

Abstract
While adaptive immune responses against hepatitis C virus (HCV) infection have been studied in great detail, the role of innate immunity in protection against HCV infection and immune evasion is only partially understood. Interferon-induced transmembrane proteins (IFITMs) are innate effector proteins restricting host cell entry of many enveloped viruses, including HCV. However, the clinical impact of IFITMs on HCV immune escape remains to be determined. Here, we show that IFITMs promote viral escape from the neutralizing antibody response in clinical cohorts of HCV-infected patients. Using pseudoparticles bearing HCV envelope proteins from acutely infected patients, we show that HCV variants isolated pre-seroconversion are more sensitive to the antiviral activity of IFITMs than variants from patients isolated during chronic infection post-seroconversion. Furthermore, HCV variants escaping neutralizing antibody responses during liver transplantation exhibited a significantly higher resistance to IFITMs than variants that were eliminated post-transplantation. Gain-of-function and mechanistic studies revealed that IFITMs markedly enhance the antiviral activity of neutralizing antibodies and suggest a cooperative effect of human monoclonal antibodies and IFITMs for antibody-mediated neutralization driving the selection pressure in viral evasion. Perturbation studies with the IFITM antagonist amphotericin B revealed that modulation of membrane properties by IFITM proteins is responsible for the IFITM-mediated blockade of viral entry and enhancement of antibody-mediated neutralization.
Conclusion:
Our results identify IFITM proteins as a previously unknown driver of viral immune escape and antibody-mediated HCV neutralization in acute and chronic HCV infection. These findings are of clinical relevance for the design of urgently needed HCV B cell vaccines and might help to increase the efficacy of future vaccine candidates.
Keywords: neutralizing antibody, cell entry, HCV, escape, vaccine
Introduction
It is estimated that more than 71 million patients are chronically infected with hepatitis C virus (HCV) (1). HCV infection is a leading cause of liver disease and cancer worldwide. The development of direct acting antivirals markedly improved the outcome of antiviral treatment with cure of the majority of treated patients (2). However, several challenges remain (3). High treatment costs prevent or limit access of patients to therapy in resource-poor countries and may lead to selective use even in industrialized countries. Moreover, in the majority of cases HCV infection remains undiagnosed or is diagnosed at a late stage due to the limited efficacy of current HCV screening programs. Furthermore, direct acting antivirals will not cure virus-induced end-stage liver disease such as hepatocellular carcinoma and certain patient subgroups do not respond to or cannot tolerate direct acting antiviral-based treatment strategies (4, 5). Finally, reinfection remains possible, making control of HCV infection difficult in people at risk, such as drug abusers. These unmet medical needs warrant the development of an effective vaccine, protecting from chronic HCV infection as a means to impact the epidemic on a global scale (3).
Both cellular and humoral immune responses have been suggested to play a key role in protection against infection in humans and nonhuman primates. Thus, vaccine development has focused on eliciting both B and T cell responses (3). Indeed, a B cell vaccine consisting of recombinant E1E2 viral envelope glycoprotein was shown to provide partial protection against chronic HCV infection (6), to induce virus neutralizing antibodies and to be safe in healthy volunteers (7). Furthermore, broadly virus neutralizing antibodies have been shown to confer protection against HCV in humanized mouse models (8, 9) and are considered a promising strategy to fight emerging infectious diseases (10). While adaptive immune responses have been studied in great detail, the role of innate immune responses in HCV infection is only partially understood.
The innate immune response constitutes the first line of defense against viral infections. Interferons stimulate the expression of a set of more than 300 interferon-stimulated genes, several of which have been shown to exert antiviral activity against HCV (11). A family of these genes, the interferon-induced transmembrane (IFITM) proteins are potent inhibitors of host cell entry of a broad range of enveloped viruses, including HCV (12–15). While IFITM1 is primarily located at the cytoplasmic membrane and restricts HCV entry by interacting with the HCV co-receptor CD81 (13), IFITM2 and 3 localize to endosomal compartments and potentially restrict viral infection by blocking virus entry at the stage of hemifusion (16) or fusion pore formation (17).
While the antiviral activity of the IFITM proteins against HCV has been studied in cell culture models (13, 14, 18), the role of the IFITM proteins in viral pathogenesis during clinical HCV infection is unknown. It is unclear whether inhibition of virus entry by IFITM proteins contributes to viral clearance, whether IFITM-HCV interactions impact viral persistence in chronic infection and whether IFITM proteins and antibodies cooperate to inhibit viral entry.
Clinical cohorts for the study of acute and chronic HCV infection have been a valuable tool to investigate the mechanisms of HCV persistence and escape. These include cohorts comparing early and late stage infection (19, 20). Furthermore, liver graft infection is a unique model since it allows the study of HCV infection and viral escape in a very well defined timeframe and detailed patient material (21–23).
To address the clinical role of IFITMs for viral escape and B cell responses, we investigated virus-host interactions of IFITM proteins and neutralizing antibodies during HCV cell entry. For this, we used HCV pseudoparticles (HCVpp) bearing envelopes from patients with acute infection prior to seroconversion or patients undergoing liver transplantation due to chronic hepatitis C. Moreover, we employed neutralizing human monoclonal antibodies (HMAbs) derived from patients with chronic HCV infection.
Material and Methods
Human material.
Human material, including sera and liver tissues from patients undergoing surgical resection for isolation of human hepatocytes and followed at Strasbourg University Hospital, was obtained with informed consent from all patients. The protocol was approved by the Ethics Committee of Strasbourg University Hospital (CPP 10–17).
Cell lines and primary human hepatocytes.
293T cells, Huh7.5.1, Huh7.5.1-NTCP and HepG2-CD81 cells were isolated and cultured as described previously (18, 19, 24). Primary human hepatocytes (PHH) were isolated from liver resections as described previously (25).
Antibodies.
The anti-E2 HMAbs (CBH-20, CBH-7, CBH-22, HC84.26.WH.5DL) and human anti-HCV sera have been described previously (19, 24, 26–29). The antibodies directed against IFITM1, IFITM2/3 (Proteintech) and β-actin (Sigma) and the protocols for detection of IFITM proteins by western blot and immune fluorescence have been described in (30).
Plasmids.
The plasmids for the generation of HCVpp and cell culture derived HCV (HCVcc) (Jc1 (genotype 2a) chimera Luc-Jc1 and Con1 (genotype 1b) chimera Con1R2A) have been described in (21), The plasmids coding for the envelope proteins and HCVcc chimera bearing envelope proteins isolated from patients undergoing liver transplantation were described in (21). The plasmids encoding the envelope variants of acute patients have been described in (31) (accession numbers KU285163, KU285164 and KU285165). Plasmid UKN1a.16.16 (accession number MK124622) was generated as described in (32). The plasmids encoding the envelope proteins of the chronic variants are described in (32) (UKN1A14.38 and UKN3A1.28.), (33) (HCV-J) and in (34) (gt3SXB1). The full-length chimeric clone incorporating the UKN1.5.3 E1E2 genes was generated in the H77/JFH-1 virus background, using previously described methods (35).
Statistics.
Data are shown as mean ± SEM if n ≥3. Representative experiments are shown as mean ± SD. Normality was assessed using the Shapiro-Wilk test. The 1-tailed Student’s t test was used for single comparisons. A p-value of less than 0.05 was considered statistically significant.
Vector production, transduction and selection of stable cells.
Retroviral vectors for transduction were generated by transfection of 293T cells as described previously (30) using the CMV-Gag-Pol MLV (mouse leukemia virus) packaging construct, a vesicular stomatitis virus-G-encoding plasmid and plasmids coding for the IFITM proteins (pQCXIP) or empty vector as a control (30). For transduction, cells were seeded at subconfluent density and spin-inoculated with the retroviral vectors at 4000 x g for 30 min. Cells were then incubated at 37°C for 48 hours to allow efficient transgene expression. Cells stably expressing IFITM proteins were subsequently selected with 1.8 µg/mL puromycin.
