

Abstract
Background:
Studies have reported mitophagy activation in renal tubular epithelial cells (RTECs) in acute kidney injury (AKI). Phosphatase and tensin homolog-induced putative kinase 1 (PINK1) and E3 ubiquitin-protein ligase Parkin are involved in mitophagy regulation; however, little is known about the role of PINK1-Parkin mitophagy in septic AKI. Here we investigated whether the PINK1-Parkin mitophagy pathway is involved in septic AKI and its effects on cell apoptosis in vitro and on renal functions in vivo.
Methods:
Mitophagy-related gene expression was determined using Western blot assay in human RTEC cell line HK-2 stimulated with bacterial lipopolysaccharide (LPS) and in RTECs from septic AKI rats induced by cecal ligation and perforation (CLP). Autophagy-related ultrastructural features in rat RTECs were observed using electron microscopy. Gain- and loss-of-function approaches were performed to investigate the role of the PINK1-Parkin pathway in HK-2 cell mitophagy. Autophagy activators and inhibitors were used to assess the effects of mitophagy modulation on cell apoptosis in vitro and on renal functions in vivo.
Results:
LPS stimulation could significantly induce LC3-II and BECN-1 protein expression (LC3-II: 1.72 ± 0.05 vs. 1.00 ± 0.05, P < 0.05; BECN-1: 5.33 ± 0.57 vs. 1.00 ± 0.14, P < 0.05) at 4 h in vitro. Similarly, LC3-II, and BECN-1 protein levels were significantly increased and peaked at 2 h after CLP (LC3-II: 3.33 ± 0.12 vs. 1.03 ± 0.15, P < 0.05; BECN-1: 1.57 ± 0.26 vs. 1.02 ± 0.11, P < 0.05) in vivo compared with those after sham operation. Mitochondrial deformation and mitolysosome-mediated mitochondria clearance were observed in RTECs from septic rats. PINK1 knockdown significantly attenuated LC3-II protein expression (1.35 ± 0.21 vs. 2.38 ± 0.22, P < 0.05), whereas PINK1 overexpression markedly enhanced LC3-II protein expression (2.07 ± 0.21 vs. 1.29 ± 0.19, P < 0.05) compared with LPS-stimulated HK-2 cells. LPS-induced proapoptotic protein expression remained unchanged in autophagy activator-treated HK-2 cells and was significantly attenuated in PINK1-overexpressing cells, but was remarkably upregulated in autophagy inhibitor-treated and in PINK1-depleted cells. Consistent results were observed in flow cytometric apoptosis assay and in renal function indicators in rats.
Conclusion:
PINK1-Parkin-mediated mitophagy might play a protective role in septic AKI, serving as a potential therapeutic target for septic AKI.
Keywords: Sepsis, Acute kidney injury, Autophagy, Mitophagy, Phosphatase and tensin homolog-induced putative kinase 1, E3 ubiquitin-protein ligase Parkin
Introduction
Sepsis-associated acute kidney injury (AKI) is one of the leading causes of sepsis-related death in intensive care units, accounting for 50% of all AKI cases.[1,2] Septic AKI may cause prolonged hospitalization and increased mortality in critically ill patients.[2,3] Current evidence suggests that the pathogenesis of septic AKI differs from that of non-septic AKI.[4,5] It has been reported that hemodynamic changes might play a minor role in sepsis-induced renal dysfunction.[6,7] Our previous studies showed that stressful conditions such as oxidant injury and mitochondrial dysfunction contribute to the pathogenesis of septic AKI.[8,9] However, the exact pathophysiology of septic AKI remains poorly understood. A better understanding of these mechanisms may provide new therapeutic strategies for septic AKI.
Autophagy is an evolutionarily conserved pathway to maintain intracellular homeostasis through lysosomal degradation and recycling of damaged organelles and macromolecules.[10] Cellular stresses, including sepsis, hypoxia, cell starvation, oxidant injury, and insults, can trigger autophagy.[11,12] Increasing evidence suggests that autophagy can prevent kidney injury from sepsis,[13–15] but its regulation remains unclear. The phosphatase and tensin homolog-induced putative kinase 1 (PINK1)-E3 ubiquitin protein ligase Parkin pathway has been extensively studied in Parkinson's disease for its association with mitophagy, a process of removing dysfunctional mitochondria by autophagy.[16,17] Previous studies revealed an essentially protective role of the PINK1-Parkin mitophagy pathway against renal tubular epithelial cell (RTEC) injury in AKI induced by cisplatin, contrast, or ischemia-reperfusion injury.[18–21] However, little is known about the role of this pathway in septic AKI. Recent studies showed that mitochondrial dysfunction occurs during sepsis, and PINK1-Parkin mitophagy can protect cardiomyocytes and macrophages in response to septic insults.[22–25] We therefore hypothesized that PINK1-Parkin mitophagy might exert a protective function against sepsis-induced renal injury.
In this study, we stimulated human RTEC cell line HK-2 with lipopolysaccharide (LPS) in vitro and established a cecal ligation and perforation (CLP)-induced septic AKI model in rats. Gain- and loss-of-function methods were performed to investigate the role of PINK1-Parkin in HK-2 cell mitophagy. Autophagy activators and inhibitors were used to assess the effects of mitophagy modulation on cell apoptosis in vitro and on renal functions in vivo. Our results suggest that PINK1-Parkin-modulated mitophagy is a potential therapeutic target in septic AKI treatment.
Methods
Ethical approval
This study was conducted in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health, Bethesda, MD, USA). The protocol was approved by the Committee on Ethics in Animal Experiments of Southern Medical University (Guangzhou, China).
