

Abstract
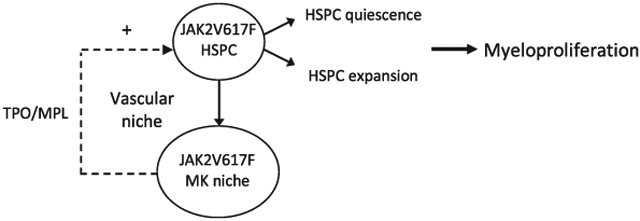
The myeloproliferative neoplasms (MPNs) are stem cell disorders characterized by hematopoietic stem/progenitor cell (HSPC) expansion and overproduction of mature blood cells. The acquired kinase mutation JAK2V617F plays a central role in these disorders. The mechanisms responsible for HSPC expansion in MPNs are not fully understood, limiting the effectiveness of current treatments. One hallmark feature of the marrow in patients with MPNs is megakaryocyte (MK) hyperplasia. Previously, we reported that JAK2V617F-bearing MKs cause a murine myeloproliferative syndrome with HSPC expansion. Here we show that JAK2V617F MKs promote MPN stem cell function by inducing HSPC quiescence with increased repopulating capacity. In addition, we demonstrate that thrombopoietin and its receptor MPL are critical for the JAK2V617F-bearing MK-induced myeloproliferation, both by directly affecting the quantity and quality of MKs and by altering the MK-endothelial interaction and vascular niche function. Therefore, targeting HSPC niche-forming MKs and/or their interactions within the vascular niche could provide novel, more effective therapeutic strategies in patients with MPNs.
Keywords: Myeloproliferative neoplasm, Megakaryocyte, JAK2, Thrombopoietin, MPL
Graphical Abstract
INTRODUCTION
The chronic Philadelphia chromosome (Ph[1]) negative myeloproliferative neoplasms (MPNs) are stem cell disorders characterized by hematopoietic stem/progenitor cell (HSPC) expansion and overproduction of mature blood cells. The acquired kinase mutation JAK2V617F plays a central role in these disorders. However, the mechanisms responsible for the malignant HSPC expansion in MPNs are not fully understood, limiting the effectiveness of current treatment. Although the etiology of dysregulated hematopoiesis has been mainly attributed to the molecular alterations within the stem cell compartment, abnormalities of the marrow microenvironment are beginning to be recognized as an important factor in the development of MPNs.[1-5]
The cellular composition of the hematopoietic niche includes both marrow stromal cells and hematopoietic cells. Vascular endothelial cells (ECs) are a major component of the HSPC niche (the “vascular niche”) and provide many key factors that are required for HSPC maintenance.[6] Megakaryocytes (MK) are rare polyploid hematopoietic cells that give rise to blood platelets. They are often located adjacent to marrow sinusoids, an anatomy required in order for the cells to release platelets directly into the sinusoidal vascular lumen.[7] Very recent evidence, including that from our own studies, has implicated MKs in regulating HSPC activity, presumably mediated by the many cytokines and extracellular matrix components produced by these cells.[8-15]
MK hyperplasia is a hallmark feature of all three chronic Ph[1] negative MPNs.[16] Previously, we have shown that JAK2V617F-bearing MKs cause a murine myeloproliferative syndrome with HSPC expansion. We also found that the JAK2V617F-bearing MKs stimulate EC angiogenesis in vitro and are associated with increased sinusoidal vascular density in vivo, suggesting that the JAK2V617F-bearing MKs may expand the sinusoidal vascular niche, which in turn could contribute to the panhematopoietic expansion phenotype displayed by the Pf4+FF1+ mice.[15] In the present work, we investigated the physiological effects and molecular mechanisms by which JAK2V617F-bearing MKs promote HSPC expansion in MPNs. We report that the JAK2V617F-bearing MK niche promotes MPN stem cell function by inducing HSPC quiescence with enhanced repopulating capacity. In addition, we demonstrate that thrombopoietin (TPO)/MPL signaling is critical for the JAK2V617F-bearing MK-induced myeloproliferation, both by directly affecting the quantity and quality of MKs and by affecting the MK-endothelial interaction and vascular niche function.
MATERIALS AND METHODS
Experimental mice
JAK2V617F Flip-Flop (FF1) mice17 and Pf4-Cre mice18 were provided by Radek Skoda (University Hospital Basal, Switzerland). FF1 mice were crossed with Pf4-Cre mice to generate MK cell lineage-specific human JAK2V617F knock-in mouse lines (Pf4+FF1+) as we previously described.[15,19] MPL knockout mice [20] was provided by Warren Alexander (Melbourne, Australia) and TPO knock-out mice [21] by Fred de Sauvage at Genentech (San Francisco, CA). TPO or MPL was ablated in Pf4+FF1+ by crossing Pf4+FF1+ mice with TPO knock-out mice or MPL knockout mice to generate TPO−Pf4+FF1+ mice and MPL−Pf4+FF1+ mice. CD45.1+ congenic mice (B6.SJL) were purchased from Taconic Inc (Albany, NY, USA). No randomization or blinding was used to allocate experimental groups. Animal experiments were performed in accordance with the guidelines provided by the Institutional Animal Care and Use Committee at Stony Brook University.
Competitive marrow transplantation assays
3×105 CD45.2 donor marrow cells from 28-wk old Pf4+FF1+ mice or Pf4-cre WT control mice were injected intravenously together with 3×105 competitor CD45.1 WT marrow cells into lethally irradiated (950 cGy) CD45.1 WT recipients. Peripheral blood was obtained every 4 weeks after transplantation and CD45.2 percentage chimerism and complete blood counts were measured.
