

Abstract
Although improvements in genetic analysis have greatly enhanced our understanding of the mechanisms behind pancreatitis, it continues to afflict many families for whom the hereditary factors remain unknown. Recent evaluation of a patient with a strong family history of pancreatitis prompted us to reexamine a large kindred originally reported over 50 years ago with an autosomal-dominant inheritance pattern of chronic pancreatitis, diabetes, and pancreatic adenocarcinoma. Whole-exome sequencing analysis identified a rare missense mutation in the gene encoding pancreas-specific protease elastase 3B (CELA3B) that cosegregates with disease. Studies of the mutant protein in vitro, in cell lines, and in CRISPR-Cas9–engineered mice indicate that this mutation causes translational upregulation of CELA3B, which, upon secretion and activation by trypsin, leads to uncontrolled proteolysis and recurrent pancreatitis. Although lesions in several other pancreatic proteases have been previously linked to hereditary pancreatitis, to our knowledge, this is the first known instance of a mutation in CELA3B and a defect in translational control contributing to this disease.
Keywords: Cell Biology, Gastroenterology
Keywords: Genetic diseases
Introduction
Malfunction or injury to the pancreas, primarily by its own digestive enzymes, results in the inflammation and abdominal pain typical of acute pancreatitis. Repeated injury and inflammation lead to chronic pancreatitis (CP), which is characterized by calcification of the pancreas, scarring and obstruction, and loss of exocrine and endocrine function (1). This results in a spectrum of clinical manifestations such as malnutrition, increased risk for diabetes and pancreatic cancer, poor quality of life, and reduced survival (2).
There have been several mutations linked to hereditary CP, including in PRSS1, the gene for cationic trypsinogen, which prevents proper autoinactivation of the enzyme (3). Other causative lesions for inherited CP have been identified in genes that regulate trypsin inhibition and inactivation (SPINK1, CTRC) (4, 5) and that are involved in clearance of trypsin from the pancreas (CFTR) (6). Several trypsin-independent genetic causes, such as mutations in carboxypeptidase A1 (CPA1), have also been reported (7). Furthermore, alcoholic pancreatitis, the most common cause of sporadic CP, is contingent on genetic and environmental factors (8). Despite these advances, the molecular basis of hereditary pancreatitis remains unknown for at least 30% of affected families (9).
Results and Discussion
In 1968, investigators at UCSF and affiliated hospitals published one of the earliest documented cases of familial pancreatitis in the US, affecting a kindred of 71 individuals centered in Northern California (10). In the affected kindred, abdominal pain manifested between 5 and 15 years of age, with onset of pancreatitis in late childhood to early adulthood that was severe and required surgical intervention for several of the individuals. In multiple cases, pancreatitis was confirmed by biopsy. Pancreatitis was accompanied by a high prevalence of diabetes and pancreatic cancer in adulthood, and there was no reported association with smoking or alcohol consumption. The data indicated that the causative variant was autosomal dominant, with an extremely high penetrance. However, without advanced genetic study methods, the cause of the disease remained undetermined.
Recently, a patient with a similar history of CP presented at the UCSF Diabetes Clinic and revealed a connection to the family reported in 1968 (Figure 1A). Like other family members, she had onset of abdominal pain in late childhood and a diagnosis of CP in early adulthood. In addition, she had no history of smoking or alcohol consumption. Sanger sequencing revealed no mutations in PRSS1 (Supplemental Figure 1, Supplemental Table 1, and Supplemental Methods; supplemental material available online with this article; https://doi.org/10.1172/JCI129961DS1), the most common cause of hereditary CP (11). Whole-exome sequencing on DNA samples from the patient (Figure 1A, member IV-9), her affected daughter (V-1), and unaffected brother (IV-10) and son (V-2) revealed no mutations in known pancreatitis-associated genes, but revealed 3 novel variants that cosegregated with disease and were predicted to be both damaging and rare (Supplemental Figure 2). Recruitment and Sanger sequencing of an unaffected sister (IV-11) ruled out 1 variant, leaving 2 candidate variants. A G→A substitution at c.959 (c.959G>A) resulted in a substitution of arginine for glutamine at codon 320 (p.R320Q) in FOXN1, which encodes a transcription factor important for thymic epithelial cells. Homozygous mutations in FOXN1 cause T cell immunodeficiency, congenital alopecia, and nail dystrophy (TIDAND) (12). A C→T substitution at c.268 (c.268C>T) resulted in a substitution of arginine for cysteine at codon 90 (p.R90C) in CELA3B (Table 1), which encodes chymotrypsin-like elastase family member 3B and is exclusively expressed in the exocrine pancreas (13) (Supplemental Figure 3). Sanger sequencing confirmed that the CELA3B and FOXN1 variants were present in affected family members and were absent in unaffected family members. As FOXN1 is not expressed in the pancreas and heterozygous FOXN1 mutations are not a cause of pancreatitis (12, 13), we hypothesized that the CELA3B p.R90C variant is the likely cause of CP in this family and therefore performed functional studies.
