

Abstract
Background
Programmed epigenetic modifications occurring at early postnatal brain developmental stages may have a long-lasting impact on brain function and complex behavior throughout life. Notably, it is now emerging that several genes that undergo perinatal changes in DNA methylation are associated with neuropsychiatric disorders. In this context, we envisaged that epigenetic modifications during the perinatal period may potentially drive essential changes in the genes regulating brain levels of critical neuromodulators such as d-serine and d-aspartate. Dysfunction of this fine regulation may contribute to the genesis of schizophrenia or other mental disorders, in which altered levels of d-amino acids are found. We recently demonstrated that Ddo, the d-aspartate degradation gene, is actively demethylated to ultimately reduce d-aspartate levels. However, the role of epigenetics as a mechanism driving the regulation of appropriate d-ser levels during brain development has been poorly investigated to date.
Methods
We performed comprehensive ultradeep DNA methylation and hydroxymethylation profiling along with mRNA expression and HPLC-based d-amino acids level analyses of genes controlling the mammalian brain levels of d-serine and d-aspartate. DNA methylation changes occurring in specific cerebellar cell types were also investigated. We conducted high coverage targeted bisulfite sequencing by next-generation sequencing and single-molecule bioinformatic analysis.
Results
We report consistent spatiotemporal modifications occurring at the Dao gene during neonatal development in a specific brain region (the cerebellum) and within specific cell types (astrocytes) for the first time. Dynamic demethylation at two specific CpG sites located just downstream of the transcription start site was sufficient to strongly activate the Dao gene, ultimately promoting the complete physiological degradation of cerebellar d-serine a few days after mouse birth. High amount of 5′-hydroxymethylcytosine, exclusively detected at relevant CpG sites, strongly evoked the occurrence of an active demethylation process.
Conclusion
The present investigation demonstrates that robust and selective demethylation of two CpG sites is associated with postnatal activation of the Dao gene and consequent removal of d-serine within the mouse cerebellum. A single-molecule methylation approach applied at the Dao locus promises to identify different cell-type compositions and functions in different brain areas and developmental stages.
Electronic supplementary material
The online version of this article (10.1186/s13148-019-0732-z) contains supplementary material, which is available to authorized users.
Keywords: Neuroepigenetics, Brain DNA methylation, 5-Hydroxymethylcytosine, DNA methylation in psychiatric disorders, d-amino acids
Background
Embryonic and postnatal brain development is a continuous process that involves well-orchestrated changes, including age- and cerebral region-specific gene expression programs. The perinatal period is the main temporal window in which the timely occurrence of programmed molecular changes is critical for correct neural functioning throughout the remainder of life. Epigenetic mechanisms favoring brain development and neuronal plasticity may underlie these fundamental events [1–5]. Accordingly, neuroepigenetics is a growing research field since DNA methylation and chromatin modifications influence gene programs, neuronal plasticity, synaptogenesis and complex behaviors, and epigenetic marks are altered in several neuropsychiatric conditions [6–14]. Notably, from recent epigenome-wide studies, it emerged that several genes that undergo perinatal changes in DNA methylation are associated with neuropsychiatric disorders [15–17]. However, the mammalian brain is a very heterogeneous structure composed of functionally different areas and a variety of cell populations [18], which may exhibit unique DNA methylation profiles, whose diversity has been poorly investigated to date. In this regard, epigenetic modifications during the perinatal period may potentially drive critical changes in the genes regulating brain levels of d-amino acids [19, 20]. In particular, free d-serine (d-Ser) and d-aspartate (d-Asp) have been identified as critical regulators of mammalian glutamatergic neurotransmission by acting as a co-agonist and agonist of N-methyl-d-aspartate receptors (NMDARs), respectively. These atypical amino acids show a unique regional and temporal pattern of emergence in the mammalian brain [21–23]. On the day of birth, a substantial amount of d-Ser is present in the mouse cortex, striatum, and cerebellum and then increases selectively in the cortex and striatum, whereas the cerebellar concentration of this d-amino acid declines drastically to trace levels around day 18 after birth [24]. Similarly, d-Asp content significantly decreases in the whole mouse brain a few days after birth, and such degradation is ultimately associated with an age-dependent increase in d-aspartate oxidase (Ddo) expression [19, 20, 25, 26]. Although the specific molecular mechanisms involved are still unclear, the precise spatiotemporal regulation of the genes involved in d-Ser synthesis (serine racemase, Srr) or degradation (d-amino acid oxidase, Dao) and d-Asp degradation (Ddo) is thought to be critical for regulating the correct regional concentrations of these d-amino acids during pre- and postnatal brain development [27]. Accordingly, the expression and activity of the enzymes encoded by these genes have been shown to vary across different brain areas and different perinatal times in humans and rodents [24, 26, 28, 29]. Since d-Asp and d-Ser are believed to play a direct role in regulating NMDAR-related synaptic plasticity, morphology, and function [22, 30, 31], the expression program of the genes controlling the metabolism of these d-amino acids must be rigorously orchestrated in mammalian brain development, and epigenetic mechanisms are the major candidate directors.
In this work, we provide a comprehensive analysis of mRNA expression and DNA methylation changes occurring at genes regulating the concentrations of d-Ser and d-Asp in three mouse brain areas during postnatal development. Here, we report striking spatiotemporal epigenetic modifications at the Dao gene during neonatal cerebellar development occurring selectively in the astrocytes for the first time. Notably, ultradeep methylation analysis allowed us to track the dynamic evolution of Dao epialleles (specific methyl-CpG arrangements) during ontogenesis in different brain areas and in specific cell types.
Results
Occurrence of free d-Ser and d-Asp in the hippocampus, cortex, and cerebellum during postnatal life
We hypothesized that dynamic epigenetic modifications may drive changes in gene expression programs ultimately leading to area-specific perinatal shifts in d-amino acid concentrations. As a first step, we wished to validate our study system in terms of changes in d-Ser and d-Asp levels in different brain areas during mouse brain postnatal development. To this end, we quantified the ratio of the amounts of the d- and total (d +l) forms of both Ser and Asp by HPLC analysis at different postnatal time points (P1-P15-P30-P60) and in different brain regions, including the cortex (CX), hippocampus (HIPP), and cerebellum (CB) (Fig. 1). We found that the d-Ser/total serine ratio significantly increased from P1 to P15 in both the hippocampus and cortex and that a significantly high ratio persisted throughout the following postnatal phases (one-way ANOVA, hippocampus: F(3, 9) = 166.7, p < 0.0001; cortex: F(3, 7) = 64.26, p < 0.0001) (Fig. 1a, b). In striking contrast to what occurs in the hippocampus and cortex, in the cerebellum, we observed a significant progressive age-dependent reduction in the d-Ser/total serine ratio during the same postnatal development stages (one-way ANOVA, F(3,8) = 20.75, p = 0.0002; Fig. 1c). At the same postnatal time points in the same brain structures, we also investigated the ratio between d-Asp and total Asp levels by HPLC analysis (Fig. 1d–f). Consistent with previous work [20], the d-Asp/total aspartate ratio dramatically decreased after P1 in the hippocampus, cortex and cerebellum and remained very low in all the following postnatal stages (one-way ANOVA, hippocampus: F(3, 9) = 53.24, p < 0.0001; cortex: F(3, 7) = 28.12, p = 0.0003; cerebellum: F(3,9) = 476.3, p < 0.000; Fig. 1d–f).
Fig. 1.