Animal model
A total of 66 specific pathogen-free Sprague-Dawley rats (male or female; 180–220 g) were obtained from the Experimental Animal Center at Southern Medical University (Guangzhou, China). The rats were housed in plastic cages under controlled conditions (temperature, 25°C; humidity, 50%–55%; and 12:12 h light/dark cycle), with free access to food and water. All rats were anesthetized with ketamine and xylazine (0.1 mL/100 g) intraperitoneally before the operation. CLP was performed as previously described.[8,9] Briefly, the cecum was ligated near the junction of ileum and colon using a 4–0 silk ligature, punctured in two locations using an 18-gauge needle, and gently compressed until the feces were extruded. The bowel was then returned to the abdomen and the incision closed. Rats in the sham group suffered the same surgical procedures, but the cecum was neither ligated nor punctured. At the end of the operation, all rats were subcutaneously resuscitated with normal saline (20 mL/kg). All rats were deprived of food but had free access to water after surgery.
Thirty rats were randomly divided into five groups (one sham operation group and four CLP groups for different time points, n = 6 in each group) to explore the time course of CLP-induced autophagy. Kidney tissues were obtained for RTEC isolation in the sham operation group or at 2, 4, 8, and 16 h after CLP to examine mitochondrial morphology and autophagy-related protein expression.
To explore the effects of mitophagy activators and inhibitors on kidney function in septic AKI, the remaining rats were randomly divided into six groups (n = 6 in each group): control, CLP + vehicle, CLP + 3-methyladenine (3-MA), CLP + chloroquine (CQ), CLP + brefeldin A (BFA), and CLP + rapamycin (RAPA) groups. Following anesthesia, rats in the control group underwent sham operation without any other treatment, whereas other rats were given dimethyl sulfoxide (DMSO, 0.3 mL), 3-MA (0.2 mL, 20 mg/kg), CQ (0.2 mL, 60 mg/kg), BFA (0.2 mL, 4 mg/kg), or RAPA (0.2 mL, 4 mg/kg) 30 min (for DMSO) or 2 h (for other drugs) prior to CLP. These rats were sacrificed at 24 h after surgery, and the blood urea nitrogen (BUN) and serum creatinine levels were measured.
RTEC isolation
RTECs were isolated from the renal tissue of each rat as previously described.[8,9] Briefly, the cortex was cut into fragments, and cells were dissociated by incubation with 1 mg/mL type-I collagenase for 30 min at 37°C. Red blood cells were removed by lysis. RTECs were then separated using Percoll gradient density centrifugation. The purity of the RTECs was determined by immunostaining with cytokeratin-18 and Hoechst dye.
Cell culture and treatment
HK-2 cells were obtained from Sigma-Aldrich (St. Louis, MO, USA) and cultured in Dulbecco's modified Eagle's medium/F12 media supplemented with 10% fetal bovine serum at 37°C in a humidified atmosphere of 5% CO2. To explore the time course of LPS-induced mitophagy, HK-2 cells were harvested at 0, 2, 4, 8, 16, and 24 h following LPS (10 μmol/L; Sigma-Aldrich; Merch Millipore, Darmstadt, Germany) treatment for determination of autophagy-related gene expression. To explore the role of the PINK1-Parkin pathway in mitophagy, HK-2 cells were treated with LPS 10 μmol/L) for 4 h, followed by transient transfection with control short interfering RNA (siRNA), PINK1 siRNA, control plasmid Ad-lacZ, or Ad-PINK1. To explore the effects of autophagy on apoptosis, HK-2 cells were stimulated with LPS (10 μmol/L) for 4 h, followed by treatment with 0.5 mL of normal saline, 3-MA (5 nmol/L), CQ (50 μmol/L), BFA (1 μg/mL), or RAPA (100 nmol/L) for 2 h.
PINK1 overexpression or knockdown
HK-2 cells were transfected with Ad-PINK1 (Addgene, Cambridge, MA, USA) or siRNA against human PINK1 (Santa Cruz Biotechnology, CA, USA) in 6-well plates using Lipofectamine 2000 (Invitrogen, Life Technology, Carlsbad, CA, USA) following the manufacturer's instructions. An empty vector Ad-lacZ (Addgene) and scrambled siRNA (Santa Cruz Biotechnology) were used as negative controls, respectively.
Electron microscopy
Kidney tissues were fixed with 2.5% glutaraldehyde for 24 h at room temperature and stained with cacodylate-buffered osmium tetroxide for 4 h at 4°C. Sections (60 nm thickness) were cut and examined under an electron microscope (H-7500; Hitachi, Tokyo, Japan) at ×10,000 magnification.[26]
Western blot assay
HK-2 cells or rat RTECs were centrifuged at 14,000 × g for 10 min after homogenization in radioimmunoprecipitation assay lysis buffer. Total protein concentrations in the supernatants were determined using the bicinchoninic acid method. Equal amounts of protein (30 μg) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride membranes using wet transfer at 100 V for 90 min at 4°C. Nonspecific binding sites were blocked using 1% bovine serum albumin in 0.05% Tween-20 Tris-buffered saline for 1 h, followed by incubation with primary antibodies recognizing microtubule-associated protein 1 light chain 3 alpha (LC3II; Sigma-Aldrich), p62 (sequestosome 1; Sigma-Aldrich), BECN-1 (Abcam, Cambridge, UK), PINK1 (Abcam), Parkin (Abcam), cleaved caspase-3 (Sigma-Aldrich), BAX (Sigma-Aldrich), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Cell Signaling Technology, Beverly, MA, USA) overnight at 4°C. The membranes were then incubated with corresponding secondary antibodies. The gray values of the immunoreactive protein bands of interest were quantified using Image Lab software (Bio-Rad, Hercules, CA, USA). Each measurement was performed at least three times.
