

Abstract
We hypothesized that the store-operated calcium entry (SOCE) channel, Orai1, participates in the activation of Th17 cells and influences renal injury. In rats, following renal ischemia/reperfusion (I/R), there was a rapid and sustained influx of Orai1+ CD4 T cells and IL-17 expression was restricted to Orai1+ cells. When kidney CD4+ cells of post–acute kidney injury (post-AKI) rats were stimulated with angiotensin II and elevated Na+ (10–7 M/170 mM) in vitro, there was an enhanced response in intracellular Ca2+ and IL-17 expression, which was blocked by SOCE inhibitors 2APB, YM58483/BTP2, or AnCoA4. In vivo, YM58483/BTP2 (1 mg/kg) attenuated IL-17+ cell activation, inflammation, and severity of AKI following either I/R or intramuscular glycerol injection. Rats treated with high-salt diet (5–9 weeks after I/R) manifested progressive disease indicated by enhanced inflammation, fibrosis, and impaired renal function. These responses were significantly attenuated by YM58483/BTP2. In peripheral blood of critically ill patients, Orai1+ cells were significantly elevated by approximately 10-fold and Th17 cells were elevated by approximately 4-fold in AKI versus non-AKI patients. Further, in vitro stimulation of CD4+ cells from AKI patients increased IL-17, which was blocked by SOCE inhibitors. These data suggest that Orai1 SOCE is a potential therapeutic target in AKI and CKD progression.
Keywords: Inflammation, Nephrology
Keywords: Calcium signaling, Fibrosis
Introduction
Acute kidney injury (AKI) results from events such as renal ischemia, nephrotoxicity, and/or sepsis. AKI increases the risk of death in the intensive care unit (ICU), and mortality rates in this setting range between 15%–60% (1–3). Survival from AKI is dependent on recovery of renal function following injury, the success of which has been suggested to be dependent on the efficiency of adaptive repair processes (4, 5). Progression of chronic kidney disease (CKD) and end-stage kidney disease are recognized as possible outcomes for AKI patients, and it has been suggested that incomplete or maladaptive repair may predispose progression of CKD following AKI (4, 5).
Immune cell activity may contribute to renal injury or may enhance renal recovery. In the setting of renal ischemia reperfusion (I/R) injury, renal CD4+ Th1 or Th17 cells are thought to exacerbate renal injury while T regulatory cells have been implicated in renal repair (6, 7). Following recovery from I/R injury in rats, subsequent exposure to high-salt diet was shown to hasten the development of interstitial fibrosis, inflammation, proteinuria, and hypertension. These parameters of CKD progression were significantly attenuated by immunosuppression with mycophenolate (8, 9), suggesting that lymphocyte activity also modulates the AKI-to-CKD transition.
Naive CD4+ cells differentiate into effector T helper cells in the ischemic milieu, where they are exposed to different antigens and proinflammatory cytokines. T helper cells secrete various cytokines and are thought to orchestrate the adaptive immune response (6). Our group demonstrated that Th17 cells, which secrete the cytokine IL-17, are the prominent lymphocyte population found in rat kidney following I/R injury (10). These cells have been implicated in a variety of autoimmune diseases such as asthma, psoriasis, inflammatory bowel disease, and lupus erythematosus (11). There is a significant expansion of Th17 cells in kidney within the first 3 days of I/R injury in rats, whereas Th17 levels resolve to near sham-operated control values within 7 days as renal function recovers. However, subsequent exposure of rats to high-salt diet (4%) strongly reactivates Th17 cell expression in post-ischemic kidney (10). This reactivation may contribute to CKD, since an IL-17R antagonist attenuated renal interstitial fibrosis and neutrophil infiltration in post I/R rats exposed to high-salt diet (12).
The basis for activation of Th17 cells in response to renal injury and exposure to high-salt diet remains to be elucidated. Th17 cell differentiation is dependent on the activity of the transcription factor RORγT, and inhibitors of this factor can alleviate the pathological activation of Th17 cells (13). Activation of these cells by high-salt diet has also been demonstrated in a mouse model of autoimmune encephalitis and associated with the activity of serum and glucocorticoid regulated kinase (SGK-1) and nuclear factor of activated T cells 5 (NFAT5) (14). Elevation of extracellular Na+ to 170 mM enhanced differentiation from naive CD4+ cells to Th17 cells in vitro in a process dependent on SGK-1 (15).
Using an in vitro stimulation assay, we demonstrated that CD4+ T cells from post-ischemic kidney manifest enhanced expression of IL-17 in response to Ang II and elevated extracellular Na+ in vitro, while no response was observed from sham-operated control-derived CD4+ cells (12). The mechanisms mediating the enhanced IL-17 activation in CD4+ cells after AKI are not known. Previous studies have demonstrated that Orai1, the pore-forming subunit of Ca2+ release-activated Ca2+ channels (CRAC), is required for Th17 cell differentiation in vitro, due in part to NFAT activity (16, 17). Interestingly, Orai1 mutant mice or inhibitors of Orai1 show impaired T cell receptor (TCR) activation and reduced IL-17 production, and are resistant to autoimmune disorders (17). Therefore, we tested the hypothesis that renal I/R enhances lymphocyte Orai1-mediated Ca2+ signaling that drives Th17 cell expression and, in turn, modulates AKI and AKI-to-CKD progression.
Results
Previous studies demonstrated that Th17 cells are rapidly induced following renal I/R and that IL-17 contributes to AKI (10, 18). To investigate the potential that Orai1 participates in AKI, Orai1 expression was measured in Th17 cells from kidneys of rats 2 days following sham or I/R injury (Study I). Orai1 was detected in Th17 cells and the number of these cells was increased following I/R relative to sham (Figure 1A). When accounting for influx, the total number of CD4+/Orai1+ cells and the number of triple-positive CD4+/IL-17+/Orai1+ cells in kidney were markedly elevated by I/R injury (Figure 1, B and C). In CD4+ cells, Orai1 was associated with increased IL-17 signal (Figure 1D) and IL-17+ cells were found almost exclusively in Orai1+ cells in both sham and post-I/R groups (Figure 1E). Orai1 expression was also observed in CD8+, B cells, NK cells, and macrophages but the percentage of these populations was modest when compared with CD4+ cells (Supplemental Table 2; supplemental material available online with this article; https://doi.org/10.1172/JCI126108DS1). Orai1 has 2 homologs, referred to Orai2 and Orai3, which have been suggested to modulate lymphocyte responses (19, 20). However, neither Orai2 nor Orai3 was significantly affected by I/R and neither Orai2+ nor Oria3+ cells coexpressed IL-17 (Supplemental Figure 2).
Figure 1. Coexpression of Orai1 and IL-17 in CD4+ T cells following renal I/R.