d-Ser and d-Asp contents in the mouse hippocampus, cortex, and cerebellum during postnatal ontogenesis. Average ratio (expressed as %) of d-Ser/total Ser at different postnatal stages (P1, P15, P30, and P60) of C57BL/6 J mice in the a hippocampus (n = 4 P1, n = 3 P15/ P30/ P60), b cortex (n = 4 P1, n = 3 P15, n = 2 P30/ P60), and c cerebellum (n = 4 P1; n = 3 P15/P30/P60). Average ratio (expressed as %) of d-Asp/total Asp at different postnatal stages (P1, P15, P30, and P60) of C57BL/6 J mice in the d hippocampus (n = 4 P1, n = 3 P15/ P30/ P60), e cortex (n = 4 P1, n = 3 P15, n = 2 P30/ P60), and f cerebellum (n = 4 P1; n = 3 P15/ P30/P60). The d- and l-forms of Asp and Ser were quantified by HPLC analysis. *p < 0.05, **p < 0.01, ***p < 0.0001, compared with P1 (Fisher’s post hoc). Data are expressed as the mean ± SEM
Dynamic demethylation of two specific relevant CpG sites at the Dao promoter determines programmed Dao gene activation and a decrease in d-Ser levels in the cerebellum
Here, we investigated the DNA methylation and mRNA expression of the Dao gene in the same brain regions and postnatal stages for which HPLC detection was performed. Methylation analysis was performed by targeted bisulfite sequencing with high coverage (approximately 100,000 reads per sample). First, we analyzed the average methylation level at the Dao promoter. We first focused on a region of 389 nucleotides of the Dao promoter (region PR1, Fig. 2a) that contains four CpG sites (+ 7; + 101; + 217; + 334), all of which are located downstream of the transcriptional start site (TSS). The average methylation levels at P1, P15, P30, and P60 in all analyzed brain regions were compared (Fig. 2b). No significant differences were found in HIPP and CX during development, and these structures displayed a high (approximately 75%) nearly stable degree of methylation during the different stages. In contrast, remarkable differences in the average DNA methylation were observed in CB. We found a significantly (one-way ANOVA; p ≤ 0.001) higher degree of methylation at postnatal day 1 (71.5% ± 0.03) compared to P15 (43.5% ± 0.02), P30 (44.8% ± 0.004), and P60 (53.1% ± 0.003). No significant differences were observed in the comparison between P15, P30, and P60. Additionally, we analyzed mRNA expression levels in HIPP, CX, and CB during brain development (Fig. 2c). Our data indicated very low Dao mRNA levels in both HIPP and CX, which showed no significant changes during ontogenesis. Conversely, a significant (one-way ANOVA; p ≤ 0.001) increase in Dao mRNA expression was found in the cerebellum during brain development (Fig. 2c). We then evaluated the methylation at a single-CpG resolution during ontogenesis in HIPP, CX, and CB (Fig. 2d). In the CB area in particular, a striking significant (one-way ANOVA; p ≤ 0.001) decrease in the degree of methylation was observed specifically at the CpG + 7 and + 101 sites from P1 to P15 (from 52.8 ± 0.06 to 8.2 ± 0.02%), and this low level of methylation persisted at P30 and P60. In contrast, the average methylation at CpG + 217 and CpG + 334 was elevated and remained rather constant over time in all brain areas except at the CpG + 217 site in CB, which showed a transient decrease at P15, switching back to high levels (above 80%) of methylation at P30 and P60. To validate the above results and determine whether other CpG sites located upstream of the TSS were subjected to similar dynamic demethylation in CB, we extended the bisulfite analysis to the PR2 region (− 234/+ 137 nts, Fig. 2a). This region encompasses the TSS and includes both the abovementioned + 7 and + 101 sites and five additional CpG sites located upstream (− 208, − 177, − 162, − 129, and − 102; Fig. 2a) in a single amplicon. The results confirmed the decrease in methylation at the + 7 and + 101 sites and showed that the methylation drift was essentially limited to these sites just downstream of the TSS (Fig. 2e, f). Thus, the very large selective reduction of DNA methylation observed in the cerebellum at the + 7 and + 101 sites (Fig. 2d, e) was functionally consistent with the robust activation of the Dao gene and ultimately with the dramatic reduction of the d-Ser concentration detected in the mouse cerebellum at P30 (Fig. 1c).
Fig. 2.

DNA methylation and mRNA expression of the Dao gene. a Dao promoter region structure showing the position of the analyzed CpGs. The numbers of the CpG sites refer to the putative transcriptional start site (TSS), indicated with + 1. White box indicates the putative upstream regulatory region; black box indicates the first exon; and gray box indicates the first intron. Black arrows at the top of the map specify the position of the primers used for bisulfite amplification. PR1 denotes the promoter region downstream of the TSS analyzed in all brain areas. PR2 indicates the promoter region upstream of the TSS explored in the cerebellum area. The Dao sequence was retrieved by Ensembl with the following accession number: ENSMUSG00000042096. b Average Dao methylation (%) is shown for HIPP, CX, and CB at the P1, P15, P30, and P60 developmental stages (n = 3 mice/stage). Comparisons between developmental stages in each area were performed using one-way ANOVA followed by Tukey’s multiple comparison post-hoc test. *p ≤ 0.001. c mRNA expression levels are reported for each analyzed brain area during brain development. Dao mRNA expression was normalized to the mean of two housekeeping genes and is expressed as 2–∆Ct values, * p ≤ 0.001 (one-way ANOVA followed by Tukey’s multiple comparison post-hoc test). d Graphs represent the average methylation at single CpG sites in promoter region 1 (PR1), downstream of the TSS, in HIPP, CX, and CB during ontogenesis. e Methylation levels at CpG sites located in promoter region 2 (PR2, see a) are shown. The methylation trend of the seven CpG sites is indicated at each developmental stage. f A comprehensive view of single CpG methylation levels upstream and downstream of the Dao promoter region in the cerebellum during the transition from P1 to P15. Nine CpG sites are indicated, and their position is referred to as the TSS. CpGs with average methylation < 30% are shown in green, while CpG sites with average methylation > 50% are indicated in red
Single-CpG resolution analysis of 5-mCpG and 5-hmCpG dynamics at the Dao regulatory region argues for the involvement of active DNA demethylation
DNA methylation and demethylation dynamics depend on the competitive opposite activities of DNA methyltransferases (DNMTs) and Ten-eleven translocation (TET) enzymes [32]. 5-hmC represents an intermediate product of TET-mediated active DNA demethylation [33, 34]. However, eventual presence of 5-hydoxymethylcytosine (5-hmC) could not be detected by bisulfite analysis. Therefore, we evaluated the levels of 5-hmC at the Dao gene in CB at P1 and P15 by oxidative bisulfite sequencing [35]. Results, shown in Fig. 3, revealed the presence of high amount of hydroxymethylation at CpG + 7 and CpG + 101. Consistently, these sites underwent substantial demethylation at later stages (Fig. 2d). Although CpG + 217 exhibited high levels of 5-hmC, at this specific site 5-mC levels increased at P15 while lower levels of 5-hmC were detected at this stage. These findings suggest that during the brain development, the activity of TET enzymes prevailed over DNMTs activity at the + 7 and + 101 CpGs while the opposite dynamics occurred at the + 217 CpG. Strikingly, at the + 334 CpG, 5-methylcytosine was very stable and did not undergo either hydroxymethylation or demethylation over time. Overall, our data strongly support the existence of an active demethylation process at selected sites and poor dynamics of 5-hmC at surrounding stably methylated CpG site (+ 334).