HCVpp production, infection, and neutralization.
HCVpp were generated by transfection of 293T cells as described previously (19), To study HCV entry, HCVpp were added to IFITM-transduced Huh7.5.1, Huh7.5.1-NTCP cells or PHH in triplicate and incubated for 72 h at 37°C. HCV entry was determined by analysis of luciferase reporter gene expression as described previously (24). For the study of antibody-mediated neutralization, HCVpp were mixed with autologous anti-HCV serum, control serum, anti-E2 HMAbs or irrelevant isotype control IgG, preincubated for 30 min at 37°C and added to Huh7.5.1, Huh7.5.1-NTCP cells or PHH in triplicate for 72 h at 37°C (21, 24). To assess the effect of amphotericin B on the cooperative inhibition of HCV entry by IFITM proteins and neutralizing antibodies, Huh7.5.1 cells were treated with 5 µmol/L amphotericin B (Sigma Aldrich) for 1 hour at 37 °C prior to infection with antibody-treated HCVpps.
HCVcc production, infection, and neutralization.
Plasmids for cell culture-derived HCV (HCVcc) production of Jc1 and Con1 chimera with luciferase reporter (Luc-Jc1 and Con1-R2A) have been described previously (22, 36–39). HCVcc were produced in Huh7.5.1 cells as described previously (39). Infectivity was quantified by luciferase activity, or by determining the tissue culture infectious dose 50% (TCID50) (22). HCVcc neutralization using patient serum, IgG, and mAbs was analyzed as described previously (22).
Results
IFITM proteins inhibit cell entry of HCVpp and HCVcc.
To characterize the role of IFITMs in clinical HCV infection, we first investigated inhibition of viral entry into cells. For this, we transduced Huh7.5.1 or Huh7.5.1-NTCP cells with retroviral vectors encoding the antivirally active IFITM proteins (IFITM1, IFITM2 and IFITM3) and then infected the cells with HCVpp bearing the envelope proteins of HCV genotype 1b. Huh7.5.1-NTCP cells were used since NTCP has been described to have a functional role in regulation of interferon stimulated gene expression (18). Since no differences in IFITM antiviral activity on HCV entry and infection were observed between Huh7.5.1 and Huh7.5.1-NTCP cells, when IFITMs were exogenously expressed, Huh7.5.1 cells were then used for all subsequent experiments. Entry of HCVpp was restricted by all three IFITMs, with IFITM2 and 3 showing a slightly higher restriction than IFITM1 (Figure 1A). Pseudoparticles bearing the envelope protein of the IFITM resistant retrovirus MLV (40) were used as negative control (Figure 1B). Entry driven by the MLV-envelope protein was not modulated by IFITM proteins, as expected. To analyze the impact of cell polarization, which might affect IFITM activity due an altered subcellular localization of IFITM1 in hepatocytes, as reported by Wilkins et al (13), we studied the effect of IFITMs on HCVpp entry in polarized HepG2-CD81 cells in side-by-side experiments. The inhibition pattern observed upon IFITM expression was very similar to that seen for nonpolarized Huh7.5.1 cells (Figure 1C and Figure S1), suggesting that polarization appears not to modulate the ability of IFITM proteins to block HCV entry. This is in line with the finding that IFITM-expression did not alter CD81 surface expression (Figure S2) or distribution (Figure S3). We next assessed the effect of IFITM proteins on HCV entry in the context of authentic virus using infectious HCVcc. The sensitivity of HCVcc infection to inhibition by IFITM proteins was assessed in Huh7.5.1 cells stably expressing IFITM1, 2 or 3. Similar to results observed for HCVpp, infection of Huh7.5.1 cells by HCVcc was inhibited by all three IFITMs (Figure 1D). Expression of IFITM proteins was confirmed by immunoblot (Figure 1E).
Figure 1. Directed IFITM expression inhibits HCV infection in cell culture.
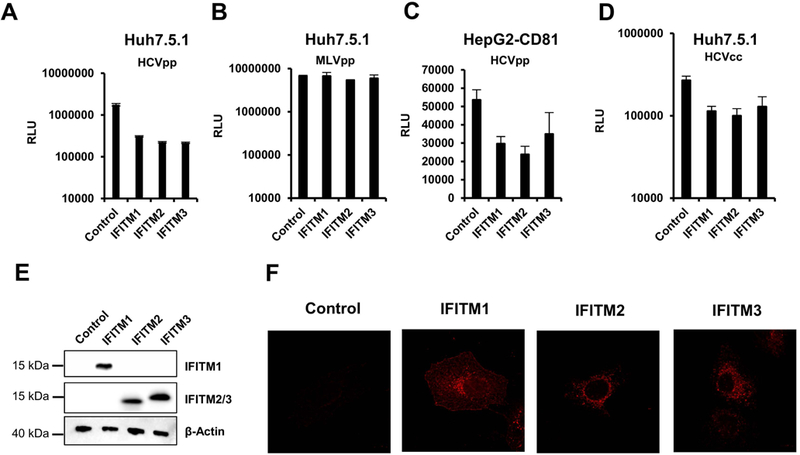
Huh7.5.1 (A and B) or HepG2-CD81 (C) cells were transduced with vectors encoding the indicated IFITM proteins or were transduced with empty vector as control. Transduced cells were then infected with HCVpp of GT1b (A), GT1a (C) or with MLVpp (B). Infection was assessed after 72 h by measuring luciferase activity. Individual representative experiments are shown. Error bars represent SD. Similar results were obtained in more than three independent experiments. (D) Huh7.5.1 cells stably expressing IFITMs were infected with HCVcc Luc-Jc1. Infection efficiency was assessed after 72 h by measuring luciferase activity. Shown is a representative of three independent experiments, error bars indicate SD. (E) Expression of IFITM proteins in Huh7.5.1 cells was assessed by immunoblot using IFITM-specific antibodies. β-Actin is used as loading control. One representative western blot is shown (F). Huh7.5.1 cells transduced with IFITM1–3 or a control vector were stained with a mouse anti-IFITM1 or a rabbit anti-IFITM2 antibody (IFITM2 and 3).
Finally, we studied the subcellular localization of IFTMs in Huh7.5.1 cells. Immunohistochemistry studies (Figure 1F, Figure S3) showed that IFITM1 was located at the plasma membrane, as shown by colocalization with SYFP tagged with a membrane-targeting signal, while IFITM2 and 3 were found in endosomal compartments, as shown by partial colocalization with the endosomal marker Rab7a (Figure S3). These observations are similar to previous results observed in Huh7 cells (14). In summary, these results demonstrate that infection of Huh7.5.1 with HCVpp or HCVcc is a suitable model to study the molecular mechanisms of inhibition of HCV infection by IFITM proteins.
Clinical variants isolated during acute HCV infection before seroconversion were more sensitive to anti-viral activity of IFITMs than variants from chronic infection.