(A) Contour plot of renal lymphocytes stained with antibodies for CD4 and IL-17 to identify Th17 cells (left). Orai1 staining in gated Th17 cells of sham (middle) and 2 days after I/R (right). (B) The number of Orai1+/CD4+ cells in sham and post-I/R rat kidneys. (C) The number of Orai1+/CD4+/IL-17+ cells in sham and post-I/R rat kidneys. (D) Representative histogram of Orai1+ cells in kidney CD4+ fraction (left) 2 days after I/R, and the distribution of IL-17 expression as a function of Oria1 (right). (E) The percentage of IL-17+ cells as a function of Orai1 expression in CD4+ cells following sham surgery or 2 days after I/R. (F) Sustained expression of Orai1 in CD4+ cells following 7 days of recovery from I/R surgery is shown as a representative histogram (left); total number of Orai1+/CD4 cells (right). In B, C, E, and F, data are mean ± SE of 4–5 rats per group. *P < 0.05 for sham versus post-AKI by Student’s t test (B, C, F) and for sham versus I/R (E); †P < 0.05 in Orai1– versus Orai1+ cells, by 1-way ANOVA and Tukey’s post hoc test.
Kidney Th17 levels return to sham-operated control values within approximately 7 days of I/R (10). Despite the reduction of Th17 cells, Orai1 expression was maintained in CD4+ cells 7 days after I/R (Figure 1F). Post-AKI rat kidneys also demonstrate a greater percentage of CD4+ cells expressing the IL-17 transcription factor, RORγT (Supplemental Figure 3A). When placed in culture, these AKI-primed CD4+ cells (7 days after I/R), but not sham CD4+ cells, increase IL-17 mRNA expression following in vitro stimulation with Ang II and elevated Na+ (10–7 M/170 mM) (Supplemental Figure 3B) (10). This treatment also significantly increases the percentage of IL-17–expressing cells from approximately 12% to approximately 49% as detected by FACS (Figure 2, A and B). This response specifically requires elevated Na+, since increasing osmolality to a similar degree with either mannitol or choline chloride does not induce IL-17 mRNA in the presence of Ang II (Supplemental Figure 3B). The IL-17+ cells induced following treatment coexpress RORγT, suggesting activation of a predominately Th17 phenotype (Supplemental Figure 3C).
Figure 2. Orai1 activity contributes to IL-17 expression in CD4+ lymphocytes primed by renal ischemia/reperfusion injury.

(A) Representative FACS showing increased IL-17 expression in CD4+ cells from 7-day post-AKI rats following stimulation in vitro with 170 mM Na+ and Ang II versus control media. (B) Percentage of IL-17+ cells in CD4+ cells isolated 7 days after sham or AKI and stimulated in vitro. (C) IL-17 mRNA, expressed as 2–ΔΔCT of kidney-derived CD4+ cells, isolated 7 days after I/R surgery and stimulated in vitro. In B and C, control refers to AKI-primed CD4+ cells stimulated with 170 mM Na+ and Ang II (10–7 M), and SOCE inhibitors are included as labeled. (D) Fura-2 fluorescence imaging of intracellular Ca2+ in CD4+ lymphocytes in response to increased Na+ (170 mM) plus Ang II (10–7 M), as indicated in the timeline and expressed as the ratio of fluorescence using 340/380 nm excitation. Shown are representative tracings of CD4+ cells from kidney following sham surgery (black) or I/R injury (red), or from I/R injury with coincubation with AnCoA4 (blue). The inset illustrates representative visual field of multiple fura-2–loaded cells. (E) Percentage of cells manifesting an increase in Ca2+ response relative to baseline following in vitro stimulation with increased Na/Ang II. Data are mean ± SE from 4–5 rats per group per assay; *P < 0.05 versus unstimulated cells (i.e., no Ang II and normal Na, data not shown, see Supplemental Figure 3); †P < 0.05 inhibitors versus stimulated post-AKI cells by 1-way ANOVA and Tukey’s post hoc test.
Kidney-derived CD4+ cells were examined further for markers of effector memory T cells (CD44+/CD62L–) 7 days following I/R injury. There was an approximately 4-fold increase in such cells from post-I/R rats versus sham (1.85% ± 0.01 % vs 7.65% ± 1.23 %; P < 0.05). Stimulation with Ang II and elevated Na+ did not affect the percentage of CD44+ effector memory T cells, suggesting this population is not responsive to stimulation that promotes IL-17 expression (Supplemental Figure 3C).
To evaluate a potential role for Orai1 in the IL-17 response, AKI-primed CD4+ cells were stimulated with Ang II and elevated Na+ in the presence or absence of different SOCE inhibitors. Both 2-ABP and YM58483/BPT2 completely blocked the increase of IL-17 mRNA as well as the increase in IL-17+ cells (Figure 2, B and C). In addition, AnCoA4, an inhibitor considered to be highly specific for Orai1 due to its binding to stromal interaction molecule 1 (STIM1), thereby inhibiting gating of the Orai1 channel (21), also completely blocked the induction of IL-17 mRNA and protein.
To further evaluate Orai1 activity in AKI-primed CD4+ cells, intracellular free Ca2+ responses were evaluated with the fluorescent indicator fura-2. Representative tracings of sham-operated and AKI-primed CD4+ cells are shown in (Figure 2D). When the superfusate was changed to a buffer containing Ang II and elevated Na+, a rapid and sustained increase in cytosolic Ca2+ was observed in a significant percentage of AKI-primed lymphocytes when compared with lymphocytes derived from sham-operated controls (Figure 2, D and E). The addition of either AnCoA4 or YM58483/BPT2 significantly attenuated the percentage of Ca2+-responding cells to levels similar to sham.
To evaluate if SOCE influences Th17 cells in AKI, rats were fed YM58483/BPT2 approximately 2 hours prior to 40 minutes of I/R (Study II). YM58483/BPT2 significantly attenuated the level of renal injury 24 hours after reperfusion as indicated by the level of plasma creatinine (Figure 3A) and mRNA expression of kidney injury marker-1 (Kim-1) (Figure 3B). YM58483/2-BPT also attenuated the infiltration of total CD4+ T cells, B cells, and dendritic cells following I/R (Figure 3, C–F). Total IL-17–expressing cells were significantly reduced by approximately 78% in YM58483/BPT2-treated rats relative to vehicle (Figure 3G). This reduction of IL-17+ was primarily observed in the CD4+ population (Figure 3H), whereas YM58483/BPT2 did not significantly effect CD8+ cells or CD8+ IL-17+ cells (Figure 3, D and I). Also, YM58483/BPT2 did not significantly influence either Th1 (IFN-γ; Figure 3J) or Th2 (IL-4+; data not shown) cells after ischemia.