Complete blood counts and colony assays
Blood was collected from mice via submandibular bleeds into Microtainer tubes with K2EDTA (Becton Dickinson, NJ, USA). Complete blood counts were performed on a Hemavet 950FS hematology system (Drew Scientific, FL, USA). It should be noted that the platelet levels obtained using this device are routinely lower than what we previously seen when using Antech Diagnostics® (Lake Success, NY) by laser flow cytometry.[15] As all samples reported here were obtained with the single instrument, they are internally consistent. Hematopoietic colony formation assays were performed as we previously described.[15]
Flow cytometry
All samples were analyzed by flow cytometry using a FACSAriaTM III (BD biosciences, San Jose, CA, USA). CD45 (Clone 104) (Biolegend, San Diego, CA, USA), CD45.1 (Clone A20) (BD Biosciences), CD45.2 (Clone 104) (Biolegend), EPCR (CD201) (Clone eBio1560, eBioscience, San Diego, CA, USA), CD150 (Clone mShad150, eBioscience), and CD48 (Clone HM48–1, Biolegend) antibodies were used to enumerate CD45+EPCR(CD201)+CD48−CD150+ (E-SLAM) cells.[22,23] MKs were sorted on the basis of CD41 expression (Clone MWReg30, Biolegend) and cell size.[15] CD45 (Clone 104) (Biolegend) and CD31 (Clone 390, BD biosciences) antibodies were used to isolate CD45−CD31+ marrow ECs.[24]
Cell cycle analysis
For HSPC cell cycle analysis, marrow cells were stained with CD150 and CD48 antibodies as described above, washed, and then stained with Hoechst33342 and Pyronin Y (Sigma, St. Louis, MO) at 37°C for 45 minutes.[25] Cells were kept on ice until analysis using a LSR II flow cytometer (BD Biosciences). Data were analyzed using FlowJo software (Tree Star, Ashland, OR, USA).
Histology and marrow MK-sinusoid interaction
Femurs from WT control (Pf4-cre), Pf4+FF1+, TPO− control (TPO−Pf4+), TPO−Pf4+FF1+, MPL− control (MPL−Pf4+), and MPL−Pf4+FF1+ mice were fixed in cold 4% PFA in PBS (Affymetrix) for 6 hours while shaking. Femurs were then washed with PBS to remove PFA and decalcified. Paraffin sections (5-μm thickness) were stained with hematoxylin and eosin (H&E). Images were taken using an Olympus IX70 microscope.
Marrow sinusoids were identified as red blood cell engorged vessels lined by a single layer of flat endothelial cells.[15,26] MKs are defined as large marrow cells with a multilobulated nucleus. About 100 Marrow MKs from 9–18 non-overlapping 20× fields were studied and categorized as MKs in direct contact with the sinusoids or MKs not in direct contact with the sinusoids (n = 2–3 mice in each group).
Isolation of murine ECs
Primary murine EC isolation was performed as we previously described27. Briefly, mice were euthanized, the lung, which is an organ with hematopoietic function especially thrombopoiesis,[28] or spleen tissue was collected and minced finely with scissors. The tissue fragments were digested in DMEM medium containing 1 mg/mL Collagenase D (Roche, Switzerland), 1 mg/mL Collagenase/Dispase (Roche) and 25 U/mL DNase (Sigma) at 37°C for 1 hr (for spleen) or 2 hr (for lung) with shaking, after which the suspension was homogenized by triturating. The homogenate was filtered through a 70μm nylon mesh (BD Biosciences, San Jose, CA) and pelleted by centrifugation (400 g for 5 min). Cells were first depleted for CD45+ cells (Miltenyi Biotec, SanDiego, CA) and then positively selected for CD31+ cells (Miltenyi Biotec) using magnetically labeled microbeads according to the manufacturer’s protocol. Isolated ECs (CD45−CD31+) were cultured in EC culture medium with no medium change for the first 72 hrs to allow EC attachment followed by medium change every 2–3 days. Cells were re-selected for CD31+ cells when they reach >70–80% confluence (usually after 3–4 days of culture).
Assays to examine EC function in vitro
ECs (passage 2–4) were cultured on 1% gelatin coated 24-well plate in complete EC medium (which has 20% fetal bovine serum (FBS)) as previously described.[15] To test the effect of TPO on the growth of vascular ECs in vitro, cells were treated with EC medium containing 2%FBS together with 0, 10, 100, or 1000 ng/ml rhTPO on Day 0. On Day 3, viable cell numbers were counted in a hemocytometer using trypan blue dye to exclude the dead cells.
To assess the effects of TPO on EC migratory function, we used an in vitro scratch assay as we previously described.[27] Briefly, WT murine spleen ECs (passage 2–3) were seeded in 1% gelatin coated 24-well plates and cultured at 37 °C to near confluence before the creation of a scratch wound. After the scratch, ECs were cultured with EC medium containing 2%FBS together with 0, 10, 100 ng/ml rhTPO at 37°C in a humidified 5% CO2 atmosphere. Images were captured with a phase-contrast microscope (AMEX-1200, AMG) at different time points after wound creation. Wound closure was measured by software Image J (National Institute of Health, Bethesda, MD).
Polymerase Chain Reaction
The TaqMan® Gene Expression Assay (ThermoFisher Scientific, Waltham, MA) was used for real-time quantitative polymerase chain reaction (qPCR) to verify differential expression of MPL, Chemokine (C-X-C motif) ligand 12 (CXCL12), VE-cadherin, ZO-1, and PECAM1 (platelet endothelial cell adhesion molecule) on an Applied Biosystems 7300 Real Time PCR System (ThermoFisher Scientific). Values obtained were normalized to the housekeeping Actin beta (Actb) gene expression and relative expression compared to control samples was calculated by the 2ΔΔCT method. All assays were performed in triplicate.
Human JAK2-specific primers GAGCAAGCTTTCTCACAAGC and AATTCTGCCCACTTTGGTGC (the same primers used for FF1 genotyping) were used to detect the expression of human JAK2V617F using reverse transcription polymerase chain reaction (RT-PCR). The primers amplify a 530-bp fragment which would be detected on 2% agarose gel.
Statistical Analysis
Differences between two groups were statistical analyzed using Student’s unpaired, 2-tailed t tests using Excel software (Microsoft). Correlation analyses between marrow MK cell numbers and E-SLAM cell numbers were performed by software PRISM (GraphPad Software, Inc. La Jolla, CA). A p value of less than 0.05 was considered significant. For all bar graphs, data are presented as mean ±standard error of the mean (SEM). All experiments were conducted and confirmed in at least two replicates.