Fig. 3.

Analysis of 5-mCpG and 5-hmCpG content at single-CpG resolution of the Dao regulatory region in the cerebellum. Bar plots represent methylation (black) and hydroxymethylation (line pattern) levels at individual CpG site in Dao regulatory region at the P1 to P15 stages. Average 5-hmC levels was calculated by subtracting values obtained from sequence analysis of DNAs treated with an oxidant reagent plus sodium bisulfite to the average methylation values obtained from DNA treated with bisulfite only. For each time point, results are indicated as the mean of n = 3 mice
Dao demethylation in the cerebellum at postnatal day 15 mostly occurs in astrocytes
Previous immunohistochemical analyses demonstrated that during postnatal stages, Dao activation occurs prevalently in cerebellar astrocytes and to a lesser extent in neurons [36, 37]. However, during prenatal and neonatal stages, the mouse brain undergoes radical changes in the cell type composition, and these modifications are distinct and characteristic for each brain area [5, 18]. To verify whether the Dao gene undergoes demethylation in cerebellar astrocytes, we performed cell separation experiments from cerebellum tissue at the P1 and P15 time points. Then, we analyzed Dao promoter (PR1 region; Fig. 2a) methylation in purified astrocytes, neurons and a fraction containing microglia, oligodendrocytes, and endothelial cells (MOE fraction) (Fig. 4a). The results showed a decrease in the degree of methylation between days P1 and P15 mainly at the + 7 and + 101 CpGs and in astrocytes compared to neurons and other cell types (Fig. 4b). Because at these postnatal stages astrocytes are likely active proliferating cells [38], these data do not exclude passive demethylation. However, oxidative bisulfite sequencing analysis performed in whole cerebellum tissue (Fig. 3) makes us confident that a process of active demethylation is mainly responsible for the methylation decrease occurring at the + 7 and + 101 CpGs in cerebellar astrocytes.
Fig. 4.

DNA methylation of the Dao promoter region in different cerebellum cell types at the P1 and P15 stages. a Average Dao methylation (%) in astrocytes, neurons, and a fraction containing microglia, oligodendrocytes, and endothelial cells (MOE fraction) at postnatal days 1 (black) and 15 (gray). b Degree of single-CpG methylation in astrocytes, neurons and a fraction containing microglia, oligodendrocytes, and endothelial cells (MOE fraction) during the transition from P1 to P15
Epiallele distribution analysis in HIPP, CX, and CB during brain development reveals area- and stage-specific epialleles at the Dao promoter
We recently provided proof of concept that studying DNA methylation at specific loci at single-molecule resolution (epiallele distribution analysis) allows one the tracking of the spatiotemporal evolution of cell-to-cell methylation differences in a given cell population [19, 39, 40]. By using this method, requiring a large number of sequence reads per sample, it is possible to quantitatively and qualitatively analyze the arrangements of all methylated CpG at a specific locus at single-molecule level. Each specific arrangement is defined here as an “epiallele.” We applied epiallele analysis to investigate whether epialleles at the Dao gene locus evolve in specific selected patterns depending on the brain developmental stage. After ultradeep bisulfite sequencing, an average of 100,000 sequences/sample were obtained and analyzed via the ampliMethProfiler pipeline (https://sourceforge.net/projects/amplimethprofiler) [41], which generated a BIOM format table containing the counts of Dao epialleles for all samples. The BIOM file was subjected to a rarefaction procedure to avoid bias due to different numbers of reads per sample and was used to perform the further analyses. Considering the Dao region including four CpG sites (PR1, Fig. 2a), the number of potential epialleles types was 16 (24). All the possible epialleles (16) were found to be present in each brain area and developmental stage but in different proportions. For each brain region, we performed principal component analysis (PCA) to verify the presence of patterns associated with various developmental stages (Fig. 5a). Strikingly, although the average levels of methylation in HIPP and CX were found to be almost identical during development (Fig. 2b), principal component analysis of epialleles showed clear grouping of the different developmental stages (Fig. 5a). In both HIPP and CX, a highly heterogeneous epiallelic composition was observed on postnatal day 1, which converged to relatively homogeneous epiallele types during brain development (Fig. 5a). For the cerebellum, the same analysis gave rise to more distinct grouping of each developmental stage, especially at P30 and P60, which was clearly influenced by the quantitative variation at these stages. Together, these data revealed non-stochastic, well-orchestrated, DNA methylation remodeling that occurs in the cells of different brain areas during development. We then performed PCA by integrating all brain areas and developmental time points (Fig. 5b). We confirmed that the evolution of the epiallelic distribution in the cerebellum was distinct compared to that in HIPP and CX (Fig. 5b). Interestingly, in very early stages (P1), the cerebellum shared epiallelic patterns with HIPP and CX, whereas at later stages, the epiallelic profiles evolved in a distinct manner (Fig. 5b). None of the above information could be retrieved from average standard methylation analyses alone, demonstrating the high potential of epiallelic distribution analysis.
Fig. 5.

Epiallelic distribution profiling of the Dao gene during brain development in HIPP, CX, and CB. a Principal component analysis (PCA) plots show the epigenetic correlations of the Dao epiallele distribution during development in HIPP, CX, and CB. The score plots and loading plots of each PCA plot are merged to obtain biplots. The biplots are derived from the qualitative and quantitative influence of each of the 16 epialleles (indicated as a string of 1 s and 0 s) displayed by the three mice at P1 (red), P15 (blue), P30 (yellow), and P60 (green) in each analyzed brain area. The direction and length of the vectors (black lines) indicate how each epiallele and its abundance influence the sample position in the plot. Adjacent samples share similar epiallelic composition. Principal components 1 and 2 are reported for each brain area. b The overall PCA analysis of brain region differences based on the epiallelic composition is shown, including all developmental stages. On the left, the score plot displays the epigenetic correlation of Dao epialleles between HIPP (green), CX (blue), and CB (red). On the right, the loading plot of the PCA displays the impact of each of 16 Dao epialleles on the brain area distribution
DNA methylation and mRNA expression analyses of the Srr gene in HIPP, CX, and CB at different developmental times
We next performed DNA methylation analysis and mRNA expression analysis of the Srr gene, encoding the d-Ser-synthesizing enzyme [42–44]. The Srr promoter region includes a very dense CpG island. We analyzed a region of 350 bp in length including 30 CpG sites located just downstream of the TSS (Fig. 6a). Very low levels of CpG methylation were detected in all analyzed brain areas and developmental stages, with average levels between 0.14% and 0.32%, (Fig. 6b, c). This finding was expected given the presence of a dense CpG island in the Srr promoter. No significant differences were found between different areas or during postnatal brain development. We then performed an mRNA expression analysis of Srr (Fig. 6d). In both HIPP and CB, we found significant (one-way ANOVA; p ≤ 0.001) differences in mRNA levels between the P1 and P30 stages. In CX, a significant (one-way ANOVA; p ≤ 0.001) increase in Srr mRNA levels was found in the comparison between P1 and the other developmental times. We conclude that the observed increase in mRNA levels is not controlled by a change in the CpG methylation state, at least in the analyzed region. Due to the low degree of methylation detected at each CpG site, epiallele analysis was considered uninformative and was therefore not performed. Interestingly, the increase in Srr mRNA levels (Fig. 6d) and stable low Dao expression (Fig. 2c) correlated well with the increase in d-Ser levels measured by HPLC in CX (and transiently in HIPP) over time (Fig. 1a, b). More strikingly, in CB, the increase in Srr levels (Fig. 6d) over time was counterbalanced by a simultaneous robust increase in Dao expression (Fig. 2c), resulting in a net decrease in d-Ser levels (Fig. 1c).
