

Abstract
Background
Drug-resistant tuberculosis (TB) is a major public health concern threathing the success of TB control efforts, and this is particularily problematic in Central Asia. Here, we present the first analysis of the population structure of Mycobacterium tuberculosis complex isolates in the Central Asian republics Uzbekistan, Tajikistan, and Kyrgyzstan.
Methods
The study set consisted of 607 isolates with 235 from Uzbekistan, 206 from Tajikistan, and 166 from Kyrgyzstan. 24-loci MIRU-VNTR (Mycobacterial Interspersed Repetitive Units - Variable Number of Tandem Repeats) typing and spoligotyping were combined for genotyping. In addition, phenotypic drug suceptibility was performed.
Results
The population structure mainly comprises strains of the Beijing lineage (411/607). 349 of the 411 Beijing isolates formed clusters, compared to only 33 of the 196 isolates from other clades. Beijing 94–32 (n = 145) and 100–32 (n = 70) formed the largest clusters. Beijing isolates were more frequently multidrug-resistant, pre-extensively resistant (pre-XDR)- or XDR-TB than other genotypes.
Conclusions
Beijing clusters 94–32 and 100–32 are the dominant MTB genotypes in Central Asia. The relative size of 100–32 compared to previous studies in Kazakhstan and its unequal geographic distribution support the hypothesis of its more recent emergence in Central Asia. The data also demonstrate that clonal spread of resistant TB strains, particularly of the Beijing lineage, is a root of the so far uncontroled MDR-TB epidemic in Central Asia.
Keywords: Molecular typing, MIRU, MDR-TB, Beijing, Cluster
Background
Drug-resistant tuberculosis (TB) is a public health concern threathing the success of TB control efforts. This is particularily problematic in the Central Asian Republics (CARs) Uzbekistan, Tajikistan, and Kyrgyzstan where multidrug-resistant (MDR)-TB rates reach 24%, 22%, 27%, and 63%, 45% and 60% among new and previously treated cases, respectively [1].
Little is known about the population structure of drug resistant Myocbacterium tuberculosis (MTB) strains in Central Asia. Available reports are limited by either outdated genotyping techniques, sampling of special populations and/or small sample sizes [2–4]. Here, we provide a snapshot of the population structure of MTB strains based on World Health Organization resistance surveys performed in Kyrgyzstan, Tajikistan, and Uzbekistan representing two thirds of the Central Asian population.
For genotyping we combined 24-loci MIRU-VNTR typing (Mycobacterial Interspersed Repetitive Units - Variable Number of Tandem Repeats) with spoligotyping. MIRU-VNTR provides high-resolution discrimination of strains for epidemiological studies and phylogenetic classification [5]. MIRU-VNTR data allow also to generate parsimonious phylogenetic networks such as minimum-spanning trees (MST) [6]. These results were combined with phenotypic drug susceptibility data to determine the presence of highly resistant dominant clones.
Methods
Seven hundred and seven clinical MTB isolates (306 from Uzbekistan, 216 from Tajikistan, and 185 from Kyrgyzstan) were collected in the framework of cross-sectional national drug resistance surveys (DRS) [7, 8] and included in the study. The study population was biased towards resistant strains since only isolates that required re-testing for quality control purposes as per the WHO DRS protocol which was valid at the time were included [8]. This approach artificially increased the proportion of MDR-,pre-extensively drug-resistant (pre-XDR) and XDR-TB strains. Ethical approvals for the DRSs were granted by the respective national ethical committees.
DNA was extracted by heat lysis of bacteria from solid culture on LJ medium. 24-loci MIRU-VNTR genotyping and spoligotyping was performed as previously described [5, 9]. Genotypes were identified on the MIRU-VNTRplus web database (www.miruvntrplus.org) [5]. Spoligotyping data were used to confirm strain relationships and for genotype classification. Typing data was analysed with Bionumerics (v7.5; Applied Maths, Sint-Martens-Latem, Belgium). MST analysis based on MIRU-VNTR data was performed using the categorical coefficient. For each 24-loci MIRU-VNTR pattern a unique multiple loci VNTR analysis (MLVA) MtbC15–9 haplotype was assigned by using the MIRU-VNTRplus nomenclature [10]. A cluster was defined as a minimum of two isolates harboring identical genotyping patterns from different patients. Patients with mixed molecular typing patterns were excluded. Drug susceptibility testing was performed by the resazurin microtiter assay [11].