To understand the role of IFITMs in the acute phase of infection, we analyzed the IFITM-sensitivity of HCVpp expressing viral envelopes of three HCV variants isolated from the same patient at three different time points post infection (UKNP1.5.1 pre-seroconversion; UKNP1.5.2 acute phase, two months later; UKNP1.5.3 chronic phase, 7 months later; Table 1). These variants vary at key residues, including residues near or within the CD81 binding sites (aa312; 439; 500; 536; 626; 742) (31) (Figure 2E). As shown in Figure 2A and Figure S4, HCVpp expressing envelopes of all variants were comparably susceptible to inhibition by all three tested IFITM proteins. Interestingly, the analysis of the HCVpp bearing sequential HCVpp envelope proteins revealed a marked and significant decrease of IFITM-sensitivity over time (Figure 2A). This decrease was not due to differences in the relative infectivities of the HCVpp (Figure 4A). Thus, infection mediated by the envelope proteins of variant UKNP1.5.1, isolated before seroconversion, and variant UKNP1.5.2, isolated right after seroconversion, was inhibited by 98% and 85% respectively. In contrast, transduction driven by the envelope proteins of variant UKNP1.5.3, which was isolated six months after seroconversion during the chronic phase of infection, was inhibited by only 60% upon directed expression of IFITM proteins (Figure 2A). The results obtained for UKNP1.5.3 were confirmed using an HCVcc chimera (Figure 2B), whereas the infectivity of HCVcc derived from the two other strains was too low to obtain conclusive results. Next, we investigated whether the differential IFITM-sensitivity of HCV envelope proteins obtained pre-seroconversion and during chronic infection could be confirmed with a larger panel of samples. For this, we analyzed the envelope proteins from six different early acute HCV patients and four variants derived from chronically infected HCV patients. Among the pre-seroconversion isolates, the highest susceptibility was observed for UKNP1.3.1 with more than 98–99 % inhibition of entry upon IFITM protein expression (Figure S4). UKNP1.6.1 was the most resistant with about 85 % inhibition (Figure S4), which correlated with the sensitivity to neutralizing antibodies that was published previously (31). The neutralization sensitivity of the E1E2 proteins of this cohort was shown to be consistent in HCVpp and HCVcc models of infection (31), indicating the same holds true for their IFITM-sensitivity. When we compared the entry of HCVpp expressing pre-seroconversion envelope glycoproteins to entry of HCVpp bearing envelope glycoproteins derived from independent chronic samples of the same genotypes, we observed a significant and unexpected difference in IFITM-susceptibility. The HCVpp bearing envelopes from variants isolated from chronic infection post-seroconversion were much more resistant to inhibition by IFITM proteins (Figure 2C, 2D and Figure S4), independent of the genotype of the variants. Taken together, these results suggest that IFITMs may pose significant selective pressure on HCV during the acute phase of infection that can result in viral evasion. The identification of mutations unique to variants during chronic infection suggest their possible involvement in these interactions.
Table 1: HCV strains used in the study.
Strain name, genotype, clinical setting, phenotype and reference are shown. Abbreviations: LT – liver transplantation. N. T. not tested.
| Name | Genotype | Acute/chronic | Neutralization Escape | Citation |
|---|---|---|---|---|
| P1VA | 1b | chronic/LT | − | (22) |
| P1VC | 1b | chronic/LT | − | (22) |
| P1VI | 1b | chronic/LT | − | (22) |
| P1VK | 1b | chronic/LT | − | (22) |
| P1VL | 1b | chronic/LT | + | (22) |
| P2VA | 1b | chronic/LT | − | (22) |
| P2VD | 1b | chronic/LT | − | (22) |
| P2VH | 1b | chronic/LT | + | (22) |
| P2VI | 1b | chronic/LT | + | (22) |
| P2VJ | 1b | chronic/LT | + | (22) |
| P3VA | 1b | chronic/LT | − | (22) |
| P3VB | 1b | chronic/LT | − | (22) |
| P3VC | 1b | chronic/LT | + | (22) |
| P5VD | 1b | chronic/LT | + | (22) |
| P5VE | 1b | chronic/LT | + | (22) |
| P5VF | 1b | chronic/LT | + | (22) |
| P6VD | 1b | chronic/LT | − | (22) |
| P6VI | 1b | chronic/LT | + | (22) |
| P6VH | 1b | chronic/LT | + | (22) |
| UKNP1.3.1 | 1a | acute | N. T. | (32) |
| UNKP1.4.1 | 1a | acute | N. T. | (32) |
| UKNP1.5.1 | 1a | acute | N. T. | (32) |
| UKNP1.5.2 | 1a | acute | N. T. | (32) |
| UKNP1.5.3 | 1a | chronic | N. T. | (32) |
| UKNP1.6.1 | 1a | acute | N. A, | (32) |
| UKN1a.16.16 | 1a | acute | N. T. | This study |
| UKNP3.2.1 | 3 | acute | N. T. | (32) |
| HCV-J | 1b | chronic | N. T. | (33) |
| UKN1A14.38 | 1a | chronic | N. T. | (34) |
| UKN3A1.22 | 3a | chronic | N. T. | (34) |
| 3a SXB | 3a | chronic | N. T. | (35) |
Figure 2. Differential sensitivity of acute and chronic HCV variants to inhibition by IFITM proteins.
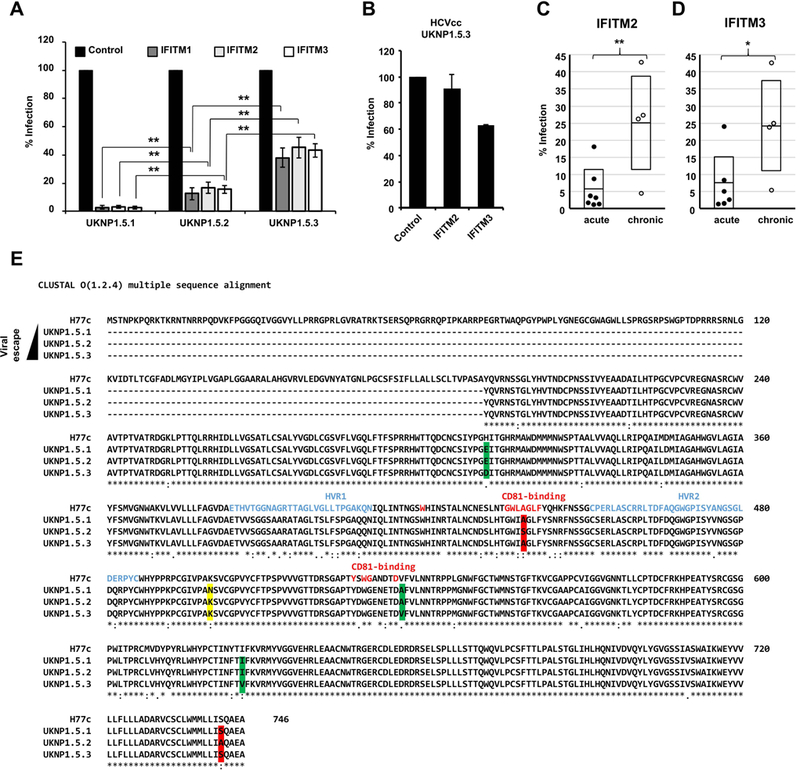
Huh7.5.1 cells were transduced to express the indicated IFITM proteins and then infected with HCVpp bearing envelope proteins derived from acute or chronic patients. (A) Analysis of the IFITM-sensitivity of sequential envelope variants (UKNP1.5.1 pre-seroconversion; UKNP1.5.2 acute phase, two months later; UKNP1.5.3 chronic phase, 7 months later), isolated from a single HCV patient. (B) TCID50 analysis of UNKP1.5.3 HCVcc infection of transduced IFITM-expressing Huh7.5.1 cells. Shown are means of two experiments performed in sextuplicates. Error bars represent SD. (C) and (D) E1E2 patient variants isolated from patients with acute or chronic HCV infection of genotype 1A and 3. Each dot represents the result for a single envelope variant and IFITM2- (C) or IFITM3-expressing (D) Huh7.5.1 cells. Shown are the means of three experiments conducted in triplicates. Error bars represent SEM. Control was set to 100%. *P < 0.05, ** P < 0.01 using one-sided students T-test. (E) Clustal O Alignment of the protein sequences of H77c, UKNP1.5.1, UKNP1.5.2 and UKNP1.5.3. The sequences were obtained from GenBank. Changes highlighted in yellow are unique for UKNP1.5.1, changes in green are only present in UKNP1.5.2 and changes highlighted in red are unique for UKNP1.5.3. HVR1 and 2 are indicated in blue, red letters mark key positions for CD81-binding.