Flow cytometry
Flow cytometry assay was performed to examine HK-2 cell apoptosis using a fluorescein isothiocyanate (FITC)-Annexin V apoptosis detection kit (BD Biosciences, San Jose, CA, USA). Cells were washed twice with phosphate-buffered saline and resuspended in 1× binding buffer at a concentration of 1 × 105 cells/mL prior to incubation with FITC-Annexin V (5 μL) and propidium iodide (PI; 10 μL of 50-μg/mL; Sigma-Aldrich–Merck Millipore) for 20 min in the dark at room temperature. The fluorescence was determined using a flow cytometer (Becton Dickinson FACScan; BD Biosciences).
Measurements of BUN and serum creatinine levels
Blood samples were collected from rats prior to sacrifice to measure the BUN and serum creatinine levels using a FUJIFILM PRI-CHEM 3500S system (Fujifilm, Tokyo, Japan).
Statistical analysis
Data were expressed as the mean ± standard deviation. Statistical analysis between groups was performed using one-way analysis of variance in SPSS 22.0 statistical software (version 22.0; SPSS Inc., Chicago, IL, USA). A P < 0.05 was considered statistically significant.
Results
Septic insults induce mitophagy in RTECs both in vitro and in vivo
To explore whether mitophagy is involved in the pathogenesis of septic AKI, we assessed autophagy-related gene expression and ultrastructural features in LPS-stimulated HK-2 cells in vitro and in CLP-induced septic AKI in vivo. As shown in Figure 1A, LC3-II and BECN-1 protein levels were significantly increased in HK-2 cells within the first 4 h after LPS stimulation and were gradually decreased thereafter. Especially, LPS stimulation induce LC3-II and BECN-1 protein expression (LC3-II: 1.72 ± 0.05 vs. 1.00 ± 0.05, P < 0.05; BECN-1: 5.33 ± 0.57 vs. 1.00 ± 0.14, P < 0.05) at 4 h compared with 0 h. We therefore used 4 h LPS stimulation in the following in vitro experiments.
Figure 1.
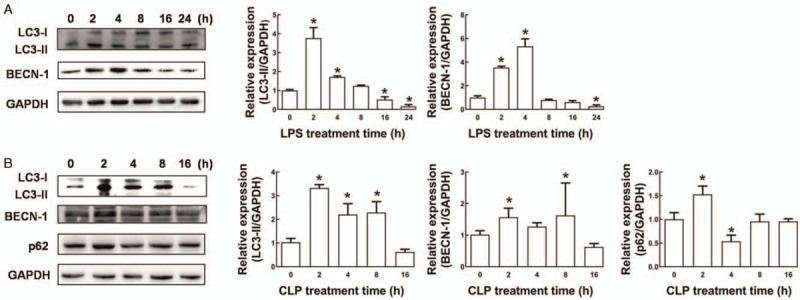
Dynamic expression analysis of autophagy proteins in response to septic insults. (A) Western blot analysis was performed to quantify LC3-II and BECN-1 levels. HK-2 cells were harvested at 0, 2, 4, 8, 16, and 24 h after LPS (10 μmol/L) treatment. (B) Western blot analysis was performed to quantify LC3-II, BECN-1, and p62 protein expression. Thirty rats were randomly divided into five groups (one sham operation group and four CLP groups for different time points, n = 6 in each group). Kidney tissues were obtained for RTEC isolation from the sham-operated rats (0 h) and from CLP rats at 2, 4, 8, and 16 h after CLP. Data are expressed as the mean ± standard deviation. ∗P < 0.05 vs. 0 h. BECN-1: Beclin1; CLP: Cecal ligation and perforation; LC3: Microtubule-associated protein light chain 3; LPS: Lipopolysaccharide; RTEC: Renal tubular epithelial cell.
Similarly, in rat RTECs, LC3-II, BECN-1, and p62 protein levels were consistently and significantly increased and peaked at 2 h after CLP compared with those after sham operation (LC3-II: 3.33 ± 0.12 vs. 1.03 ± 0.15, P < 0.05; BECN-1: 1.57 ± 0.26 vs. 1.02 ± 0.11, P < 0.05; p62 1.51 ± 0.28 vs. 1.01 ± 0.23, P < 0.05; Figure 1B). Furthermore, electron microscopy showed that, compared with RTECs from sham-operated rats, RTECs from CLP-treated rats exhibited pronounced accumulation of swollen and malformed mitochondria and mitophagosome formation that began at 2 h and peaked at 4 h after CLP [Figure 2A–2C]. We also observed that lysosome membrane fused with mitophagosomes to form mitolysosomes at 2 h after CLP [Figure 2B]. The mitolysosomes gradually disappeared thereafter and were replaced by irregularly shaped, swollen, and disrupted mitochondria with poorly defined cristae and electron-lucent matrices [Figure 2D and 2E]. Taken together, these data suggest that septic insults can induce mitophagy in RTECs both in vitro and in vivo and that mitophagy might be involved in the pathogenesis of septic AKI.
Figure 2.
