.
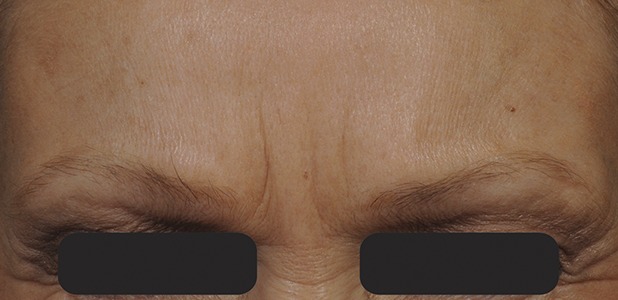
Glabellar lines at maximum frown at Day 120 after treatment with 20-U prabotulinumtoxinA.
Positive outcomes were also observed when subjects were evaluated for overall aesthetic improvement and level of satisfaction (Figures 9–12). Marked differences between prabotulinumtoxinA and placebo groups were evident from the first post-treatment assessment visit, Day 2. Based on the GAIS, at the Day 7, 14, and 30 visits, more than 90% of prabotulinumtoxinA subjects were assessed by both the investigator and subject as either “improved” or “much improved” (Figures 9 and 10). By the Day 150 or early termination visit, 53.6% and 50.2% of prabotulinumtoxinA subjects in EV-001, and 43.0% and 57.0% of prabotulinumtoxinA subjects in EV-002, continued to report this degree of improvement, based on investigator and subject assessment, respectively. Similarly, at study end on the Day 150 or early termination visit, 70.0% of prabotulinumtoxinA subjects in EV-001 and 71.3% of prabotulinumtoxinA subjects in EV-002 continued to be “satisfied” or “very satisfied” with the outcome of their treatment (Figures 11 and 12).
Figure 9.
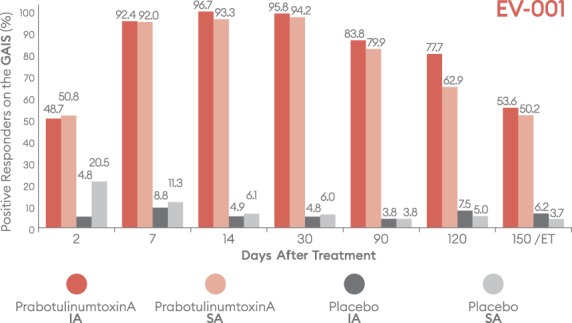
Percentage of subjects with a positive response (improved/much improved) on the Global Aesthetic Improvement Scale (GAIS) by visit—EV-001, ITT population. ET = early termination; IA, investigator assessment; ITT, intent-to-treat; SA, subject assessment.
Figure 12.
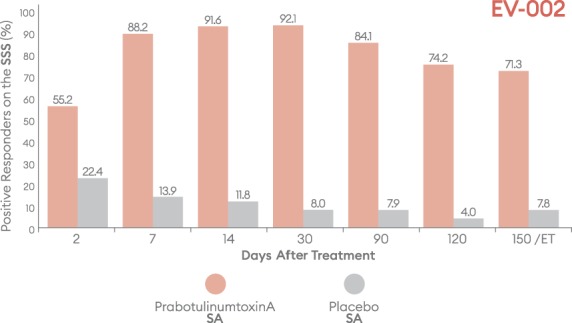
Percentage of subjects with a positive response (satisfied/very satisfied) on the Subject Satisfaction Scale (SSS) by visit—EV-002, ITT population. ET, early termination; ITT, intent-to-treat; SA, subject assessment.
Figure 10.
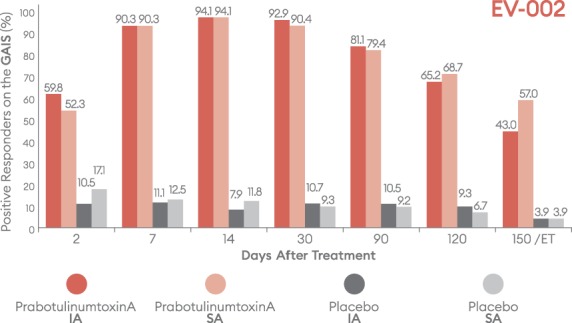
Percentage of subjects with a positive response (improved/much improved) on the Global Aesthetic Improvement Scale (GAIS) by visit—EV-002, ITT population. ET, early termination; IA, investigator assessment; ITT, intent-to-treat; SA, subject assessment.
Figure 11.