Figure 1. Identification of a pathogenic mutation in a family with hereditary pancreatitis.

(A) Pedigree of the study family. Squares indicate males; circles indicate females. Shapes were partitioned and colored to indicate conditions (green: pancreatitis, blue: diabetes, red: pancreatic cancer). Generations I–IV were previously reported (10), whereas generation V is newly reported here. Teal branches indicate family members who participated in this study to identify the CELA3B p.R90C mutation, and black branches indicate previously reported individuals. The proband is indicated by an arrow and labeled with a “P.” The mutation status confirmed by sequencing is indicated below the shapes and labeled with “C/C,” indicating WT individuals, or “C/T,” indicating individuals who harbor the CELA3B c.268C>T (p.R90C) variant. (B) Map of human CELA3B showing known domains and key amino acid residues.
Table 1. CELA3B mutation.
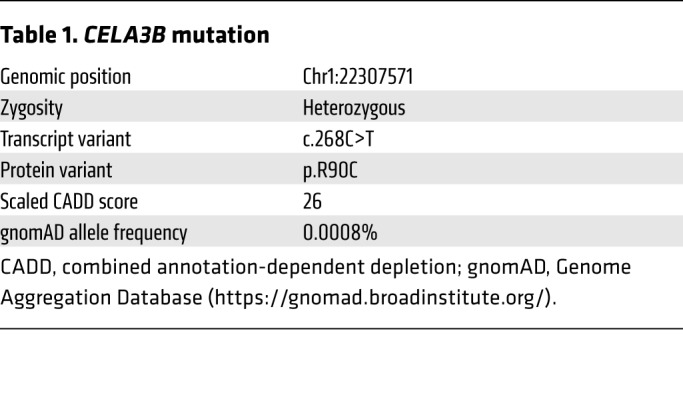
CELA3B belongs to a family of 6 elastases, 4 of which (2A, 2B, 3A, and 3B) are only produced in and secreted by pancreatic acinar cells (13). Structurally, it consists of a signal peptide (residues 1–15), a propeptide (residues 16–28), and a catalytic domain (residues 29–270) (Figure 1B). Upon secretion, CELA3B is cleaved at R28 by trypsin, converting it from zymogen to active protease. No pathogenic mutations of CELA3B have been reported (14). Sequence alignments of elastases revealed that R90 is highly conserved among CELA3Bs, whereas other elastase family members typically have a leucine or isoleucine at this position (Figure 2A and Supplemental Figure 4A), suggesting that position 90 plays a crucial regulatory, structural, or functional role.
Figure 2. Functional characterization of CELA3B mutations at arginine 90.

(A) Sequence alignment of the 6 human elastases centered at position 90 in CELA3B. Arginine (R) residues are labeled in red; leucine (L) residues are labeled in blue. (B) Immunoblots of whole-cell lysates (intracellular) and conditioned media (secreted protein) from 293T cells transfected with empty vector (Mock) or the indicated CELA3B variant. (C) Quantification from the 9 experiments in B. Values represent the mean ± SD. (D) Autoradiographs of His-tagged CELA3B variants purified from brefeldin A–treated 293T cells pulsed with 35S-labeled methionine and cysteine for the indicated durations. The first (control) lane is the same in the WT and R90C images. (E) Quantification from the 4 experiments in D, showing data points normalized to WT levels at 90 minutes and their corresponding lines of best fit (linear regression). Values represent the mean ± SD. **P < 0.01 among all samples. (F) Western blots of CELA3B variants lacking a signal peptide (ΔSP). Samples are whole-cell lysates from 293T cells transfected with empty vector (Mock) or vector containing ΔSP-CELA3B variants. Blots were probed with the indicated antibodies. (G) Quantification of data from the 4 experiments in F, with protein levels normalized to ΔSP-WT CELA3B. Values represent the mean ± SD. (H) Efficiency of catalytic activation of CELA3B variants in conditioned media after limited activation with trypsin. Raw catalytic activity was quantified as a change in absorbance due to cleavage of a colorimetric CELA3B substrate. Raw values were normalized to total CELA3B protein levels and are shown as the mean ± SD for 3 experiments. Multiplicity-adjusted P values were determined by matched 2-way ANOVA (C) or matched 1-way ANOVA (panels G and H) with Tukey’s multiple comparisons test. **P < 0.01, ***P < 0.001, and ****P < 0.0001.