RESULTS
The JAK2V617F-bearing MKs promote MPN stem cell function
To study the effects of the JAK2V617F-bearing MK niche on MPN stem cell function, we crossed mice that bear a Cre-inducible human JAK2V617F gene (FF1) with mice that express Cre under the promoter of platelet factor 4 (Pf4-Cre). This generated the Pf4+FF1+ mice that express JAK2V617F restricted to MK lineage as previously described.[15,18,19,29,30] Levels of the human JAK2V617F expression was ~ 10% of that of the endogenous murine JAK2 in MKs isolated from the Pf4+FF1+ mice.[19] As expected, the Pf4+/FF1+ mice developed a modest thrombocytosis, splenomegaly, and greatly increased marrow megakaryopoiesis at 28 weeks of age.[15] Surprisingly, these mice also developed a significant increase in CD45+EPCR+CD48−CD150+ (E-SLAM) cells,[15] a highly purified long-term repopulating HSPC population.[22,23] To be certain that JAK2V617F did not directly influence the numbers of E-SLAM cells because the Pf4 promoter was “leaky”,[31,32] we tested purified cells by FF1 RT-PCR.[17,19] Consistent with previous reports,[9,15,18,30] human JAK2 gene expression was detected in MKs but not in E-SLAM cells, marrow endothelial cells (ECs), or plucked colony-forming unit granulocyte/macrophage (CFU-GM) colonies from Pf4+FF1+ mice.
Previously we demonstrated that the expression of TPO receptor MPL, which is associated with both HSPC quiescence and HSPC repopulating activity,[33,34] was increased on expanded E-SLAM cells from the Pf4+FF1+ mice compared to WT control mice.[15] These observations led us to hypothesize that JAK2V617F-bearing MKs promote MPN stem cell function by inducing HSPC quiescence and enhancing its repopulating capacity. To test this hypothesis, we isolated marrow CD150+CD48− cells, of which ~ 20% display long-term repopulating capacity,[22] from 28weeks old (an age we have been using to analyze the myeloproliferative phenotype in Pf4+FF1+ mice15) Pf4+FF1+ mice and their age-matched littermate controls (n = 4 in each group) and analyzed their cell cycle status using Hoechst33342 and Pyronin Y staining.[25] We found that the percentage of CD150+CD48− cells in the G0 phase was moderately increased in the Pf4+FF1+ mice compared to control mice (43% vs 31%, P = 0.047, n = 4), suggesting that the CD150+CD48− HSPCs in the JAK2V617F-bearing MK niche are more quiescent than in the WT MK niche.[35] (Figure 1).
Figure 1.

HSPCs in the JAK2V617F-bearing MK niche are more quiescent than in the WT MK niche. Cell cycle analysis of marrow CD150+CD48− HSPCs from WT control mice and Pf4+FF1+ mice. Data are presented as mean ±SEM; n = 4 mice per group. *P < 0.05.
Next, we tested the effects of JAK2V617F-bearing MK niche on HSPC function by performing a competitive repopulation assay. CD45.2 donor marrow cells from 28weeks old Pf4+FF1+ mice or age-matched littermate control mice were injected intravenously together with 12weeks old CD45.1 wild-type competitor marrow cells into lethally irradiated (950 cGy) CD45.1 recipients. During a 16-wk follow up, recipients of Pf4+FF1+ marrow cells displayed a higher peripheral blood donor (CD45.2) chimerism than recipients of the control mice (CD45.2 chimerism 64% vs 22% at 16 weeks post transplant). (Figure 2A-B) Marrow cell colony assays demonstrated a moderate increase of CD45.2+ hematopoietic progenitors in Pf4+FF1+ marrow recipients compared to controls (1.5-fold, p = 0.0025). (Figure 2C) Marrow cell flow cytometry analysis revealed that, although there was no significant difference in the total CD41+ MK cell numbers between the two groups, CD45.2+ MKs were significantly increased (3.3-fold, P = 0.012) while CD45.1+ MKs were significantly decreased (3.4-fold, P = 0.024) in the Pf4+FF1+ marrow recipients compared with controls. (Figure 2D) Similarly, although the total numbers of marrow E-SLAM cells were not changed between the two groups, CD45.2+ E-SLAM cells were significantly increased (3.5-fold, P = 0.012) while CD45.1+ E-SLAM cells were significantly decreased (5.4-fold, P = 0.024) in the Pf4+FF1+ marrow recipient compared to the controls. (Figure 2E) These results indicate that both HSPC number and function (i.e. repopulating capacity) are increased in the Pf4+FF1+ mice compared to WT controls. JAK2V617F expression was not detected in pooled CFU-GM colonies from recipient of the Pf4+FF1+ marrow, suggesting that there was no activation of the JAK2V617F at the HSPC or committed myeloid progenitor cell level during marrow reconstitution. (data not shown) No significant difference in blood cell count, spleen weight, or total femoral cell count was observed between the Pf4+FF1+ marrow recipients and control marrow recipients.[35]
Figure 2.

The JAK2V617F-bearing MK niche promotes the expansion/engraftment of JAK2V617F HSPCs. (A) Competitive marrow transplantation scheme. (B) Following the competitive transplantation experiment, recipients of the Pf4+FF1+ marrow (n = 5) displayed a higher peripheral blood donor (CD45.2) chimerism than the recipients of the control marrow (n = 5). (C) There was a moderate increase of CD45.2+ hematopoietic progenitors in Pf4+FF1+ marrow recipients compared to control marrow recipients. (D) CD45.2+ MKs were significantly increased while CD45.1+ MKs were significantly decreased in the Pf4+FF1+ marrow recipients compared to controls. (E) CD45.2+ E-SLAM cells were significantly increased while CD45.1+ E-SLAM cells were significantly decreased in recipients of Pf4+FF1+ marrow compared to controls. Data are presented as mean ±SEM. *P < 0.05.
We noticed that the control mice displayed lower donor (CD45.2) chimerism than expected (e.g. ~20% at 12-16weeks), probably due to the age difference between the CD45.2 donors (28weeks) and CD45.1 competitors (12weeks), which was the same for both the experiment group and control group and therefore would not alter the conclusion that marrow cells from the Pf4+FF1+ mice have higher engraftment rates than cells from the WT control.