Figure 4. The antiviral effect of the IFITMs is independent of virus infectivity.
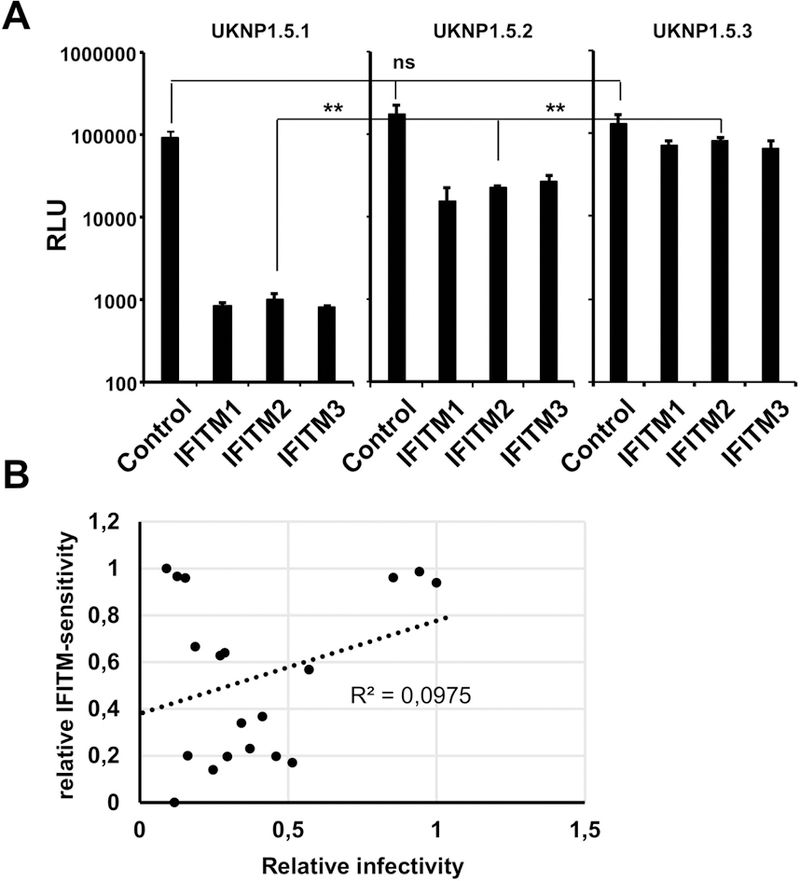
Huh7.5.1 cells were transduced to express IFITM proteins and infected with HCVpp bearing the envelope proteins of the indicated variants. (A) Shown are three individual datasets of the experiments that are featured in figure 2A. (B) Correlation of relative infectivity (highest infectivity set to 1) and relative IFITM-sensitivity (highest IFITM-sensitivity set to 1, lowest to 0). Each dot represents one of the escape variants shown in Figure 3C and 3D. Shown are the results of one representative experiment performed in triplicate (n=9).
Clinical HCV variants associated with viral immune escape during liver transplantation are more resistant to inhibition by IFITM proteins than non-escape variants.
To investigate the contribution of IFITM proteins to viral escape in chronic HCV infection, we took advantage of a well-characterized clinical cohort of patients undergoing liver transplantation with de novo infection of the liver graft (21, 22) (Table 1). In this cohort, variants selected post-transplantation are characterized by more efficient viral entry and escape from neutralizing antibodies (21, 22). We produced HCVpp bearing the full length E1/E2 proteins of variants differing in sensitivity to neutralizing antibodies and subsequently infected transiently transduced IFITM-expressing Huh7.5.1 cells. We observed that all patient-derived envelope proteins were sensitive to inhibition by IFITM proteins. However, variants that were characterized by escape from the neutralizing antibody (nAb) response were less affected by expression of IFITM proteins than those that were sensitive to neutralizing antibodies, as shown for variants derived from two different patients (Figure 3A). This was confirmed by TCID50 analyses on IFITM2 and 3 expressing cells using HCVcc chimeras expressing the envelope proteins of two representative variants (variant VL with nAb escape phenotype and variant VA with nAb sensitivity) isolated from the same patient (Figure 3B) (22). Next, we extended our analysis to 19 envelope variants (nine non-nAb escape and ten nAb escape-variants (Figure S5)) derived from five different patients and observed a significantly higher sensitivity to inhibition by IFITM2 (Figure 3C) and IFITM3 (Figure 3D) for non-nAb escape variants and a significantly higher resistance of nAb escape variants to inhibition by IFITM proteins. The direct comparison of entry efficiency and IFITM-sensitivity revealed no apparent correlation of these two variables (Figure 4B). This shows that indeed selection for IFITM-sensitivity post-liver transplantation and not just more efficient entry of the escape variants is responsible for the differential inhibition by IFITM proteins.
Figure 3. IFITMs differentially restrict HCV variants isolated from patients undergoing liver transplantation.
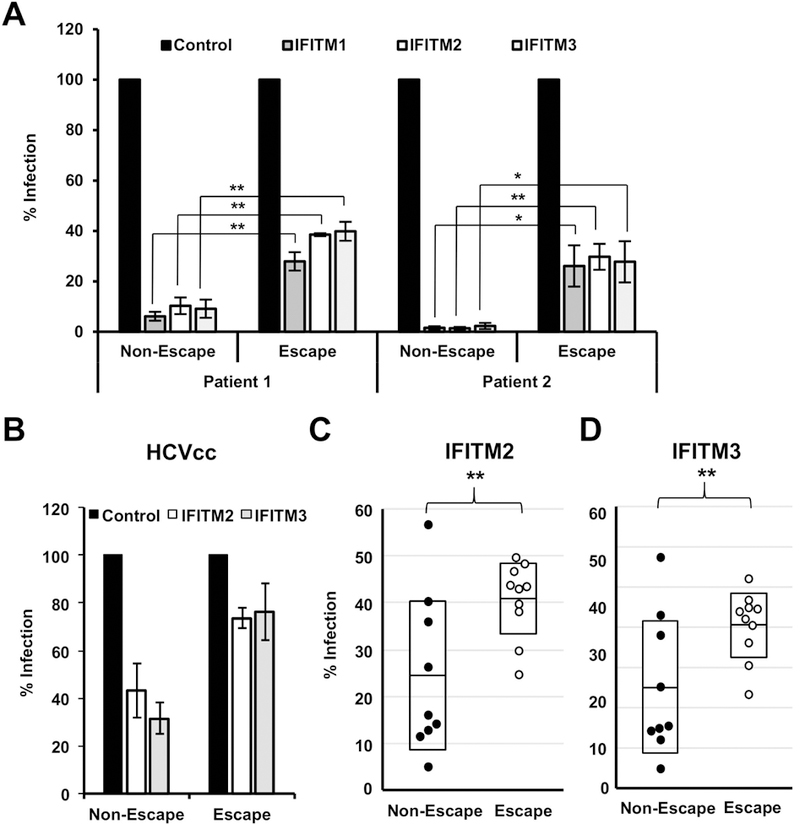
Huh7.5.1 cells were transduced to express the indicated IFITM proteins and then infected with HCVpp pseudotyped with HCV E1E2 patient variants isolated from patients undergoing liver transplantation. Infection was assessed after 72 h by measuring luciferase activity. (A) Results for variants from two patients are expressed as means ± SEM percentage HCVpp infection compared to control cells (set at 100%) from three independent experiments performed in triplicate. (B) TCID50 analysis of HCVcc infection of IFITM-expressing Huh7.5.1 cells. Shown are means of three experiments performed in sextuplicates. Error bars represent SEM. Control was set to 100 %. (C and D) Each dot represents the result for a single variant. Results for % Infection of IFITM2 (C) and IFITM3 (D) positive cells compared to the control of HCV variants from five different patients. *P < 0.05; ** P < 0.01 one-sided Students T-test. Shown are the means of three experiments performed in triplicates.