We explored the functional divergence of WT, R90C (patient mutation), and R90L (evolutionary substitution) variants in 293T cells. Transfection assays revealed that the R90C and R90L substitutions led to elevated levels of intracellular and secreted CELA3B (Figure 2, B and C), despite equal mRNA transcript levels (Supplemental Figure 4B). Introduction of reverse mutations (L90C and L90R) into CELA3A, the closest relative of CELA3B, reduced its expression (Supplemental Figure 4, C–E), indicating an evolutionarily conserved role for this residue in regulating elastase levels.
Pulse-chase analysis revealed that the R90 substitutions do not appreciably impact CELA3B stability or secretory rates (Supplemental Figure 4, F–I), though they significantly increased translation rates in correlation with their steady-state levels (Figure 2, D and E). Conversely, we found that CELA3B variants lacking the signal peptide (ΔSP) were translated into the cytosol at equal levels (Figure 2, F and G), thereby linking differences in expression of the full-length transcripts to cotranslational translocation at the ER membrane.
In addition to regulation of expression, mutations in proteases can affect their intrinsic catalytic capabilities and their activation and inactivation by regulatory factors. Michaelis-Menten enzyme kinetics of purified WT, R90C, and R90L variants of CELA3B suggest that R90 does not grossly affect the catalytic efficiency of the protease (Supplemental Table 2) (15). We detected no differences in activation efficiency of the R90 variants (unpublished observations), though trypsin treatment of unmodified media containing secreted elastases yielded much more rapid activation of the R90C and R90L variants than did WT CELA3B (Figure 2H). Additionally, CELA3B binds and impedes premature activation of carboxypeptidases 1 and 2 (CPA1 and CPA2) (16, 17). Therefore, we tested whether mutations in CELA3B might disrupt its binding to CPA1 and CPA2 and result in excessive protease activation. However, we detected normal binding of R90C CELA3B and R90L CELA3B to both CPA1 and CPA2, whereas a known binding-deficient variant (A241G) CELA3B was incapable of interacting with either CPA1 or CPA2 (Supplemental Figure 5, A and B). Taken together, these data suggest that the R90C mutation removes a molecular brake to increase translation and secretion of the CELA3B protease, thereby enhancing its proteolytic activation by trypsin.
To bridge the gap between our molecular and patient data, we used CRISPR-Cas9–directed genome editing to knock in the homologous mutations (R89C and R89L) at the endogenous Cela3b locus in mice. Similar to other familial pancreatitis mouse models, homozygous Cela3b-mutant mice (both R89C/R89C and R89L/R89L) did not spontaneously develop pancreatitis by 40 weeks of age (18, 19). Current research supports a 2-hit theory, in which CP-causing mutations increase the susceptibility of the pancreas to environmental insults, leading to the development of pancreatitis (20). We provided a “second hit” by injecting the cholecystokinin analog caerulein, a chemical agent used to induce pancreatitis in rodents. Under the dose and regimen of caerulein used, control mice typically make a full recovery within 7 days (21, 22), while pathology is exaggerated in mouse models that are genetically predisposed to pancreatitis (19).
In response to caerulein, homozygous Cela3b R89C and R89L animals developed more severe pancreatitis than did WT control animals, as characterized by increased immune infiltration, acinar dedifferentiation, and loss of lobular integrity (Figure 3, A–C). Moreover, the R89L animals showed even greater pancreatic injury than did the R89C animals, consistent with the effects of these 2 variants on CELA3B protein expression (Figure 3C).
Figure 3. Cela3b-mutant mice show increased pancreatic injury following induction of acute pancreatitis.