Fig. 6.

DNA methylation at the Srr locus and Srr gene expression. a The analyzed CpG island in the Srr promoter region is shown. White box represents the putative upstream regulatory region; black box indicates the first exon; and gray box indicates the first intron. The analyzed Srr amplicon is located between the black arrows on the top, indicating the forward and reverse primers used for bisulfite amplification. The positions of the analyzed CpGs refer to the TSS (+ 1). The Srr sequence was retrieved by Ensembl with the accession number: ENSMUSG00000001323. b Average methylation (%) at single-CpG level is indicated for each brain region during ontogenesis. c The average methylation (%) of the Srr gene in all brain regions at all analyzed developmental stages is reported. Statistical analyses were performed using one-way ANOVA followed by Tukey’s multiple comparison post-hoc test. Alpha was considered significant at ≤ 0.001. d The spatiotemporal distribution of Srr mRNA levels is reported in histograms. Srr mRNA expression is normalized to the mean values for two housekeeping genes and expressed as 2–∆Ct values. Statistical analyses were performed using one-way ANOVA followed by Tukey’s multiple comparison post-hoc test. * p ≤ 0.001
DNA methylation and mRNA expression of the Ddo gene in different brain areas at different developmental stages
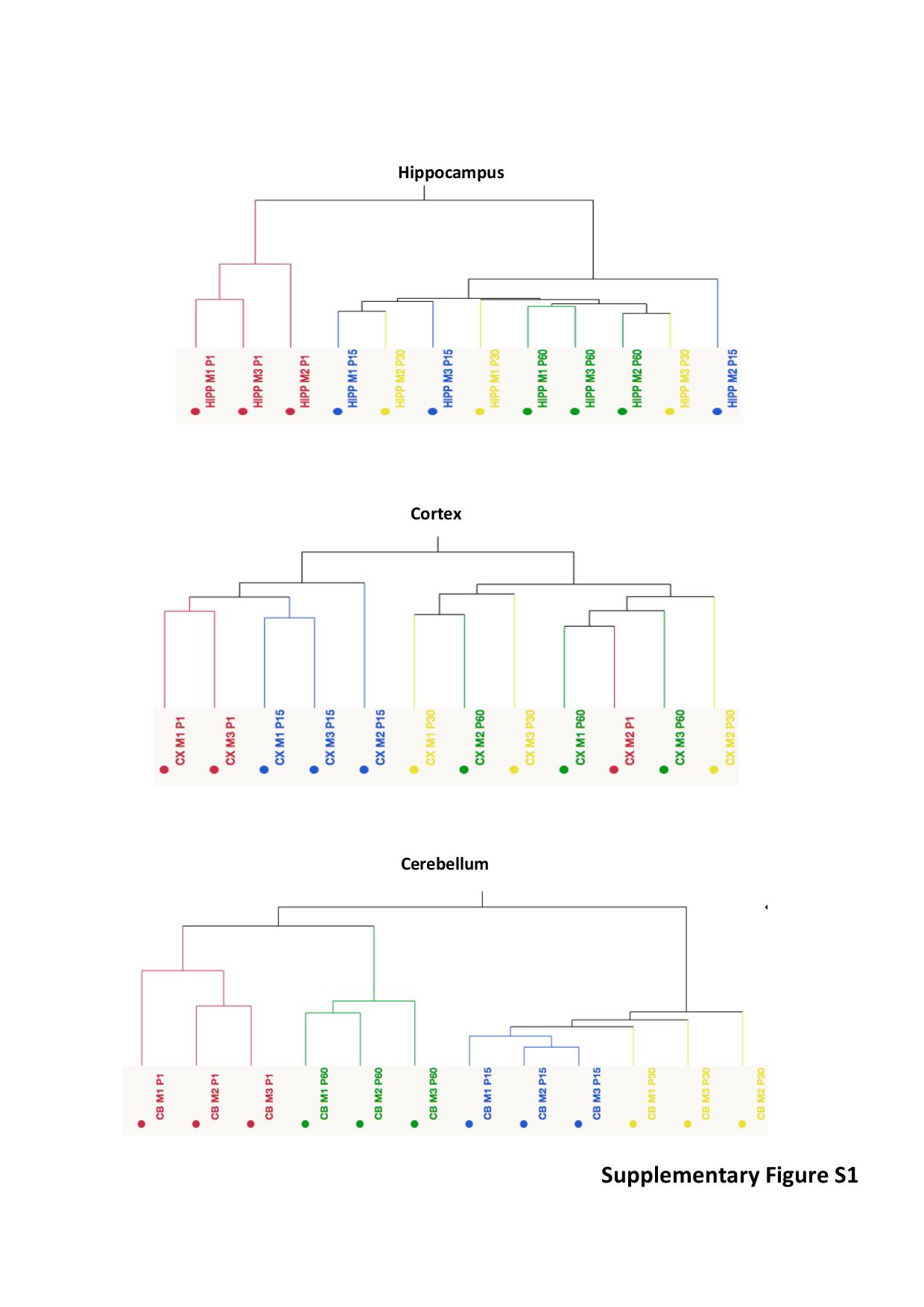
We previously reported that demethylation of Ddo in whole brain leads to increased Ddo mRNA expression and downregulation of cerebral d-Asp levels [19, 20]. Here, we further investigated whether regional Ddo demethylation and changes in the d-Asp concentration occur in specific brain areas and postnatal stages. We analyzed the DNA methylation state and mRNA expression of the Ddo gene in HIPP, CX, and CB at P1, P15, P30, and P60. A region of 405 bp upstream of the TSS, including six CpG sites (− 363, − 330, − 318, − 242, − 175, − 125), was analyzed (Fig. 7a). In CX, neither the average Ddo methylation level nor single-CpG methylation within the Ddo promoter changed at any of the examined developmental stages (Fig. 7b, c). However, a slight increase in mRNA expression was found during development (Fig. 7d). An evident and significant decrease (one-way ANOVA; p = 0.001) in average methylation was found in HIPP during the transition from P1 to P60 (Fig. 7b). Interestingly, all analyzed CpG sites underwent demethylation from P1 to P15, and the methylation level then remained low over time (Fig. 7c). Consistent with the DNA methylation data, Ddo mRNA levels significantly (one-way ANOVA; p ≤ 0.001) increased from P1 to P60 (Fig. 7d, Additional file 2: Figure S2). In CB, a tendency of the average Ddo methylation to decrease was observed from P1 to P15, which then slightly increased at P30 and P60 (Fig. 7b). Accordingly, a gradual significant (one-way ANOVA; p ≤ 0.001) increase in mRNA expression was found over time in the cerebellum area (Fig. 7d; Additional file 2: Figure S2). Interestingly, when we evaluated the average single-CpG methylation of Ddo, we found that all six CpG sites presented a trend of decreased methylation from the P1 to P15 stages, and the methylation level then remained consistently low at the − 125 and − 175 CpG sites (Fig. 7c). Conversely, all the other distal CpGs underwent a slight increase in methylation (Fig. 7c), possibly explaining the apparent tendency of higher average methylation levels observed from developmental stages P15 to P60 (Fig. 7b). These data highlight a decisive role of the degree of methylation at the − 125 and − 175 CpG sites in the control of Ddo expression during brain development for the first time. Consistent with the observed Ddo mRNA increase in HIPP, CX, and CB during development (Fig. 7d; Additional file2: Figure S2), a considerable decrease in d-Asp levels was observed in all analyzed brain areas in the temporal window of P1–P15 (Fig. 1d–f). We previously reported a Ddo epiallele distribution analysis of the developing whole brain [19]. Here, we performed Ddo epiallele analysis of specific areas over time. Hierarchical cluster analysis (HCA) showed that developmental times were distinguishable by their epiallelic compositions mainly in cerebellum (Additional file 1: Figure S1).