In summary, these results suggest that IFITM proteins are important determinants for viral escape and that escape from IFITM proteins is associated with resistance to antibody-mediated neutralization.
Neutralizing antibodies and IFITM proteins cooperatively block HCV entry.
The differential inhibition of antibody escape and non-escape HCV strains by IFITM proteins in chronic infection as well as the enhanced IFITM-sensitivity of viral strains in the acute phase prior to antibody development prompted us to analyze the interplay between the antiviral activities of IFITM proteins and the neutralizing B cell response in detail. For this, HCVpp were incubated with low concentrations of neutralizing sera prior to infection of transiently transduced IFITM2 expressing cells. Treatment with low concentrations of neutralizing patient serum (1:200) did not significantly reduce viral entry into IFITM-negative control cells (Figure 5A). IFITM2 expression reduced virus entry in the absence of neutralizing sera by ten-fold for the non-escape variant and five-fold for the escape variant, respectively (Figure 5A). When serum-treated HCVpp were used to infect IFITM2-expressing cells, we observed a marked increase in neutralization. The neutralization was about three- to four-fold higher compared to the control-treated HCVpp, although the same serum-treatment had no effect on IFITM-negative control cells. Furthermore, the increase in neutralization was significantly and markedly higher for the non-escape variants compared to the escape variants (Figure 5A), suggesting a potential role of the IFITM-mediated enhancement of neutralization as a determinant for viral escape. Titration of the neutralizing serum corroborated our finding that neutralizing antibodies and IFITM proteins cooperatively block virus entry. Inhibition correlated with the concentration of the neutralizing serum, as shown by the slope of the regression curves for neutralization on control or IFITM2-expressing cells (Figure 5B). The slope on IFITM2-expressing cells was more than ten-fold higher as compared to the IFITM-negative control cells confirming a marked enhancement of neutralization by IFITM2. Furthermore, the IFITM-mediated enhancement of neutralization was confirmed using HCVcc of genotype 1b (Con1). As shown in Figure 5C, expression of IFITM2 enhanced the neutralization of HCVcc Con1 by a weakly neutralizing heterologous serum (1:100 dilution) from less than two-fold to 60-fold (Figure 5C).
Figure 5. IFITM2 enhances antibody-mediated neutralization of HCV cell entry.
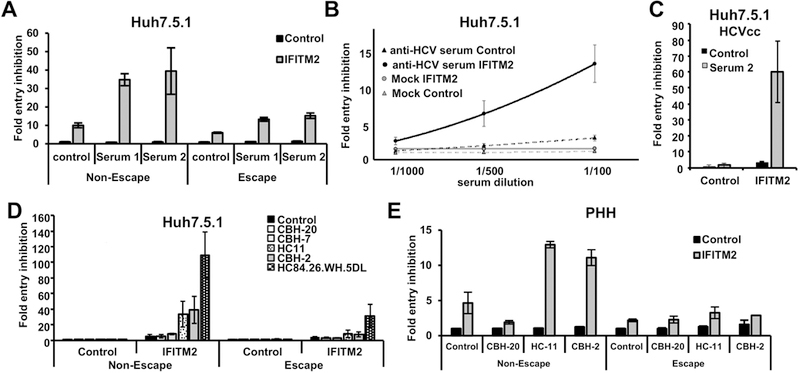
Huh7.5.1 cells (A, B, C, D) or primary human hepatocytes (E) were transduced by retroviral vectors coding for IFITM2 or empty vector as control. Forty-eight hours after transduction cells were infected with HCVpps expressing the envelope of variants selected during liver graft infection associated or not-associated with viral escape (A,B,D,E), or with HCVcc of genotype 1b (Con1) (C). Before infection, the particles were coincubated with serum derived from chronically HCV infected patients (A, B, C) or 15 µg/ml of the patient-derived HMAbs CBH-20 CBH-7, HC11, CBH-2 and HC84.26.WH.5DL or control antibody R04 (D, E) at 37 °C for 1 h. Entry of HCVpp was assessed 72 h post infection by measuring luciferase activity. Results are shown as fold inhibition of virus entry. Inhibition of entry by control vector in combination with the control antibody R04 or with control serum was set to 1. (B) The equation of the regression curve of anti-HCV serum and IFITM2 expressing cells was calculated as 48748 ×2+ 640 x + 1.9 The corresponding curve for anti-HCV serum on naïve cells was 4380 ×2+ 149 x + 1.1 (C) Infection with HCVcc was analyzed 72 h post infection by measuring luciferase activity. The graph represents means of three (D) or four (A, B) experiments that were performed in triplicates. Error bars represent SEM. (C) Shows a representative experiment performed in triplicates (n=6). (E) Represents a single experiment performed in PHH. Error bars show SD.
To assess which serum component was responsible for the enhanced neutralizing capacity of the sera in the presence of IFITM proteins we used HMAbs directed against different epitopes of the HCV E2 protein, some of which overlap with the polymorphic sites in acute patient variants. Similar as in experiments using sera, the HMAbs were used at sub-neutralizing concentrations (15 µg/mL) that in our model only had a low effect on virus entry inhibition (at maximum about 40 % or 1.67 fold inhibition by HC84.26.WH.5DL, lower for the other HMAbs) (Figure 5D). When IFITM2-expressing cells where infected with the HMAb-treated HCVpp we again observed a marked cooperative effect that directly correlated with the neutralizing properties of the antibody. Indeed, while the non-neutralizing HMAb CBH-20 did not exert a cooperative effect, the affinity matured anti-E2 antibody HC84.26.WH.5DL with potent neutralizing properties increased the inhibition of virus entry following IFITM2 expression to more than 100-fold (Figure 5D). The antibodies CBH-7, HC11 and CBH-2 had intermediate effects (Figure 5D).
Next, we confirmed these findings in the most physiologically relevant cell culture system: infection of PHH (Figure 5E). Similar to the Huh7.5.1 cells, treatment of the HCVpp with low concentrations of antibodies only had a minor influence on virus entry into naive PHH. As shown for Huh7.5.1 cells, IFITM2 expression blocked HCV entry into PHH, with the non-escape variant being more susceptible. Treatment with the neutralizing antibodies HC-11 and CBH-2 increased the neutralization of the non-escape variant on IFITM-expressing cells by about 3-fold. Again, the increase in neutralization was markedly lower in the escape variants (Figure 5E), confirming the results that were obtained with Huh7.5.1 cells. Taken together, these data show that innate and adaptive immune responses targeting viral entry cooperate to inhibit HCV infection and drive viral immune evasion in acute and chronic HCV infection.
Cooperative inhibition of HCV entry by IFITM proteins and neutralizing antibodies is attenuated by treatment with amphotericin B.
