

Abstract
During biliary disease, cholangiocytes become activated by various pathological stimuli, including transforming growth factorβ (TGFβ). The result is an epigenetically-regulated transcriptional program leading to a pro-fibrogenic microenvironment, activation of hepatic stellate cells (HSC), and progression of biliary fibrosis. This study evaluated how TGFβ signaling intersects with epigenetic machinery in cholangiocytes to support fibrogenic gene transcription. We performed RNA seq in cholangiocytes with or without TGFβ. Ingenuity pathway analysis identified “HSC Activation” as the highly upregulated pathway, including overexpression of fibronectin 1 (FN), connective tissue growth factor, and other genes. Bioinformatics identified enhancer of zeste homologue 2 (EZH2) as an epigenetic regulator of the cholangiocyte TGFβ response. EZH2 overexpression suppressed TGFβ induced FN protein in vitro, suggesting FN as a direct target of EZH2-based repression. ChIP assays identified a FN promoter element in which EZH2-mediated tri-methylation of lysine 27 on histone 3 (H3K27me3) is diminished by TGFβ. TGFβ also caused a 50% reduction in EZH2 protein levels. Proteasome inhibition rescued EZH2 protein and led to reduced FN production. Immunoprecipitation followed by mass-spectrometry identified ubiquitin protein ligase E3 component N-recognin 4 (UBR4) in complex with EZH2, which was validated by western blotting in vitro. Ubiquitin mutation studies suggested K63-based ubiquitin linkage and chain elongation on EZH2 in response to TGFβ. A deletion mutant of EZH2, lacking its N-terminal domain, abrogates both TGFβ stimulated EZH2 degradation and FN release. In vivo, cholangiocyte-selective knockout of EZH2 exacerbates bile duct ligation induced fibrosis while MDR2−/− mice are protected from fibrosis by the proteasome inhibitor, bortezomib.
Conclusion:
TGFβ regulates proteasomal degradation of EZH2 through N-terminal, K63-linked, ubiquitination in cholangiocytes and activates transcription of a fibrogenic gene program that supports biliary fibrosis.
Keywords: cholangiocyte, fibrosis, epigenetics, TGFβ, EZH2, ubiquitin, proteasome
Cholestatic fibrogenesis is a pathobiological process in which cumulative injury to the bile ducts coincides with progressive biliary fibrosis (1). This phenomenon is clinically relevant to several cholangiopathies, such as congenital hepatic fibrosis (2), primary biliary cholangitis , and primary sclerosing cholangitis (PSC) (3). During biliary fibrosis, diseased cholangiocytes become highly secretory, releasing a variety of paracrine signaling molecules that subsequently activate portal fibroblasts (PF) and/or hepatic stellate cells (HSC) (4, 5) through epithelial/mesenchymal cross talk (6). Activated myofibroblasts subsequently deposit the dense, peri-portal, collagen-based extracellular matrix (ECM), that typifies biliary fibrosis (7, 8). The pathobiologic mechanisms regulating peri-portal fibrogenesis and to what extent these processes contribute to the progression of disease remain poorly understood (3). What is becoming clear, based on genome wide association studies, is that genetic causes alone are not sufficient to fully explain the pathobiology (9). Rather, it appears that the pathogenesis of biliary fibrosis is a complex process involving factors such as specific environmental exposures in genetically susceptible individuals as well as complex interactions with the microbiome and the immune system (3). It is likely that these diverse signals are ultimately integrated at the level of the epigenome and yet very few epigenetic studies have been performed (10). Thus, it is critical to elucidate the epigenetic events within cholangiocytes that coordinate cholestatic fibrogenesis in order to identify pathways that can be targeted therapeutically.
TGFβ levels increase markedly in chronic biliary diseases (11, 12), and TGFβ signaling plays a prominent role in the development and progression of liver fibrosis . While TGFβ signaling is well-studied in non-parenchymal cells of the liver, such as HSC and portal myofibroblasts, the signaling events in liver epithelia are less clear. In particular, the mechanisms by which cholangiocyte TGFβ stimuli are transduced into gene transcription events through epigenetic regulators are unknown. Therefore, this study extends our understanding of an epigenetic silencing complex that maintains cholangiocyte homeostasis and, when altered during TGFβ signaling, contributes to biliary fibrosis.
Modification of histones is a critical epigenetic regulatory mechanism that is unexplored in biliary disease (10). The polycomb group protein complexes play a critical role in gene silencing in many biological contexts by facilitating the formation and maintenance of facultative heterochromatin (13). As the primary catalytic protein of the polycomb repressive complex 2 (PRC2), EZH2 (14) is a key epigenetic regulatory protein that silences gene expression across a diverse array of cellular processes in a cell-type-specific and context-dependent manner (15). EZH2 enzymatically mediates the H3K27me3 repressive mark via its intrinsic histone methyltransferase activity and thus EZH2 is a key writer of the histone code (14). Prior studies have demonstrated that biliary cells respond to TGF-β by acquiring a pro-fibrogenic phenotype (6). In this project, we mechanistically characterize a novel epigentic pathway in which TGFβ leads to proteasomal degradation of EZH2, loss of the H3K27me3 repressive mark, and thereby facilitates the activation of a transcriptional program in cholangiocytes. Since biliary cirrhosis is untreatable without liver transplantation, the identification of druggable epigenetic targets would represent an important advance. These advances improve our understanding of cholangiocyte pathobiology and peri-portal scar formation and should allow for the optimization of future anti-fibrotic protocols. This is particularly appealing in the era of epigenetic pharmacology, an approach that is already reaching clinical trials for various diseases and allowing for better precision in molecular targeting (16).
Experimental Procedures
Cell Culture and Transfections
The H69 cholangiocyte cell line was provided by Nicholas LaRusso, M.D. (Mayo Clinic). Human Intrahepatic Biliary Epithelial Cells (HiBEC) and Human Hepatic Stellate Cells (hHSC) were purchased from ScienCell (Catalog #5100 and #5300, respectively). The RGF-1 portal fibroblast cell line was provided by Jonathan Dranoff, M.D. (University of Arkansas) (17). Cells were grown in DMEM/F12 supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, adenine, insulin, epinephrine, T3-T, hydrocortisone, and epidermal growth factor. Cells were serum starved in basal DMEM containing 1% penicillin/streptomycin for 4 hours or overnight before treatment with 10 ng/mL recombinant TGFβ (R&D Systems #240-B). In some experiments, cells were incubated with 10µM MG132 (Cell Signaling Technology #2194), 10 µM E64D (Sigma Aldrich #E8440), or 10 µM Bafilomycin A1 (Sigma-Aldrich #B1793), one hour prior to TGFβ. For some experiments, cells at 70% confluence were transfected using the Lipofectamine 3000 kit (Invitrogen #L3000–015). The plasmids used for transfection were from Addgene; HA-Ubiquitin (WT, #17608, K48, # 17605, K63, #17606), myc-EZH2 WT was obtained from William Faubion, M.D. (Mayo Clinic).
RNA Sequencing
RNA sequencing and bioinformatics analysis was conducted in collaboration with the Mayo Medical Genomics Facility as previously described (18). Quality of RNA was assessed by the Mayo Gene Expression Core using Agilent Bioanalyzers. RNA sequencing was performed as described before (19). Differential expression analyses between samples were computed using an edgeR version 3.3.8 algorithm. Data was deposited in the NCBI Gene Expression Omnibus (GSE125577). Heatmap was created using the Heatmapper web server as described elsewhere (20). Enrichr evaluation (http://amp.pharm.mssm.edu/Enrichr/) of gene enrichment was analyzed using the Fisher exact test, as described elsewhere (21, 22).
Chromatin Immunoprecipitation (ChIP)
ChIP assays were performed using Magna-ChIP™ A kit (Millipore Corp. #MAGNA0001). The nuclear extract was sonicated and immunoprecipitated with an antibody against H3K27me3 (Abcam #ab6002) or EZH2 (Cell Signaling, #5246). Primer sets targeting the FN promoter were designed for quantitative PCR analysis. We utilized 1% of input for all ChIP assays and to normalize the samples we employed the following standard equation: 1% input = 2 ^ [(mean Ct input − 6.64) − mean Ct ChIP] * 100.