TPO/MPL signaling is critical for the JAK2V617F-bearing MK niche function
Our finding that the JAK2V617F-bearing MK niche only promoted the expansion of its own precursor JAK2V617F HSPCs in the competitive transplantation assay suggests that there are certain locoregional interactions/associations between the MK niches and HSPCs. Therefore, we examined the relationship between marrow MK cell numbers and E-SLAM cell numbers in 28-wk old WT control mice and Pf4+FF1+ mice. We found that, while there is no correlation between MKs and E-SLAM cells in the WT control mice, there is a strong correlation between marrow MK cell numbers and E-SLAM cell numbers in the Pf4+FF1+ mice (R2 = 0.71, P = 0.017). (Figure 3) Taken together with our previous report that there was altered cytokine expression in Pf4+FF1+ MKs compared with control MKs, these results suggest that the JAK2V617F mutation not only increases MK quantity but also alters its quality to promote HSPC expansion in the Pf4+FF1+ mice.
Figure 3.

Marrow E-SLAM cell numbers correlate strongly with marrow MK cell numbers in the Pf4+FF1+ mice, but not in the WT control mice. Marrow MK and E-SLAM cell frequencies were measured by flow cytometry analysis. Left: WT control mice (n = 6); Right: Pf4+FF1+ mice (n = 7).
TPO acting through its receptor MPL are key regulators of both megakaryopoiesis and HSPC activity.[34,37-39] To determine the role(s) of TPO/MPL signaling in the development of JAK2V617F MK-induced myeloproliferation and HSPC expansion, we ablated TPO or MPL in Pf4+FF1+ by crossing the Pf4+FF1+ mice with TPO−/− mice or MPL−/− mice to generate TPO−Pf4+FF1+ mice and MPL− Pf4+FF1+ mice. In contrast to what we have observed in the Pf4+FF1+ mice, although the TPO−Pf4+FF1+ mice demonstrated a mild increase in hemoglobin and platelet count compared to the TPO− control mice, neither the TPO−Pf4+FF1+ mice nor the MPL−Pf4+FF1+ mice developed any significant thrombocytosis as the Pf4+FF1+ mice during more than 1-year follow-up (Figure 4A-B). Most importantly, no MK or E-SLAM cell expansion was observed in the TPO−Pf4+FF1+ mice or MPL−Pf4+FF1+ mice as in the Pf4+FF1+ mice despite the presence of the JAK2V617F-bearing MKs in all of them. (Figure 4C-D).
Figure 4.

TPO/MPL signaling is required for the development of JAK2V617F MK-induced myeloproliferation. (A-B) Peripheral blood cell counts in WT control (Pf4+FF1−), Pf4+FF1+, TPO− control (TPO−Pf4+FF1−), TPO−Pf4+FF1+, MPL− control (MPL−Pf4+FF1−), and MPL−Pf4+FF1+ mice at 28 weeks of age (A) and 1 year of age (B). Data are presented as mean ±SEM; n = 6–11 mice per group. (C-D) At 28 weeks of age, marrow MKs (C) and marrow E-SLAM cells (D) were significantly increased in the Pf4+FF1+ mice compared to WT controls, but not in the TPO−Pf4+FF1+ mice or MPL−Pf4+FF1+ mice. Data are presented as mean ±SEM; n = 4–8 mice per group. (E-F) There was no correlation between Marrow MK and E-SLAM cell frequencies in TPO−Pf4+FF1+ (n = 4) (E) or MPL−Pf4+FF1+ (n = 5) mice (F). Data are presented as mean ±SEM. *P < 0.05.
In addition, in contrast to what we have seen in the Pf4+FF1+ mice (Figure 3), when we ablated TPO (TPO−Pf4+FF1+ mice) or MPL (MPL−Pf4+FF1+ mice) in the Pf4+FF1+ mice, no correlation between marrow MKs and E-SLAM cells was observed. (Figure 4E) Taken together, these data suggest that TPO/MPL signaling is required for the development of JAK2V617F MK-induced myeloproliferation and HSPC expansion phenotype we have observed in the Pf4+FF1+ mice, probably through its effects on both the quantity and quality of the niche megakaryocytes.
TPO/MPL signaling contributes to the JAK2V617F-bearing MK-induced myeloproliferation via its effect on the vascular niche function
In addition to their roles in megakaryopoiesis and HSPC activity, MPL is also expressed on several types of endothelium and TPO can stimulate angiogenesis.[40-42] Whether the EC MPL receptor can affect the vascular niche function, and contribute to the critical role of TPO/MPL signaling in HSPC maintenance, is not known.
Previously, using an in vitro competitive growth assay in which both JAK2WT and JAK2V617F HSPCs were cultured together in the presence of conditioned medium collected from WT or MPL−/− murine ECs, we found that there were significantly more JAK2WT cells than JAK2V617F cells in the presence of MPL−/− EC conditioned medium compared to in WT EC conditioned medium, suggesting that the EC MPL receptor is important for the maintenance/expansion of the JAK2V617F cells over JAK2WT cells.[43] These observations led us to hypothesize that some secreted factors from vascular ECs contribute to the JAK2V617F clonal expansion in MPNs, and that TPO/MPL signaling is critical for this vascular niche function. Therefore, we measured the expression levels of the key vascular niche factor CXCL12 [44,45] in marrow ECs isolated from WT mice, Pf4+FF1+, MPL−/− mice, and TPO−/− mice. Although there is no difference in CXCL12 expression between WT marrow ECs and Pf4+FF1+ marrow ECs, CXCL12 is significantly downregulated in TPO−/− or MPL−/− marrow ECs compared to WT marrow ECs. (Figure 5A).
Figure 5.