It is known that IFITM proteins restrict virus entry at the stage of hemifusion (16) or fusion pore formation (17) by altering curvature and fluidity of host cell membranes through direct or indirect mechanisms, which render virus-host cell fusion less energetically favorable (41, 42). Notably, the antiviral effect of IFITM2 and 3 on influenza virus infection is attenuated by incubation of host cells with amphotericin B, an amphiphilic antifungal drug that integrates into endosomal membranes (43), which can be regarded as an IFITM antagonist. Mechanistic studies revealed that the compound decreases the curvature and increases the fluidity of the endosomal membrane, which counteracts the IFITM-mediated antiviral effects within the endosomal membrane in an indirect manner (43), as illustrated in Figure 6D. We thus used amphotericin B to analyze whether IFITM-mediated modulation of membrane properties and the resulting inhibition of viral entry is required for the cooperative antiviral activity of antibodies and IFITM proteins. Treatment of cells with amphotericin B reduced HCVpp entry into the host cells by about three-fold (Figure 6A) and markedly reduced the antiviral activity of all IFITM proteins (Figure 6B). Moreover, amphotericin B treatment largely abrogated the cooperation of IFITMs and neutralizing sera in inhibition of HCVpp cellular entry (Figure 6C), indicating that IFITM-mediated modulation of cellular membranes and the resulting antiviral activity are responsible for the cooperative inhibition of virus entry by IFITM proteins and antibodies (illustrated in Figure 6D). Interestingly, a similar enhancement of neutralization was observed using interferon-treatment of Huh7 cells (Figure S6) supporting our conclusion that the IFITM-mediated antiviral effect and not a direct interaction with the IFITM proteins is responsible for the enhancement of neutralization.
Figure 6. Cooperative inhibition of HCV entry by IFITM proteins and neutralizing antibodies can be attenuated by treatment with amphotericin B.
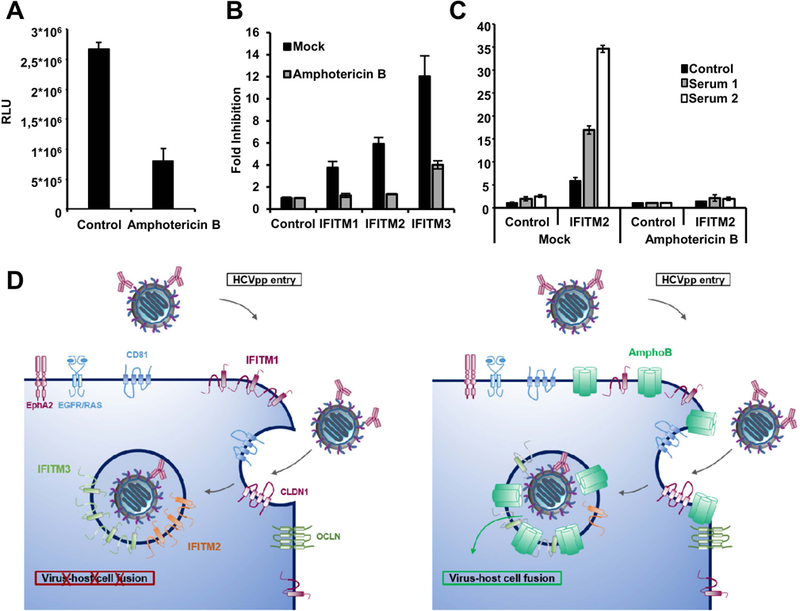
Huh7.5.1 cells were transduced by retroviral vectors coding for an empty control vector (A), with vectors coding for IFITM1, 2 and 3 and empty vector as control (B) or only IFITM2 plus control (C) Forty-eight h after transduction cells were treated with vehicle control or 5 µg/ml amphotericin B for 1 h. Afterwards, the cells were infected with HCVpp expressing the envelope of a variant not associated with viral escape and sensitive to antibody-mediated neutralization (P1VA). (C) Cells were infected with HCVpp pretreated with serum derived from chronically HCV infected patients or with control serum at 37 °C for 1 h. Entry of HCVpp was assessed 72 h post infection by measuring luciferase activity. (A) Results are shown in RLU. (B,C) Results were normalized for the vector control and are shown as fold inhibition compared to the respective controls. Shown are the means of representative experiments performed in triplicates (n=6) ± SD. (D) Model of cooperative inhibition of HCV entry by IFITMs and neutralizing antibodies and the antagonistic effect of amphotericin B. The interaction between infectious particles and cell surface receptors triggers endocytosis. Entry is blocked by IFITM proteins and neutralizing antibodies. Amphotericin B (AmphoB) is believed to rescue virus entry by antagonizing the IFITM-mediated increase of membrane rigidity and curvature.
Discussion
In this study we provide conclusive evidence that IFITM proteins are important determinants of viral escape from antiviral B cell responses in patients. This is supported by our finding that viral envelope proteins obtained from acute pre-seroconversion patients showed an increased IFITM-sensitivity as compared to envelope proteins obtained from chronic patients. Furthermore, the functional analysis of HCV variants from patients escaping viral neutralizing responses during liver transplantation compared with variants that are eliminated post-transplantation revealed a direct correlation of escape from neutralizing responses and resistance to inhibition by IFITM proteins.
Our finding that HCV variants of acute patients isolated pre-seroconversion were more susceptible to inhibition by IFITM proteins than variants derived from chronically-infected patients indicates that IFITM proteins drive immune evasion. This is supported by our analysis of sequentially isolated envelope proteins from one patient. IFITM resistance increased over time, with the envelope proteins isolated during chronic infection showing the highest IFITM resistance. Furthermore, the acquisition of mutations within epitope II and the CD81 binding domain that are targeted by neutralizing antibodies suggests that the IFITMs might modulate interactions with the adaptive immune response. Indeed, the analysis of post-transplant variants revealed a direct association of resistance to inhibition by IFITM proteins and escape from the nAb response. Escape of the virus from host responses is critical for viral spread and survival (44). In part, these findings could explain the low efficacy of innate immune activation in chronic HCV-infected patients (45). Escape from innate responses does not only prevent the immune system from clearing the viral infection but also limits the response to interferon-based therapies (44). Furthermore, the finding that variants selected post-liver transplantation and characterized by viral escape were significantly more resistant to inhibition by IFITM proteins than variants that were eliminated post-transplantation (with sensitivity to antibody-mediated neutralization) could also explain the rapid selection of these resistant variants. In addition, the distribution of the IFITM-sensitivity of the escape variants appeared to be less dispersed than that of the non-escape variants, potentially reflecting a bottleneck during the selection process. Differences in IFITM expression levels between host and graft tissue might drive the selection of highly infectious IFITM resistant variants, subsequently leading to reinfection of the graft, as universally observed.
How can mutations present in HCV in chronic patients confer relative IFITM resistance? For one, HCV variants that escape immune control frequently exhibit enhanced binding to the HCV co-receptor CD81 (22) and IFITM1 has been suggested to exert antiviral activity in part by interacting with CD81 (13). However, there are no reports of direct interactions of IFITM2 and 3 with CD81 and other HCV (co-)receptors (14), although IFITM3 seems to partially colocalize with CD81 (14). Additionally, IFITM resistant HCV, like IFITM resistant influenza A viruses (46), might exhibit an altered pH-optimum for virus entry thereby avoiding the need to fuse with IFITM-rich internal membranes. Finally, IFITM-sensitivity might by linked to the number of viral glycoproteins incorporated into the viral membrane, as previously demonstrated for simian immunodeficiency virus (47) or to the composition of the viral membrane itself, however our previous characterization of HCV envelope glycoprotein variants proteins in liver transplantation indicate that an increased amount of incorporation of envelope proteins is most likely not responsible for the observed changes in IFITM-sensitivity (21). It remains to be determined whether IFITM-induced changes in membrane composition or altered interaction with lipoproteins contribute to the IFITM-mediated escape from the nAb response.
The proteins of the IFITM family are potent inhibitors of host cell entry of a wide range of enveloped viruses (11), including HCV (13, 14, 18). A single-cell analysis of clinical human liver samples by laser capture microdissection and qRT-PCR revealed that IFITM3 expression and HCV RNA were largely mutually exclusive (48), indicating an important role of the IFITM proteins in HCV cell tropism. A very recent publication indicates that stem cells, that do not respond to interferon, express high levels of interferon stimulated genes, including IFITM3, to protect them from viral infection (49). Constitutive expression of interferon stimulated genes is lost upon differentiation into hepatocyte-like cells (49) but becomes interferon-inducible, highlighting an important contribution of the IFITM proteins to the innate defenses against HCV and other pathogens.