Animal Models
KRT19-Cre ERT LSLTdTomato mice were obtained from Stuart Forbes, Ph.D. (University of Edinburgh). EZH2fl/fl mice were obtained from Raul Urrutia, M.D. (Medical College of Wisconsin). These mice were crossed to obtain EZH2fl/fl/KRT19-CreERTLSLTdTomato offspring. At 6–8 weeks of age, EZH2fl/fl (controls) and the EZH2fl/fl/KRT19-CreERTLSLTdTomato (experimental) mice (males and females) were injected with Tamoxifen for 5 days. Two days later BDL surgery or sham surgery was performed as previously described (23). Two weeks later, animals were sacrificed. Liver tissue was harvested for mRNA and hydroxyproline assay as well as paraffin embedded for histology. Fibrosis was analyzed by Sirius Red and Masson’s Trichrome staining. Mouse serum was collected for liver biochemistry.
MDR−/− mice were obtained from Nicholas LaRusso, M.D. (Mayo Clinic). Six week old MDR−/− mice (males and females) were treated with bortezomib or vehicle three times per week for four weeks by intraperitoneal injection. At four weeks mice were sacrificed and livers were harvested. All animal experiments followed protocols approved by Mayo Clinic Institutional Animal Care and Use Committee.
Patient Samples
Patient liver explant tissues were obtained and analyzed under Mayo Clinic institutional review board–approved protocols. Demographic details in Supplementary table 3.
Statistical Analysis
Data represents typical experiments reproduced at least three times. The data were analyzed using analysis of variance (ANOVA) with Bonferroni post-test or paired t -test using GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA). The difference was considered significant when P < 0.05. The results are presented as mean ± SEM. Enrichr analyses calculated enrichment using the Fisher exact test as described elsewhere (22, 23).
Please refer to Supplementary information for other methods and detailed information.
Results
TGFβ Drives Expression of an HSC-Activating Gene Program in Cholangiocytes:
TGFβ is one of the most prominently upregulated growth factors in the context of liver fibrosis (11, 12). Therefore, in order to identify pathological gene targets downstream of TGFβ, we performed genome-wide RNA-seq from cholangiocytes treated with and without TGFβ. We observed 923 genes that were upregulated 3-fold or greater by TGFβ (Figure 1A). We noted that many of these genes encode proteins that are known activators of HSCs. In fact, ingenuity pathway analysis indicated that “Integrin Signaling” and “HSC Activation” were the most highly upregulated pathways (Figure 1B). Indeed, the upregulated gene program included prominent overexpression of several genes that are well-known to be involved in HSC activation including FN, connective tissue growth factor (CTGF), vascular endothelial growth factor (VEGF), and platelet-derived growth factor (PDGF), among others (Figure 1B, inset). These results corroborate prior reports of TGFβ-induced upregulation of genes such as CTGF, VEGF, and PDGF (6, 24). We confirmed upregulation of these target genes by RT-PCR analysis (Figure 1C). FN was ultimately chosen as a prototype for more in-depth analyses owing to its prominent overexpression, its role as an important early molecule in liver fibrosis (8), and its ability to recruit and activate HSCs (25, 26). We observed a greater than 25-fold increase in FN transcription by quantitative RT-PCR (Figure 1C). In contrast to FN, we observed a less-prominent upregulation of certain mesenchymal markers, both in our RNA-seq data and by RT-PCR, (Supplementary Figure 1A and B), consistent with previous observations (27). In order to gain insight into potential novel epigenetic mechanisms that mediate this aspect of the cholangiocyte TGFβ response, subsequent bioinformatics used the Enrichr platform (amp.pharm.mssm.edu/Enrichr) to cross-reference gene enrichment in our RNA-seq dataset to the ENCODE database (www.genome.gov). ENCODE includes a compendium of publicly available ChIP-seq datasets from various cell types. This analysis identified EZH2 as a potential epigenetic regulator of the cholangiocyte TGFβ response (Figure 1D). Therefore, we hypothesized that TGFβ signaling can intersect with this epigenetic regulatory pathway in cholangiocytes to initiate a fibrogenic program of gene transcription that subsequently supports HSC activation.
Figure 1.
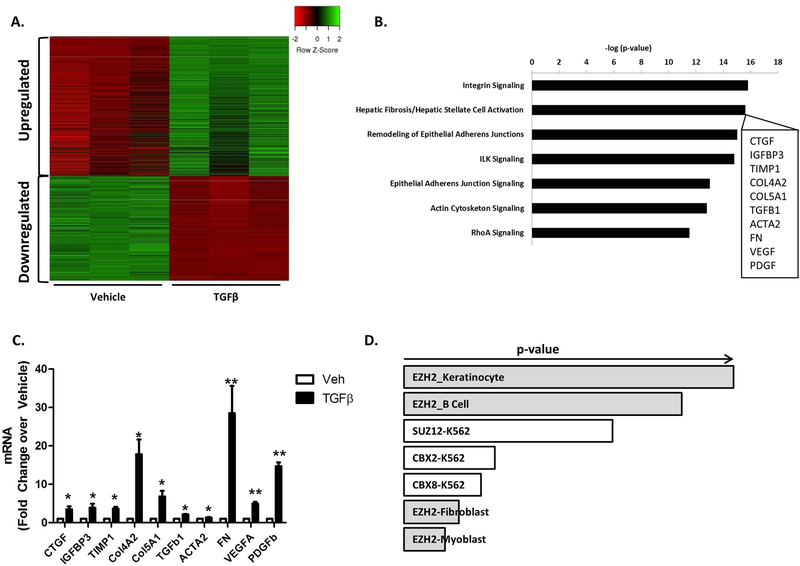
TGFβ Drives Expression of an HSC-Activating Gene Program in Cholangiocytes.
A. Heat Map representing the gene changes in a cholangiocytes cell line (HIBEC) treated with 10ng/ml TGFβ for 48 hours (green: upregulated gene set; red: downregulated gene set). n=3
B. Ingenuity Pathway Analysis (IPA) shows Integrin signaling and Hepatic Fibrosis/Hepatic Stellate Cell Activation as the most significantly upregulated pathways (inset shows 10 representative genes from the Heaptic Stellate Cell Activation Pathway).
C. RT-PCR in cholangiocytes treated with 10ng/ml TGFβ for 24 hours confirms upregulation of HSC activation genes. *p<0.01 and **p<0.001. All error bars are SEM, n=3
D. EnrichR analysis identified EZH2 as an epigenetic regulator of the cholangiocyte TGFβ response (p-vaue represents Fisher exact test).
Cholangiocytes Upregulate FN in Response to TGFβ:
In order to explore the specificity of the FN response to TGFβ, we used Western blot to evaluate FN protein levels in response to several agonists that are relevant to biliary disease. In addition to TGFβ, these included the TGR5 agonist taurolithocholic acid (TLCA), lipopolysaccharide (LPS), and the inflammatory cytokine interleukin-6 (IL-6). These studies indicated that while TGFβ drives robust upregulation of FN protein (8.47-fold), the other agonists had minimal effects on FN production (Figure 2A), supporting specificity of the TGFβ response. We confirmed a direct inhibitory effect of EZH2 on FN using adenoviral overexpression of EZH2 which blocked the TGF- β induced increase in FN from 4.8-fold to 2-fold (Figure 2B). We confirmed in cultured cholangiocytes that TGFβ leads to increased FN protein production, but we also observed a coinciding decrease in levels of EZH2 (Figure 2C). Densitometry confirmed a significant increase in FN as well as a 50% reduction in EZH2 protein following TGFβ. mRNA analysis with RT-PCR showed an increase in FN transcript with TGFβ treatment. However, there was no detectable change in the EZH2 mRNA levels (Figure 2D), suggesting a potential mechanism involving protein degradation. In order to maintain a focus on biological relevance, we confirmed in isolated mouse cholangiocytes that TGFβ also leads to approximately 40% loss of EZH2 protein (Figure 2E) and that this corresponds to about 1.7-fold increase in FN mRNA levels (Figure 2F).
Figure 2.
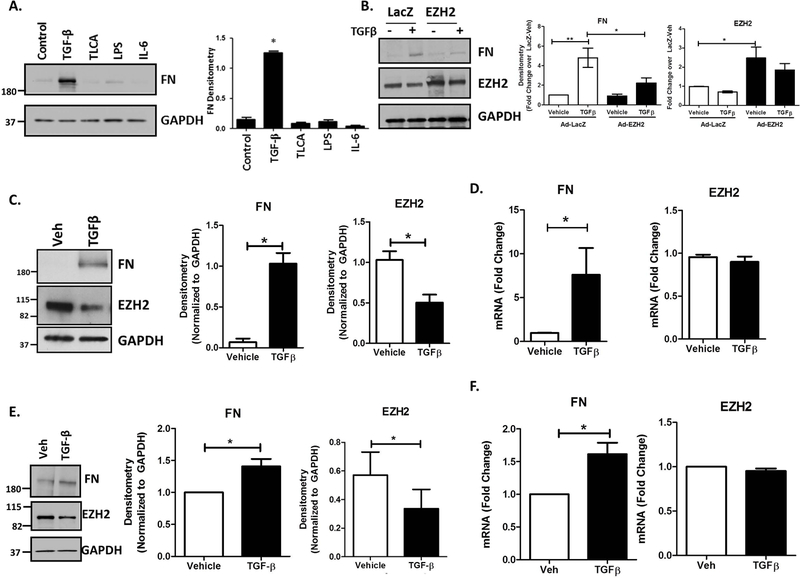
Cholangiocytes Upregulate FN in Response to TGFβ.