TPO and MPL are important for vascular niche function. (A) The expression levels of CXCL12 in marrow ECs isolated from WT ctrl, Pf4+FF1+, TPO−/−, and MPL−/− mice. Gene expression is shown as the relative fold-change compared to WT ctrl marrow EC expression which was set as ‘1’. (B) Representative images of H&E sections of femoral marrow of Pf4-cre WT ctrl mice, Pf4+FF1+ mice, TPO−Pf4+FF1+ mice and MPL−Pf4+FF1+ mice. Yellow arrows indicate examples of MKs physically associated with marrow sinusoids. Black arrows indicate examples of MKs not in direct contact with marrow sinusoids. Magnification: x20 (C) Quantification of MKs physically associated with marrow sinusoids in WT ctrl mice (n = 3), Pf4+FF1+ mice (n = 3), TPO− ctrl mice (n = 2), TPO−Pf4+FF1+ mice (n = 2), MPL− ctrl mice (n = 2), and MPL−Pf4+FF1+ mice (n = 2). About 100 marrow MKs from 9–18 non-overlapping fields were studied. (D) TPO did not affect primary murine spleen EC cell growth in vitro. (E) TPO stimulated primary murine spleen EC cell migration in vitro. A lesion was produced across the primary murine spleen EC monolayer in the presence of different TPO concentrations. The distances from one side of the scratch wound to the other side were measured using ImageJ software (National Institute of Health, Bethesda, MD, USA) at six different locations for each culture condition. The distance of wound closure at time 24, 48, and 72 h was compared with the distance at time 0 h which was set as 1. The results were expressed as the mean ±s.e.m. (n = 6). Data are from one of two independent experiments that gave similar results. (F) The expression levels of VE-cadherin, ZO1, and PECAM1 in WT murine lung EC treated with different TPO concentrations. Data are presented as mean ±SEM. *P < 0.05.
CXCL12 is important in directing MK migration toward the vascular niche and promoting MK maturation and platelet release. [46,47] The decreased CXCL12 expression in TPO-ablated and MPL-ablated marrow ECs could impair the interactions between MKs and ECs in the vascular niche and contribute to the phenotype we have observed in the TPO−Pf4+FF1+ and MPL−Pf4+FF1+ mice (Figure 3). Indeed, histological examination of the marrow revealed that in TPO−Pf4+FF1+ and MPL−Pf4+FF1+ mice, not only there was a paucity of MKs compared to the Pf4+FF1+ mice, but also the MKs were less likely to be in direct contact with the sinusoidal vessels compared to MKs in the Pf4+FF1+ mice, even when controlled for MK number. (Figure 5B-C).
Recently, it has been reported that marrow vascular barrier integrity plays an important role in HSPC regulation. A leaky endothelium promotes HSPC differentiation, while less permeable blood vessels provide a microenvironment promoting HSPC quiescence and maintenance.[24] Although we did not detect any significant effect of TPO on EC cell proliferation in vitro as assayed by direct cell count, TPO significantly stimulated EC cell migration in a scratch assay (as a measure of in vitro EC migration). (Figure 5D-E) In addition, endothelial junction molecules ZO-124 and PECAM1[48] are both upregulated in TPO-treated ECs compared to untreated ECs. (Figure 5F) These observations suggest that the TPO/MPL signaling may also regulate vascular integrity to promote HSPC quiescence and maintenance.
Taken together, results from our studies suggest that the TPO/MPL signaling has important role(s) in the hematopoietic vascular niche and can contribute to the myeloproliferation and HSPC expansion in the Pf4+FF1+ mice by regulating MK-endothelial interactions in the vascular niche.
DISCUSSION
In contrast to non-hematopoietic niche cells (e.g. endothelial cells, mesenchymal stromal cells, CXCL12-abundant reticular (CAR) cells), niche MKs provide direct feedback to their precursor HSPCs, many of which are located adjacent to MKs in vivo,[9,10] and therefore play important roles in malignant HSPC clone expansion during neoplastic hematopoiesis. Recently, we reported that the JAK2V617F-bearing MKs cause a murine myeloproliferative syndrome with increased marrow angiogenesis and HSPC expansion.[49] In this study, we explored the physiological effects and molecular mechanism(s) by which the JAK2V617F-bearing MKs promote HSPC function.
TPO receptor MPL is associated with both HSPC quiescence and HSPC repopulating activity.[33,34] Consistent with the increased MPL expression on E-SLAM cells from the Pf4+FF1+ mice compared to WT controls, we have found that the CD150+CD48− HSPCs from the Pf4+FF1+ mice (with JAK2V617F-bearing MKs) are more quiescent than those from the WT control mice (with JAK2WT MKs). In addition, using a competitive repopulation assay, we confirmed that marrow cells from the Pf4+FF1+ mice have higher engraftment capacity than cells from WT control mice. Since quiescence is required for long-term HSPC function and expansion,[38,50] maintenance of HSPC quiescence would be important in MPNs to prevent HSPC exhaustion and contribute to eventual HSPC expansion in these disorders.
In our study, a strong correlation between marrow MK cell numbers and E-SLAM cell numbers was observed in the JAK2V617F-bearing Pf4+FF1+ mice but not in the WT control mice. (Figure 3) In the competitive repopulation assay where both JAK2V617F marrow cells and JAK2WT marrow cells were transplanted together, while there was no significant difference in the total CD41+ MK cell numbers or E-SLAM cell numbers between the experiment group and control group, CD45.2+ MKs and E-SLAM cells were significantly increased while CD45.1+ MKs and E-SLAM cells were significantly decreased in recipients of the Pf4+FF1+ marrow compared to recipients of control marrow. (Figure 2) Two conclusions can be drawn from these data to guide future investigation. First, these data indicate that the JAK2V617F mutation alters both MK quantity and quality to promote the expansion of HSPCs in the Pf4+FF1+ mice. Second, these observations suggest that the JAK2V617F-bearing MK niche promotes the expansion of its own precursor HSPC while suppressing wild-type hematopoiesis. Therefore, our study provides a possible mechanism for the JAK2V617F HSPC clonal expansion seen in animal models and patients with MPNs – that is, a JAK2V617F-mutant HSPC gives rise to a mutant MK niche which feeds back to expand the disease clone itself, probably through certain locoregional interactions between the JAK2V617F-bearing MKs and its precursor HSPCs. Such a positive feed-back loop would provide the MPN stem cells a competitive advantage over normal stem cells. Contrary to what we observed previously in a direct transplantation experiment in which Pf4+FF1+ marrow cells were transplanted into WT recipients and thrombocytosis developed 8 weeks after transplantation15, no significant difference in blood cell count, spleen weight, or total femoral cell count was observed between the Pf4+FF1+ marrow recipients and control marrow recipients in the competitive transplantation experiment performed here. Such different phenotypes between direct19 and competitive [51] marrow transplantation were also observed in another JAK2V617F-positive MPN murine model in which the JAK2V617F mutation is expressed in all hematopoietic cells (including HSPCs) and ECs. Considering that competition between normal cells and cancer cells is a major factor for cancer initiation,[52] the difference in Pf4+FF1+ marrow donor chimerism (i.e. ~60% in the competitive transplant assay reported here vs. ~95% in the direct transplant assay15) could contribute to the different phenotype we have observed. These observations further indicate that the microenvironment plays important roles in MPN clonal expansion and disease development.