Perturbation studies with amphotericin B that acts as an IFITM-antagonist in the context of influenza virus infections (43) revealed that modulated membrane-properties are responsible for the IFITM-mediated enhancement of neutralization, indicating that other innate entry effectors, as shown for pretreatment with interferon-alpha, might exploit similar inhibitory mechanisms to block virus entry.
Interestingly, a recent study has suggested a different role for IFITMs in clinical HIV-1 infection: While transmitted founder viruses where almost resistant to inhibition by IFITM proteins, the virus became more susceptible over time, as it escaped from the nAb response (50). This observation could reflect different roles of innate immune responses in HCV and HIV infections with our findings supporting a much more prominent role of the IFITMs during acute HCV infection compared to HIV. Furthermore, the increased IFITM-sensitivity could in part explain the high susceptibility of acute HCV infection to interferon treatment. On a mechanistic point of view, IFITM-sensitivity of HIV was associated with receptor-usage, with CCR5-tropic viruses being generally more resistant to IFITM proteins than CXCR4-tropic viruses (50). This suggests that the differential sensitivity of HIV to inhibition by the IFITM proteins might be due to receptor-mediated targeting to subcellular compartments with differential IFITM expression, or due to changes in envelope structure and electrochemical properties due to the switch of receptor tropism from CCR5 to CXCR5, which was not observed for HCV infection.
Taken together, our findings show that IFITMs are important drivers of viral immune evasion in acute and chronic HCV infection by enhancing antibody—mediated neutralization. Harnessing these effects will help to facilitate the design of protective B cell HCV vaccines.
Supplementary Material
Acknowledgements.
The authors thank F. Chisari (The Scripps Research Institute, La Jolla, CA) for the gift of Huh7.5.1 cells, R. Bartenschlager (University of Heidelberg), C. Rice (Rockefeller University), T. Wakita (University of Tokyo), for plasmids for HCVcc and HCVpp production. We would like to thank Sarah Durand (Inserm U1110) and Sabine Gärtner (German Primate Center) for excellent technical support. We thank Dr. Eloi Verrier (Inserm U1110) for helpful discussions.
Funding sources: This work was supported in part by National Institutes of Health grants U19-AI123862 (TFB, SKHF) and R01-AI132213 (SKHF), by the ARC-IHU TheraHCC program (TFB), by the European Union (ERC-AdG-2014–671231-HEPCIR and H2020–2015-667273-HEP-CAR to TFB), ANRS (ECTZ87384 to TFB and G. L.) and by the German research foundation (DFG - 395783133 to FW). This work has been published under the framework of the LABEX ANR-10-LABX-0028_HEPSYS (TFB, MBZ, CS) and benefits from funding from the state managed by the French National Research Agency as part of the Investments for the Future Program.
List of abbreviations:
- HCV
hepatitis C virus
- IFITM
interferon-induced transmembrane protein
- HCVpp
HCV pseudoparticle
- HMAb
human monoclonal antibody
- HCVcc
cell culture derived HCV
- MLV
murine leukemia virus
- TCID50
tissue culture infectious dose 50 %
- nAb
neutralizing antibody
Footnotes
Conflict of interest. The authors declare no conflict of interest.
References
Author names in bold designate shared first authorship
- 1.World Health Organization. Hepatitis C fact sheet https://www.who.int/news-room/fact-sheets/detail/hepatitis-c; accessed April 6 2019.
- 2.Chung RT, Baumert TF. Curing chronic hepatitis C--the arc of a medical triumph. N Engl J Med 2014;370:1576–1578. [DOI] [PubMed] [Google Scholar]
- 3.Baumert TF, Fauvelle C, Chen DY, Lauer GM. A prophylactic hepatitis C virus vaccine: a distant peak still worth climbing. J Hepatol 2014;61:S34–44. [DOI] [PubMed] [Google Scholar]
- 4.Ferenci P Treatment of hepatitis C in difficult-to-treat patients. Nat Rev Gastroenterol Hepatol 2015;12:284–292. [DOI] [PubMed] [Google Scholar]
- 5.Hezode C, Fontaine H, Dorival C, Larrey D, Zoulim F, Canva V, de Ledinghen V, et al. Triple therapy in treatment-experienced patients with HCV-cirrhosis in a multicentre cohort of the French Early Access Programme (ANRS CO20-CUPIC) - NCT01514890. J Hepatol 2013;59:434–441. [DOI] [PubMed] [Google Scholar]
- 6.Choo QL, Kuo G, Ralston R, Weiner A, Chien D, Van Nest G, Han J, et al. Vaccination of chimpanzees against infection by the hepatitis C virus. Proc Natl Acad Sci U S A 1994;91:1294–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stamataki Z, Coates S, Abrignani S, Houghton M, McKeating JA. Immunization of human volunteers with hepatitis C virus envelope glycoproteins elicits antibodies that cross-neutralize heterologous virus strains. J Infect Dis 2011;204:811–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Law M, Maruyama T, Lewis J, Giang E, Tarr AW, Stamataki Z, Gastaminza P, et al. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat Med 2008;14:25–27. [DOI] [PubMed] [Google Scholar]
- 9.de Jong YP, Dorner M, Mommersteeg MC, Xiao JW, Balazs AB, Robbins JB, Winer BY, et al. Broadly neutralizing antibodies abrogate established hepatitis C virus infection. Sci Transl Med 2014;6:254ra129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marston HD, Paules CI, Fauci AS. Monoclonal Antibodies for Emerging Infectious Diseases - Borrowing from History. N Engl J Med 2018;378:1469–1472. [DOI] [PubMed] [Google Scholar]
- 11.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011;472:481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yao L, Dong H, Zhu H, Nelson D, Liu C, Lambiase L, Li X. Identification of the IFITM3 gene as an inhibitor of hepatitis C viral translation in a stable STAT1 cell line. J Viral Hepat 2011;18:e523–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilkins C, Woodward J, Lau DT, Barnes A, Joyce M, McFarlane N, McKeating JA, et al. IFITM1 is a tight junction protein that inhibits hepatitis C virus entry. Hepatology 2013;57:461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Narayana SK, Helbig KJ, McCartney EM, Eyre NS, Bull RA, Eltahla A, Lloyd AR, et al. The Interferon-induced Transmembrane Proteins, IFITM1, IFITM2, and IFITM3 Inhibit Hepatitis C Virus Entry. J Biol Chem 2015;290:25946–25959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bailey CC, Zhong G, Huang IC, Farzan M. IFITM-Family Proteins: The Cell’s First Line of Antiviral Defense. Annu Rev Virol 2014;1:261–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li K, Markosyan RM, Zheng YM, Golfetto O, Bungart B, Li M, Ding S, et al. IFITM proteins restrict viral membrane hemifusion. PLoS Pathog 2013;9:e1003124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Desai TM, Marin M, Chin CR, Savidis G, Brass AL, Melikyan GB. IFITM3 restricts influenza A virus entry by blocking the formation of fusion pores following virus-endosome hemifusion. PLoS Pathog 2014;10:e1004048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verrier ER, Colpitts CC, Bach C, Heydmann L, Zona L, Xiao F, Thumann C, et al. Solute Carrier NTCP Regulates Innate Antiviral Immune Responses Targeting Hepatitis C Virus Infection of Hepatocytes. Cell Rep 2016;17:1357–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pestka JM, Zeisel MB, Blaser E, Schurmann P, Bartosch B, Cosset FL, Patel AH, et al. Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc Natl Acad Sci U S A 2007;104:6025–6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Osburn WO, Fisher BE, Dowd KA, Urban G, Liu L, Ray SC, Thomas DL, et al. Spontaneous control of primary hepatitis C virus infection and immunity against persistent reinfection. Gastroenterology 2010;138:315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fafi-Kremer S, Fofana I, Soulier E, Carolla P, Meuleman P, Leroux-Roels G, Patel AH, et al. Viral entry and escape from antibody-mediated neutralization influence hepatitis C virus reinfection in liver transplantation. J Exp Med 2010;207:2019–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fofana I, Fafi-Kremer S, Carolla P, Fauvelle C, Zahid MN, Turek M, Heydmann L, et al. Mutations that alter use of hepatitis C virus cell entry factors mediate escape from neutralizing antibodies. Gastroenterology 2012;143:223–233 e229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fauvelle C, Felmlee DJ, Crouchet E, Lee J, Heydmann L, Lefèvre M, Magri A, et al. Apolipoprotein E Mediates Evasion From Hepatitis C Virus Neutralizing Antibodies. Gastroenterology 2016;150:206–217 e204. [DOI] [PubMed] [Google Scholar]