A. H69 cells treated for 24 hours with several agonists prominent in biliary disease show that only TGFβ stimulates FN production. TGFβ: 10ng/ml, TLCA: 20uM, LPS: 200ng/ml, IL-6: 50ng/ml. Western blot for FN and EZH2 with GAPDH as a loading control. These expression levels were quantified by densitometry (right). *p<0.01. All error bars are SEM, n=3.
B. Western blot showing repression of TGFβ–stimulated FN by overexpression of EZH2 using adenoviral constructs (AdEZH2) (left). Quantification of the blot for EZH2 and FN normalized to LacZ-vehicle. *p<0.01 and **p<0.001. All error bars are SEM, n=4.
C. TGFβ treatment for 24 hours stimulates FN production in cholangiocytes and a reduction in total EZH2 levels. Immunoblotting for FN and EZH2 on H69 cells treated with TGFβ for 24 hours (left). These were quantified by densitometry for FN and EZH2 (right) *p<0.01 compared to vehicle. All error bars are SEM, n=3.
D. RT-PCR analysis on H69 cells treated with TGFβ for 24 hours shows an increase in FN gene expression with no change in EZH2 gene expression. *p<0.01 compared to vehicle. All error bars are SEM, n=3.
E. Cholangiocytes isolated from WT miee were treated with 10ng/ml TGFβ for 24 hours. Western blotting for FN and EZH2 show TGFβ -stimulated FN production with a concurrent decrease in EZH2 protein (left). Densitometry from 3 different experiments shows an increase in FN and a decrease in EZH2 protein (right). *p<0.01 compared to vehicle. All error bars are SEM, n=3.
F. RT-PCR analysis on mouse cholangiocytes treated with TGFβ shows an approximately 2-fold increase in FN gene transcription with no change in EZH2 gene transcription. *p<0.01 compared to vehicle. All error bars are SEM, n=3.
TGFβ Stimulation Leads to EZH2 Protein Degradation in Cholangiocytes:
A temporal analysis of TGFβ treated cholangiocytes confirmed an initial decrease in EZH2 protein levels starting at 12 hours with approximately 50% of EZH2 protein being degraded by 24 hours (Figure 3A). We also observed increased FN production beginning at 12 hours as well as its release into the culture media, particularly at 24 hours (Figure 3A). A corresponding decrease in SUZ12, another canonical member of the PRC2 repressive complex, suggested that the EZH2 degradation occurs in coordination with other PRC2 complex members. While FN mRNA was highly upregulated (30-fold), the loss of EZH2 protein appeared despite no changes in EZH2 mRNA (Figure 3B), again supporting the idea of post-transcriptional regulation at the level of protein degradation. Furthermore, upon treatment with cycloheximide (to inhibit synthesis of new protein), we observed enhanced degradation of EZH2 in the presence of TGFβ (Figure 3C). In order to evaluate for potential routes of EZH2 degradation, we treated cholangiocytes with TGFβ in the presence of MG132, an inhibitor of the ubiquitin proteasome system, E64D (a cysteine protease inhibitor), or bafilomycin (an autophagy inhibitor). MG132 increased basal levels of EZH2 and also significantly rescued EZH2 from the degradation induced by TGFβ. This was associated with reduced levels of both cellular and secreted FN (Figure 3D), while E64D and bafilomycin had no effect. Overall, these data suggest that the ubiquitin proteasome system is the primary system for EZH2 degradation in this context.
Figure 3.
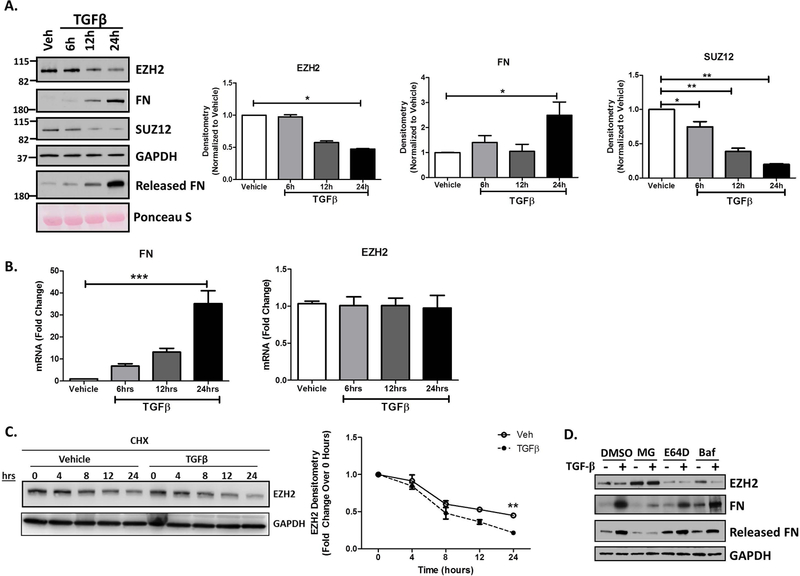
TGFβ Stimulation Leads to EZH2 Protein Degradation in Cholangiocytes.
A. Western blotting on H69 cells treated with TGFβ for 6, 12, and 24 hours show a time-dependent increase in FN production as well as its release into the media. This was seen in conjunction with a decrease in EZH2 and SUZ12 protein at 12 hours with a 50% decrease at 24 hours (left). Densitometry for FN, EZH2 and SUZ12 show statistically significant differences compared to vehicle at the 24 hour time point. * p<0.01. All error bars are SEM, n=3.
B. RT-PCR analysis for FN and EZH2 gene transcription in H69 cells treated with TGFβ. FN gene transcription increases with time after TGFβ treatment with no change in EZH2 gene transcription. ***p<0.0001. All error bars are SEM, n=3.
C. Western blot for EZH2 in cholangiocytes treated with cycloheximide (CHX: 40µg/ml) for the indicated time (hours) in the presence of vehicle or TGFβ (left). Right shows the rate of EZH2 protein turnover with CHX in the presence of vehicle or TGFβ. **p<0.001. All error bars are SEM, n=4
D. Western blotting on lysates of H69 cells treated with vehicle or TGFβ in presence of various inhibitors (MG-132, E64D, and Bafilomycin with DMSO as control). Only inclusionof MG-132 rescued EZH2 levels with concurrent decrease in FN levels.
EZH2 Degradation Occurs via the Ubiquitin Proteasome System Downstream of TGFβ:
In order to confirm and quantify the degree to which EZH2 degradation occurs via the ubiquitin proteasome system, we inhibited the proteasome with the two small molecule inhibitors, MG132 or bortezomib. In response to MG132, the TGFβ induced loss of EZH2 was rescued by 2-fold and this led to a 7-fold reduction in FN levels, both within the cells and in the culture media (Figure 4A). We confirmed this effect using the more clinically-relevant proteasome inhibitor, bortezomib, which is currently in clinical use for the treatment of lymphoma (28) (Figure 4B). This agent showed a corresponding 4-fold increase in EZH2 levels and and 3-fold reduction in FN. Chromatin IP (ChIP) assays identified a specific FN promoter element in which EZH2 association as well as the catalyzed H3K27me3 is diminished by TGFβ (Figure 4C and Supplementary Figure 2A). Based on this data, we postulated that under basal conditions EZH2 epigenetically silences FN transcription through its intrinsic histone methyltransferase activity and that TGFβ disrupts this cascade through proteasomal degradation of the EZH2 enzyme. Supporting this view, ChIP assays on cholangiocytes treated with or without TGFβ and in the presence or absence of MG132 show a reduction in the H3K27me3 silencing mark at the FN promoter upon TGFβ treatment which is restored in presence of MG132 (Figure 4D). The reduction in the repressive mark correlates with an increase in FN mRNA transcription and rescuing the H3K27me3 mark with MG132 also suppresses FN transcription (Figure 4E). Collectively, these results strongly suggest that EZH2 is degraded by the ubiquitin proteasome system in response to TGFβ and this results in derepression of FN.
Figure 4.