Figure 3. YM58483 pretreatment attenuates renal ischemia reperfusion injury.

(A) Assessment of renal function by plasma creatinine 24 hours after I/R in Sprague-Dawley rats pretreated with YM-58483 or vehicle. Values for sham rats were 0.4 ± 0.1 mg/dL and are not shown on graph. (B) Kim-1 mRNA levels from total kidney RNA 24 hours after sham or I/R are shown. ND, not detectable. Total kidney-derived CD4+ and CD8+ cells are shown in C and D, respectively; total B cells (RT1B+) and dendritic/macrophage cells (DC/Mᶲ; CD11b/c+) are shown in E and F, respectively. The total number of IL-17+, CD4+/IL-17+ and CD8+/IL-17+ cells are shown in G, H, and I respectively. CD4+/IFN-γ+ cells are shown in J. Data are mean ± SE; n = 11 rats/group in A and n = 7–8 rats in B–J. *P < 0.05 I/R vehicle versus sham; †P < 0.05 vehicle versus YM58483 by 1-way ANOVA and Tukey’s post hoc test.
Orai1 is present in immune cells, but may also be present in other cell types such as vascular cells. We sought to determine if the effect of YM58483/BPT on renal injury was primarily due to its effects on Th17 activation. To address this, rats were subjected to bilateral renal I/R and treated with either YM58483/BPT2, the IL-17Rc receptor antagonist, or both. Consistent with our previous data, YM58483/BPT2 treatment alone significantly reduced plasma creatinine as compared with vehicle controls in post-ischemic rats; however, IL-17Rc+YM58483/BPT2 treatment did not have any additional effect (Table 1). Levels of kidney CD4+ and CD4+/IL-17+ cells were significantly reduced versus vehicle-treated rats by a similar degree in both YM58483/BPT- and IL-17Rc–treated rats (Table 1). When rats were treated with a combination of YM58483/BPT and IL-17Rc, the reduction in renal injury was similar to that observed with either YM58483/BPT or IL-17Rc alone (Table 1). The lack of an additional effect of IL-17Rc suggests that the primary effect of YM58483/BPT is due to inhibition of Th17 cells in the early post-ischemic period.
Table 1. Effect of SOCE inhibitor YM58483 and IL-17Rc on renal injury and renal lymphocyte content 24 hours following ischemia reperfusion.

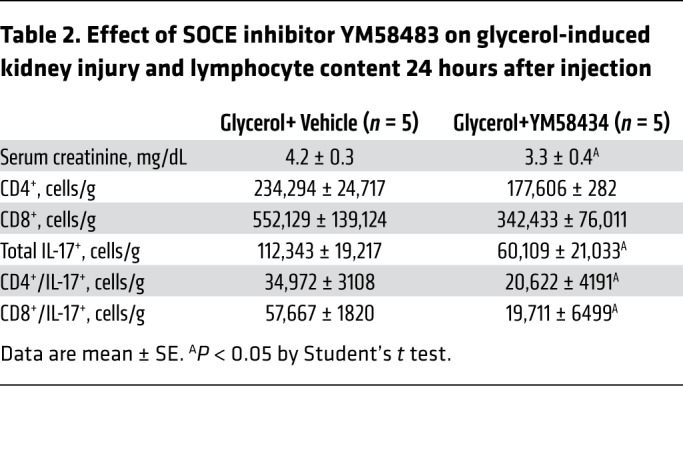
To further explore whether SOCE affects Th17 differentiation and renal injury, YM58483/BPT2 was tested in a different model of AKI associated with rhabdomyolysis. Intramuscular injection of glycerol into water-deprived rats resulted in severe renal injury (Table 2). In this model, renal CD8+ cells, including CD8+/IL-17+ cells, appeared to be the predominant lymphocyte population, as opposed to the more prevalent CD4+ lymphocyte response following I/R. Nevertheless, YM58483/BPT2 significantly attenuated the rise in serum creatinine as well as the total number of IL-17–expressing cells (Table 2).
Table 2. Effect of SOCE inhibitor YM58483 on glycerol-induced kidney injury and lymphocyte content 24 hours after injection.
Exposure of rats to high-salt diet at 5 weeks following recovery from I/R restimulates renal Th17 cell activity that is thought to contribute to CKD progression (10). The effect of SOCE on Th17 activation by high-salt diet following AKI was evaluated in rats subjected to unilateral renal I/R followed by contralateral nephrectomy and transition to high-salt diet (Study III). Post-AKI rats treated with vehicle during high-salt diet treatment manifested a significant infiltration of kidney CD4+ and CD8+ cells and IL-17–expressing cells. YM58483/BPT2 treatment significantly attenuated the increase in CD4+ and CD8+ cells and the increased expression of IL-17+ cells (Figure 4, A–D). YM58483/BPT2 also significantly attenuated the increase in B cells and Mᶲ/dendritic cells in post-AKI rats fed high-salt diet (Figure 4, E and F). Creatinine clearance at the end of the study period (i.e., 9 weeks after I/R) was significantly reduced in vehicle-treated I/R rats relative to sham-operated controls, but creatinine clearance was not reduced in YM58483/BPT2-treated rats relative to sham (Figure 4G). Post-ischemic rats treated with vehicle also showed significant alterations in other parameters related to CKD, including urinary albumin excretion, the development of interstitial fibrosis, and the expression of Kim-1; these parameters were all significantly attenuated in YM58483/BPT2-treated rats (Figure 4, H and I; and Supplemental Figure 4).
Figure 4. YM58483 attenuates inflammation and progressive renal damage following I/R and exposure to high-salt diet.

Shown are levels of renal CD4+ (A), CD8+ T cells (B), IL-17+ expressing cells (C), and CD4+/IL-17+ cells (D). Levels of B cells (E) and dendritic cells/Mᶲ (F) are shown. (G) Creatinine clearance from 24-hour urine collections at day 62–63 and (H) urinary albumin/creatinine ratio. (I) Representative picrosirius red–stained sections through renal outer medulla from sham, I/R vehicle, or I/R plus YM58483-treated rats. (J) Quantification of stained area. Data are mean ± SE; n = 5–6 rats/group. *P < 0.05 I/R vehicle vs sham; †P < 0.05 vehicle versus YM58483 by 1-way ANOVA and Tukey’s post hoc test.