Results
Twenty four-loci MIRU-VNTR typing and spoligotyping were successfully performed for 607 isolates, while 76 isolates (62 from Uzbekistan, seven from Tajikistan, and seven from Kyrgyzstan) yielded indeterminate results. For 23 isolates (9 from Uzbekistan, three from Tajikistan, and 11 from Kyrgyzstan), mixed genotyping patterns were observed. One isolate from Kyrgyzstan had incomplete phenotypic data.
The final study set consisted of 607 isolates of which 235 (38.7%) originated from Uzbekistan, 206 (33.9%) from Tajikistan, and 166 (27.3%) from Kyrgyzstan (Table 1). The male to female ratio of patients was 1.5 and ages ranged from 12 to 85 years with a mean of 37.9 years (SD ±15.7). Across the three countries, 333 (54.9%) were new and 221 (36.4%) previously treated cases (for 53 patients information regarding previous treatments was missing; Table 1). Thirty five isolates (5.8%) were pan-susceptible, 293 (48.3%) MDR-, pre-XDR, or XDR-TB, while the remaining 279 isolates (46.0%) had any other resistance pattern. MDR- and XDR-TB were more prominent among Kyrgyz (33%) and Tajik (35%) than among Uzbek isolates (20%), which was mainly attributable to different re-testing algorithms in the DRS protocols.
Table 1.
Demographic data, genotypes, haplotypes and phenotypes of the Central Asian M. tuberculosis complex study population
| All countries | Uzbekistan | Tajikistan | Kyrgystan | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. strains | % | SD | No. strains | % | SD | No. strains | % | SD | No. strains | % | SD | |
| Samples | 607 | 235 | 38.7 | 206 | 33.9 | 166 | 27.3 | |||||
| Gender | ||||||||||||
| Male | 359 | 59.1 | 139 | 59.1 | 120 | 58.3 | 100 | 60.2 | ||||
| Female | 239 | 39.4 | 96 | 40.9 | 85 | 41.3 | 58 | 34.9 | ||||
| Unknown | 9 | 0 | 1 | 8 | ||||||||
| Age a | ||||||||||||
| Mean | 37.94 | 15.67 | 42.91 | 16.6 | 34.53 | 14.36 | 34.18 | 13.49 | ||||
| Treatment | ||||||||||||
| New | 333 | 54.9 | 156 | 66.4 | 93 | 45.1 | 84 | 50.6 | ||||
| Previous | 221 | 36.4 | 79 | 33.6 | 113 | 54.9 | 29 | 17.5 | ||||
| Unknown | 53 | 0 | 0 | 53 | ||||||||
| MIRU lineage | ||||||||||||
| Beijing | 411 | 67.7 | 136 | 57.9 | 154 | 74.8 | 121 | 72.9 | ||||
| Dehli/CAS | 6 | 1 | 6 | 2.6 | ||||||||
| H37Rv-like | 60 | 9.9 | 31 | 13.2 | 12 | 5.8 | 17 | 10.2 | ||||
| LAM | 36 | 5.9 | 22 | 9.4 | 5 | 2.4 | 9 | 5.4 | ||||
| NEW-1 | 31 | 5.1 | 16 | 6.8 | 10 | 4.9 | 5 | 3 | ||||
| URAL | 28 | 4.6 | 10 | 4.3 | 11 | 5.3 | 7 | 4.2 | ||||
| Haarlem | 26 | 4.3 | 7 | 3 | 13 | 6.3 | 6 | 3.6 | ||||
| S-type | 3 | 0.5 | 2 | 0.9 | 1 | 0.6 | ||||||
| TUR | 1 | 0.2 | 1 | 0.5 | ||||||||
| X-type | 1 | 0.2 | 1 | 0.4 | ||||||||
| Undefined | 4 | 0.7 | 4 | 1.7 | ||||||||
| MLVA MtbC15–9 | ||||||||||||
| 94–32 (Beijing) | 145 | 23.9 | 58 | 24.7 | 49 | 23.8 | 38 | 22.9 | ||||
| 100–32 (Beijing) | 70 | 11.5 | 12 | 5.1 | 51 | 24.8 | 7 | 4.2 | ||||
| Resistance pattern | ||||||||||||
| Pan-susceptible | 35 | 5.8 | 21 | 8.9 | 2 | 1 | 12 | 7.2 | ||||
| Other resistances | 279 | 46 | 127 | 54 | 66 | 32 | 86 | 51.8 | ||||
| MDR-TB | 172 | 28.3 | 47 | 20 | 71 | 34.5 | 54 | 32.5 | ||||
| Pre XDR-TB | 94 | 15.5 | 34 | 14.5 | 50 | 24.3 | 10 | 6 | ||||
| XDR-TB | 27 | 4.4 | 6 | 2.6 | 17 | 8.3 | 4 | 2.4 | ||||
aUnkown for Tajikistan and 23 unkown for Kyrgystan
SD standard deviation
Based on the MIRU-VNTR profiles and spoligotyping patterns, 603 isolates were classified into previously described lineages (Table 1). 411 (67.7%) isolates belonging to lineage 2 (East-Asian; Beijing genotype) [12]. Six (1.0%) isolates belonged to lineage 3 (East-African-Indian; Delhi/CAS genotype), and 186 (30.6%) isolates belonged to lineage 4 (Euro-American) with the predominiating genotypes H37Rv-like (60; 9.9%), LAM (36; 5.9%), NEW-1 (31; 5.1%), URAL (28; 4.6%), and Haarlem (26; 4.3%). Four (0.7%) isolates could not be linked to a previously described lineage and were classified as “undefined”. An MST was calculated (Fig. 1) confirming the UPGMA (unweighted pair group method with arithmetic mean) tree-based genotype classification of MIRU-VNTRplus (data not shown).