Fig. 7.

DNA methylation and mRNA expression at the Ddo promoter region. a The Ddo gene structure with the analyzed CpG sites is shown. White box indicates the putative upstream regulatory region; black box indicates the first exon; and gray box indicates the first intron. Black arrows on the top indicate the forward and reverse primers used for bisulfite amplification. The CpG sites position refers to the TSS (+ 1). The Ddo sequence was retrieved by Ensembl with accession number: ENSMUSG00000063428. b The average Ddo methylation is presented for HIPP, CX and CB at each developmental stage. Statistical analyses were performed using one-way ANOVA followed by Tukey’s multiple comparison post-hoc test. Alpha was considered significant at ≤ 0.001. c The average methylation (%) calculated at all seven CpGs is shown for each brain region during ontogenesis. d Ddo mRNA levels are reported for all brain regions and developmental times. Ddo mRNA expression is normalized to the mean values for two housekeeping genes and expressed as 2–∆Ct values. One-way ANOVA followed by Tukey’s multiple comparison post-hoc test was used to assess statistical significance. *p ≤ 0.001
Discussion
d-Ser is a physiological endogenous co-agonist of NMDA receptors at central excitatory synapses [45]. d-Ser can influence many NMDAR-dependent functions, including brain development [46, 47], and behaviors such as cognition and social interaction. Although the role of free d-Asp has been less addressed, it has been recently shown that this d-amino acid influences NMDAR-mediated transmission [48]. In the brain, extracellular d-Asp activates NMDARs inducing considerable l-glutamate release in the prefrontal cortex of freely moving mice through the presynaptic activation of NMDA, AMPA/kainate, and mGlu5 receptors [49]. d-Aspartate is enriched in the embryonic brain of rodents and humans and its concentration strongly decreases after birth [50]. The proper control of brain d-amino acid levels has a clinical implication in both pathogenesis and therapeutic approach of different clinical conditions. For instance, serine deficiency syndromes include epilepsy, spasticity, and neurocognitive symptoms; these symptoms may partially be ameliorated by serine supplementation [51] In addition, d-serine deregulation is involved in the positive, negative, and cognitive symptoms of schizophrenia [52] and the inhibition of DAO appears to be a viable strategy to increase d-serine level and to have therapeutic potential in schizophrenia [53].
Based on the basic and translational interest of free d-amino acids in brain physiology and pathology, we have here investigated the spatiotemporal changes in the DNA methylation and expression of genes involved in the modulation of d-Ser and d-Asp, along with contextual changes in the levels of these neurochemicals. Methylation analysis was performed via a single-molecule approach (ultradeep methylation analysis), as a proxy for single-cell methylation analysis, in three different brain regions (HIPP, CX, and CB) at postnatal days P1, P15, P30, and P60. We found programmed selective demethylation at two specific CpG sites in genes involved in d-Ser and d-Asp metabolism during specific developmental stages and in specific brain areas. Dynamic demethylation phenomena resulted in progressive activation of these genes and a dramatic area-specific modulation of d-Ser and d-Asp levels during postnatal brain development. Environmentally induced alterations in physiological DNA methylation/demethylation turnover at specific genes in the brain during the perinatal period have been linked to the subsequent onset of neuropsychiatric diseases [54, 55]. Pioneering studies demonstrated the crucial role of postnatal programmed demethylation of a single or few CpG sites in the glucocorticoid receptor gene (exon 17 promoter). The absence of this demethylation phenomenon, caused by environmental sources, leads to persistent alteration of the HPA axis, resulting in the transmission of the adaptive response across generations [14, 56]. It has been proposed that effects on DNA methylation such as those described in the present work, even at single CpG sites, serve as an intermediate mechanism that imprints dynamic environmental experiences upon the fixed genome, resulting in stable alterations in phenotype [56].
In this work, we found that the Dao gene underwent programmed demethylation selectively in CB in a specific time window (P1–P15). The concomitant Dao activation and consequent postnatal d-Ser degradation likely represent physiological events required for correct postnatal cerebellum development. Accordingly, it has been shown that the major impact on d-Ser levels observed in mice lacking the Dao gene is limited to the cerebellum, where d-Ser levels remain high until adult life [57]. Dynamic demethylation of the Dao gene occurred specifically at only two CpG sites (+ 7 and + 101) located just downstream of the TSS, and this phenomenon supports gene activation. The possibility that the astrocyte fraction (assuming that these cells exhibit demethylated + 7 and + 101 CpG sites from P1 onward) may prevail over time in the cerebellum, providing false evidence of demethylation, was considered. Following cell separation, a decreased degree of methylation was still observed in purified cerebellar astrocytes collected at different stages. Therefore, our findings indicate that loss of methylation, rather than simple astrocyte number expansion, was mainly responsible for the observed demethylation. The upstream signaling mechanisms as well as the mechanisms driving specificity of targeted methylation and demethylation events remain unknown. Nevertheless, it is now established that mammalian cells, including neural cells, harbor the complete enzymatic machinery for not only methylating CpGs (and non-CpGs) but also actively demethylating DNA [58, 59]. In this work, we provide evidence that hydroxymethylcytosine, an intermediate compound of TET-mediated demethylation, is specifically enriched at the CpG sites undergoing demethylation at later developmental stages. In contrast, poor methylation dynamics was detected at the + 334 CpG, where the 5-methylcytosine was very stable and did not undergo either hydroxymethylation or demethylation over time. Our data strongly argue for the occurrence of an active demethylation process at Dao gene in cerebellum during development.
Through epiallele distribution analysis, we also investigated how specific methylation patterns are generated in different stages and brain areas during development. The evolution of Dao epialleles was distinct between different stages and different brain areas in mice. This phenomenon was often not associated with changes in average methylation levels but was instead likely due to the reconfiguration of methylation patterns among the cells of each area at each developmental time point. Our recent epiallele analysis of the human Dao gene based on methylation classes performed in different areas of postmortem brain [40] showed that the epiallele distribution at Dao is distinct in the cerebellum compared to other areas, indicating the conservative nature of epiallele evolution across species.
To obtain a complete picture of the methylation events occurring at d-Ser-modulating genes, we also performed methylation and expression analysis of the Srr gene in selected mouse brain regions for the first time in this work. Consistent with a recent human study [40], no changes in Srr gene methylation (stably hypomethylated) were found in mouse brain, although substantial regionally and temporally specific expression modifications were detected. On the other hand, increasing Srr mRNA levels and stable low Dao expression correlated well with the progressive increases in d-Ser levels observed in CX and HIPP over time. Interestingly, in the cerebellum, the increase in Srr mRNA levels over time was counterbalanced by a simultaneous robust increase in Dao expression, resulting in a net decrease in d-Ser levels. For Ddo, the d-Asp-degrading enzyme, in a recent study conducted on the whole brain, we found that this gene is strongly activated at early postnatal stages by contextual demethylation of a large (approximately 400 bp) region surrounding the TSS, accompanied by a postnatal decrease in d-Asp levels [19, 20]. Here, we have investigated separate brain areas and found that demethylation occurred in HIPP and CB but, surprisingly, not in CX. In CX, the observed slight increase in Ddo mRNA levels can likely be attributed to mechanisms other than DNA methylation changes. Of particular interest, strong Ddo gene activation was prominent in CB, where only two CpG sites (− 125 and − 175) remained demethylated over time, suggesting a critical role of these two specific sites.