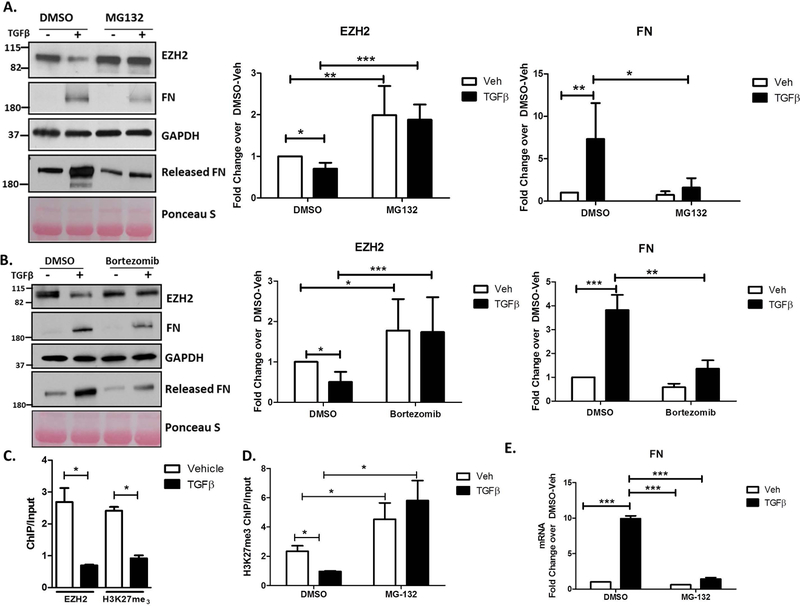
EZH2 Degradation Occurs via the Ubiquitin Proteasome System Downstream of TGFβ.
A. The lysates of H69 cells treated with vehicle or TGFβ in the presence of DMSO or MG-132 were blotted for EZH2, FN, and GAPDH (loading control) and the cell media was blotted for released FN with Ponceau Stain as a loading control for the media (left). Densitometry for EZH2 and FN show rescue of EZH2 levels in presence of MG-132 and concomitant decrease in FN production (right). * p<0.01, **p<0.001, ***p<0.0001. All error bars are SEM, n=4.
B. Protein lysates from H69 cells treated with vehicle or TGFβ in the presence of the clinically-relevent proteasome inhibitor, Bortezomib or DMSO were immunoblotted for EZH2, FN, and GAPDH (loading control) and the cell media was blotted for released FN with Ponceau Stain as loading control for the media (left). Densitometric quantification reveals recue of EZH2 protein levels with Bortezomib treatment and a concurrent decrease in FN levels (right). * p<0.01, **p<0.001, ***p<0.0001. All error bars are SEM, n=3.
C. Cells treated with vehicle or TGFβ were analyzed by ChIP for the H3K27me3 mark and EZH2 occupancy on the FN promoter. The assay demonstrates reduced levels of the H3K27me3 mark consistent with reduced EZH2 occupancy on the FN promoter. * p<0.01. All error bars are SEM, n=3.
D. H69 treated with vehicle or TGFβ in the presence of DMSO or MG-132 were analyzed by ChIP for the H3K27me3 mark on the FN promoter. MG-132 rescues TGFβ-mediated reduction in H3K27me3. * p<0.01. All error bars are SEM, n=3.
E. RT-PCR analysis for FN on H69 cells treated with vehicle or TGFβ in the presence of DMSO or MG-132. The TGFβ-mediated increase in FN gene transcription is abrogated in the presence of MG-132. ***p<0.0001. All error bars are SEM, n=3.
The N-Terminus of EZH2 is Poly-Ubiquitinated by Through K63-Based Ubiquitin Linkage:
Since proteasomal degradation is largely driven by post-translational ubiquitination, we hypothesized that EZH2 is ubiquitinated by an E3 ubiquitin ligase downstream of TGFβ to target its degradation at the proteasome. In order to evaluate whether TGFβ leads to ubiquitination of EZH2, cholangiocyte lysates treated with vehicle or TGFβ were subjected to Tandem Ubiquitin Binding Entity (TUBE) assays. Upon pull-down of all ubiquitinated proteins using TUBEs, immunoblotting for EZH2 indicates about 2-fold increase in pull-down of EZH2 from TGFβ treated cells, confirming ubiquitination of EZH2 (Figure 5A). Additionally, EZH2 contains an N-terminal WD repeat domain which is a known ubiquitin ligase binding motif (29, 30). Using an EZH2 N-terminal truncation mutant that lacks the first 100 amino acids (including the WD repeat domain), we found that the TGFβ induced overexpression of FN is markedly diminished (5-fold increase to 1.5-fold), suggesting that FN transcription is repressed when EZH2 is protected from degradation through N-terminal truncation (Figure 5B). Furthermore, while we detected degradation of EZH2 following TGFβ treatment in the wild-type condition, EZH2 was not degraded in the N-terminal deletion group (Figure 5B). Subsequently, we used transfected ubiquitin mutants to define whether the poly-ubiquitination of EZH2 occurs through K63-based or K48-based ubiquitin linkage. The K48 ubiquitin construct lacks all lysine resiudes except K48 while the K63 construct lacks all lysine residues except K63. We found that both the wild-type and the K63 constructs led to poly-ubiquitination of EZH2, while the K48 construct was unable to poly-ubiquitinate EZH2 (Figure 5C). This suggested that the poly-ubiquitination of EZH2 occurs through K63-based ubiquitin linkage.
Figure 5.
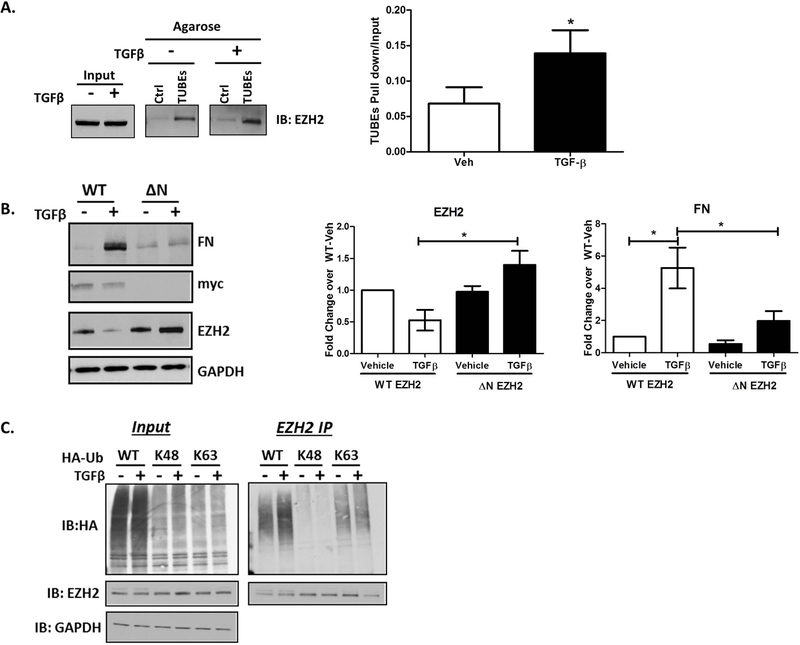
The N-Terminus of EZH2 is Poly-Ubiquitinated by Through K63-Based Ubiquitin Linkage.
A. Lysates of vehicle or TGFβ treated H69 cells were incubated with control (Ctrl) or TUBEs agarose beads. Immunoblotting for EZH2 on the beads reveals increased EZH2 pull-down in the presence of TGFβ (left). Densitometry on the right show a 1.5-fold increase in EZH2 binding to TUBEs agarose in presence of TGFβ. * p<0.01. All error bars are SEM, n=3.
B. H69 cells were transfected with N-terminal, myc-tagged, full-length EZH2 (WT) or N-terminal deletion mutant of EZH2 (ΔN). Lysates of transfected cells treated with vehicle or TGFβ were immunoblotted for FN, myc, EZH2, and GAPDH (loading control). Densitometry reveals that TGFβ-induced loss of WT EZH2 protein was abolished with deletion of the N-terminal 100 residues, which further blocked TGFβ-stimulated FN production. * p<0.01. All error bars are SEM, n=3.
C. H69 cells transfected with WT, K48, or K63 HA-Ubiquitin (Ub) were treated with either vehicle or TGFβ. Lysates were immunoprecipitated with EZH2 antibody and immunoblotted for HA and EZH2. HA blot on immunoprecipitates demonstrate EZH2 ubiquitination with WT HA-Ub and K63 HA-Ub.