We previously demonstrated increased circulating Th17 cells in rat blood following I/R, suggesting that blood may be used as source of activated lymphocytes in the setting of AKI (22). To investigate the potential that SOCE influences Th17 differentiation in human AKI, peripheral blood samples were obtained from critically ill patients with and without AKI. Samples were collected within 24–48 hours of AKI diagnosis for AKI cases or within 24–48 hours of ICU admission for frequency-matched (age, sex, baseline eGFR) controls without AKI (Table 3). In isolated blood mononuclear cells, the percentages of total IL-17+ cells and CD4+/IL-17+ cells were significantly higher in AKI patients versus non-AKI patients (Figure 5, A–C). Moreover, the percentage of Orai1+ cells was also prominently increased from approximately 3% in non-AKI patients to approximately 30% in patients with AKI. Similar to studies in rat kidney, Th17 cells were predominantly found within Orai1-expressing cells versus Orai1– cells (Figure 5, D–F).
Table 3. Characteristics of patients according to AKI status.

Figure 5. Elevated Th17 and Orai1 expression and effect of SOCE on IL-17 responses in CD4+ blood lymphocytes in critically ill patients with or without AKI.

(A) Representative FACS analysis of total peripheral blood cells in non-AKI (left, n = 8) vs AKI patients (right, n = 9) demonstrating the increase in CD4+ IL-17+–expressing cells. Quantification of the percentages of total IL-17+ cells (B) and CD4+/IL-17+ cells (C) in AKI versus non-AKI patients are shown. Representative dot plots showing (D) an increase in Orai-1+ cells in AKI patients as compared with non-AKI patients and (E) the percentage of Orai-1+ cells in AKI vs non-AKI patients. (F) Percentage of CD4+/IL-17+ cells gated on Orai1+ and Orai1– fractions (arrows). (G) IL-17 response to stimulation with elevated sodium (170 mM) and Ang II is shown for CD4+ cells isolated from AKI (n = 5) or non-AKI (n = 5) patients. Data are expressed as mean ± SE. *P < 0.05 by Student’s t test in AKI versus non-AKI patients (B–D), in Orai1+ versus Orai1– cells (F), or versus unstimulated (i.e., 140 mM and no Ang II) (G). †P < 0.05 inhibitors versus stimulated using 1-way ANOVA and Tukey’s post hoc test.
We next examined whether AKI enhanced IL-17 responses in circulating human CD4 cells and if these responses depend on SOCE activity. Human CD4+ cells responded in vitro to elevated extracellular sodium by increasing IL-17 expression in samples from AKI patients, but not in those from patients without AKI (Figure 5G). In contrast to rat kidney CD4+ cells, human blood CD4+ cells responded to elevated extracellular Na+ alone and did not require the addition of Ang II. Importantly, the IL-17 response appeared dependent on SOCE activity, since both YM58483/BPT2 and AnCoA4 inhibited this response (Figure 5G).
Discussion
In this study, we propose a novel regulatory pathway related to the activation of lymphocytes in the setting of acute and chronic kidney injury based on the role of intracellular calcium signaling and the differentiation of Th17 cells. Our studies demonstrate that the store-operated Ca2+ channel Orai1 is prominently induced in renal T cells in the setting of kidney injury. Moreover, blockade of this channel attenuated Th17 cell induction and renal damage in response to ischemia/reperfusion injury as well as subsequent exposure to high-salt diet. Thus, Orai1 may represent a therapeutic target to attenuate AKI or immune-mediated renal fibrosis and hypertension, which may occur secondary to AKI.
Th17 cells were originally described as a distinct T helper subset that secretes the cytokine IL-17 and is a major factor in autoimmune disorders. Th17 cells play an important role in host defense. However, in models of asthma, inflammatory bowel disease, psoriasis, or autoimmune encephalitis, Th17 cells aggravate inflammation by recruitment of other immune cells (such as neutrophils), which express the IL-17RA receptor (23). Th17 cells have also received significant attention in the setting of renal inflammatory disorders, including anti–neutrophil cytoplasmic antibody–associated (ANCA-associated) vasculitis (24), crescentic glomerular nephritis (25), and nephrotic syndrome (26, 27). Following renal transplant, there is an increased prevalence of Th17 cells in patients with chronic allograft nephropathy (28).
Th17 cells have recently been examined in the setting of AKI. Studies using Il17-null mice suggest that Th17 cells contribute to the severity of renal injury in response to I/R (29) or cisplatin (18). Work from our group demonstrated a biphasic Th17 response in rats, with an early transient phase of expression peaking between 1–3 days following injury and the second peak induced when rats are provided high-salt diet. Th17 cells were the predominant lymphocyte population activated by high-salt diet, whereas no significant effect was observed on Th1 or Th2 cells (12). The exposure to high-salt diet exacerbates inflammation, fibrosis, and hypertension, and can be attenuated by mycophenolate or an IL-17 antagonist (8, 12). Therefore, the mechanisms mediating IL-17 expression in response to I/R and high-salt intake are of interest. An in vitro model using CD4+ cells from kidneys 7 days after I/R revealed that stimulation by Ang II and elevated extracellular sodium increased IL-17 expression while having no effect on Th1 or Th2 responses (12). This priming of CD4+ cells by AKI provided an opportunity to investigate altered lymphocyte signaling leading to Th17 differentiation in response to renal injury, and it was the basis of the current study.
Kleinewietfeld et al. investigated the potential mechanism by which Na+ may directly influence Th17 cell differentiation. These authors demonstrated that naive T cells, when cultured for 4 days with TGF-β and IL-6, differentiate into Th17 cells. This response was potentiated by elevating the concentration of Na+ in the culture media. In addition, the response was abrogated by inhibition of p38/MAPK, NFAT5, and SGK-1. It was suggested that the increase in Na+ concentration by up to an additional 40 mM might be observed in skin under high-salt diet conditions and potentially influence T cell function. Indeed, Th17 induction and severity of autoimmune encephalopathy was enhanced when mice were placed on a high-salt diet (14).
TCR stimulation invokes an increase in intracellular Ca2+ via Ca2+ release–activated Ca2+ channels (CRAC). The result of this activity is thought to be calcineurin-mediated dephosphorylation of NFAT, which translocates to the nucleus and activates transcriptional programs. Genome-wide RNAi screens helped to identify Orai1 as the pore-forming subunit of CRAC channels (16, 30). Activation of Orai1 is mediated by the activity of STIM1, an endoplasmic reticulum (ER) membrane spanning protein, which senses Ca2+ depletion from the ER secondary to TCR stimulation. Interaction between STIM1 and Orai1 increases CRAC activity and results in sustained increases in intracellular Ca2+ (31). Mutations in either ORAI1 or STIM1 result in a severe combined immunodeficiency (SCID) phenotype (32). For Th17 cell differentiation, TCR stimulation in combination with various other cytokines represents an important driver of differentiation, which is considered more inflammatory than Th1 or Th2 phenotypes. Interestingly, chemical library screening recently identified putative Orai1 inhibitors, which showed greater selectivity in abrogating Th17 differentiation versus Th1 or Th2 differentiation. Moreover, Orai1 inhibition reduced the nuclear accumulation of NFAT and RORγT, critical transcriptional regulators of Th17 differentiation (16).