Taken together, our data highlight the importance of post-natal changes of DNA methylation and hydroxymethylation at few CpG sites in the proper control of d-Ser and d-Asp levels with potential clinical implications.
Conclusion
Our data strongly encourage the investigation in the near future of whether failure of timely programmed active demethylation at relevant CpG sites may affect the physiological decreases in d-Ser and d-Asp levels thought to be required for correct brain development [22, 27, 45, 60–64]. Coherently, altered levels of d-Ser and d-Asp have been found in different neuropsychiatric conditions [50, 65–68]. It will be very important to investigate possible environmental factors affecting correct demethylation at the Dao and Ddo genes and whether altered methylation of the Dao and Ddo genes may be associated with neuropsychiatric conditions.
In conclusion, we hypothesize that eventual dysfunction of postnatal changes of DNA methylation/hydroxymethylation dynamics at few CpG sites, indirectly establishing the proper d-Ser and D-Asp brain levels, may have profound clinical impact in the etio-pathogenesis and treatment of neurodevelopmental disorders.
Materials and methods
Animals
All experiments were performed on male animals. C57BL/6 J mice were purchased from The Jackson Laboratory. Mice were housed in groups (n = 4 or 5) in standard cages (29 × 17.5 × 12.5 cm) at constant temperature (21–24 °C) and maintained on a 12/12 h light/dark cycle, with food and water ad libitum. All research involving animals was performed in accordance with the European directive 86/609/EEC governing animal welfare and protection, which is acknowledged by the Italian Legislative Decree no. 26 (March 14, 2014). Animal research protocols were also reviewed and approved by the local animal care committee at University of Naples “Federico II.” All efforts were made to minimize the animal’s suffering.
Mouse tissue collection
Hippocampus, cortex, and cerebellum were collected from C57BL/6 J mice at different developmental stages, including the following time points: post natal day (P) P1 (n = 3), P15 (n = 3), P30 (n = 3), and P60 (n = 3). Mice were killed, and all brain regions were dissected out within 20 s on an ice-cold surface. All tissue samples were pulverized in liquid nitrogen and stored at 80 °C for subsequent processing.
High-performance liquid chromatography analysis
Brain tissue samples were analyzed as previously reported [69] with minor modifications [20, 70]. Samples were homogenized in 1:10 (w/v) 0.2 M TCA, sonicated (3 cycles, 10 s each), and centrifuged at 13,000 g for 20 min. The precipitated protein pellets were stored at − 80 °C for protein quantification, whereas the supernatants were neutralized with NaOH and subjected to precolumn derivatization with o-phthaldialdehyde (OPA)/N-acetyl-l-cysteine in 50% methanol. Enantiomer derivatives were then resolved on a Simmetry C8 5 m reversed-phase column (Waters, 4.6 × 250 mm), in isocratic conditions (0.1 M sodium acetate buffer, pH 6.2, 1% tetrahydrofuran, 1 mL/min flow rate). A washing step in 0.1 M sodium acetate buffer, 3% tetrahydrofuran, and 47% acetonitrile was performed after every single run. Identification and quantification of d-Ser and d-asp was based on retention times and peak areas, compared with those associated with external standards. The identity of d-Asp peak was confirmed by adding known amount of external standards, and by the selective degradation catalyzed by an active hDDO recombinant enzyme (kind gift by Dr. Homma and Dr. Katane). The samples were added with 10 μg of the enzyme, incubated at 30 °C for 4 h and then derivatized. Total protein content of homogenates was determined using the Bradford assay method after resolubilization of the TCA precipitated protein pellets. The detected total levels of the amino acids in homogenates were normalized by the total protein content. The content of free d-Ser and d-Asp was expressed as ratio between d- and total (d + l) amount of both Ser and Asp.
Isolation of neurons, astrocytes, and enriched microglia/oligodendrocyte/endothelial cells
Cerebral cortex and cerebellum were dissected from six to eight C57/BL6 mice at P1 and P15 and dissociated into a single-cell suspension using the Neural Tissue Dissociation Kit Postnatal Neurons (Milteny Biotec), according to the manufacturer’s protocol, with minor modifications [71, 72]. Briefly, after carefully removing the meninges, brain tissue was weighed and cut into small pieces in ice-cold Hank’s buffered salt solution (HBSS, Gibco). The minced tissue were then dissociated enzymatically in a solution of Enzyme Mix 1 for 25 min at 37 °C and then mechanically after adding Enzyme Mix 2. Finally, the sample was applied to a cell strainer, and the resulting single-cell suspension was centrifuged at 300×g for 10 min. Mouse astrocytes, neurons, and other non-neuronal cells were sequentially separated from the same brain samples with a magnetic activated cell sorting approach (MACS®). ACSA2-expressing astrocytes were enriched by positive selection using antibody-conjugated magnetic beads (Milteny Biotec). Neuronal cells were enriched by a negative depletion of non-neuronal cells using the (mouse) Neuron Isolation Kit (Milteny Biotec). Briefly, up to 107 dissociated cells were resuspended in 80 μL of cold Dulbecco’s phosphate-buffered saline containing 0.5% bovine serum albumin (DPBS-BSA buffer), incubated with 10 μL FcR Blocking Buffer for 10 min, and then with 10 μL ACSA-2 MicroBeads for 15 min, always at 4 °C. After washing, cells were centrifuged to remove excess beads from the solution. The pellet was suspended with 500 μL of DPBS-BSA buffer and the suspension was applied to the appropriate MACS column fitted in MACS Midi magnetic cell separator. The flow-through containing the unlabeled negative fraction was collected for subsequent cell separation. Following column removal from the magnetic separator, bound astrocytes were flushed out with 1 mL of DPBS-BSA, centrifuged, and the pellet stored at − 80 °C for further analysis. Cell number in the negative fraction was determined and up to 107 dissociated cells were resuspended in 80 μL DPBS-BSA. Then, they were first incubated with 20 μL of Non-Neuronal Cell Biotin-Antibody Cocktail for 5 min and then with 20 μL di Anti-Biotin MicroBeads for 10 min, always at 4 °C. After resuspension in 500 μL buffer, the solution was eluted on MACS column. The flow-through containing the negative fraction of unlabeled neuronal cells was centrifuged and the pellet stored at − 80 °C. Following column removal from the magnetic separator, bound non-neuronal cells (MOE fraction: mainly microglia, oligodendrocytes, and endothelial cells) were eluted in 1 mL buffer, centrifuged and stored. Cell identity and purity of astrocyte and neuronal fractions was confirmed in immunocytochemical experiments (data not shown).
DNA extraction from tissues
DNA was extracted using Dneasy Blood & Tissue Kit (Qiagen, Hilden, Germany), following the manufacturer’s instructions. DNA was quality checked using NanoDrop 2000, (Thermo Scientific) and quantified using Qubit 2.0 Fluorometer with the dsDNA broad range assay kit (Invitrogen, Q32850).