We examined the molecular mechanism(s) by which the JAK2V617F-bearing MK niche enhances HSPC function. We focused on TPO and its receptor MPL which are key regulators of both megakaryopoiesis and HSPC activity. To determine the role(s) of TPO and MPL in the development of JAK2V617F MK-induced myeloproliferation in vivo, we ablated TPO or MPL in the Pf4+FF1+ mice. We found that, in contrast to the Pf4+FF1+ mice which developed thrombocytosis and a substantial expansion of MKs and HSPCs compared to age-matched WT controls, TPO−Pf4+FF1+ mice and MPL−Pf4+FF1+ mice did not display any of these features.[35] (Figure 4) These data indicate that the TPO/MPL signaling is critical for the enhanced JAK2V617F MK niche function seen in the (TPO and MPL) wild type animals. The finding that platelet counts are higher in the TPO−Pf4+FF1+ mice compared to TPO−/− mice suggests that the JAK2V617F-bearing MKs promote thrombocytosis through some TPO-independent pathway, although it is likely that the mutant JAK2 signaling is enhanced with the presence of TPO compared to without TPO. In contrast, there was no difference in blood counts between MPL−Pf4+FF1+ mice and MPL−/− mice, indicating that MPL is required for the JAK2V617F-bearing MK niche-induced myeloproliferation. These results are consistent with recent reports that MPL is essential for the development of MPNs,[53-57] likely by acting as the cell surface anchor of the mutant kinase.
In addition to its roles in megakaryopoiesis and HSPC function, MPL is also expressed on several types of ECs and TPO can affect EC angiogenesis.[40-42,58] Results from our previous studies and current work provide the first evidence that TPO and MPL have important role(s) in the vascular niche function and altered TPO/MPL signaling can contribute to JAK2V617F-bearing MK-induced HSPC expansion via its effect on the vascular niche function. First, we demonstrated that the EC MPL receptor is important for the maintenance/expansion of the JAK2V617F clone over the JAK2WT clone in vitro. [43] Second, we showed that TPO and MPL are important for the expression of key vascular niche factor CXCL12 in marrow ECs in vivo. Third, considering that most HSPCs reside close to a marrow sinusoid, the interactions between MKs and the vascular ECs in the hematopoietic vascular niche are positioned to play an important role in modulating HSPC function, and by extrapolation, might be deregulated in disease states. We found that the JAK2V617F-bearing MKs are less likely to be in direct contact with the sinusoidal vessels in the TPO-ablated TPO−Pf4+FF1+ mice or MPL-ablated MPL−Pf4+FF1+ mice compared to the Pf4+FF1+ mice, suggesting that TPO/MPL signaling can affect the MK-endothelial interactions in the vascular niche. Fourth, TPO stimulates EC migratory function in vitro in a dose-dependent fashion and endothelial junction molecules ZO-1 and PECAM1 are both upregulated in TPO-treated ECs compared to untreated ECs, suggesting that the TPO/MPL signaling may also regulate vascular integrity to regulate HSPC activity.[24] Although data from our studies demonstrated that the TPO/MPL signaling can affect marrow EC CXCL12 expression and MK-endothelial interactions in the vascular niche, we did not detect any difference in CXCL12 expression between WT marrow ECs and Pf4+FF1+ marrow ECs. In addition, our previous study did not reveal any difference in CXCL12 or CXCR4 expression between JAK2WT MKs (from WT control mice) and JAK2V617F MKs (from Pf4+FF1+ mice).[15] Therefore, further investigation will be required to understand how the JAK2V617F mutation alters MK-endothelial interactions in the vascular niche in Pf4+FF1+ mice.
In summary, our study demonstrates that JAK2V617F-bearing MKs are an important part of the hematopoietic niche in MPNs and can promote both MPN stem cell expansion and MPN stem cell function. In addition, we showed that the TPO/MPL signaling is critical for the JAK2V617F-bearing MK-induced myeloproliferation, both by directly affecting the quantity and quality of MKs and by altering the MK-endothelial interaction and vascular niche function. Therefore, targeting the JAK2V617F-bearing MK niche and/or the altered TPO/MPL signaling could provide more effective therapeutic strategies for patients with MPNs.
ACKNOWLEDGEMENTS
The authors thank Todd Rueb and Rebecca Connor (Flow Cytometry Core Facility, Stony Brook University, NY, USA) for their assistance with the flow cytometry experiments. We would also like to thank Dr. Yupo Ma (Stony Brook University, NY) for his continuous support throughout this work. Y.Z. is supported by the State Scholarship Fund from Chinese Scholarship Council. This research was supported by the Veterans Affairs Career Development Award IK2BX001559 (H.Z.) and National Heart, Lung, and Blood Institute grant R01 HL134970 (H.Z.).
Footnotes
Publisher's Disclaimer: This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process which may lead to differences between this version and the Version of Record. Please cite this article as doi:10.1002/stem.2888
CONFLICT OF INTERST
The authors declare no conflict of interest.
CONFLICT-OF-INTEREST DISCLOSURE
The authors declare no competing financial interests.