Data from the current study provide compelling evidence for an essential role of Orai1 in the differentiation of Th17 cells following renal I/R. IL-17–expressing CD4+ cells are rapidly expanded following renal injury, and using FACS analysis we show that Orai1 expression is also prominent in kidney CD4+ cells following renal injury. Measurements from circulating blood of critically ill patients with AKI also demonstrated a profound enhancement of Ora1+ IL-17+ cells, indicating that this pathway is activated in human AKI.
Importantly, IL-17 expression was almost exclusive to cells expressing Orai1, and was essentially absent in Oria1– cells. Published reports suggest that Orai1 mutations result in impaired production of Th17 cells. The data from the current study indicate that Orai1 is persistently expressed following the resolution of AKI (7 days after I/R). Given that the reexpression of IL-17 following in vitro stimulation is inhibited by SOCE antagonist, the results suggest that Orai1-mediated SOCE channel is required for Th17 differentiation following I/R.
The potential ability to target IL-17 has already shown promise in the setting of autoimmune disease, and several therapeutic agents are in use to target this pathway in diseases such as psoriasis (33). IL-17 could also represent a target for both acute and chronic kidney disease since both have been shown to be ameliorated by IL-17 blockade or Il17 gene knockout strategies (9, 18). Similarly, we confirmed that a SOCE antagonist showed a significant degree of protection using a standard model of AKI in rats induced by bilateral renal I/R. We also extended this observation to a model of AKI secondary to rhabdomyolysis. The protection was associated with a clear reduction in the genesis of Th17 cells in response to injury. However, since Orai1 may be expressed in endothelial cells or smooth muscle cells (34), we cannot exclude the possibility that the effects observed may be independent of IL-17 production. Nevertheless, in the current study, IL-17Rc blockade provided no additional protection over YM58483/BPT2 alone, suggesting that the primary activity of Orai1 in the early post-I/R period is to promote Th17 activation.
The transition from acute to chronic kidney disease has been the subject of significant research, and maladaptive repair responses predispose CKD progression (4). Since immune suppression strongly attenuates the AKI to CKD transition, persistent inflammation in the kidney in response to AKI represents a potential maladaptive response. It is therefore noteworthy that Orai1 expression remains persistently elevated in CD4+ cells despite the recovery of kidney function and the decline in IL-17 expression. The degree and duration of sustained Orai1 expression following recovery from AKI remains unclear, but it is worth noting that reactivation of Th17 cells by high-salt diet was attenuated by SOCE inhibition 35–63 days after injury. Whether Orai1 persists in activated T cells, memory T cells, or other leukocyte populations that secrete IL-17 remains to be determined. Our studies demonstrated an increase in effector memory T cells 7 days after I/R. This population was not affected by in vitro stimulation, so it is not yet clear whether this population contributes directly to the IL-17 response after injury. Nevertheless, we suggest that sustained Orai1 expression may represent the basis for susceptibility to reactivation of Th17 cells and therefore represents an important link predisposing to salt-sensitive CKD progression following AKI.
In addition to providing a potential link between AKI and CKD, there is also clear evidence that CKD predisposes to AKI (35). It could be suggested that sustained expression of Orai1 in CKD may enhance sensitivity to AKI by promoting a greater inflammatory response to a given insult. It is also reasonable to suggest that increased Orai1 expression enhances the IL-17 response to other inputs. For example, in the current study, IL-17 expression was elevated in response to Ang II and elevated Na+ in vitro and was dependent on SOCE activity. Recent studies have shown that Ang II–dependent hypertension is, in part, dependent on IL-17 activity (36, 37); whether Orai1 modulates Ang II–dependent Th17 responses in these models remains to be determined.
Taken together, the results from the current study suggest that Orai1 could be considered a novel pathway to target inflammatory renal disease associated with the Th17 phenotype. Novel inhibitors targeting this pathway are currently in development (38) and could represent therapies for inflammatory diseases associated with Th17 cells, including autoimmune disease as well as AKI, CKD, and salt-sensitive hypertension.
Methods
Animal study protocols.
All studies used male Sprague-Dawley rats (250–300 g) that were purchased from Envigo. Rats were anesthetized with a cocktail of ketamine (100 mg/kg, KetoVed, Vedco Inc) and xylazine (50 mg/kg, AnaSed, Lloyd Inc). Rats were placed on a heated surgical table to maintain body temperature. A midline incision and either unilateral (left) or bilateral renal ischemia (as indicated in Results) was induced by applying micro-aneurism clamps on the renal pedicles for a period of 40 minutes. Reestablishment of perfusion was verified by visual examination following removal of the clamps. Rats were provided post-operative analgesia using buprenorphine-SR (1 mg/kg).
Rats were allowed to recover for various periods of time as described in Results. In Study I, lymphocytes were studied in vitro after recovery for either 2 or 7 days following I/R or sham surgery for evaluation of function in vitro or for FACS analysis. In Study II, the effect of SOCE on kidney injury was studied with the inhibitor YM58483/BPT2 (N-[4-[3,5-Bis(trifluoromethyl)-1H-pyrazol-1-yl]phenyl]-4-methyl-1,2,3-thiadiazole-5-carboxamide; Tocris/Bio-Techne). Rats were pretreated p.o. 2–3 hours prior to surgery with YM58483.BPT2 (1 mg • kg−1) in sugar-free chocolate pudding, following dilution in 100% EtOH, at a dose previously shown to affect T cell activation in rats (39). In some experiments, rats were also treated with the IL-17Rc (150 ng/day i.p.; R&D Systems). In addition, one experiment utilized a model of rhabdomyolysis, induced by injection of 50% glycerol (10 mL/kg) into the hindlimb muscle of rats following 16 hours of water restriction as previously described (40).
Study III was designed to investigate SOCE on progression of CKD induced by high-salt diet following recovery from I/R injury using a model of AKI-to-CKD described previously (41). Rats acclimated to a standard diet (AIN 76A, Dyets) containing 0.4% NaCl were subjected to left unilateral I/R or sham surgery and allowed to recover for approximately 5 weeks. Rats were then subjected to right unilateral nephrectomy and subsequently exposed to elevated dietary Na+ (AIN76A plus 4% NaCl) for an additional 4 weeks (Supplemental Figure 4). Sham-control rats were not subjected to renal pedicle clamping, but were subjected to unilateral nephrectomy. Simultaneous with exposure to high-salt diet, rats were randomly assigned to vehicle or YM58483 treatment (1 mg • kg−1 • day−1; p.o.). At the end of all studies, rats were deeply anesthetized with 50–100 mg/kg pentobarbital (Fatal Plus, Vortech) and kidneys harvested for analysis.