(A) H&E-stained images of pancreata from WT, Cela3b R89C/R89C, and Cela3b R89L/R89L C57BL/6 mice 7 days after 2 consecutive days of 8 hourly i.p. injections with vehicle (PBS) or 75 μg/kg caerulein. Scale bars: 100 μm. (B) Stacked bar graphs classifying pancreatic damage of 13 to 30 caerulein-treated mice per genotype. Values from 0 to 5 were assigned for immune infiltration (infiltrate), acinar dedifferentiation (ADM), and lobular integrity (integrity), with the mean ± SD for each separate classification. (C) Combined scores from B, with the mean ± SD. P values were determined by ordinary 2-way ANOVA (B) or ordinary 1-way ANOVA (C) with Tukey’s multiple comparisons test. *P < 0.05 and ****P < 0.0001.
Although this family was originally described over 50 years ago, the molecular basis underlying their autosomal-dominant inherited syndrome of pancreatitis, diabetes, and pancreatic adenocarcinoma had remained a mystery. Using next-generation sequencing, we have identified the likely causative lesion in CELA3B: a single amino acid mutation of arginine 90 to cysteine. Intriguingly, this mutation does not alter the catalytic profile of CELA3B, as typically anticipated for a protease, but enhances its rate of translation, thereby increasing the total amount of active enzyme and the risk of pancreatitis in response to pancreatic insult.
Precisely how these mutations increase expression is unclear, but the answer may lie in the regulation of translation elongation, a growing field of research with numerous correlations to disease (23). While codon optimization is an obvious candidate, we observed no differences in CELA3B expression when using alternate codons for R90, R90C, and R90L (unpublished observations); instead, the translation rate appears to depend on the amino acid residue at this position. Codon optimization also failed to account for our observation that CELA3B expression was dependent on cotranslational translocation at the ER. However, translational elongation can be mediated by stress chaperones such as HSP70 (23), which raises the possibility that ER-resident chaperones, like BiP, could mediate the translation rate. Regardless, this mechanism of action clearly sets this mutation apart from other causative lesions, which typically regulate activation and inactivation of trypsin or lead to ER stress.
Moreover, although the position of CELA3B downstream of the canonical trypsin regulatory mechanism makes it an unexpected candidate for triggering uncontrolled pancreatic proteolysis, our mouse model clearly demonstrates that this elastase is important for propagating pancreatitis, and in vitro assays showed that excessive expression allows it to be rapidly activated by trypsin. Given the severe pancreatitis phenotype in patients, the possibility remains that mutant CELA3B has undiscovered catalytic properties or targets that enhance its pathogenicity. The severity of this disease also suggests that CELA3B may play a prominent role in other forms of CP. As such, it would be interesting to test whether CELA3B-knockout animals are resistant to pancreatitis, particularly in the presence of CP-causing trypsin mutations. Support for this notion comes from a recent report that a different variant (c.643-7G>T) in CELA3B is associated with a small protective effect against alcoholic CP (17). Furthermore, pharmacologic inhibition of elastase family members has shown efficacy in preclinical models of pancreatitis (24).
Importantly, many kindred are still afflicted by hereditary pancreatitis with unknown genetic causes, and future studies will need to determine whether they also carry mutations in CELA3B. As shown here, a coordinated approach using modern human genetics, CRISPR-based mouse models, and molecular biology and biochemistry will open new opportunities to strongly link rare variants to disease risk.
Methods
Additional details on the methods used in this study are provided in the Supplemental Methods.
Human subjects.
The original pedigree was previously described in 1968 (10), and we corroborated and expanded it with the help of the proband (Figure 1).
Study approval.
The study was approved by the UCSF Committee on Human Research. All participating family members provided written informed consent to participate in the study. All mouse experiments complied with the Animal Welfare Act and NIH guidelines for the ethical care and use of animals in biomedical research and were performed under the approval of the IACUC of UCSF. Animals were maintained in a specific pathogen–free animal facility on a 12-hour light/12-hour dark cycle at an ambient temperature of 21°C. The animals were given free access to water and food.
Statistics.
All data are expressed as the mean ± SD, and significance was defined as a P value of less than 0.05. Data sets with 1 factor and multiple groups were compared by ordinary or matched (paired) 1-way ANOVA with Tukey’s multiple comparisons test; sets with 2 factors were compared by ordinary or matched (paired) 2-way ANOVA with Tukey’s multiple comparisons test. Slopes in Figure 2E were calculated by linear regression and were determined to be different (nonparallel) with a P value of 0.0011. Curves in Supplemental Figure 4G were calculated by exponential decay (1-phase decay) with rate constants, K, determined to differ from the WT control with P value of less than 0.0001. All statistical analyses were performed using GraphPad Prism, version 6.0 (GraphPad Software).