REFERENCES
- 1.Walkley CR, Olsen GH, Dworkin S et al. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell 2007;129(6):1097–1110. 10.1016/j.cell.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schepers K, Pietras EM, Reynaud D et al. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell 2013;13(3):285–299. 10.1016/j.stem.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arranz L, Sanchez-Aguilera A, Martin-Perez D et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature 2014;512(7512):78–81. 10.1038/nature13383. [DOI] [PubMed] [Google Scholar]
- 4.Mager LF, Riether C, Schurch CM et al. IL-33 signaling contributes to the pathogenesis of myeloproliferative neoplasms. The Journal of clinical investigation 2015;125(7):2579–2591. 10.1172/JCI77347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walkley CR, Shea JM, Sims NA et al. Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell 2007;129(6):1081–1095. 10.1016/j.cell.2007.03.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature 2014;505(7483):327–334. 10.1038/nature12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Junt T, Schulze H, Chen Z et al. Dynamic visualization of thrombopoiesis within bone marrow. Science 2007;317(5845):1767–1770. 10.1126/science.1146304. [DOI] [PubMed] [Google Scholar]
- 8.Heazlewood SY, Neaves RJ, Williams B et al. Megakaryocytes co-localise with hemopoietic stem cells and release cytokines that up-regulate stem cell proliferation. Stem Cell Res 2013;11(2):782–792. 10.1016/j.scr.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 9.Zhao M, Perry JM, Marshall H et al. Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat Med 2014;20(11):1321–1326. 10.1038/nm.3706. [DOI] [PubMed] [Google Scholar]
- 10.Bruns I, Lucas D, Pinho S et al. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat Med 2014;20(11):1315–1320. 10.1038/nm.3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakamura-Ishizu A, Takubo K, Kobayashi H et al. CLEC-2 in megakaryocytes is critical for maintenance of hematopoietic stem cells in the bone marrow. The Journal of experimental medicine 2015;212(12):2133–2146. 10.1084/jem.20150057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakamura-Ishizu A, Takubo K, Fujioka M et al. Megakaryocytes are essential for HSC quiescence through the production of thrombopoietin. Biochemical and biophysical research communications 2014;454(2):353–357. 10.1016/j.bbrc.2014.10.095. [DOI] [PubMed] [Google Scholar]
- 13.Malara A, Currao M, Gruppi C et al. Megakaryocytes contribute to the bone marrow-matrix environment by expressing fibronectin, type IV collagen, and laminin. Stem Cells 2014;32(4):926–937. 10.1002/stem.1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao M, Ross JT, Itkin T et al. FGF signaling facilitates postinjury recovery of mouse hematopoietic system. Blood 2012;120(9):1831–1842. 10.1182/blood-2011-11-393991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhan H, Ma Y, Lin CH et al. JAK2(V617F)-mutant megakaryocytes contribute to hematopoietic stem/progenitor cell expansion in a model of murine myeloproliferation. Leukemia 2016;30(12):2332–2341. 10.1038/leu.2016.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ciurea SO, Merchant D, Mahmud N et al. Pivotal contributions of megakaryocytes to the biology of idiopathic myelofibrosis. Blood 2007;110(3):986–993. 10.1182/blood-2006-12-064626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tiedt R, Hao-Shen H, Sobas MA et al. Ratio of mutant JAK2-V617F to wild-type Jak2 determines the MPD phenotypes in transgenic mice. Blood 2008;111(8):3931–3940. 10.1182/blood-2007-08-107748. [DOI] [PubMed] [Google Scholar]
- 18.Tiedt R, Schomber T, Hao-Shen H et al. Pf4-Cre transgenic mice allow the generation of lineage-restricted gene knockouts for studying megakaryocyte and platelet function in vivo. Blood 2007;109(4):1503–1506. 10.1182/blood-2006-04-020362. [DOI] [PubMed] [Google Scholar]
- 19.Etheridge SL, Roh ME, Cosgrove ME et al. JAK2V617F-positive endothelial cells contribute to clotting abnormalities in myeloproliferative neoplasms. Proc Natl Acad Sci U S A 2014;111(6):2295–2300. 10.1073/pnas.1312148111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alexander WS, Roberts AW, Nicola NA et al. Deficiencies in progenitor cells of multiple hematopoietic lineages and defective megakaryocytopoiesis in mice lacking the thrombopoietic receptor c-Mpl. Blood 1996;87(6):2162–2170. [PubMed] [Google Scholar]
- 21.de Sauvage FJ, Carver-Moore K, Luoh SM et al. Physiological regulation of early and late stages of megakaryocytopoiesis by thrombopoietin. J Exp Med 1996;183(2):651–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kiel MJ, Yilmaz OH, Iwashita T et al. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 2005;121(7):1109–1121. 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 23.Kent DG, Copley MR, Benz C et al. Prospective isolation and molecular characterization of hematopoietic stem cells with durable self-renewal potential. Blood 2009;113(25):6342–6350. 10.1182/blood-2008-12-192054. [DOI] [PubMed] [Google Scholar]
- 24.Itkin T, Gur-Cohen S, Spencer JA et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature 2016;532(7599):323–328. 10.1038/nature17624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shapiro HM. Flow cytometric estimation of DNA and RNA content in intact cells stained with Hoechst 33342 and pyronin Y. Cytometry 1981;2(3):143–150. 10.1002/cyto.990020302. [DOI] [PubMed] [Google Scholar]
- 26.Kopp HG, Avecilla ST, Hooper AT et al. The bone marrow vascular niche: home of HSC differentiation and mobilization. Physiology 2005;20:349–356. 10.1152/physiol.00025.2005. [DOI] [PubMed] [Google Scholar]
- 27.Zhan H, Ma Y, Lin CH et al. JAK2V617F-mutant megakaryocytes contribute to hematopoietic stem/progenitor cell expansion in a model of murine myeloproliferation. Leukemia 2016;30(12):2332–2341. 10.1038/leu.2016.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lefrancais E, Ortiz-Munoz G, Caudrillier A et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 2017;544(7648):105–109. 10.1038/nature21706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hitchcock IS, Fox NE, Prevost N et al. Roles of focal adhesion kinase (FAK) in megakaryopoiesis and platelet function: studies using a megakaryocyte lineage specific FAK knockout. Blood 2008;111(2):596–604. 10.1182/blood-2007-05-089680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ng AP, Kauppi M, Metcalf D et al. Mpl expression on megakaryocytes and platelets is dispensable for thrombopoiesis but essential to prevent myeloproliferation. Proc Natl Acad Sci U S A 2014;111(16):5884–5889. 10.1073/pnas.1404354111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chagraoui H, Kassouf M, Banerjee S et al. SCL-mediated regulation of the cell-cycle regulator p21 is critical for murine megakaryopoiesis. Blood 2011;118(3):723–735. 10.1182/blood-2011-01-328765. [DOI] [PubMed] [Google Scholar]