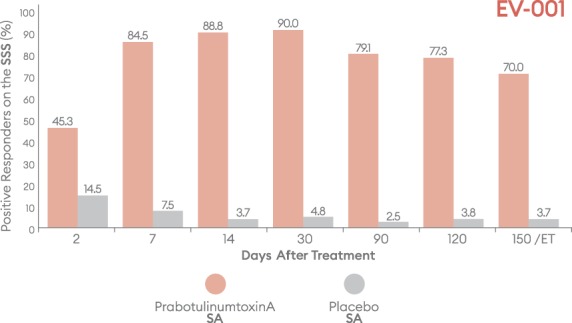
Percentage of subjects with a positive response (satisfied/very satisfied) on the Subject Satisfaction Scale (SSS) by visit—EV-001, ITT population. ET, early termination; ITT, intent-to-treat; SA, subject assessment.
Safety
All randomized subjects received a complete treatment that included 5 intramuscular injections of 0.1 mL each, in the glabellar region. All prabotulinumtoxinA subjects received a total of 20 U of botulinum toxin Type A reconstituted with saline; placebo subjects received saline only. In both studies, prabotulinumtoxinA and placebo groups were similar in the percentages of subjects who experienced: 1 or more AEs, most common AEs, and AEs assessed as possibly, probably, or definitely study drug related (Table 4). Three prabotulinumtoxinA subjects (3/246, 1.2%) in EV-001 experienced a serious AE: 1 malignant melanoma, 1 uterine cancer, and 1 intracranial aneurysm. Four prabotulinumtoxinA subjects (4/246, 1.6%) in EV-002 experienced a serious AE: 1 breast cancer, 1 stress-induced cardiomyopathy, 1 femur fracture, and 1 transient ischemic attack (TIA). No serious AE in either study was assessed as study drug related. The subject who experienced a TIA discontinued for this reason; no other subject in either study experienced an AE that led to study discontinuation.
TABLE 4.
Summary of Adverse Events—Safety Population

By preferred term, a total of 6 AEs in each study occurred in 1% or more prabotulinumtoxinA subjects—that is, in 3 or more prabotulinumtoxinA subjects. In EV-001, these included headache (14.6% vs 16.7% placebo), paresthesia (1.2% vs 0.0% placebo), upper respiratory tract infection (3.3% vs 0.0% placebo), eyelid ptosis (1.6% vs 0.0% placebo), increased white blood cell count (2.4% vs 0.0% placebo), and seasonal allergy (1.2% vs 0.0% placebo). In EV-002, these included headache (8.5% vs 9.0% placebo), nasopharyngitis (1.2% vs 1.3% placebo), upper respiratory tract infection (2.0% vs 1.3% placebo), urinary tract infection (1.2% vs 0.0% placebo), eyelid ptosis (1.6% vs 0.0% placebo), and injection site bruising (1.2% vs 0.0% placebo).
Few AEs were assessed as both of special interest and as study drug related (Table 4); all resolved. None were reported for placebo subjects, and the incidence among prabotulinumtoxinA subjects was low. Three prabotulinumtoxinA subjects (3/246, 1.2%) in EV-001 experienced 4 study drug–related AESIs: 1 eyebrow ptosis (0.4%), 2 eyelid ptosis (0.8%), and 1 blurred vision (0.4%), which was secondary to eyelid ptosis. Four prabotulinumtoxinA subjects (4/246, 1.6%) in EV-002 experienced 7 study drug–related AESIs: 2 eyebrow ptosis (1 left, 1 right) in 1 subject (0.4%); 3 eyelid ptosis (1.2%); and 1 blurred vision (0.4%) and 1 diplopia (0.4%) in subjects who also experienced eyelid ptosis assessed as study drug related.
In both studies, no subject who was negative for the presence of botulinum toxin antibodies at baseline tested positive on any of the repeat tests taken at Days 30, 90, and end of study/early termination. No particularly noteworthy difference between prabotulinumtoxinA and placebo groups was seen in any of the laboratory measures and findings, vital sign measures, ECG findings, physical examination results, or in the use of concomitant medications.
Discussion
The primary analysis of the primary efficacy end point establishes the effectiveness of prabotulinumtoxinA for the treatment of moderate to severe glabellar lines and its superiority over placebo at Day 30. For the composite end point where both the investigator and subject independently reached the assessment that a ≥2-point improvement had occurred on the GLS at maximum frown from Days 0 to 30, the absolute differences in the percentages of responders in the prabotulinumtoxinA and placebo groups were 66.3% in EV-001 and 69.1% in EV-002 (both p < .001). Statistically significant differences between prabotulinumtoxinA and placebo groups were still evident for the composite end points even at the Day 150 or early termination visit.