Author contributions
PCM and JTC are co–first authors, with PCM listed first because of his major contributions regarding molecular mechanism and manuscript preparation. PCM, JTC, MSA, and SAO conceptualized the study. PCM, JTC, CEC, AC, JLB, MSA, and SAO designed the study methodology. PCM, JTC, and CEC were responsible for data validation. PCM, JTC, and CEC conducted formal analyses. PCM, JTC, CEC, AC, JLB, and JP performed experiments. DA, ACB, ZQ, AC, and MHC provided resources. PCM and SAO wrote the original draft of the manuscript. PCM, JTC, CEC, DA, ACB, ZQ, AC, MHC, MSG, MSA, and SAO reviewed and edited the manuscript. PCM, JTC, and CEC organized data presentation/visualization. MSG, MSA, and SAO supervised the study. MSG, MSA and SAO were responsible for funding acquisition.
Supplementary Material
Acknowledgments
This work was supported in part by the NIH (P30DK63720, R01CA219815, R01EY027810, and U01DK108332); the Helmsley Charitable Trust; the Bern Schwartz Family Foundation; and a Larry L. Hillblom Foundation grant (2018-D-0006-NET). We would especially like to thank Miklos Sahin-Toth (UCLA) for his generous donation of elastase and carboxypeptidase constructs and for guidance on purification and analysis of these enzymes.
Version 1. 08/01/2019
In-Press Preview
Version 2. 09/23/2019
Electronic publication
Version 3. 11/01/2019
Print issue publication
Footnotes
Conflict of interest: MSG holds stock in Viacyte Inc. and Encellin Inc. MA holds stock in Medtronic and Merck. SAO is a scientific co-founder of, equity holder in, and consultant for OptiKira LLC.
Copyright: © 2019, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2019;129(11):4676–4681.https://doi.org/10.1172/JCI129961.
Contributor Information
Paul C. Moore, Email: paul.moore@ucsf.edu.
Jessica T. Cortez, Email: jessica.cortez@ucsf.edu.
Chester E. Chamberlain, Email: Chester.Chamberlain@ucsf.edu.
Diana Alba, Email: diana.alba@ucsf.edu.
Amy C. Berger, Email: amy.berger@ucsf.edu.
Zoe Quandt, Email: zoe.quandt@ucsf.edu.
Alice Chan, Email: alice.chan1@ucsf.edu.
Mickie H. Cheng, Email: mickie.cheng@ucsf.edu.
Jhoanne L. Bautista, Email: jhoanne.bautista@ucsf.edu.
Justin Peng, Email: justin.peng@ucsf.edu.
Michael S. German, Email: mgerman@diabetes.ucsf.edu.
Scott A. Oakes, Email: scott.oakes@ucsf.edu.
References
- 1.Muniraj T, Aslanian HR, Farrell J, Jamidar PA. Chronic pancreatitis, a comprehensive review and update. Part I: epidemiology, etiology, risk factors, genetics, pathophysiology, and clinical features. Dis Mon. 2014;60(12):530–550. doi: 10.1016/j.disamonth.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 2.Yadav D, Timmons L, Benson JT, Dierkhising RA, Chari ST. Incidence, prevalence, and survival of chronic pancreatitis: a population-based study. Am J Gastroenterol. 2011;106(12):2192–2199. doi: 10.1038/ajg.2011.328. [DOI] [PubMed] [Google Scholar]
- 3.Whitcomb DC, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet. 1996;14(2):141–145. doi: 10.1038/ng1096-141. [DOI] [PubMed] [Google Scholar]
- 4.Rosendahl J, et al. Chymotrypsin C (CTRC) variants that diminish activity or secretion are associated with chronic pancreatitis. Nat Genet. 2008;40(1):78–82. doi: 10.1038/ng.2007.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Witt H, et al. Mutations in the gene encoding the serine protease inhibitor, Kazal type 1 are associated with chronic pancreatitis. Nat Genet. 2000;25(2):213–216. doi: 10.1038/76088. [DOI] [PubMed] [Google Scholar]
- 6.Sharer N, et al. Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. N Engl J Med. 1998;339(10):645–652. doi: 10.1056/NEJM199809033391001. [DOI] [PubMed] [Google Scholar]
- 7.Issa Y, et al. Treatment options for chronic pancreatitis. Nat Rev Gastroenterol Hepatol. 2014;11(9):556–564. doi: 10.1038/nrgastro.2014.74. [DOI] [PubMed] [Google Scholar]