Fig. 1.

Minimum spanning tree based on the 24-loci MIRU-VNTR typing data of 607 MTB isolates from Uzbekistan, Tajikistan and Kyrgyzstan. The size of each circle is proportional to the number of MIRU-VNTR types belonging to a particular complex. Classification of the isolates into the different phylogenetic lineages and resistance patterns is visualized by color coding
Cluster analysis revealed that 382 (62.9%) of the 607 isolates shared a genotyping pattern with at least one other isolate. They were grouped in 38 clusters ranging from two to 145 isolates. The two largest clusters had haplotypes 94–32 (n = 145; 23.9%) and 100–32 (n = 70; 11.5%) (Fig. 1). The prevalence of 94–32 was equally distributed over the CARs whereas cluster 100–32 was particularly prominent in Tajikistan (51 strains; 72.9%). 349 (84.9%) of the 411 Beijing isolates formed clusters, compared to only 33 (16.8%) of the 196 isolates from other clades.
Beijing strains were more frequently MDR-, pre-XDR- or XDR-TB (265/411; 64.5%) than strains of other genotypes (28/196; 14.3%; no XDR-TB) (Fig. 2). Beijing cluster 94–32 showed the same drug resistance pattern as other Beijing strains, whereas cluster 100–32 was almost completely (66/70; 94.3%) MDR, pre-XDR, or XDR.
Fig. 2.
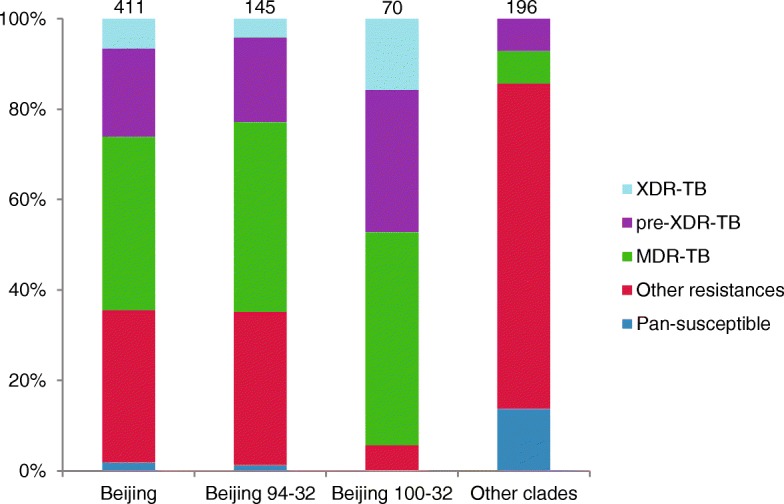
Proportions of isolates based on drug resistance patterns. The number of isolates with a given genotype/MLVA MtbC15–9 haplotype is given above the corresponding column
Discussion
High prevalence of Beijing strains has previously been reported from Central Asia [4]. The star-like shape of the lineage 2/Beijing population in Fig. 1 with two central clones surrounded by layers of single and multi-locus variants is a typical pattern for emerging and expanding MTB populations [13]. The two largest clusters 94–32 and 100–32 were found in all three Central Asian countries and represented more than a third of all study isolates indicating strong population expansion across borders. Our findings confirm the results of a global Beijing population study which reported that 94–32 and 100–32 were predominantly prominent in Uzbekistan, Russia and Eastern European countries and had particularly high clustering rates [13]. The smaller size of 100–32 and its unequal distribution to the three CARs suggest however its more recent arrival in Central Asia and a shorter regional expansion time than of 94–32. This hypothesis is further supported by Mokrousov [14] and Skibe et al. [15] who have already reported 100–32 as minor group (< 4%) of Beijing isolates in Central Asia while it reached proportions of over 10% and over 20% in Eastern Europe and Russia, respectively, and while Skibe et al. observed 94–32 as dominant cluster in Kazakhstan. The extent of cross-border spread and locally restricted transmisison networks can however not be ultimately resolved with MIRU-VNTR cluster data and require further investigation with higher resolution.