Human studies.
We designed a prospective case-control study to examine peripheral blood from AKI patients (n = 9) and matched controls without AKI (n = 8) admitted to the ICU at the University of Kentucky Hospital. AKI was defined by KDIGO criteria (42) using both serum creatinine (SCr) and urine output data. Only patients with AKI stage 2 or greater were included in the study as cases. Controls were frequency-matched by age (10-year intervals), sex, and 2-category baseline estimated glomerular filtration rate (eGFR, calculated using CKD EPI equation, ≥90 and 60–89 mL/min/1.73 m2). Baseline SCr was defined as the most recent SCr within the 6-month period before ICU admission. Inclusion criteria were as follows: adults ≥18 years of age, admission to the ICU, and baseline eGFR ≥60 mL/min/1.73 m2. Exclusion criteria consisted of prior kidney or any other solid organ transplant, end-stage kidney disease, evidence of AKI before ICU admission, or the presence of uroepithelial tumors.
Single-timepoint peripheral whole blood samples were obtained 24–48 hours after AKI diagnosis (cases) or ICU admission (controls). Standardized techniques for blood collection, transport, and storage were employed.
Measurements of renal function.
To measure creatinine, tail blood was collected in heparin-containing tubes and centrifuged to collect plasma. Plasma creatinine was measured using a Pointe Scientific Analyzer and Creatinine Assay reagents using methods outlined by the manufacturer. Urine was collected for 24 hours by placing rats in metabolic cages, and urine volume was determined gravimetrically. Urine creatinine was measured using a colorimetric assay adopted for microplate readers, as previously described (43). Creatinine clearance was measured using UCr*V/PCr.
Renal histology, immunohistochemistry, and RNA expression.
At the time of tissue harvest, kidneys were bisected and one half was fixed by immersion in 10% formalin, embedded in paraffin, and 5-μm sections stained with Picrosirus red to assess fibrosis. For quantitative analysis, 5 random images of renal outer medulla were obtained using Leica DMLB (Scientific Instruments) microscope with a ×20 objective. The percent area of Picrosirus red stain was scored in a blinded fashion with Image J (NIH) using procedures described previously (10).
Total RNA was obtained from kidney using Trizol and the Zymogen RNA extraction kit and cDNA was prepared using MMLV enzyme (Invitrogen). Quantitative real-time PCR (qPCR) using gene-specific primers was performed using ABI 7500 (Applied Biosystems). mRNA values were calculated using 2–ΔΔCt. Specific primers sequences for IL-6 (catalog Rn01410330_m1), Kim-1 (catalog Rn00597703_m1), and IL-17 (catalog Rn01757168_m1) were purchased from Thermo Fisher Scientific.
Rat CD4+ isolation and FACS analysis.
Freshly harvested kidneys were minced and digested in liberase (2 μg/mL; Roche) for 15 minutes at 37°C using Gentle MACs (Miltenyi). The digested tissue was filtered through a 100-μm mesh and washed with RPMI containing 10% fetal bovine serum (Invitrogen). Mononuclear cells were isolated using Percol (MilliporeSigma) density gradient centrifugation.
All antibodies, their sources, and concentrations used are listed in Supplemental Table 1. To evaluate protein expression of IL-17 or Orai1, Orai2, or Orai3, cells were restimulated with PMA and ionomycin for 6 hours in the presence of monensin (Golgistop, 1 μg/mL; BD Biosciences) permeabilized with saponin (10%) and stained with relevant antibody. Cells were scanned using flow cytometry (FACSCalibur, BD Biosciences) and scans were analyzed using Flowjo software (Tree Star). Lymphocyte gating strategy was described previously (10) and a representative example of the gating strategy used in these studies is shown in Supplemental Figure 1.
Functional T cell analysis (rat).
CD4+ T cells were isolated using the MACS Pan-T cell microbead separation kit (Miltenyi). T cells were stimulated with plate bound anti-CD3 (precoated with 2 μg/mL) and soluble anti-CD28 (1 μg/mL). Cells (2.5 × 105 in 0.25 mL) were incubated 12–14 hours at 37°C in RPMI medium supplemented with 10% FBS (Invitrogen) in a 48-well plate. Cells were challenged with Ang II (MilliporeSigma, 10–7 M), raising the extracellular Na+ from 140 mM to 170 mM using a 1M NaCl solution. Calcium channel inhibitors 2ABP (10 μM, MilliporeSigma), AnCoA4 (10 μM, Millipore), and YM58483/BTP2 (10 μM) were included to evaluate effects on IL-17.
To assess Ca2+ responses, fura2 imaging was performed similar to methods previously described (44). Briefly, isolated CD4+ T cells were loaded with fura-2AM (2.5 μM, MilliporeSigma) for 45 minutes, washed, placed on poly-lysine (MilliporeSigma) coated coverslips, and placed in a superfusion chamber with physiological salt solution (PSS) containing 2 mM Ca2+. The chamber was mounted on an inverted epifluorescence microscope and signal measured with alternating excitation at 340 nm and 380 nm and emission at 510 nm using the InCa2+-imaging system (Intracellular Imaging Systems). Data were acquired at 1.5 Hz and representative tracings were smoothed to the 10 nearest neighbor points using GraphPad Prism. For analysis, an increase in signal intensity of more than 1 standard deviation from baseline was considered a positive response. Frequency of responding cells was determined from an average 53 ± 19 cells for each animal. Cells that did not respond to the Ca2+ ionophore ionomycin (1 μM) at the conclusion of the study were excluded from analysis.
Human CD4+ isolation and FACS analysis.
Fresh blood cells from patients were washed with PBS twice and stimulated with PMA, Iono, and monensin for 4–6 hours. Cells were stained for antibodies against IL-17, CD4+, and Orai-1 (Supplemental Table 2). The samples were initially blinded and diagnosis (AKI or non AKI) revealed after measurements of all samples were completed.
For in vitro stimulation of human blood cells, primary T cells were isolated from fresh blood using Straight whole blood CD4 kit from Miltenyi Biotech according to manufactures` protocol. CD4+T cells were plated at a density of 1 × 106 cells/mL in RPMI medium supplemented with FBS. Cells were stimulated with human anti-CD3/CD28 dynabeads (Gibco; catalog 11161D) along with labeled treatment overnight (~12 hours). T cells were harvested and incubated with monensin for 6 hours, prior to staining for IL-17 (Supplemental Table 2).
Statistics.
All data are expressed as mean ± SE or SD or median [IQ1-IQ3]. For experimental data, differences in means were established by 1-tailed Student’s t test or 1-way ANOVA with Tukey’s multiple comparison test as indicated in the figure legends. For clinical data, comparisons were done using Fisher’s exact test for categorical variables and Mann-Whitney U test for continuous variables. Analysis was done with the aid of Graph Pad Prism software or SAS 9.4 (SAS Institute). P<0.05 was considered significant.