EZH2 is Ubiquitinated by the E3 Ubiquitin Ligase, UBR4:
In order to gain further insight into the EZH2 ubiquitination mechanism, we treated cholangiocytes with vehicle or TGFβ for 6 hours and EZH2 complexes were isolated from these cells by immunoprecipitation. The immunoprecipitates were subjected to SDS-PAGE, coomassie-stained, gel extracted, trypsin digested, and identified by liquid chromatography tandem mass spectroscopy (LC-MS/MS). Using this approach, we confirmed the presence of SUZ12 and EED, canonical members of the PRC2 silencing complex (Figure 6A) in our cells. EnrichR pathway analysis of the proteins in complex with EZH2 identified the “Ubiquitin Proteasomal System” as the most highly represented pathway (Figure 6B). Indeed, we identified that EZH2 binds several proteins involved in the ubiquitination process (Figure 6C). Among the co-immunoprecipitated proteins, we identified ubiquitin protein ligase E3 component N-Recognin 4 (UBR4), an E3 ubiquitin ligase that marks proteins with ubiquitin at their N-termini for subsequent proteasomal degradation. Western blotting of EZH2 immunoprecipitates validated the presence of an EZH2/UBR4 complex that is enhanced by 60% in response to TGFβ (Figure 6D). Similarly, immunoprecipitation of UBR4 also showed enrichment of EZH2 in the pull-downs after TGFβ (Figure 6E). These studies are clinically important because drug targeting of ubiquitin ligases is gaining wider appeal (31).
Figure 6.
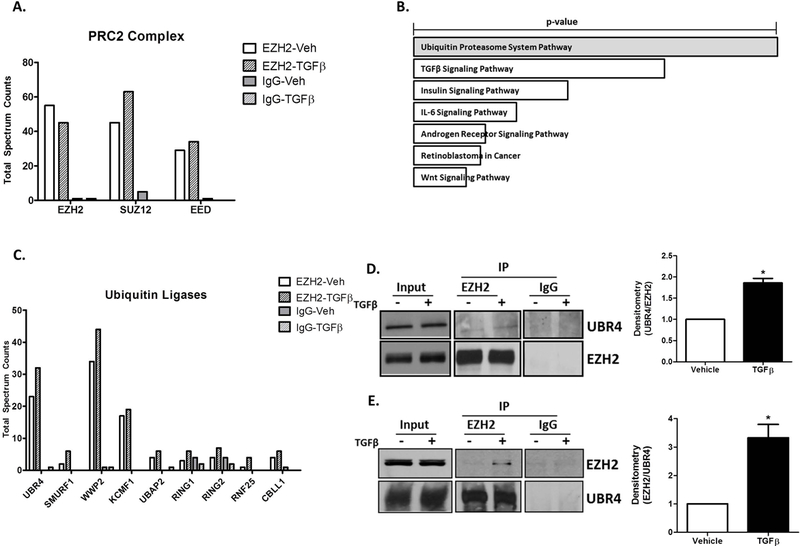
EZH2 is Ubiquitinated by the E3 Ubiquitin Ligase, UBR4.
A. Total Spectrum Counts of PRC2 complex components; EZH2, EED, and SUZ12 identified by mass spectrometry in EZH2 and IgG immunoprecipitates from vehicle and TGFβ treated H69 cells.
B. EnrichR analysis on proteins co-immunoprecipitated with EZH2 identified the Ubiquitin Proteasome System and TGFβ Signaling as top pathways (p-vaue represents Fisher exact test).
C. Total Spectrum Counts of the Ubiquitin ligases co-immunoprecipitated with EZH2 and IgG (control) from H69 cells treated with vehicle or TGFβ. UBR4: Ubiquitin Protein Ligase E3 Component N-Recognin 4, SMURF1: SMAD Specific E3 Ubiquitin Protein Ligase 1, WWP2: WW Domain Containing E3 Ubiquitin Protein Ligase 2, KCMF1: Potassium Channel Modulatory Factor 1, UBAP2: Ubiquitin Associated Protein 2, RING1: Ring Finger Protein 1, RING2: Ring Finger Protein 2, RNF25: Ring Finger Protein 25, CBLL1: Cbl Proto-Oncogene Like 1.
D. Western blotting on EZH2 and IgG immunoprecipitates from H69 cells treated with vehicle or TGFβ confirms the presence of UBR4 (left). Densitometry on the right shows a1.6-fold increase in UBR4 co-immunoprecipitated with EZH2. *p<0.01. All error bars are SEM, n=4.
E. Western blotting on UBR4 and IgG immunoprecipitates from H69 cells treated with vehicle or TGFβ confirms EZH2 in complex with UBR4 (left). Ddensitometric quantification demonstrates a 3-fold increase in EZH2 co-immunoprecipitated with UBR4 after TGF β. *p<0.01. All error bars are SEM, n=4.
Cholangiocyte Conditioned Media Supports Hepatic Stellate Cell Activation:
In order to investigate cholangiocyte - myofibroblast communication, we incubated human HSCs (hHSC) or portal fibroblasts (RGF-1) with conditioned media (CM) from cholangiocytes treated with vehicle or TGFβ in the presence or absence of MG-132. CM from TGFβ-treated cholangiocytes activated the HSCs as assessed by collagenα1 and α-SMA levels (Supplementary Figure 3A). This increase was attenuated by CM from MG-132 treated cholangiocytes, supporting a role for proteasomal degradation of EZH2. Furthermore, inclusion of the RGD peptide (which interferes with the interaction of FN and β1 integrin) also attenuated HSC activation, supporting a role for the FN-integrin interaction. Interestingly, these effects were much less prominent in the portal fibroblast cell line, RGF-1 (Supplementary Figure 3B), supporting the specificity of this particular mechanism.
Cholangiocyte-Selective Knockout of EZH2 Exacerbates Biliary Fibrosis:
In order to evaluate the effects of cholangiocyte EZH2 during biliary fibrosis in vivo, we generated a cholangiocyte-selective EZH2 knockout animal and subjected the animals to BDL. We crossed the EZH2fl/fl mouse (provided by Raul Urrutia, M.D.; Medical College of Wisconsin) with a mouse harboring a tamoxifen-inducible Cre recombinase driven by a cholangiocyte-selective promoter (32), KRT19-CreERTLSLTdTomato (provided by Stuart Forbes, Ph.D.; University of Edinburgh), to generate EZH2fl/fl/KRT19-CreERTLSLTdTomato mice. In addition to tamoxifen-inducible, cholangiocyte-selective knockout of EZH2 (Supplementary Figure 4A), this mouse has permanent expression of TdTomato (RFP) in Cre-positive cells to facilitate identification in histological sections (Supplementary Figure 4B). EZH2fl/fl animals (control group) or EZH2fl/fl/KRT19-CreERTLSLTdTomato animals (experimental group) were injected with tamoxifen daily for five days followed by sham or BDL surgery. After two weeks, control mice subjected to BDL showed peri-portal fibrosis as assessed by picro Sirius red (Figure 7A) and Masson’s trichrome staining (Figure 7B). Deletion of EZH2 in cholangiocytes exacerbated the fibrosis induced by BDL. Quantitatively, there was a 1.5-fold increase in Sirius red positve area and a 2.5-fold increase in Trichrome positive area in the EZH2-deleted animals (Figure 7B). mRNA analysis on the whole liver tissue confirmed increases in collagen1A1 and α-SMA gene transcription in EZH2 knockout mice compared to control mice upon BDL surgery (Figure 7C). Furthermore, laser capture microdissection of bile ducts followed by RT-PCR confirmed that cholangiocyte FN expression is significantly increased following EZH2 knockout (Figure7D). Immunofluorescence for FN and CK19 also confirmed increased FN with BDL in EZH2 deleted mice (Supplementary Figure 5A). Hydroxyproline assays on whole liver tissue confirmed increased collagen following BDL in EZH2-deleted animals (Figure7D). Liver biochemistires (ALT and total bilirubin) were mildly elevated in animals lacking EZH2 in cholangiocytes (Supplementary Figure 5B). Additional analysis showed increases in CTGF tissue staining as well as inflamatory transcripts for IL-1β and MCP-1 (but not CD68 and F4/80). This suggests the possibility of increased monocyte recruitment upon EZH2 deletion (Supplementary Figure 5C and 5D).
Figure 7.