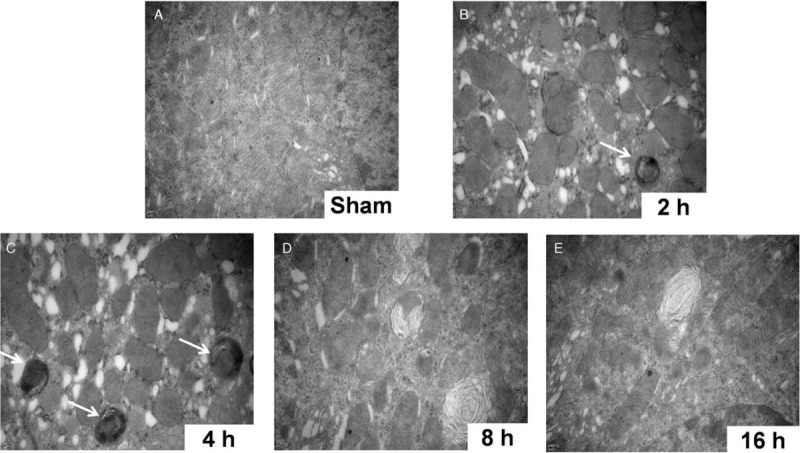
Electron microscopy imaging of mitochondria in renal tubular epithelial cells from septic rats induced by cecal ligation and perforation (original magnification ×10,000). Thirty rats were randomly divided into five groups (one sham operation group and four CLP groups for different time points, n = 6 in each group). (A) Well-developed cristae and electron-dense matrices of healthy mitochondria in sham-operated rats. (B and C) Swollen and malformed mitochondria containing many autophagosomes (white arrows) after CLP (2 and 4 h). (D and E) Irregular, swollen, and disrupted mitochondria with poorly defined cristae and electron-lucent matrices after CLP (8 and 16 h). CLP: cecal ligation and perforation.
The PINK1-Parkin pathway is essential for LPS-induced LC3-II upregulation in HK-2 cells
To explore the role of the PINK1-Parkin pathway in LPS-induced mitophagy protein expression, we performed loss- and gain-of-function experiments in HK-2 cells. As shown in Figure 3A, knockdown of PINK1 significantly reduced the protein expression of downstream Parkin (0.13 ± 0.03 vs. 0.82 ± 0.13, P < 0.05) and effectively reversed LPS-induced upregulation of LC3-II expression compared with control siRNA (1.39 ± 0.14 vs. 1.68 ± 0.19, P < 0.05), whereas overexpression of PINK1 showed the opposite effects (Parkin: 1.98 ± 0.16 vs. 0.85 ± 0.13, P < 0.05; LC3-II: 1.98 ± 0.17 vs. 1.29 ± 0.19, P < 0.05; Figure 3B). These results suggest that the PINK1-Parkin pathway might play an essential role in sepsis-related mitophagy in RTECs.
Figure 3.
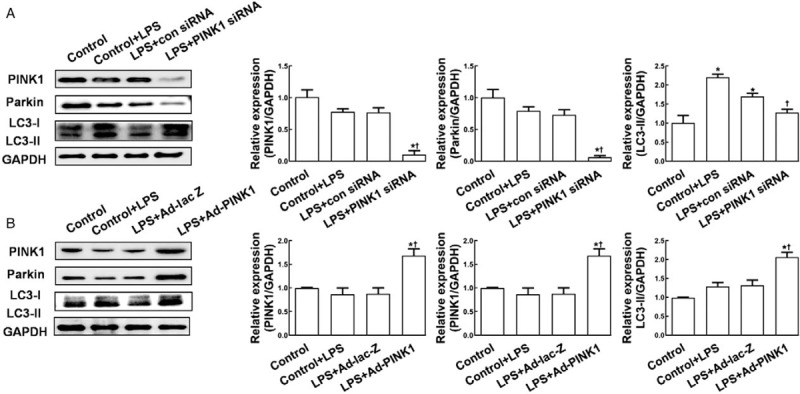
Phosphatase and tensin homolog-induced putative kinase 1 modulated autophagy-related protein expression in HK-2 cells. (A) Western blot analysis was performed to determine PINK1, Parkin, and LC3-II protein expression, cells were transfected with control siRNA or PINK1 siRNA. (B) Western blot analysis was performed to determine PINK1, Parkin, and LC3-II protein expression, cells were transfected with control plasmid Ad-lacZ or Ad-PINK1. HK-2 cells were treated with LPS (10 μmol/L) for 4 h, and collected at 48 h after transfection. GAPDH was used as an internal control. Data are expressed as the mean ± standard deviation. ∗P < 0.05 vs. control; †P < 0.05 vs. control + LPS treatment. GAPDH: glyceraldehyde-3-phosphate dehydrogenase; LC3: microtubule-associated protein light chain 3; LPS: lipopolysaccharide; PINK1: Phosphatase and tensin homolog-induced kinase 1; siRNA: short interfering RNA.
Mitophagy protects LPS-stimulated HK-2 cells from apoptosis
To explore the roles of mitophagy and the PINK1-Parkin pathway in RTECs exposed to septic environment, we assessed apoptosis in LPS-stimulated HK-2 cells. As shown in Figure 4A, LPS significantly enhanced the protein expression of proapoptotic cleaved caspase-3 and BAX in HK-2 cells in a generally time-dependent manner, suggesting that sepsis may induce apoptosis in RTECs. In LPS-stimulated HK-2 cells, the protein levels of cleaved caspase-3 and BAX remained unchanged in response to autophagy activators RAPA (cleaved caspase-3: 0.98 ± 0.09 vs. 0.99 ± 0.04, P > 0.05; BAX: 1.17 ± 0.09 vs. 0.99 ± 0.05, P > 0.05) and BFA (cleaved caspase-3: 1.01 ± 0.19 vs. 0.99 ± 0.04, P > 0.05; BAX: 0.91 ± 0.03 vs. 0.99 ± 0.05, P > 0.05), but were significantly increased in response to autophagy inhibitors 3-MA (cleaved caspase-3: 2.63 ± 0.39 vs. 0.99 ± 0.04, P < 0.05; BAX: 2.78 ± 0.13 vs. 0.99 ± 0.05, P < 0.05) and CQ (cleaved caspase-3: 2.45 ± 0.11 vs. 0.99 ± 0.04, P < 0.05; BAX: 3.01 ± 0.13 vs. 0.99 ± 0.05, P < 0.05; Figure 4B). Consistently, flow cytometry analysis revealed an increased apoptotic rate in LPS-stimulated HK-2 cells in response to autophagy inhibitors (3-MA: 41.28 ± 2.38%; CQ: 43.45 ± 4.38%) compared with that in cells exposed to DMSO (0.05 ± 0.04%; P < 0.05) or autophagy activators (3-MA: 0.75 ± 0.08%; CQ: 1.25 ± 0.07%; P < 0.05, Figure 4C). These results suggest that autophagy activation might protect RTECs from sepsis-induced apoptosis whereas autophagy inhibition might exacerbate apoptosis.