Study approval.
Rats were maintained in accordance with the policies of the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All studies were approved by IACUC at Indiana University School of Medicine, Indianapolis, IN. For human studies, the protocol was approved by the IRB of the University of Kentucky, Lexington, KY. Written, informed consent was obtained for all study participants.
Author contributions
PM and DPB were responsible for the design and analysis of all rat studies described. JAN participated in the design of studies involving human samples, the screening, selection, and matching of patients, and obtaining samples used in the study. MS participated in the experimental design, training, and interpretation of Ca2+ imaging studies and interpretation of studies using Ca2+ inhibitors. PM conducted all experimental procedures and collected all data. DPB assisted with animal surgeries. DPB and PM were responsible for writing the manuscript. MS and JAN reviewed and edited the manuscript.
Supplementary Material
Acknowledgments
This work was supported by NIH DK063114 (to DPB) and by the Ralph W. and Grace M. Showalter Research Trust Fund (PM). Additional support was provided by Fortune Fry Funding from the Indiana University Research Foundation (to DPB and MS). JAN is currently supported by an Early Career Pilot Grant from the National Center for Advancing Translational Sciences (NCATS), NIH, through grant UL1 TR001998. The authors would like to thank Riley O’Shaughnessy and the Life Health Internship program and Indiana University Purdue University-Indianapolis. The authors appreciate the assistance of Stacy Dineen Rodenbeck with calcium measurements. The authors would like to thank Victor Ortiz-Soriano and Madona Elias for their help with human sample collection and transport.
Version 1. 08/15/2019
In-Press Preview
Version 2. 10/14/2019
Electronic publication
Version 3. 11/01/2019
Print issue publication
Footnotes
Conflict of interest: DPB, PM, and MS are authors on a patent application “Methods to treat renal disorders using calcium channel inhibitors,” US provisional application serial number 62/741,302, filed October 4, 2018. MS is cofounder and Chief Scientific Officer of Corvus Biomedical LLC.
Copyright: © 2019, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2019;129(11):4951–4961.https://doi.org/10.1172/JCI126108.
See the related Commentary at Orai1: CRACing the Th17 response in AKI.
Contributor Information
Purvi Mehrotra, Email: pmehrotr@iupui.edu.
Michael Sturek, Email: msturek@iupui.edu.
Javier A. Neyra, Email: javier.neyra@uky.edu.
David P. Basile, Email: dpbasile@iupui.edu.
References
- 1.Basile DP, Anderson MD, Sutton TA. Pathophysiology of acute kidney injury. Compr Physiol. 2012;2(2):1303–1353. doi: 10.1002/cphy.c110041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chertow GM, Burdick E, Honour M, Bonventre JV, Bates DW. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol. 2005;16(11):3365–3370. doi: 10.1681/ASN.2004090740. [DOI] [PubMed] [Google Scholar]
- 3.Singbartl K, Kellum JA. AKI in the ICU: definition, epidemiology, risk stratification, and outcomes. Kidney Int. 2012;81(9):819–825. doi: 10.1038/ki.2011.339. [DOI] [PubMed] [Google Scholar]
- 4.Basile DP, et al. Progression after AKI: Understanding maladaptive repair processes to predict and identify therapeutic treatments. J Am Soc Nephrol. 2016;27(3):687–697. doi: 10.1681/ASN.2015030309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chawla LS, Eggers PW, Star RA, Kimmel PL. Acute kidney injury and chronic kidney disease as interconnected syndromes. N Engl J Med. 2014;371(1):58–66. doi: 10.1056/NEJMra1214243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jang HR, Rabb H. Immune cells in experimental acute kidney injury. Nat Rev Nephrol. 2015;11(2):88–101. doi: 10.1038/nrneph.2014.180. [DOI] [PubMed] [Google Scholar]
- 7.Kinsey GR, Sharma R, Okusa MD. Regulatory T cells in AKI. J Am Soc Nephrol. 2013;24(11):1720–1726. doi: 10.1681/ASN.2013050502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pechman K, Basile DP, Lund H, Mattson DL. Immune suppression blocks sodium sensitive hypertension following recovery from acute renal failure. Am J Physiol Regulatory Integrative. 2008;294(4):R1234–R1239. doi: 10.1152/ajpregu.00821.2007. [DOI] [PubMed] [Google Scholar]
- 9.Mehrotra P, Collett JA, McKinney S, Stevens J, Ivancic CM, Basile DP. IL-17 mediates neutrophil infiltration and renal fibrosis following recovery from ischemia reperfusion:compensatory role of natural killer cells in athymic rats. Am J Physiol Renal Physiol. 2017;312(3):F385–FF97. doi: 10.1152/ajprenal.00462.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mehrotra P, Patel JB, Ivancic CM, Collett JA, Basile DP. Th-17 cell activation in response to high salt following acute kidney injury is associated with progressive fibrosis and attenuated by AT-1R antagonism. Kidney Int. 2015;88(4):776–784. doi: 10.1038/ki.2015.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang J, Sundrud MS, Skepner J, Yamagata T. Targeting Th17 cells in autoimmune diseases. Trends Pharmacol Sci. 2014;35(10):493–500. doi: 10.1016/j.tips.2014.07.006. [DOI] [PubMed] [Google Scholar]
- 12.Mehrotra P, Collett JA, McKinney SD, Stevens J, Ivancic CM, Basile DP. IL-17 mediates neutrophil infiltration and renal fibrosis following recovery from ischemia reperfusion: compensatory role of natural killer cells in athymic rats. Am J Physiol Renal Physiol. 2017;312(3):F385–F397. doi: 10.1152/ajprenal.00462.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guendisch U, et al. Pharmacological inhibition of RORγt suppresses the Th17 pathway and alleviates arthritis in vivo. PLoS One. 2017;12(11):e0188391. doi: 10.1371/journal.pone.0188391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kleinewietfeld M, et al. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature. 2013;496(7446):518–522. doi: 10.1038/nature11868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu C, et al. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature. 2013;496(7446):513–517. doi: 10.1038/nature11984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim KD, et al. Calcium signaling via Orai1 is essential for induction of the nuclear orphan receptor pathway to drive Th17 differentiation. J Immunol. 2014;192(1):110–122. doi: 10.4049/jimmunol.1302586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feske S. Calcium signalling in lymphocyte activation and disease. Nat Rev Immunol. 2007;7(9):690–702. doi: 10.1038/nri2152. [DOI] [PubMed] [Google Scholar]