- 32.Calaminus SD, Guitart AV, Sinclair A et al. Lineage tracing of Pf4-Cre marks hematopoietic stem cells and their progeny. PloS one 2012;7(12):e51361 10.1371/journal.pone.0051361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Solar GP, Kerr WG, Zeigler FC et al. Role of c-mpl in early hematopoiesis. Blood 1998;92(1):4–10. [PubMed] [Google Scholar]
- 34.Yoshihara H, Arai F, Hosokawa K et al. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell Stem Cell 2007;1(6):685–697. 10.1016/j.stem.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 35.Zhang Y LC, Kaushansky K, Zhan H. The JAK2V617F-bearing Megakaryocytes Promote Hematopoietic Stem/Progenitor Cell [DOI] [PMC free article] [PubMed]
- 36.Expansion through the Thrombopoietin/MPL signaling. Blood 2017;130(Suppl. 1). [Google Scholar]
- 37.Fox N, Priestley G, Papayannopoulou T et al. Thrombopoietin expands hematopoietic stem cells after transplantation. J Clin Invest 2002;110(3):389–394. 10.1172/JCI15430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Qian H, Buza-Vidas N, Hyland CD et al. Critical role of thrombopoietin in maintaining adult quiescent hematopoietic stem cells. Cell Stem Cell 2007;1(6):671–684. 10.1016/j.stem.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 39.Kaushansky K, Lok S, Holly RD et al. Promotion of megakaryocyte progenitor expansion and differentiation by the c-Mpl ligand thrombopoietin. Nature 1994;369(6481):568–571. 10.1038/369568a0. [DOI] [PubMed] [Google Scholar]
- 40.Cardier JE, Dempsey J. Thrombopoietin and its receptor, c-mpl, are constitutively expressed by mouse liver endothelial cells: evidence of thrombopoietin as a growth factor for liver endothelial cells. Blood 1998;91(3):923–929. [PubMed] [Google Scholar]
- 41.Brizzi MF, Battaglia E, Montrucchio G et al. Thrombopoietin stimulates endothelial cell motility and neoangiogenesis by a platelet-activating factor-dependent mechanism. Circulation research 1999;84(7):785–796. [DOI] [PubMed] [Google Scholar]
- 42.Eguchi M, Masuda H, Kwon S, Shirakura K, Shizuno T, Ito R et al. Lesion-targeted thrombopoietin potentiates vasculo-genesis by enhancing motility and enlivenment of transplanted endothelial progenitor cells via activation of Akt/mTOR/p70S6kinase signaling pathway. J Mol Cell Cardiol 2008;45(5):661–669. doi: 10.1016/j.yjmcc.2008.08.002 [DOI] [PubMed] [Google Scholar]
- 43.Lin CH, Kaushansky K, Zhan H. JAK2V617F-mutant vascular niche contributes to JAK2V617F clonal expansion in myeloproliferative neoplasms. Blood Cells Mol Dis 2016;62:42–48. 10.1016/j.bcmd.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 2013;495(7440):231–235. 10.1038/nature11885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Greenbaum A, Hsu YM, Day RB et al. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 2013;495(7440):227–230. 10.1038/nature11926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Niswander LM, Fegan KH, Kingsley PD et al. SDF-1 dynamically mediates megakaryocyte niche occupancy and thrombopoiesis at steady state and following radiation injury. Blood 2014;124(2):277–286. 10.1182/blood-2014-01-547638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Avecilla ST, Hattori K, Heissig B et al. Chemokine-mediated interaction of hematopoietic progenitors with the bone marrow vascular niche is required for thrombopoiesis. Nat Med 2004;10(1):64–71. 10.1038/nm973. [DOI] [PubMed] [Google Scholar]
- 48.Privratsky JR, Newman PJ. PECAM-1: regulator of endothelial junctional integrity. Cell Tissue Res 2014;355(3):607–619. 10.1007/s00441-013-1779-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhan H, Ma Y, Lin CH et al. JAK2V617F-mutant megakaryocytes contribute to hematopoietic stem/progenitor cell expansion in a model of murine myeloproliferation. Leukemia 2016;30:2332–2341. 10.1038/leu.2016.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Passegue E, Wagers AJ, Giuriato S et al. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. The Journal of experimental medicine 2005;202(11):1599–1611. 10.1084/jem.20050967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhan H, Lin CHS, Segal Y et al. The JAK2V617F-bearing vascular niche promotes clonal expansion in myeloproliferative neoplasms. Leukemia 2017;32:462–469. 10.1038/leu.2017.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wagstaff L, Kolahgar G, Piddini E. Competitive cell interactions in cancer: a cellular tug of war. Trends Cell Biol 2013; 23(4):160–167. e-pub ahead of print 2012/12/12; doi: 10.1016/j.tcb.2012.11.002 [DOI] [PubMed] [Google Scholar]
- 53.Sangkhae V, Etheridge SL, Kaushansky K et al. The thrombopoietin receptor, MPL, is critical for development of a JAK2V617F-induced myeloproliferative neoplasm. Blood 2014;124(26):3956–3963. 10.1182/blood-2014-07-587238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Araki M, Yang Y, Masubuchi N et al. Activation of the thrombopoietin receptor by mutant calreticulin in CALR-mutant myeloproliferative neoplasms. Blood 2016;127(10):1307–1316. 10.1182/blood-2015-09-671172. [DOI] [PubMed] [Google Scholar]
- 55.Chachoua I, Pecquet C, El-Khoury M et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood 2016;127(10):1325–1335. 10.1182/blood-2015-11-681932. [DOI] [PubMed] [Google Scholar]
- 56.Elf S, Abdelfattah NS, Chen E et al. Mutant Calreticulin Requires Both Its Mutant C-terminus and the Thrombopoietin Receptor for Oncogenic Transformation. Cancer Discov 2016;6(4):368–381. 10.1158/2159-8290.CD-15-1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marty C, Pecquet C, Nivarthi H et al. Calreticulin mutants in mice induce an MPL-dependent thrombocytosis with frequent progression to myelofibrosis. Blood 2016;127(10):1317–1324. 10.1182/blood-2015-11-679571. [DOI] [PubMed] [Google Scholar]
- 58.Jin DK, Shido K, Kopp HG et al. Cytokine-mediated deployment of SDF-1 induces revascularization through recruitment of CXCR4+ hemangiocytes. Nat Med 2006;12(5):557–567. 10.1038/nm1400. [DOI] [PMC free article] [PubMed] [Google Scholar]