- 8.Whitcomb DC, et al. Common genetic variants in the CLDN2 and PRSS1-PRSS2 loci alter risk for alcohol-related and sporadic pancreatitis. Nat Genet. 2012;44(12):1349–1354. doi: 10.1038/ng.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Whitcomb DC, et al. Multicenter approach to recurrent acute and chronic pancreatitis in the United States: the North American Pancreatitis Study 2 (NAPS2) Pancreatology. 2008;8(4-5):520–531. doi: 10.1159/000152001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davidson P, Costanza D, Swieconek JA, Harris JB. Hereditary pancreatitis. A kindred without gross aminoaciduria. Ann Intern Med. 1968;68(1):88–96. doi: 10.7326/0003-4819-68-1-88. [DOI] [PubMed] [Google Scholar]
- 11.Kleeff J, et al. Chronic pancreatitis. Nat Rev Dis Primers. 2017;3:17060. doi: 10.1038/nrdp.2017.60. [DOI] [PubMed] [Google Scholar]
- 12.Vigliano I, et al. FOXN1 mutation abrogates prenatal T-cell development in humans. J Med Genet. 2011;48(6):413–416. doi: 10.1136/jmg.2011.089532. [DOI] [PubMed] [Google Scholar]
- 13.Uhlén M, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 14.Naruse S, et al. Fecal pancreatic elastase: a reproducible marker for severe exocrine pancreatic insufficiency. J Gastroenterol. 2006;41(9):901–908. doi: 10.1007/s00535-006-1884-0. [DOI] [PubMed] [Google Scholar]
- 15.Szabó A, Sahin-Tóth M. Determinants of chymotrypsin C cleavage specificity in the calcium-binding loop of human cationic trypsinogen. FEBS J. 2012;279(23):4283–4292. doi: 10.1111/febs.12018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szabó A, Pilsak C, Bence M, Witt H, Sahin-Tóth M. Complex formation of human proelastases with procarboxypeptidases A1 and A2. J Biol Chem. 2016;291(34):17706–17716. doi: 10.1074/jbc.M116.743237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Párniczky A, et al. Genetic Analysis of Human Chymotrypsin-Like Elastases 3A and 3B (CELA3A and CELA3B) to Assess the Role of Complex Formation between Proelastases and Procarboxypeptidases in Chronic Pancreatitis. Int J Mol Sci. 2016;17(12):2148. doi: 10.3390/ijms17122148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Athwal T, et al. Expression of human cationic trypsinogen (PRSS1) in murine acinar cells promotes pancreatitis and apoptotic cell death. Cell Death Dis. 2014;5:e1165. doi: 10.1038/cddis.2014.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Archer H, Jura N, Keller J, Jacobson M, Bar-Sagi D. A mouse model of hereditary pancreatitis generated by transgenic expression of R122H trypsinogen. Gastroenterology. 2006;131(6):1844–1855. doi: 10.1053/j.gastro.2006.09.049. [DOI] [PubMed] [Google Scholar]
- 20.Whitcomb DC. Genetic risk factors for pancreatic disorders. Gastroenterology. 2013;144(6):1292–1302. doi: 10.1053/j.gastro.2013.01.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lugea A, Nan L, French SW, Bezerra JA, Gukovskaya AS, Pandol SJ. Pancreas recovery following cerulein-induced pancreatitis is impaired in plasminogen-deficient mice. Gastroenterology. 2006;131(3):885–899. doi: 10.1053/j.gastro.2006.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ulmasov B, Oshima K, Rodriguez MG, Cox RD, Neuschwander-Tetri BA. Differences in the degree of cerulein-induced chronic pancreatitis in C57BL/6 mouse substrains lead to new insights in identification of potential risk factors in the development of chronic pancreatitis. Am J Pathol. 2013;183(3):692–708. doi: 10.1016/j.ajpath.2013.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richter JD, Coller J. Pausing on polyribosomes: make way for elongation in translational control. Cell. 2015;163(2):292–300. doi: 10.1016/j.cell.2015.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamano M, Miyata K, Yamada T. Protective effect of a pancreatic elastase inhibitor against a variety of acute pancreatitis in rats. Jpn J Pharmacol. 1998;77(3):193–203. doi: 10.1254/jjp.77.193. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.