- 24.Haberstroh A, Schnober EK, Zeisel MB, Carolla P, Barth H, Blum HE, Cosset FL, et al. Neutralizing host responses in hepatitis C virus infection target viral entry at postbinding steps and membrane fusion. Gastroenterology 2008;135:1719–1728 e1711. [DOI] [PubMed] [Google Scholar]
- 25.Krieger SE, Zeisel MB, Davis C, Thumann C, Harris HJ, Schnober EK, Mee C, et al. Inhibition of hepatitis C virus infection by anti-claudin-1 antibodies is mediated by neutralization of E2-CD81-claudin-1 associations. Hepatology 2010;51:1144–1157. [DOI] [PubMed] [Google Scholar]
- 26.Owsianka A, Tarr AW, Juttla VS, Lavillette D, Bartosch B, Cosset FL, Ball JK, et al. Monoclonal antibody AP33 defines a broadly neutralizing epitope on the hepatitis C virus E2 envelope glycoprotein. J Virol 2005;79:11095–11104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hadlock KG, Lanford RE, Perkins S, Rowe J, Yang Q, Levy S, Pileri P, et al. Human monoclonal antibodies that inhibit binding of hepatitis C virus E2 protein to CD81 and recognize conserved conformational epitopes. J Virol 2000;74:10407–10416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keck ZY, Li TK, Xia J, Gal-Tanamy M, Olson O, Li SH, Patel AH, et al. Definition of a conserved immunodominant domain on hepatitis C virus E2 glycoprotein by neutralizing human monoclonal antibodies. J Virol 2008;82:6061–6066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Keck ZY, Xia J, Wang Y, Wang W, Krey T, Prentoe J, Carlsen T, et al. Human monoclonal antibodies to a novel cluster of conformational epitopes on HCV E2 with resistance to neutralization escape in a genotype 2a isolate. PLoS Pathog 2012;8:e1002653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wrensch F, Winkler M, Pohlmann S. IFITM proteins inhibit entry driven by the MERS-coronavirus spike protein: evidence for cholesterol-independent mechanisms. Viruses 2014;6:3683–3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Urbanowicz RA, McClure CP, Brown RJ, Tsoleridis T, Persson MA, Krey T, Irving WL, et al. A Diverse Panel of Hepatitis C Virus Glycoproteins for Use in Vaccine Research Reveals Extremes of Monoclonal Antibody Neutralization Resistance. J Virol 2015;90:3288–3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lavillette D, Tarr AW, Voisset C, Donot P, Bartosch B, Bain C, Patel AH, et al. Characterization of host-range and cell entry properties of the major genotypes and subtypes of hepatitis C virus. Hepatology 2005;41:265–274. [DOI] [PubMed] [Google Scholar]
- 33.Kato N, Hijikata M, Ootsuyama Y, Nakagawa M, Ohkoshi S, Sugimura T, Shimotohno K. Molecular cloning of the human hepatitis C virus genome from Japanese patients with non-A, non-B hepatitis. Proc Natl Acad Sci U S A 1990;87:9524–9528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Colpitts CC, Tawar RG, Mailly L, Thumann C, Heydmann L, Durand SC, Xiao F, et al. Humanisation of a claudin-1-specific monoclonal antibody for clinical prevention and cure of HCV infection without escape. Gut 2018;67:736–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McClure CP, Urbanowicz RA, King BJ, Cano-Crespo S, Tarr AW, Ball JK. Flexible and rapid construction of viral chimeras applied to hepatitis C virus. J Gen Virol 2016;97:2187–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Merz T, Sadeghian K, Schutz M. Why BLUF photoreceptors with roseoflavin cofactors lose their biological functionality. Phys Chem Chem Phys 2011;13:14775–14783. [DOI] [PubMed] [Google Scholar]
- 37.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 2005;11:791–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reiss S, Rebhan I, Backes P, Romero-Brey I, Erfle H, Matula P, Kaderali L, et al. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe 2011;9:32–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pietschmann T, Kaul A, Koutsoudakis G, Shavinskaya A, Kallis S, Steinmann E, Abid K, et al. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc Natl Acad Sci U S A 2006;103:7408–7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang IC, Bailey CC, Weyer JL, Radoshitzky SR, Becker MM, Chiang JJ, Brass AL, et al. Distinct patterns of IFITM-mediated restriction of filoviruses, SARS coronavirus, and influenza A virus. PLoS Pathog 2011;7:e1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Amini-Bavil-Olyaee S, Choi YJ, Lee JH, Shi M, Huang IC, Farzan M, Jung JU. The antiviral effector IFITM3 disrupts intracellular cholesterol homeostasis to block viral entry. Cell Host Microbe 2013;13:452–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bailey CC, Kondur HR, Huang IC, Farzan M. Interferon-induced transmembrane protein 3 is a type II transmembrane protein. J Biol Chem 2013;288:32184–32193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lin TY, Chin CR, Everitt AR, Clare S, Perreira JM, Savidis G, Aker AM, et al. Amphotericin B increases influenza A virus infection by preventing IFITM3-mediated restriction. Cell Rep 2013;5:895–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heim MH, Thimme R. Innate and adaptive immune responses in HCV infections. J Hepatol 2014;61:S14–25. [DOI] [PubMed] [Google Scholar]
- 45.Wieland S, Makowska Z, Campana B, Calabrese D, Dill MT, Chung J, Chisari FV, et al. Simultaneous detection of hepatitis C virus and interferon stimulated gene expression in infected human liver. Hepatology 2014;59:2121–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gerlach T, Hensen L, Matrosovich T, Bergmann J, Winkler M, Peteranderl C, Klenk HD, et al. pH Optimum of Hemagglutinin-Mediated Membrane Fusion Determines Sensitivity of Influenza A Viruses to the Interferon-Induced Antiviral State and IFITMs. J Virol 2017;91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wrensch F, Hoffmann M, Gartner S, Nehlmeier I, Winkler M, Pohlmann S. Virion Background and Efficiency of Virion Incorporation Determine Susceptibility of Simian Immunodeficiency Virus Env-Driven Viral Entry to Inhibition by IFITM Proteins. J Virol 2017;91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kandathil AJ, Graw F, Quinn J, Hwang HS, Torbenson M, Perelson AS, Ray SC, et al. Use of laser capture microdissection to map hepatitis C virus-positive hepatocytes in human liver. Gastroenterology 2013;145:1404–1413 e1401–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu X, Dao Thi VL, Huang Y, Billerbeck E, Saha D, Hoffmann HH, Wang Y, et al. Intrinsic Immunity Shapes Viral Resistance of Stem Cells. Cell 2018;172:423–438 e425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Foster TL, Wilson H, Iyer SS, Coss K, Doores K, Smith S, Kellam P, et al. Resistance of Transmitted Founder HIV-1 to IFITM-Mediated Restriction. Cell Host Microbe 2016;20:429–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.