Of note, the composite 2-point or greater improvement responder definitions mandated by the FDA and used for the primary and secondary efficacy analyses are statistically robust tests well suited to regulatory objectives. Placebo-controlled Phase III trials using this type of responder definition have been reported for 2 other botulinum Type A toxins: responder rates of 48% and 60% have been reported for 20 U of incobotulinumtoxinA (Xeomin; Merz Pharmaceuticals GmbH, Frankfurt, Germany); responder rates of 52%, 55%, and 60% have been reported for 50 U of abobotulinumtoxinA (Dysport; Ipsen Biopharm Ltd., Wrexham, United Kingdom).6,7 Data of this type were not reported for the onabotulinumtoxinA Phase III trials.8 Although direct comparisons across any of these studies are not valid, these data and the authors' data serve to illustrate that responder rates based on a composite 2-point or greater improvement are generally lower than those based on other definitions typically reported in the literature for this indication. From a clinician's perspective, commonly reported metrics that may translate into more meaningful or desirable clinical outcomes for their patients include a ≥1-point improvement on the GLS at maximum frown, or a GLS score at maximum frown of 0 or 1, by each or either of investigator and subject assessment. In the EV-001 and EV-002 studies, these were included as exploratory efficacy end points (presented as supplemental digital content only), and accordingly, no statistical analyses were performed.
The safety of a single treatment of 20 U of prabotulinumtoxinA, administered as 5 injections of 0.1 mL each, for the treatment of moderate to severe glabellar lines was established in comparison with placebo treatment based on a broad range of safety outcomes. The overall incidence of AEs, the incidence of the most common event, and the incidence of study drug–related events were similar between prabotulinumtoxinA and placebo groups. No deaths were reported. No serious AE was study drug related. Only headache was reported in 5% or more subjects, with equal incidence in prabotulinumtoxinA and placebo subjects. Adverse events, both related and unrelated to study drug, occurring in 1% or more subjects were similar to those reported on label for other botulinum toxin products used for the same indication6–8; eyelid ptosis, which was observed at an incidence of 1.6% in prabotulinumtoxinA subjects in both the EV-001 and EV-002 studies, was similar to the incidence rate of between 0.2% and 3% reported for placebo-controlled trials with other toxin products.6–8
One limitation of these individual studies is the relatively small number of subjects representing various subpopulations of interest—for example, subjects' age ≥65 years, men, non-Caucasian racial groups, and individual Fitzpatrick skin types. Sample sizes for these categories were not sufficiently large to draw any conclusions about the relative efficacy of prabotulinumtoxinA. Because individual studies have not been conducted targeting specific subpopulations, pooling of data across studies would be required to draw meaningful conclusions about the efficacy and safety of prabotulinumtoxinA within any particular subgroup. A second limitation of these studies is the absence of an active comparator that would provide insight into the comparative safety and the efficacy of different toxins using the same composite end point. Although comparative data have been published between the original prabotulinumtoxinA formulation and onabotulinumtoxinA in a Korean study population,4 efficacy was not based on the more stringent FDA-mandated end point used in these 2 US studies.
Conclusions
Based on the results of these 2 identical Phase III multicenter, randomized, double-blind, placebo-controlled studies, a single treatment of 20 U of prabotulinumtoxinA was safe and effective in adult subjects for the treatment of moderate to severe glabellar lines.
Supplementary Material
Acknowledgments
The authors acknowledge the dedicated work of their coinvestigators in the EV-001 and EV-002 studies as follows; the principal investigator at each site is identified in bold type.
EV-001: Jeffrey S. Dover and Lara Kruter at Skin Care Physicians, Chestnut Hill, MA; Steven Fagien at Steven Fagien MD, Boca Raton, FL; Dee Anna Glaser, Joseph Kallini, and Timur Galperin at the St. Louis University Hospital, St. Louis, MO; Miles Graivier at the Graivier Center, Roswell, GA; Pearl E. Grimes at the Vitiligo and Pigmentation Institute of Southern California, Los Angeles, CA; Derek H. Jones and Naissan Wesley at Skin Care and Laser Physicians of Beverly Hills, Los Angeles, CA; Neil Sadick, Zeena Al-Dujaili, and Dina Began at the Sadick Research Group, New York, NY; Soheil Simzar at ATS Clinical Research, Santa Monica, CA; Stacy R. Smith and Sharon Boothe-Kepple at the California Dermatology and Clinical Research Institute, Encinitas, CA; and Robert A. Weiss, Margaret Weiss, Karen Beasley, and Christian Halvorson at The Maryland Laser Skin and Vein Institute, Hunt Valley, MD.