Figure 4.
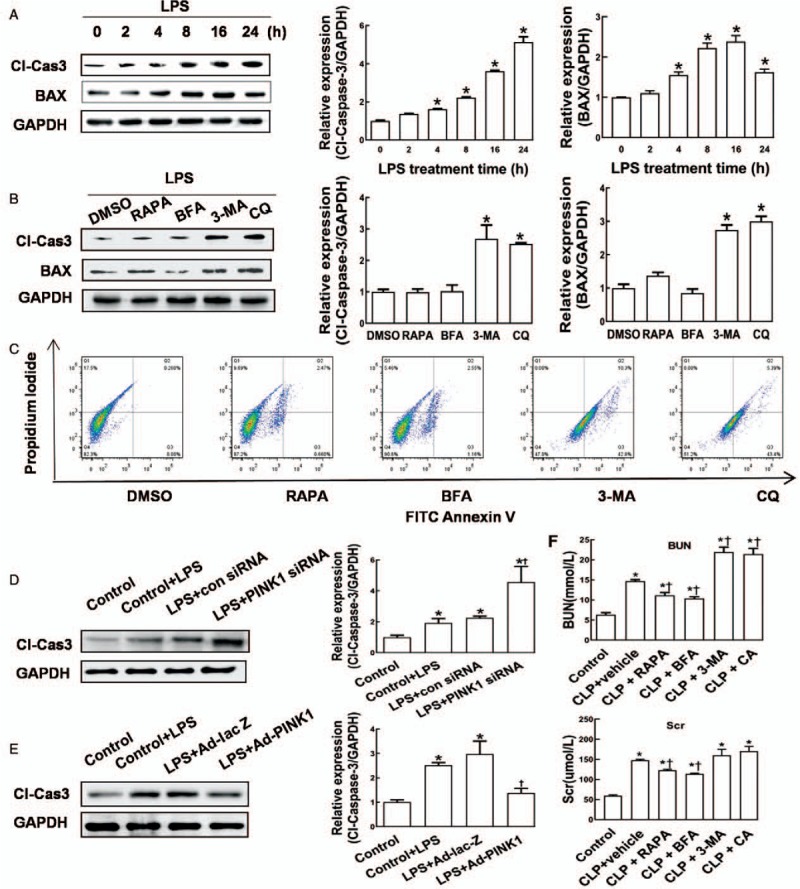
The effects of autophagy activators and inhibitors HK-2 cell apoptosis and rat renal function in septic environment. (A) Western blot analysis was performed to determine protein levels of cleaved caspase-3 and BAX. HK-2 cells were treated with LPS (10 μmol/L) for 0, 2, 4, 8, 16, or 24 h. ∗P < 0.05 vs. 0 h. (B) Western blot analysis was performed to determine protein levels of cleaved caspase-3 and BAX. HK-2 cells were stimulated with LPS (10 μmol/L) for 4 h, followed by treatment with 0.5 mL of DMSO, 3-MA (5 nmol/L), CQ (50 μmol/L), BFA (1 μg/mL), or RAPA (100 nmol/L) for 2 h. ∗P < 0.05 vs. DMSO. (C) Flow cytometry analysis was performed to assess apoptosis in HK-2 cells. (D and E) Western blot analysis was performed to determine protein level of cleaved caspase-3 at 48 h after transfection. HK-2 cells were treated with LPS (10 μmol/L) for 4 h, followed by transfection with control siRNA, PINK1 siRNA, Ad-lacZ, or Ad-PINK1. ∗P < 0.05 vs. control, †P < 0.05 vs. Control + LPS treatment. (F) The blood urea nitrogen and serum creatinine levels in rats. Rats were randomly divided into six groups (n = 6 in each group). Following anesthesia, rats in the control group underwent sham operation without any other treatment, whereas other rats were given DMSO (0.3 mL), 3-MA (0.2 mL, 20 mg/kg), CQ (0.2 mL, 60 mg/kg), BFA (0.2 mL, 4 mg/kg), or RAPA (0.2 mL, 4 mg/kg) 30 min (for DMSO) or 2 h (for other drugs) prior to CLP. These rats were sacrificed at 24 h after surgery, and the blood urea nitrogen and serum creatinine levels were measured. ∗P < 0.05 vs. the control, †P < 0.05 vs. CLP + vehicle treatment. Data are expressed as mean ± standard deviation. 3-MA: 3-methyladenine; BFA: Brefeldin A; BUN: blood urea nitrogen; Cl-Cas3: cleaved caspase-3; CQ: chloroquine; DMSO: dimethyl sulfoxide; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; LPS: lipopolysaccharide; PINK1: phosphatase and tensin homolog-induced kinase 1; RAPA: rapamycin; Scr: serum creatinine; siRNA: short interfering RNA.