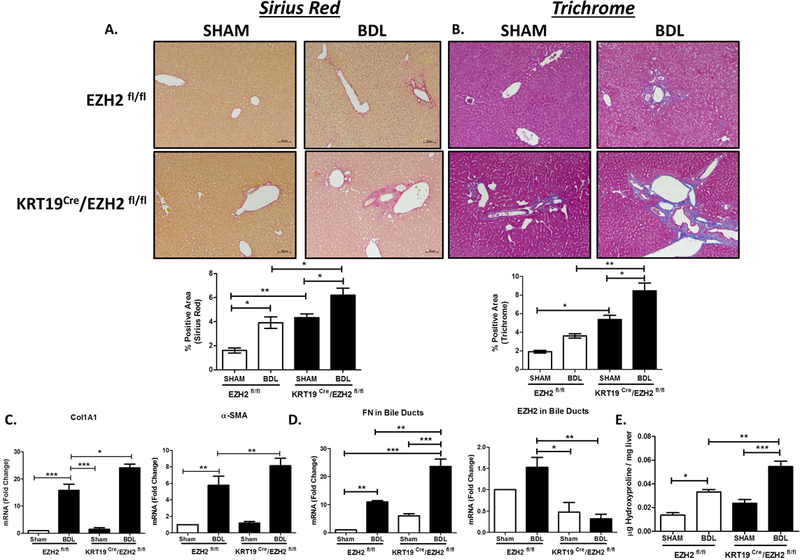
Cholangiocyte-Selective Knockout of EZH2 Exacerbates Biliary Fibrosis.
A. Liver histology shows Sirius red staining on EZH2fl/fl and KRT19CRE/EZH2fl/fl animals with sham or BDL surgery (Top). Bottom shows quantification of % positive area for Sirius red (n=4–5 animals per group). *p<0.01, **p<0.001 All error bars are SEM.
B. Liver histology showing Trichrome staining on EZH2fl/fl and KRT19CRe/EZH2fl/fl animals with sham or BDL surgery (Top). Bottom shows quantification of % positive area for Trichrome (n=4–5 animals per group). *p<0.01, **p<0.001. All error bars are SEM.
C. RT-PCR analysis for collagen1A1 and alpha-SMA gene expression in whole liver tissue from EZH2fl/fl and KRT19 Cre/EZH2fl/fl animals with Sham and BDL surgery (n=4–5 animals per group). *p<0.01, **p<0.001, ***p<0.0001. All error bars are SEM.
D. RT-PCR analysis for FN1 and EZH2 in isolated bile ducts using LCM from EZH2fl/fl and KRT19 Cre/EZH2fl/fl animals with Sham and BDL surgery (n=4–5 animals per group). *p<0.01, **p<0.001, ***p<0.0001. All error bars are SEM.
E. Hydroxyproline analysis on liver tissue from EZH2 EZH2fl/fl and KRT19 Cre/EZH2fl/fl animals with Sham and BDL surgery (n=4–5 animals per group). *p<0.01, **p<0.001, ***p<0.0001. All error bars are SEM.
Proteasome Inhibition with Bortezomib Restores EZH2 Protein and Reduces Fibrosis in Mdr−/− Mice:
To test our findings in a second in vivo model, we utilized the Mdr2 knockout mouse (Mdr2−/−). Mdr2−/− is a well-known and commonly-used murine model of biliary fibrosis in which TGFβ is prominently activated. Mdr2−/− mice spontaneously develop progressive biliary fibrosis throughout their lifetime (Supplementary Figure 2). Therefore, young mice (< 8 weeks) have limited fibrosis while older mice (> 8 weeks) have significant peri-portal fibrosis. To evaluate the role of proteasomal degradation of EZH2 in vivo, we first evaluated the levels of EZH2 protein in nuclear extracts from Mdr2−/− liver. We found that EZH2 levels were negatively correlated with fibrosis, with a 4-fold reduction in EZH2 in fibrotic versus non-fibrotic Mdr2−/− mice (Figure 8A). Next, we treated Mdr−/− mice with the proteasome inhibitor, bortezomib (1 mg / kg, i.p. for 3 weeks). Western blot analysis of the liver nuclear fraction showed that bortezomib rescued the levels of EZH2 protein (Figure 8B), but did not affect EZH2 mRNA levels (Figure 8C). In mice treated with bortezomib, there was also a 3.5-fold reduction in liver fibrosis as assessed by Sirius red and a corresponding 4-fold reduction based on trichrome staining (Figure 8D). Further supporting the pathobiological relevance of this mechanism, we also observed that fibrotic human livers from patients with primary sclerosing cholangitis demonstrate activation of TGFβ (Supplementary Figure 3A) and a corresponding loss of nuclear EZH2 (Supplementary Figure 3B) as well as it’s epigenetic silencing mark, H3K27me3 (Supplementary Figure 3C), by immunohistochemistry.
Figure 8.
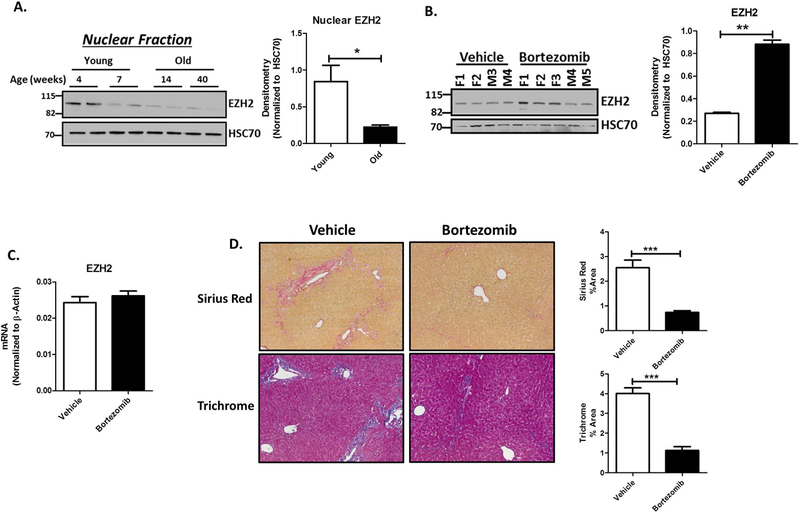
Proteasome Inhibition Restores EZH2 Protein and Reduces Fibrosis in Mdr−/− Mice.
A. Western blotting on nuclear fractions of liver lysates from young (4 and 7 weeks) and old (14 and 40 weeks) Mdr2−/− mice show decreasing levels of EZH2 protein (left) with HSC70 as a loading control. Densitometry on the right shows a 3-fold decrease in EZH2 levels. *p<0.01. All error bars are SEM, n=4.
B. Mdr2−/− mice were treated with 1mg/kg Bortezomib from 7weeks to 11 weeks of age (males and females). Immunoblotting for EZH2 on nuclear fraction of liver lysates from both vehicle and Bortezomib treated mice show an increase in EZH2 protein levels with Bortezomib treatment (left), F: female, M: Male. Densitometry on the right shows 3-fold increase EZH2 protein when normalized to HSC70 as loading control. **p<0.001. All error bars are SEM, n=3.
C. RT-PCR analysis on the liver tissue from vehicle and Bortezomib treated Mdr2−/− mice show no change in EZH2 gene expression. All error bars are SEM, n=3.
D. Liver histology from Mdr2−/− mice treated with vehicle or Bortezomib show reduced fibrosis by Sirius red and Trichrome staining (left). Right shows the quantification for the % positive area for Sirius red and trichrome staining. ***p<0.0001. All error bars are SEM, n=4–5 animals per group.
Discussion
Recent conceptual advances support an important role for epithelial / mesenchymal cross-talk between cholangiocytes and myofibroblasts as a key driver of biliary fibrosis (6, 33). However, important knowledge gaps remain as to how various pathological signaling events lead to modifications in the cholangiocyte epigenome to influence the peribiliary fibrotic process. In this context, the current study provides several novel advances: 1) TGFβ signaling induces an HSC-activating gene program in cholangiocytes via an epigenetic mechanism; 2) EZH2 protein is degraded via the ubiquitin proteasome system in response to TGFβ, leading to de-repression of key paracrine regulators of fibrosis including FN; 3) the E3 ubiquitin ligase, UBR4, ubiquitinates EZH2, marking the protein for degradation at the proteasome; and 4) inducible, cholangiocyte-selective knockout of EZH2 exacerbates biliary fibrosis while proteasomal inhibition rescues EZH2 levels in cholangiocytes and protects animals from biliary fibrosis. While our studies corroborate prior concepts related to a pro-fibrogenetic phenotype mediated by TGF-β, the primary significance of these studies lies in the novel paradigm that epigenetic regulators allow specific membrane signals in diseased cholangiocytes to re-write the histone code. These changes generate fibrogenic paracrine molecules that promote myofibroblast activation and progression of biliary fibrosis. In turn, interventions targeting these newly discovered pathways may have the capability to prevent, delay, or reverse the extracellular matrix events that drive biliary fibrosis.