EV-002: Hillary Julius at the Research Institute of the Southeast, West Palm Beach, FL; Sue Ellen Cox and Chris G. Adigun at Aesthetic Solutions, Chapel Hill, NC; John H. Joseph at Clinical Testing of Beverly Hills Inc., Beverly Hills, CA; Z. Paul Lorenc at Lorenc Aesthetic Plastic Surgery Center, New York, NY; Gary D. Monheit, James Highsmith, and Heidi Neugent at the Total Skin and Beauty Dermatology Center, Birmingham, AL; Ronald L. Moy at Moy-Fincher-Chipps Facial Plastics/Dermatology, Beverly Hills, CA; Alexander Rivkin and Robert Cohen at Westside Aesthetics, Los Angeles, CA; Harry H. Sharata and Blaine Jensen at Madison Skin and Research Inc., Madison, WI; Susan C. Taylor at Society Hill Dermatology, Philadelphia, PA; and Vernon Leroy Young, Lisa Backues, Cyndi Buchanan, Ann-Elizabeth Mohart, Michelle Schaning, and Donna Straatmann at Mercy Health Research, Washington, MO.
The authors also gratefully acknowledge the contributions of the following: the study sponsor, Evolus, Inc., of Newport Beach, CA, with special mention of Gregg Peterson and Rose Monroe of the Clinical Operations team, and Song Wang, PhD, Statistical Science Director at Pharmaceutical Product Development (PPD) of Wilmington, NC, who served as senior statistical consultant.
Footnotes
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.dermatologicsurgery.org).
A. T. Shamban and K. R. Beer served as principal clinical trial investigators for the EV-001 and EV-002 studies. All participating coinvestigators who formed the EV-001/EV-002 Study Group are listed in the acknowledgments. As sponsor of the EV-001 and EV-002 studies, Evolus, Inc., of Newport Beach, CA, was involved in the design of these studies and provided funding, study materials, equipment, and medications to all investigational sites. Evolus also provided funding to contract organizations involved in data collection, analysis, and reporting of the results. R. L. Avelar is the Head of R&D and Chief Medical Officer for Evolus, Inc., and receives compensation in salary, stock, and stock options. Before and during the time of these studies and manuscript preparation, J. E. Gross was the Chief Scientific Officer at Evolus, Inc.; he will receive royalty and milestone payments should the product be approved. Anneke Jonker of Medical Writing Associates, West Vancouver, BC, Canada, provided technical assistance with manuscript preparation and submission; she holds stock in Evolus, Inc.
References
- 1.American Society of Aesthetic Plastic Surgery. 2016 Cosmetic Surgery National Data Bank Statistics. Available from: http://www.surgery.org/sites/default/files/ASAPS-Stats2016.pdf. Accessed June 23, 2017. [DOI] [PubMed] [Google Scholar]
- 2.International Society of Aesthetic Plastic Surgeons. ISAPS International Survey on Aesthetic/Cosmetic Procedures Performed in 2015. Available from: https://www.isaps.org/Media/Default/global-statistics/2016%20ISAPS%20Results.pdf. Accessed June 23, 2017. [Google Scholar]
- 3.Dorizas A, Krueger N, Sadick NS. Aesthetic uses of the botulinum toxin. Dermatol Clin 2014;32:23–36. [DOI] [PubMed] [Google Scholar]
- 4.Won CH, Kim HK, Kim BJ, Kang H, et al. Comparative trial of a novel botulinum neurotoxin type A versus onabotulinumtoxinA in the treatment of glabellar lines: a multicenter, randomized, double-blind, active-controlled study. Int J Dermatol 2015;54:227–34. [DOI] [PubMed] [Google Scholar]
- 5.U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Guidance for Industry. Upper Facial Lines: Developing Botulinum Toxin Drug Products. Draft Guidance. Washington, DC: United States Department of Health and Human Services; 2014. [Google Scholar]
- 6.Merz Pharma GmbH & Co. Xeomin: Highlights of Prescribing Information. 2015. Available from: http://www.xeomin.com/wp-content/uploads/xeomin-full-prescribing-information.pdf. Accessed September 10, 2018. [Google Scholar]
- 7.Ipsen Biopharm Ltd. Dysport: Highlights of Prescribing Information. 2017. Available from: https://www.dysport.com/pdfs/Dysport_Full_Prescribing_Information.pdf. Accessed September 10, 2018. [Google Scholar]
- 8.Allergan, Inc. Botox Cosmetic: Highlights of Prescribing Information. 2016. Available from: https://www.allergan.com/assets/pdf/botox_cosmetic_pi.pdf. Accessed September 10, 2018. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.