To explore the effect of the PINK1–Parkin pathway on apoptosis, we determined the protein expression of cleaved caspase-3 in HK-2 cells with PINK1 depletion or overexpression. As shown in Figure 4D and 4E, compared with the control siRNA group, PINK1 depletion significantly enhanced (4.25 ± 0.83 vs. 2.02 ± 0.08, P < 0.05), whereas PINK1 overexpression markedly attenuated the protein expression of cleaved caspase-3 in LPS-stimulated HK-2 cells (1.21 ± 0.25 vs. 2.92 ± 0.69, P < 0.05). Combining its promotive role in LC3-II expression in LPS-treated HK-2 cells shown in Figure 3B, the PINK1-Parkin pathway might protect HK-2 cells from LPS-induced apoptosis possibly via activation of mitophagy.
Mitophagy improves renal functions of rats with septic AKI
Next, we examined the renal functions of rats with CLP-induced septic AKI in response to autophagy activators or inhibitors. As shown in Figure 4F, compared with sham operation, CLP induced significant increases in the BUN (RAPA: 9.98 ± 0.85 mmol/L vs. 13.24 ± 0.39 mmol/L, P < 0.05; BFA: 9.73 ± 0.37 mmol/L vs. 13.24 ± 0.39 mmol/L, P < 0.05) and serum creatinine levels (RAPA: 108.45 ± 12.21 μmol/L vs. 148.87 ± 10.16 μmol/L, P < 0.05; BFA: 101.54 ± 6.25 μmol/L vs. 148.87 ± 10.16 μmol/L, P < 0.05) that were effectively reversed by treatment with autophagy activators RAPA and BFA. In contrast, treatment with autophagy inhibitors 3-MA or CQ further increased the BUN (3-MA: 20.12 ± 2.35 mmol/L vs. 13.24 ± 0.39 mmol/L, P < 0.05; CQ: 19.99 ± 3.31 mmol/L vs. 13.24 ± 0.39 mmol/L, P < 0.05) in rats with septic AKI, However, the levels of serum creatinine remained unchanged in response to autophagy inhibitors (3-MA: 155.64 ± 23.19 μmol/L vs. 147.86 ± 6.21 μmol/L, P > 0.05; CQ: 168.54 ± 24.32 μmol/L vs. 147.86 ± 6.21 μmol/L, P > 0.05). These results suggest that autophagy activation might be a potential therapeutic strategy for septic AKI.
Discussion
Autophagy activation during AKI has been observed in various experimental models.[27] In this study, septic insults induced mitophagy-related protein expression in RTECs both in vitro and in vivo. Gain- and loss-of functional PINK1 assays suggest that the PINK1-Parkin pathway is essential for mitophagy protein expression in RTECs. We also found mitophagy activation could prevent RTEC apoptosis and improve renal functions, consistent with a recent study showing that the activated PINK1-Parkin mitophagy can prevent renal ischemia-reperfusion injury.[15,19] We therefore conclude that PINK1-Parkin-modulated mitophagy might play a protective role in septic AKI, providing PINK1-Parkin mitophagy activation as a potential therapeutic approach against septic AKI.
Autophagy, another type of programmed cell death different from apoptosis, occurs in all eukaryotic cells and contributes to the turnover of cellular components and energy homeostasis.[28] Cell death is the endpoint of apoptosis, while autophagy is a “double-edged sword” for both cell survival and cell death.[29] To some extent, autophagy and apoptosis are inseparable. Autophagy, prior to apoptosis, is activated to protect cells from apoptosis and necrosis under stress; however, excessive autophagy can promote mitochondrial damage, leading to increased apoptosis. Previous study showed that autophagy is initiated early after sepsis, rendering protection against endotoxic kidney injury.[12,15] Consistently, we observed autophagy marker protein upregulation and autophagosome formation in RTECs within 2 h after septic insults. Pathological results from patients dying of septic AKI showed that apoptosis commonly occurs in RTECs.[30] Our results showed that autophagy activators could prevent further apoptosis whereas autophagy inhibitors could exacerbate apoptosis in LPS-stimulated HK-2 cells, suggesting that autophagy activation can protect RTECs from sepsis-induced apoptosis.
Mitochondria play a critical role in oxidative phosphorylation to supply energy for cellular activities in eukaryotes.[31] Mitochondrial damage promotes the production of reactive oxygen species (ROS) and apoptotic factors, leading to cell damage and apoptosis.[32] Mitophagy, a process that selectively degrades mitochondria via autophagy, contributes to mitochondrial quality control by regulating mitochondria numbers and diminishing ROS production. PINK1-Parkin is one of the major pathways that mediate mitophagy.[33,34] Mutations in both genes disrupt mitophagy through different mechanisms.[35] Double deletion of PINK1 and Parkin impairs hepatic mitophagy and exacerbates acetaminophen-induced liver injury in mice.[36] In our study, transient knockdown and overexpression of PINK1 accompanied downregulation and upregulation of Parkin expression, respectively, in HK-2 cells. Our results revealed essential roles of the PINK1-Parkin pathway in LC3-II induction and cleaved caspase 3 suppression in LPS-stimulated HK-2 cells, suggesting that the PINK1-Parkin pathway might protect HK-2 cells from LPS-induced apoptosis possibly via activation of mitophagy. However, how PINK1 affects Parkin expression in septic RTECs remains unclear and needs to be addressed in the future study. It has been reported that both PINK1 overexpression and a loss of mitochondrial membrane potential can trigger translocation of cytosolic Parkin to mitochondria. As a putative serine/threonine kinase, PINK1 might promote Parkin translocation via phosphorylation of Parkin that thereby ubiquitinates multiple mitochondrial proteins, leading to p62 recruitment and subsequent targeting of the damaged mitochondria to LC3-positive phagophores for lysosomal clearance.[37–39] In this study, we observed significantly increased LC3-II, BECN-1, and p62 protein levels in RTECs that peaked at 2 h after CLP. Consistently, RTECs from septic rats exhibited pronounced mitochondria deformation and mitophagosome formation that began at 2 h and peaked at 4 h after CLP. Combining with the promotive effect of PINK1 overexpression on LC3-II expression, these data suggest that septic insults can rapidly induce mitophagy in RTECs possibly via the PINK1-Parkin pathway.