Our genome-wide RNA-seq experiments confirm activation of a network of genes within cholangiocytes that promote myofibroblast activation, including FN, CTGF, and others. We logically extended these results by performing tertiary bioinformatics to identify a subset of cholangiocyte gene targets that are activated by TGFβ and also regulated by EZH2 through H3K27me3. By cross-referencing large-scale expression analysis (RNA-seq) with an analysis of the epigenetic landscape using ChIP assays, we are able to dissect how TGFβ signaling is coupled to specific chromatin modifications in order to regulate distinct gene expression networks relevant to fibrosis. This is important since epigenetic pharmacology may allow targeted interventions that partially inactivate TGFβ signaling by attenuating specific gene networks that are regulated through H3K27me3. Indeed, histone modifications can represent critical portions of a biological or pathobiological pathway that affects gene transcription. As we demonstrate in this study, key epigenetic regulators, such as EZH2, whose primary role is in facultative gene repression during development, can also regulate and fine-tune pathobiological signaling cascades to drive aberrant gene programs in disease. In this case, loss of EZH2 through ubiquitination and proteasomal degradation leads to dynamically less H3K27me3 and de-repression of the downstream gene network.
Fibronectin (FN), a key constituent of the ECM, is a glycoprotein produced and secreted by several cell types in the liver and its expression is associated with hepatic fibrosis, cellular damage, differentiation, and repair (7, 8). In this context, FN production by activated epithelial cells is an important and potentially reversible step in the progression of biliary fibrosis (34, 35). Cholangiocyte-derived FN constitutes an important paracrine molecule in cirrhosis that is subsequently converted by HSC from a soluble protein into an insoluble matrix constituent (36). Indeed, FN is one of the first matrix molecules to appear in the fibrotic liver and accelerates the subsequent steps of liver fibrosis (26). Our data reiterates that the FN-integrin interaction plays an important role in HSC activation. Especially with recent evidence indicating that there is substantial reversibility of many aspects of fibrosis (37), a better understanding of how these early matrix remodeling steps progress is of major significance.
There have been a few other reports of EZH2 ubiquitination in other cell types (38–41). Several ubiquitin ligases such as β-TrCP (40), TRAF6 (41), and Smurf2 (38) have been implicated in various contexts. Our study defines the biochemical steps in cholangiocytes by which UBR4 poly-ubiquitinates the N-terminus of EZH2 through K63-based ubiquitin linkage, marking the protein for subsequent degradation at the proteasome. This process contributes to activation of a subset of pro-fibrogenic genes in cholangiocytes that support HSC activation and promote biliary fibrosis. To our knowledge, this is the first study to demonstrate UBR4-based ubiquitination of EZH2. We also found that proteasome inhibition with bortezomib prevents BDL-induced liver fibrosis in mice. As such, proteasomal degradation of EZH2 may represent a new and potentially generalizable mechanism of polycomb regulation that could be targeted clinically.
It is important to note that epigenetic regulators, including EZH2, have described roles in other liver cell types beyond cholangiocytes (42–44). In fact, our group and others have previously shown a pro-fibrotic role for EZH2 in HSCs (19, 45). EZH2 represses peroxisome proliferator-activated receptor gamma, which is a negative master regulator of HSC activation (46). Furthermore, targeted delivery of the histone methyltransferase inhibitor 3-deazaneplanocin A, selectively to HSCs, using an antibody-liposome targeting vehicle, attenuated carbon tetrachloride-induced liver fibrosis in mice (47). In contrast, the current results demonstrate that pharmacologic inhibition of EZH2 degradation is actually protective during biliary fibrosis while selective deletion of EZH2 in cholangiocytes exacerbates biliary fibrosis, consistent with our previous finding (48). EZH2 also has complex mechanisms of action within hepatocytes, including multiple roles in the development of hepatocellular carcinoma (49). This cell-type specificity with respect to epigenetic regulators is not surprising, and in fact, epigenetic differences truly define cellular identity (50). Overall, our findings suggest that the epigenetic regulator, EZH2, is selectively targetted for proteasomal degradation downstream of TGFβ in cholangiocytes. This drives transcription of a pro-fibrotic gene network that supports hepatic stellate cell activation. Ultimately, the use of epigenetic pharmacology may provide more specific anti-fibrotic therapies for biliary fibrosis with fewer off-target effects than targeting TGFβ signaling directly.
Supplementary Material
Acknowledgements
The authors acknowledge Thomas Greuter for technical assistance, and Deb Hintz for secretarial services.
Financial Support: This work was supported by grants DK100575, DK113339, DK117861, and DK084567 from the National Institutes of Health; by the Satter Foundation; and the Second Hospital of Jilin University.
List of Abbreviations:
- TGFβ
Transforming growth factor-β
- EZH2
enhancer of zeste homologue 2
- H3K27me3
tri-methylation of lysine 27 on histone 3
- FN
fibronectin 1
- UBR4
ubiquitin protein ligase E3 component N-recognin 4
- PSC
primary sclerosing cholangitis
- HSC
hepatic stellate cells
- ECM
extracellular matrix
- PRC2
polycomb repressive complex 2
- NHC
normal human cholangiocytes
- HiBEC
human intrahepatic biliary epithelial cells
- ChIP
chromatin immunoprecipitation
- CTGF
connective tissue growth factor
- VEGF
vascular endothelial growth factor
- PDGF
platelet-derived growth factor
- TLCA
taurolithocholic acid
- LPS
lipopolysaccharide
- IL-6
interleukin-6
- TUBE
tandem ubiquitin binding entity
References
- 1.Abshagen K, Konig M, Hoppe A, Muller I, Ebert M, Weng H, Holzhutter HG, et al. Pathobiochemical signatures of cholestatic liver disease in bile duct ligated mice. BMC Syst Biol 2015;9:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fabris L, Spirli C, Cadamuro M, Fiorotto R, Strazzabosco M. Emerging Concepts in Biliary Repair and Fibrosis. Am J Physiol Gastrointest Liver Physiol 2017:ajpgi 00452 02016. [DOI] [PMC free article] [PubMed]
- 3.Lazaridis KN, LaRusso NF. Primary Sclerosing Cholangitis. N Engl J Med 2016;375:2501–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones H, Hargrove L, Kennedy L, Meng F, Graf-Eaton A, Owens J, Alpini G, et al. Inhibition of mast cell-secreted histamine decreases biliary proliferation and fibrosis in primary sclerosing cholangitis Mdr2(−/−) mice. Hepatology 2016;64:1202–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pi L, Robinson PM, Jorgensen M, Oh SH, Brown AR, Weinreb PH, Trinh TL, et al. Connective tissue growth factor and integrin alphavbeta6: a new pair of regulators critical for ductular reaction and biliary fibrosis in mice. Hepatology 2015;61:678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fabris L, Strazzabosco M. Epithelial-mesenchymal interactions in biliary diseases. Semin Liver Dis 2011;31:11–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Altrock E, Sens C, Wuerfel C, Vasel M, Kawelke N, Dooley S, Sottile J, et al. Inhibition of fibronectin deposition improves experimental liver fibrosis. J Hepatol 2015;62:625–633. [DOI] [PubMed] [Google Scholar]
- 8.Liu XY, Liu RX, Hou F, Cui LJ, Li CY, Chi C, Yi E, et al. Fibronectin expression is critical for liver fibrogenesis in vivo and in vitro. Mol Med Rep 2016;14:3669–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alberts R, de Vries EMG, Goode EC, Jiang X, Sampaziotis F, Rombouts K, Bottcher K, et al. Genetic association analysis identifies variants associated with disease progression in primary sclerosing cholangitis. Gut 2017. [DOI] [PMC free article] [PubMed]
- 10.Cheung AC, LaRusso NF, Gores GJ, Lazaridis KN. Epigenetics in the Primary Biliary Cholangitis and Primary Sclerosing Cholangitis. Semin Liver Dis 2017;37:159–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gressner AM, Weiskirchen R, Breitkopf K, Dooley S. Roles of TGF-beta in hepatic fibrosis. Front Biosci 2002;7:d793–807. [DOI] [PubMed] [Google Scholar]