- 18.Chan AJ, et al. Innate IL-17A-producing leukocytes promote acute kidney injury via inflammasome and Toll-like receptor activation. Am J Pathol. 2014;184(5):1411–1418. doi: 10.1016/j.ajpath.2014.01.023. [DOI] [PubMed] [Google Scholar]
- 19.Vaeth M, et al. ORAI2 modulates store-operated calcium entry and T cell-mediated immunity. Nat Commun. 2017;8:14714. doi: 10.1038/ncomms14714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Motiani RK, Stolwijk JA, Newton RL, Zhang X, Trebak M. Emerging roles of Orai3 in pathophysiology. Channels (Austin) 2013;7(5):392–401. doi: 10.4161/chan.24960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sadaghiani AM, et al. Identification of Orai1 channel inhibitors by using minimal functional domains to screen small molecule microarrays. Chem Biol. 2014;21(10):1278–1292. doi: 10.1016/j.chembiol.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 22.Mehrotra P, Collett JA, Gunst SJ, Basile DP. Th17 cells contribute to pulmonary fibrosis and inflammation during chronic kidney disease progression after acute ischemia. Am J Physiol Regul Integr Comp Physiol. 2018;314(2):R265–R273. doi: 10.1152/ajpregu.00147.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Monin L, Gaffen SL. Interleukin 17 family cytokines: signaling mechanisms, biological activities, and therapeutic implications. Cold Spring Harb Perspect Biol. 2018;10(4):a028522. doi: 10.1101/cshperspect.a028522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nogueira E, et al. Serum IL-17 and IL-23 levels and autoantigen-specific Th17 cells are elevated in patients with ANCA-associated vasculitis. Nephrol Dial Transplant. 2010;25(7):2209–2217. doi: 10.1093/ndt/gfp783. [DOI] [PubMed] [Google Scholar]
- 25.Paust HJ, et al. Chemokines play a critical role in the cross-regulation of Th1 and Th17 immune responses in murine crescentic glomerulonephritis. Kidney Int. 2012;82(1):72–83. doi: 10.1038/ki.2012.101. [DOI] [PubMed] [Google Scholar]
- 26.Liu LL, et al. Th17/Treg imbalance in adult patients with minimal change nephrotic syndrome. Clin Immunol. 2011;139(3):314–320. doi: 10.1016/j.clim.2011.02.018. [DOI] [PubMed] [Google Scholar]
- 27.Kitching AR, Holdsworth SR. The emergence of TH17 cells as effectors of renal injury. J Am Soc Nephrol. 2011;22(2):235–238. doi: 10.1681/ASN.2010050536. [DOI] [PubMed] [Google Scholar]
- 28.Chung BH, Kim KW, Kim BM, Doh KC, Cho ML, Yang CW. Increase of Th17 cell phenotype in kidney transplant recipients with chronic allograft dysfunction. PLoS One. 2015;10(12):e0145258. doi: 10.1371/journal.pone.0145258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo L, Lee HH, Noriega ML, Paust HJ, Zahner G, Thaiss F. Lymphocyte-specific deletion of IKK2 or NEMO mediates an increase in intrarenal Th17 cells and accelerates renal damage in an ischemia-reperfusion injury mouse model. Am J Physiol Renal Physiol. 2016;311(5):F1005–F1014. doi: 10.1152/ajprenal.00242.2016. [DOI] [PubMed] [Google Scholar]
- 30.Feske S, et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441(7090):179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 31.Derler I, Jardin I, Romanin C. Molecular mechanisms of STIM/Orai communication. Am J Physiol, Cell Physiol. 2016;310(8):C643–C662. doi: 10.1152/ajpcell.00007.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feske S, Picard C, Fischer A. Immunodeficiency due to mutations in ORAI1 and STIM1. Clin Immunol. 2010;135(2):169–182. doi: 10.1016/j.clim.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chiricozzi A, Krueger JG. IL-17 targeted therapies for psoriasis. Expert Opin Investig Drugs. 2013;22(8):993–1005. doi: 10.1517/13543784.2013.806483. [DOI] [PubMed] [Google Scholar]
- 34.Xu N, Cioffi DL, Alexeyev M, Rich TC, Stevens T. Sodium entry through endothelial store-operated calcium entry channels: regulation by Orai1. Am J Physiol, Cell Physiol. 2015;308(4):C277–C288. doi: 10.1152/ajpcell.00063.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.He L, et al. AKI on CKD: heightened injury, suppressed repair, and the underlying mechanisms. Kidney Int. 2017;92(5):1071–1083. doi: 10.1016/j.kint.2017.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wade B, Petrova G, Mattson DL. Role of immune factors in angiotensin II-induced hypertension and renal damage in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol. 2018;314(3):R323–R333. doi: 10.1152/ajpregu.00044.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Madhur MS, et al. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension. 2010;55(2):500–507. doi: 10.1161/HYPERTENSIONAHA.109.145094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tian C, Du L, Zhou Y, Li M. Store-operated CRAC channel inhibitors: opportunities and challenges. Future Med Chem. 2016;8(7):817–832. doi: 10.4155/fmc-2016-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoshino T, et al. YM-58483, a selective CRAC channel inhibitor, prevents antigen-induced airway eosinophilia and late phase asthmatic responses via Th2 cytokine inhibition in animal models. Eur J Pharmacol. 2007;560(2-3):225–233. doi: 10.1016/j.ejphar.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 40.Korrapati MC, Shaner BE, Schnellmann RG. Recovery from glycerol-induced acute kidney injury is accelerated by suramin. J Pharmacol Exp Ther. 2012;341(1):126–136. doi: 10.1124/jpet.111.190249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Basile DP, Leonard EC, Tonade D, Friedrich JL, Goenka S. Distinct effects on long-term function of injured and contralateral kidneys following unilateral renal ischemia-reperfusion. Am J Physiol Renal Physiol. 2012;302(5):F625–F635. doi: 10.1152/ajprenal.00562.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.KDIGO Work Group. KDIGO clinical practice guideline for acute kidney injury. Kidney Int Suppl. 2012;17(1):1–138. doi: 10.1159/000339789. [DOI] [Google Scholar]
- 43.Basile DP, Donohoe D, Roethe K, Osborn JL. Renal ischemic injury results in permanent damage to peritubular capillaries and influences long-term function. Am J Physiol Renal Physiol. 2001;281(5):F887–F899. doi: 10.1152/ajprenal.2001.281.5.F887. [DOI] [PubMed] [Google Scholar]
- 44.Badin JK, Bruning RS, Sturek M. Effect of metabolic syndrome and aging on Ca2+ dysfunction in coronary smooth muscle and coronary artery disease severity in Ossabaw miniature swine. Exp Gerontol. 2018;108:247–255. doi: 10.1016/j.exger.2018.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.