In summary, this study showed that septic insults can activate mitophagy in RTECs in vitro and in vivo, suggesting that mitophagy is involved in septic AKI. The PINK1-Parkin pathway might play an essential role in mitophagy activation that possibly favors RTEC survival and improves renal function in the septic environment. Mitophagy activation might serve as a potential therapeutic strategy against septic AKI.
Funding
This work was supported by grants from the National Natural Science Foundation of China (Nos. 81601708, 81671960), and the Natural Science Foundation of Hunan Province, China (No. 2018JJ2014).
Conflicts of interest
None.
Footnotes
How to cite this article: Dai XG, Xu W, Li T, Lu JY, Yang Y, Li Q, Zeng ZH, Ai YH. Involvement of phosphatase and tensin homolog-induced putative kinase 1–Parkin-mediated mitophagy in septic acute kidney injury. Chin Med J 2019;132:2340–2347. doi: 10.1097/CM9.0000000000000448
Xin-Gui Dai and Wei Xu contributed equally to this work.
References
- 1.Bellomo R, Kellum JA, Ronco C, Wald R, Martensson J, Maiden M, et al. Acute kidney injury in sepsis. Intensive Care Med 2017; 43:816–828. doi: 10.1007/s00134-017-4755-7. [DOI] [PubMed] [Google Scholar]
- 2.Dai X, Zeng Z, Fu C, Zhang S, Cai Y, Chen Z. Diagnostic value of neutrophil gelatinase-associated lipocalin, cystatin C, and soluble triggering receptor expressed on myeloid cells-1 in critically ill patients with sepsis-associated acute kidney injury. Crit Care 2015; 19:223.doi: 10.1186/s13054-015-0941-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pettila V, Bellomo R. Understanding acute kidney injury in sepsis. Intensive Care Med 2014; 40:1018–1020. doi: 10.1007/s00134-014-3313-9. [DOI] [PubMed] [Google Scholar]
- 4.Morrell ED, Kellum JA, Pastor-Soler NM, Hallows KR. Septic acute kidney injury: molecular mechanisms and the importance of stratification and targeting therapy. Crit Care 2014; 18:501.doi: 10.1186/s13054-014-0501-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poston JT, Koyner JL. Sepsis associated acute kidney injury. BMJ 2019; 364:k4891.doi: 10.1136/bmj.k4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kosaka J, Lankadeva YR, May CN, Bellomo R. Histopathology of septic acute kidney injury: a systematic review of experimental data. Crit Care Med 2016; 44:e897–e903. doi: 10.1097/CCM.0000000000001735. [DOI] [PubMed] [Google Scholar]
- 7.Wan L, Bagshaw SM, Langenberg C, Saotome T, May C, Bellomo R. Pathophysiology of septic acute kidney injury: what do we really know? Crit Care Med 2008; 36:S198–S203. doi: 10.1097/CCM.0b013e318168ccd5. [DOI] [PubMed] [Google Scholar]
- 8.Xu S, Gao Y, Zhang Q, Wei S, Chen Z, Dai X, et al. SIRT1/3 activation by resveratrol attenuates acute kidney injury in a septic rat model. Oxid Med Cell Longev 2016; 2016:7296092.doi: 10.1155/2016/7296092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao Y, Chen T, Lei X, Li Y, Dai X, Cao Y, et al. Neuroprotective effects of polydatin against mitochondrial-dependent apoptosis in the rat cerebral cortex following ischemia/reperfusion injury. Mol Med Rep 2016; 14:5481–5488. doi: 10.3892/mmr.2016.5936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res 2014; 24:24–41. doi: 10.1038/cr.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 2012; 11:709–730. doi: 10.1038/nrd3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaushal GP, Shah SV. Autophagy in acute kidney injury. Kidney Int 2016; 89:779–791. doi: 10.1016/j.kint.2015.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsiao HW, Tsai KL, Wang LF, Chen YH, Chiang PC, Chuang SM, et al. The decline of autophagy contributes to proximal tubular dysfunction during sepsis. Shock 2012; 37:289–296. doi: 10.1097/SHK.0b013e318240b52a. [DOI] [PubMed] [Google Scholar]
- 14.Yang CC, Yao CA, Yang JC, Chien CT. Sialic acid rescues repurified lipopolysaccharide-induced acute renal failure via inhibiting TLR4/PKC/gp91-mediated endoplasmic reticulum stress, apoptosis, autophagy, and pyroptosis signaling. Toxicol Sci 2014; 141:155–165. doi: 10.1093/toxsci/kfu121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mei S, Livingston M, Hao J, Li L, Mei C, Dong Z. Autophagy is activated to protect against endotoxic acute kidney injury. Sci Rep 2016; 6:22171.doi: 10.1038/srep22171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nguyen TN, Padman BS, Lazarou M. Deciphering the molecular signals of PINK1/Parkin mitophagy. Trends Cell Biol 2016; 26:733–744. doi: 10.1016/j.tcb.2016.05.008. [DOI] [PubMed] [Google Scholar]
- 17.Rub C, Schroder N, Voos W. Biochemical properties of the kinase PINK1 as sensor protein for mitochondrial damage signalling. Biochem Soc Trans 2015; 43:287–291. doi: 10.1042/BST20150005. [DOI] [PubMed] [Google Scholar]