RNA extraction
Total RNA was extracted by RNeasy mini kit (QIAGEN) according to the manufacturer’s instructions. The integrity of the RNA was assessed by denaturing agarose gel electrophoresis (presence of sharp 28S, 18S, and 5S bands) and spectrophotometry (NanoDrop 2000, Thermo Scientific). Total RNA was purified to eliminate potentially contaminating genomic DNA using recombinant DNase (QIAGEN).
qRT-PCR
A total of 1 μg of total RNA of each sample was reverse-transcribed with Quanti Tect Reverse Transcription (QIAGEN) using oligo-dT and random primers according to the manufacturer’s instructions. qRT-PCR amplifications were performed using LightCycler 480 SYBR Green I Master (Roche Diagnostic) in a LightCycler480 Real Time thermocycler. The following protocol was used: 10 s for initial denaturation at 95 °C followed by 40 cycles consisting of 10 s at 94 °C for denaturation, 10 s at 60 °C for annealing, and 6 s for elongation at 72 °C temperature. The following primers were used for mouse Ddo, Dao, Srr cDNA amplification: Ddo forward 5-ACCACCAGTAATGTAGCGGC-3 and Ddo reverse 5-GGTACCGGGGTATCTGCAC-3; Dao forward 5-TTTTCTCCCGACACCTGGC-3 and Dao reverse 5-TGAACGGGGTGAATCGATCT-3; Srr forward 5-CCCTTGGTAGATGCACTGGT and Srr reverse 5-TCAGCAGCGTATACCTTCACAC-3. b-actin and PP1A were used as housekeeping genes for PCR: b-actin forward 5-CTAAGGCCAACCGTGAAAAG-3 and b-actin reverse 5-ACCAGAGGCATACAGGGACA-3, PP1A forward 5-GTGGTCTTTGGGAAGGTGAA-3 and PP1A reverse 5-TTACAGGACATTGCGAGCAG-3.
Bisulfite and oxidative bisulfite conversion and amplicon library preparation
Genomic DNA (1 μg) was converted by sodium bisulfite with EZ DNA Methylation Kit (Zymo Research) and eluted in 50 μL H2O according to the manufacturer’s instruction. Oxidative bisulfite experiments for hydroxymethylcytosine detection at single nucleotide level were performed using TrueMethyl oxBS module (Nugen, Tecan, California USA) following the manufacturer’s instruction). Quality and amount (1 μg) of genomic DNA was evaluated by Nanodrop and Qubit instruments. DNA was first purified with magnetic beads and then treated with oxidant solution. Both bisulfite- and ox-bisulfite-treated samples underwent to double amplification strategy to generate an amplicon library. The first PCR step was performed using bisulfite-specific primers. Reactions were performed in 30 μL total volumes: 3 μL 10× reaction buffer, 0.6 μL of 10 mM dNTP mix, 0.9 μL of 5 mM forward and reverse primers, 3.6 μL MgCl2 25 mM, 2–4 μL bisulfite template DNA, 0.25 μL FastStart Taq, and H2O up to the final volume. Sequences of gene-specific primers, along with individual PCR conditions, are reported in Table 1. The second step of PCR was used to add multiplexing indices and Illumina sequencing adaptors to first amplicons. Second PCR step was performed in 50 μL total volumes: 5 μL 10× reaction buffer, 1 μL dNTP mix, 5 μL forward and reverse “Nextera XT” primers (Illumina, San Diego, CA), 6 μL 25 mM MgCl2, 5 μL of first PCR product, 0.4 μL FastStart Taq, and H2O up to the final volume. After both PCR steps, a purification phase was performed using AMPure purification magnetic Beads (Beckman-Coulter, Brea, CA) following the manufacturer’s protocol. All amplicons were quantified using Qubit® 2.0 Fluorometer. A library of amplicons derived from bisulfite- and ox-bisulfite-treated genomic DNAs was obtained pooling amplicons at equimolar ratio. Library was diluted to final concentration of 8 pM. Phix control libraries (Illumina) were combined with normalized library [10% (v/v)] to increase diversity of base calling during sequencing. Amplicons’ library was subjected to sequencing using V3 reagents kits on Illumina MiSeq system (Illumina). Paired-end sequencing was performed in 281 cycles per read (281 × 2). An average of approximately 100,000 reads/sample were obtained. To estimate the rate of bisulfite conversion, fully unmethylated M13mp18 double-strand DNA (New England BioLabs) was added in representative samples.
Table 1.
Primers and amplification conditions used in the first PCR step for all genes in this study. The capital letters in the primer sequences indicate the original C or G. For all genes, the positions refer to the TSS; for M13mp18, the position refers sequence entry X02513.1 (NCBI, GenBank)
| Gene | Amplicons | Fw primer | Rv primer | Amplification conditions |
|---|---|---|---|---|
| Dao (PR1) | + 3/+ 365 | TagTTagagaagtTaggYtgYtYaYta | agattggtgaRRRaaaaaaggagaga | Denature at 95 °C for 2 min; 35 cycles of denaturing at 95 °C for 30 s, annealing at 52 °C for 40 s, and extension at 72 °C for 50 s. Final elongation at 72 °C for 6 m |
| Dao (PR2) | − 234/+ 137 | gaaYagYagTgagYtagYtgg | caccaRccaRRaatRaaacacaa | Denature at 95 °C for 2 min; 38 cycles of denaturing at 95 °C for 30 s, annealing at 54 °C for 40 s, and extension at 72 °C for 50 s. Final elongation at 72 °C for 6 m |
| Srr | + 27/+ 377 | GTaTtgggagTaaaagTattTag | tttaaactccacaatccaAAcct | Denature at 95 °C for 2 min; 35 cycles of denaturing at 95 °C for 30 s, annealing at 57 °C for 40 s, and extension at 72 °C for 50 s. Final elongation at 72 °C for 6 m |
| Ddo | − 63/− 468 | gtgtgtttTtgaggaggtgaTaTtTa | aActtaccctccattAAtccatAcc | Denature at 95 °C for 2 min; 36 cycles of denaturing at 95 °C for 30 s, annealing at 52 °C for 40 s, and extension at 72 °C for 50 s. Final elongation at 72 °C for 6 m |
| M13mp18 | 5946/6294 | Ggtgaagggtaattagttgttgtt | ccaataccaaacttacatacct | Denature at 95 °C for 2 min; 33 cycles of denaturing at 95 °C for 30 s, annealing at 57 °C for 40 s, and extension at 72 °C for 50 s. Final elongation at 72 °C for 6 m |
Sequence handling and bioinformatics analyses
Paired-end reads were obtained from Illumina Miseq sequencer platform. Using PEAR tool [73], reads were assembled together with a minimum of 40 overlapping residues as threshold. FASTQ assembled reads were then converted in FASTA format using PRINSEQ tool [74]. Sequences derived from bisulfite- and ox-bisulfite-treated DNAs were analyzed with ampliMethProfiler pipeline software (https://sourceforge.net/projects/amplimethprofiler) [36], specifically designed for deep-targeted bisulfite amplicon sequencing. ampliMethProfiler produces quality filtered FASTA files for all samples and directly extracts methylation average and methylation profiles. To determine the average 5-hmC levels, the average values at single-CpGs obtained from sequence analysis of DNAs treated with oxidant plus bisulfite were subtracted to the average methylation values obtained from DNA treated with bisulfite only. As output, the pipeline generates also a summary file with information about the number of reads passing filters, the methylation percentage of each C in CpG sites, and the bisulfite efficiency for each C in non-CpG sites. ampliMethProfiler produces a tabular format file (BIOM format) containing the number of methylation profiles (epialleles) for all samples. The BIOM table was normalized for the same number of sequence/sample through a rarefaction procedure using QIIME [75]. Rarefied BIOM table was then processed using JMP to obtain principal component analysis (PCA) and hierarchical cluster analysis (HCA) based on epialleles distribution.