- 12.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest 2005;115:209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aranda S, Mas G, Di Croce L. Regulation of gene transcription by Polycomb proteins. Sci Adv 2015;1:e1500737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O’Meara MM, Simon JA. Inner workings and regulatory inputs that control Polycomb repressive complex 2. Chromosoma 2012;121:221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grzenda A, Lomberk G, Svingen P, Mathison A, Calvo E, Iovanna J, Xiong Y, et al. Functional characterization of EZH2beta reveals the increased complexity of EZH2 isoforms involved in the regulation of mammalian gene expression. Epigenetics Chromatin 2013;6:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prachayasittikul V, Prathipati P, Pratiwi R, Phanus-Umporn C, Malik AA, Schaduangrat N, Seenprachawong K, et al. Exploring the epigenetic drug discovery landscape. Expert Opin Drug Discov 2017;12:345–362. [DOI] [PubMed] [Google Scholar]
- 17.Fausther M, Goree JR, Lavoie EG, Graham AL, Sevigny J, Dranoff JA. Establishment and Characterization of Rat Portal Myofibroblast Cell Lines. Plos One 2015;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Assuncao TM, Sun Y, Jalan-Sakrikar N, Drinane MC, Huang BQ, Li Y, Davila JI, et al. Development and characterization of human-induced pluripotent stem cell-derived cholangiocytes. Laboratory investigation; a journal of technical methods and pathology 2015;95:684–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosa Martin-Mateos TMDA, Arab Juan Pablo, Jalan-Sakrikar Nidhi, Yaqoob Usman, Greuter Thomas, Verma Vikas K., Mathison Angela J., Cao Sheng, Lomberk Gwen, Mathurin Philippe, Urrutia Raul, Huebert Robert C., Shah Vijay H. Enhancer of Zeste Homologue 2 Inhibition Attenuates TGF-β Dependent Hepatic Stellate Cell Activation and Liver Fibrosis. Cellular and Molecular Gastroenterology and Hepatology 2019;7:197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Babicki S, Arndt D, Marcu A, Liang Y, Grant JR, Maciejewski A, Wishart DS. Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res 2016;44:W147–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 2013;14:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 2016;44:W90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tag CG, Sauer-Lehnen S, Weiskirchen S, Borkham-Kamphorst E, Tolba RH, Tacke F, Weiskirchen R. Bile duct ligation in mice: induction of inflammatory liver injury and fibrosis by obstructive cholestasis. J Vis Exp 2015. [DOI] [PMC free article] [PubMed]
- 24.Sedlaczek N, Jia JD, Bauer M, Herbst H, Ruehl M, Hahn EG, Schuppan D. Proliferating bile duct epithelial cells are a major source of connective tissue growth factor in rat biliary fibrosis. Am J Pathol 2001;158:1239–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhan S, Chan CC, Serdar B, Rockey DC. Fibronectin stimulates endothelin-1 synthesis in rat hepatic myofibroblasts via a Src/ERK-regulated signaling pathway. Gastroenterology 2009;136:2345–2355 e2341–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olsen AL, Sackey BK, Marcinkiewicz C, Boettiger D, Wells RG. Fibronectin extra domain-A promotes hepatic stellate cell motility but not differentiation into myofibroblasts. Gastroenterology 2012;142:928–937 e923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McDaniel K, Meng F, Wu N, Sato K, Venter J, Bernuzzi F, Invernizzi P, et al. Forkhead box A2 regulates biliary heterogeneity and senescence during cholestatic liver injury in micedouble dagger. Hepatology 2017;65:544–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Field-Smith A, Morgan GJ, Davies FE. Bortezomib (Velcadetrade mark) in the Treatment of Multiple Myeloma. Ther Clin Risk Manag 2006;2:271–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Han Z, Xing X, Hu M, Zhang Y, Liu P, Chai J. Structural basis of EZH2 recognition by EED. Structure 2007;15:1306–1315. [DOI] [PubMed] [Google Scholar]
- 30.Jin X, Yang C, Fan P, Xiao J, Zhang W, Zhan S, Liu T, et al. CDK5/FBW7-dependent ubiquitination and degradation of EZH2 inhibits pancreatic cancer cell migration and invasion. J Biol Chem 2017;292:6269–6280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Popovic D, Vucic D, Dikic I. Ubiquitination in disease pathogenesis and treatment. Nat Med 2014;20:1242–1253. [DOI] [PubMed] [Google Scholar]
- 32.Raven A, Lu WY, Man TY, Ferreira-Gonzalez S, O’Duibhir E, Dwyer BJ, Thomson JP, et al. Cholangiocytes act as facultative liver stem cells during impaired hepatocyte regeneration. Nature 2017;547:350–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rygiel KA, Robertson H, Marshall HL, Pekalski M, Zhao L, Booth TA, Jones DE, et al. Epithelial-mesenchymal transition contributes to portal tract fibrogenesis during human chronic liver disease. Lab Invest 2008;88:112–123. [DOI] [PubMed] [Google Scholar]
- 34.Jarnagin WR, Rockey DC, Koteliansky VE, Wang SS, Bissell DM. Expression of variant fibronectins in wound healing: cellular source and biological activity of the EIIIA segment in rat hepatic fibrogenesis. J Cell Biol 1994;127:2037–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Massey VL, Dolin CE, Poole LG, Hudson SV, Siow DL, Brock GN, Merchant ML, et al. The hepatic “matrisome” responds dynamically to injury: Characterization of transitional changes to the extracellular matrix in mice. Hepatology 2017;65:969–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kadler KE, Hill A, Canty-Laird EG. Collagen fibrillogenesis: fibronectin, integrins, and minor collagens as organizers and nucleators. Curr Opin Cell Biol 2008;20:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iredale JP. Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest 2007;117:539–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu YL, Chou RH, Shyu WC, Hsieh SC, Wu CS, Chiang SY, Chang WJ, et al. Smurf2-mediated degradation of EZH2 enhances neuron differentiation and improves functional recovery after ischaemic stroke. EMBO Mol Med 2013;5:531–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu SC, Zhang Y. Cyclin-dependent kinase 1 (CDK1)-mediated phosphorylation of enhancer of zeste 2 (Ezh2) regulates its stability. J Biol Chem 2011;286:28511–28519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sahasrabuddhe AA, Chen X, Chung F, Velusamy T, Lim MS, Elenitoba-Johnson KS. Oncogenic Y641 mutations in EZH2 prevent Jak2/beta-TrCP-mediated degradation. Oncogene 2015;34:445–454. [DOI] [PubMed] [Google Scholar]
- 41.Lu W, Liu S, Li B, Xie Y, Izban MG, Ballard BR, Sathyanarayana SA, et al. SKP2 loss destabilizes EZH2 by promoting TRAF6-mediated ubiquitination to suppress prostate cancer. Oncogene 2017;36:1364–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koike H, Ouchi R, Ueno Y, Nakata S, Obana Y, Sekine K, Zheng YW, et al. Polycomb group protein Ezh2 regulates hepatic progenitor cell proliferation and differentiation in murine embryonic liver. PLoS One 2014;9:e104776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang Y, Chen XX, Li WX, Wu XQ, Huang C, Xie J, Zhao YX, et al. EZH2-mediated repression of Dkk1 promotes hepatic stellate cell activation and hepatic fibrosis. J Cell Mol Med 2017;21:2317–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bae WK, Kang K, Yu JH, Yoo KH, Factor VM, Kaji K, Matter M, et al. The methyltransferases enhancer of zeste homolog (EZH) 1 and EZH2 control hepatocyte homeostasis and regeneration. FASEB J 2015;29:1653–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang MD, Chiang YM, Higashiyama R, Asahina K, Mann DA, Mann J, Wang CC, et al. Rosmarinic acid and baicalin epigenetically derepress peroxisomal proliferator-activated receptor gamma in hepatic stellate cells for their antifibrotic effect. Hepatology 2012;55:1271–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mann J, Chu DC, Maxwell A, Oakley F, Zhu NL, Tsukamoto H, Mann DA. MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology 2010;138:705–714, 714 e701–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zeybel M, Luli S, Sabater L, Hardy T, Oakley F, Leslie J, Page A, et al. A Proof-of-Concept for Epigenetic Therapy of Tissue Fibrosis: Inhibition of Liver Fibrosis Progression by 3-Deazaneplanocin A. Mol Ther 2017;25:218–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jalan-Sakrikar N, De Assuncao TM, Lu J, Almada LL, Lomberk G, Fernandez-Zapico ME, Urrutia R, et al. Hedgehog Signaling Overcomes an EZH2-Dependent Epigenetic Barrier to Promote Cholangiocyte Expansion. PLoS One 2016;11:e0168266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu H, Liu Y, Liu W, Zhang W, Xu J. EZH2-mediated loss of miR-622 determines CXCR4 activation in hepatocellular carcinoma. Nat Commun 2015;6:8494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barrero MJ, Boue S, Izpisua Belmonte JC. Epigenetic mechanisms that regulate cell identity. Cell Stem Cell 2010;7:565–570. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.