- 18.Zhao C, Chen Z, Xu X, An X, Duan S, Huang Z, et al. Pink1/Parkin-mediated mitophagy play a protective role in cisplatin induced renal tubular epithelial cells injury. Exp Cell Res 2017; 350:390–397. doi: 10.1016/j.yexcr.2016.12.015. [DOI] [PubMed] [Google Scholar]
- 19.Lin Q, Li S, Jiang N, Shao X, Zhang M, Jin H, et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol 2019; 26:101254.doi: 10.1016/j.redox.2019.101254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang X, Yan X, Yang D, Zhou J, Song J, Yang D. Rapamycin attenuates mitochondrial injury and renal tubular cell apoptosis in experimental contrast-induced acute kidney injury in rats. Biosci Rep 2018; 38.doi: 10.1042/BSR20180876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang C, Han H, Yan M, Zhu S, Liu J, Liu Z, et al. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia-reperfusion injury. Autophagy 2018; 14:880–897. doi: 10.1080/15548627.2017.1405880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun Y, Cai Y, Zang QS. Cardiac autophagy in sepsis. Cells 2019; 8.doi: 10.3390/cells8020141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Piquereau J, Godin R, Deschenes S, Bessi VL, Mofarrahi M, Hussain SN, et al. Protective role of PARK2/Parkin in sepsis-induced cardiac contractile and mitochondrial dysfunction. Autophagy 2013; 9:1837–1851. doi: 10.4161/auto.26502. [DOI] [PubMed] [Google Scholar]
- 24.Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, et al. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem 2013; 288:915–926. doi: 10.1074/jbc.M112.411363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang X, Yuan D, Sun Q, Xu L, Lee E, Lewis AJ, et al. Calcium/calmodulin-dependent protein kinase regulates the PINK1/Parkin and DJ-1 pathways of mitophagy during sepsis. FASEB J 2017; 31:4382–4395. doi: 10.1096/fj.201601096RRR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zeng Z, Yang Y, Dai X, Xu S, Li T, Zhang Q, et al. Polydatin ameliorates injury to the small intestine induced by hemorrhagic shock via SIRT3 activation-mediated mitochondrial protection. Expert Opin Ther Targets 2016; 20:645–652. doi: 10.1080/14728222.2016.1177023. [DOI] [PubMed] [Google Scholar]
- 27.Harris RC, Cheng H. Telomerase, autophagy and acute kidney injury. Nephron 2016; 134:145–148. doi: 10.1159/000446665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cuervo AM, Bergamini E, Brunk UT, Droge W, Ffrench M, Terman A. Autophagy and aging: the importance of maintaining “clean” cells. Autophagy 2005; 1:131–140. doi: 10.4161/auto.1.3.2017. [DOI] [PubMed] [Google Scholar]
- 29.Vellai T, Toth ML, Kovacs AL. Janus-faced autophagy: a dual role of cellular self-eating in neurodegeneration? Autophagy 2007; 3:461–463. doi: 10.4161/auto.4282. [DOI] [PubMed] [Google Scholar]
- 30.Langenberg C, Wan L, Egi M, May CN, Bellomo R. Renal blood flow in experimental septic acute renal failure. Kidney Int 2006; 69:1996–2002. doi: 10.1038/sj.ki.5000440. [DOI] [PubMed] [Google Scholar]
- 31.McCreath G, Scullion MM, Lowes DA, Webster NR, Galley HF. Pharmacological activation of endogenous protective pathways against oxidative stress under conditions of sepsis. Br J Anaesth 2016; 116:131–139. doi: 10.1093/bja/aev400. [DOI] [PubMed] [Google Scholar]
- 32.Duann P, Lianos EA, Ma J, Lin PH. Autophagy, Innate Immunity and Tissue Repair in Acute Kidney Injury. International Journal of Molecular Sciences 2016; 17:pii: E662.doi: 10.3390/ijms17050662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanki T, Klionsky DJ, Okamoto K. Mitochondria autophagy in yeast. Antioxid Redox Signal 2011; 14:1989–2001. doi: 10.1089/ars.2010.3762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 2010; 12:119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 35.Springer W, Kahle PJ. Regulation of PINK1-Parkin-mediated mitophagy. Autophagy 2011; 7:266–278. doi: 10.4161/auto.7.3.14348. [DOI] [PubMed] [Google Scholar]
- 36.Wang H, Ni HM, Chao X, Ma X, Rodriguez YA, Chavan H, et al. Double deletion of PINK1 and Parkin impairs hepatic mitophagy and exacerbates acetaminophen-induced liver injury in mice. Redox Biol 2019; 22:101148.doi: 10.1016/j.redox.2019.101148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gegg ME, Schapira AH. PINK1-parkin-dependent mitophagy involves ubiquitination of mitofusins 1 and 2: Implications for Parkinson disease pathogenesis. Autophagy 2011; 7:243–245. doi: 10.4161/auto.7.2.14332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koyano F, Yamano K, Kosako H, Tanaka K, Matsuda N. Parkin recruitment to impaired mitochondria for nonselective ubiquitylation is facilitated by MITOL. J Biol Chem 2019; 294:10300–10314. doi: 10.1074/jbc.RA118.006302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Durcan TM, Fon EA. The three ’P's of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev 2015; 29:989–999. doi: 10.1101/gad.262758.115. [DOI] [PMC free article] [PubMed] [Google Scholar]