High percentages of MDR among Beijing strains has also been reported from other Eastern European countries, like Georgia, Kazakhstan, and Uzbekistan [4, 15, 16]. Higher proportions of MDR- and XDR-TB were also observed within the 100–32 genotype by Merker and colleagues [13]. The authors analyzed the full genomes of MDR-TB strains belonging to MLVA-VNTR haplotypes 94–32 and 100–32 and showed that the maximal divergences were 17 and 23 SNPs, respectively, within genomes of those clusters [13].
Our data indicate that spread of resistant MTB strains, particularly of lineage 2/Beijing, is an important root of the ongoing MDR-TB epidemic in Central Asia. MIRU-VNTR is a powerful tool for investigating population structures and transmission dynamics, however, advances in the field of whole genome sequencing have resulted in molecular methods that can give better resolution and should ideally be used to elucidate specific transmission pathways. Analyzing the full genomes of this or an unbiased and thus even more representative study population could provide an even deeper insight into the MTB population structure in CAR.
Conclusions
Clusters 94–32 and 100–32 are the dominant genotypes driving population expansion of Beijing strains in the CARs Uzbekistan, Tajikistan, and Kyrgyzstan. The data demonstrate that spread of resistant TB strains, particularly of the Beijing lineage and its clusters 94–32 and 100–32, is an important root of the ongoing MDR-TB epidemic in Central Asia. Measures to combat this epidemic need to be improved.
Acknowledgements
The authors thank Tanja Struve-Sonnenschein, Anja Lüdemann and Tanja Ubben for technical assistance.
Abbreviations
- CAR
Central Asian Republic
- DRS
Drug resistance surveys
- MDR
Multi-drug-resistant / resistance
- MIRU-VNTR
Mycobacterial Interspersed Repetitive Units - Variable Number of Tandem Repeats
- MLVA
Multiple loci VNTR analysis
- MST
Minimum-spanning tree
- MTB
Myocbacterium tuberculosis
- TB
Tuberculosis
- UPGMA
Unweighted pair group method with arithmetic mean
- XDR
Extensively drug resistant / extensive drug resistance
Authors’ contributions
SN and HH designed the study, AE performed the experiments, AE, UA, AK, GK, OK, NP, AR, ES, ZS, and HH collected data, AE, KK, SN and HH analyzed the data, AE, KK, MM, SN and HH wrote the manuscript. All authors read and approved the final manuscript.
Authors’ information
None
Funding
AE was supported by the International Postdoc Grant from the Swedish Research Council. The funding body had no role in the study design, collection of isolates, analysis, interpretation of data nor in writing the manuscript.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
Not applicable, but see please see methods section and references 8 and 9.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Stefan Niemann and Harald Hoffmann contributed equally to this work.
Contributor Information
Anna Engström, Phone: +49 (0) 4537 / 188-0, Phone: +46 (0)18 - 471 44 44, Phone: +46 (0)8-524 815 00, Email: anna.engstrom@scilifelab.se, Email: annaengstroem@gmail.com, Email: anna.lyander@gmail.com.
Uladzimir Antonenka, Phone: +49 89 85791 5401, Email: u.antonenka@imlred.de.
Abdylat Kadyrov, Phone: +996 312 59-55-61, Email: abdylat.kadyrov@gmail.com.
Gulmira Kalmambetova, Phone: +996 312 59-55-61, Email: gulmira.kalmambetova@gmail.com.
Katharina Kranzer, Phone: +49 (0)4537-188 213, Email: Katharina.Kranzer@lshtm.ac.uk.
Matthias Merker, Phone: +49 (0) 4537 / 188-0, Email: mmerker@fz-borstel.de.
Olim Kabirov, Phone: +99 2918 592630, Email: olim.kabirov@mail.ru.
Nargiza Parpieva, Phone: +998946110013, Email: nargizaparpieva@gmail.com.
Asliddin Rajabov, Phone: +99 2918 592630, Email: asliddin.81@mail.ru.
Evgeni Sahalchyk, Phone: +49 (0) 4537 / 188-0, Email: e.sahalchyk@imlred.de.
Zayniddin Sayfudtinov, Phone: +998946110013, Email: zayniddin.nrl@gmail.com.
Stefan Niemann, Phone: +49 (0) 4537 / 188-0, Email: sniemann@fz-borstel.de.
Harald Hoffmann, Phone: +49 89 85791 5401, Email: harald.hoffmann@imlred.de.
References
- 1.World Health Organization, Global tuberculosis report 2017. Vol. WHO/HTM/TB/2017.23. 2017, Geneva, Switzerland.
- 2.Mokrousov I, et al. Molecular snapshot of mycobacterium tuberculosis population structure and drug-resistance in Kyrgyzstan. Tuberculosis (Edinb) 2013;93(5):501–507. doi: 10.1016/j.tube.2013.05.008. [DOI] [PubMed] [Google Scholar]
- 3.Mokrousov I, et al. Penitentiary population of mycobacterium tuberculosis in Kyrgyzstan: exceptionally high prevalence of the Beijing genotype and its Russia-specific subtype. Infect Genet Evol. 2009;9(6):1400–1405. doi: 10.1016/j.meegid.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 4.Cox HS, et al. The Beijing genotype and drug resistant tuberculosis in the Aral Sea region of Central Asia. Respir Res. 2005;6:134. doi: 10.1186/1465-9921-6-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allix-Beguec C, et al. Evaluation and strategy for use of MIRU-VNTRplus, a multifunctional database for online analysis of genotyping data and phylogenetic identification of mycobacterium tuberculosis complex isolates. J Clin Microbiol. 2008;46(8):2692–2699. doi: 10.1128/JCM.00540-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Homolka S, et al. High genetic diversity among mycobacterium tuberculosis complex strains from Sierra Leone. BMC Microbiol. 2008;8:103. doi: 10.1186/1471-2180-8-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ulmasova DJ, et al. Multidrug-resistant tuberculosis in Uzbekistan: results of a nationwide survey, 2010 to 2011. Euro Surveill. 2013;18(42):pii: 20609. doi: 10.2807/1560-7917.ES2013.18.42.20609. [DOI] [PubMed] [Google Scholar]
- 8.World Health Organization, Guidelines for surveillance of drug resistance in tuberculosis. Vol. WHO/HTM/TB/2009.422. 2009, Geneva, Switzerland.
- 9.Kamerbeek J, et al. Simultaneous detection and strain differentiation of mycobacterium tuberculosis for diagnosis and epidemiology. J Clin Microbiol. 1997;35(4):907–914. doi: 10.1128/jcm.35.4.907-914.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weniger T, et al. MIRU-VNTRplus: a web tool for polyphasic genotyping of Mycobacterium tuberculosis complex bacteria. Nucleic Acids Res. 2010;38(Web Server issue):W326–W331. doi: 10.1093/nar/gkq351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Palomino JC, et al. Resazurin microtiter assay plate: simple and inexpensive method for detection of drug resistance in mycobacterium tuberculosis. Antimicrob Agents Chemother. 2002;46(8):2720–2722. doi: 10.1128/AAC.46.8.2720-2722.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gagneux S, Small PM. Global phylogeography of mycobacterium tuberculosis and implications for tuberculosis product development. Lancet Infect Dis. 2007;7(5):328–337. doi: 10.1016/S1473-3099(07)70108-1. [DOI] [PubMed] [Google Scholar]
- 13.Merker M, et al. Evolutionary history and global spread of the mycobacterium tuberculosis Beijing lineage. Nat Genet. 2015;47(3):242–249. doi: 10.1038/ng.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mokrousov I. Insights into the origin, emergence, and current spread of a successful Russian clone of mycobacterium tuberculosis. Clin Microbiol Rev. 2013;26(2):342–360. doi: 10.1128/CMR.00087-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skiba Y, et al. Molecular snapshot of mycobacterium tuberculosis population in Kazakhstan: a country-wide study. Tuberculosis (Edinb) 2015;95(5):538–546. doi: 10.1016/j.tube.2015.04.012. [DOI] [PubMed] [Google Scholar]
- 16.Niemann S, et al. Mycobacterium tuberculosis Beijing lineage favors the spread of multidrug-resistant tuberculosis in the republic of Georgia. J Clin Microbiol. 2010;48(10):3544–3550. doi: 10.1128/JCM.00715-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.