Statistical analyses
Methylation average data are expressed as means ± standard error. Comparisons between groups were performed using one-way ANOVA with α significance level ≤ 0.001. mRNA expression levels are reported as 2-∆Ct and analyzed by one-way ANOVA. All statistical analyses were performed using GraphPad (GraphPad Prism Software, Inc., La Jolla, CA, USA www.graphpad.com/guides/prism/7/statistics/index.htm).
Additional files
{kind=link}
Figure S1. Ddo epiallelic distribution analysis in all analyzed brain regions during ontogenesis. Hierarchical cluster based on the epiallelic composition of each developmental stage is presented for HIPP, CX and CB. Developmental stages are indicated with different colors (P1 = red; P15 = blue; P30 = yellow; P60 = green). (JPG 87 kb)
Figure S2. Regression analysis associating the degree of methylation with mRNA expression levels of the Ddo gene. Regression analysis associating DNA methylation and mRNA expression of Ddo gene is shown in each analyzed brain area during development. Ddo mRNA expression is normalized to the mean values for two housekeeping genes and expressed as 2–∆Ct values. The three mice for each time point (P1 = red; P15 = blue; P30 = yellow; P60 = green) are indicated. Statistical analyses were performed using Pearson correlation. * p ≤ 0.05 (PDF 86 kb)
Acknowledgements
We thank Dr. H. Homma and Dr. M. Katane for kindly providing us with the human recombinant Ddo enzyme. We wish to thank Dr. Rita San-Bento for precious suggestions related to hydroxymethylation experiments.
Abbreviations
- 5hmC
5-Hydroxymethylcytosine
- 5mC
5-Methylcytosine
- CB
Cerebellum
- CX
Cortex
- Dao
d-amino acid oxidase
- d-Asp
d-aspartate
- Ddo
d-aspartate oxidase
- DNMT
DNA methyltransferase
- d-Ser
d-serine
- HIPP
Hippocampus
- Srr
Serine racemase
- TET
Ten-eleven translocation enzyme
Authors’ contributions
MCu and SK performed the sequencing experiments, analyzed the data, and wrote the manuscript. MCu performed oxidative bisulfite sequencing experiments. DP performed tissue selection and collection, mRNA experiments, and analysis. EF and LCo participated in the experiments and performed the data analysis. OA and SC performed statistical and data analyses. TN performed the HPLC experiments. FB and VdR isolated cell types and performed the experiments on purified cells. FE, MCa, VEA, and AU analyzed and interpreted the data and contributed to writing the manuscript. LCh supervised the experimental plan and data analyses and wrote the main manuscript text. All authors reviewed the manuscript. All authors read and approved the final manuscript.
Funding
S.K. was supported by 2017 NARSAD Young Investigator Grant from the Brain and Behavior Research Foundation (Grant no: 26370); L. Chiariotti was supported by Epigen Flagship Project C.N.R. SP6.6. This work was supported by the POR Campania FESR 2014–2020 “SATIN” grant.
Availability of data and materials
The datasets supporting the results of this article are included within the article. All raw data have been deposited in a public database (ENA) under accession number: PRJEB28662 and no restrictions will be applied.
Ethics approval and consent to participate
All research involving animals was performed in accordance with the European directive 86/609/EEC governing animal welfare and protection, which is acknowledged by the Italian Legislative Decree no. 26 (March 14, 2014). Animal research protocols were also reviewed and approved by the local animal care committee at the University of Naples “Federico II”. All efforts were made to minimize the animals’ suffering.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Mariella Cuomo, Simona Keller and Daniela Punzo contributed equally to this work.
Contributor Information
Alessandro Usiello, Email: usiello@ceinge.unina.it.
Lorenzo Chiariotti, Email: chiariot@unina.it.
References
- 1.Bird A. Perceptions of epigenetics. Nature. 2007;447(7143):396–398. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- 2.Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, et al. Global Epigenomic reconfiguration during mammalian brain development. Science. 2013;341(6146):1237905. doi: 10.1126/science.1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Desplats PA. Perinatal programming of neurodevelopment: epigenetic mechanisms and the prenatal shaping of the brain. Adv Neurobiol. 2015;10:335–361. doi: 10.1007/978-1-4939-1372-5_16. [DOI] [PubMed] [Google Scholar]
- 4.Tognini P, Napoli D, Pizzorusso T. Dynamic DNA methylation in the brain: a new epigenetic mark for experience-dependent plasticity. Front Cell Neurosci. 2015;9:331. doi: 10.3389/fncel.2015.00331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosenberg AB, Roco CM, Muscat RA, Kuchina A, Sample P, Yao Z, et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science. 2018;360(6385):176–182. doi: 10.1126/science.aam8999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mill J, Tang T, Kaminsky Z, Khare T, Yazdanpanah S, Bouchard L, et al. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am J Hum Genet. 2008;82(3):696–711. doi: 10.1016/j.ajhg.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petronis A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature. 2010;465(7299):721–727. doi: 10.1038/nature09230. [DOI] [PubMed] [Google Scholar]
- 8.Keller S, Sarchiapone M, Zarrilli F, Videtic A, Ferraro A, Carli V, et al. Increased BDNF promoter methylation in the Wernicke area of suicide subjects. Arch Gen Psychiatry. 2010;67(3):258–267. doi: 10.1001/archgenpsychiatry.2010.9. [DOI] [PubMed] [Google Scholar]
- 9.Keller S, Errico F, Zarrilli F, Florio E, Punzo D, Mansueto S, et al. DNA methylation state of BDNF gene is not altered in prefrontal cortex and striatum of schizophrenia subjects. Psychiatry Res. 2014;220(3):1147–1150. doi: 10.1016/j.psychres.2014.08.022. [DOI] [PubMed] [Google Scholar]
- 10.Sweatt JD. The emerging field of neuroepigenetics. Neuron. 2013;8(3):624–632. doi: 10.1016/j.neuron.2013.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hannon E, Spiers H, Viana J, Pidsley R, Burrage J, Murphy TM, et al. Methylation QTLs in the developing brain and their enrichment in schizophrenia risk loci. Nat Neurosci. 2015;19(1):48–54. doi: 10.1038/nn.4182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nestler EJ, Peña CJ, Kundakovic M, Mitchell A, Akbarian S. Epigenetic basis of mental illness. Neuroscientist. 2016;22(5):447–463. doi: 10.1177/1073858415608147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christopher MA, Kyle SM, Katz DJ. Neuroepigenetic mechanisms in disease. Epigenetics Chromatin. 2017;10(1):47. doi: 10.1186/s13072-017-0150-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farrell C, Doolin K, O’Leary N, Jairaj C, Roddy D, Tozzi L, et al. DNA methylation differences at the glucocorticoid receptor gene in depression are related to functional alterations in hypothalamic–pituitary–adrenal axis activity and to early life emotional abuse. Psychiatry Res. 2018;265:341–348. doi: 10.1016/j.psychres.2018.04.064. [DOI] [PubMed] [Google Scholar]
- 15.Caldji C, Hellstrom IC, Zhang TY, Diorio J, Meaney MJ. Environmental regulation of the neural epigenome. FEBS Lett. 2011;585(13):2049–2058. doi: 10.1016/j.febslet.2011.03.032. [DOI] [PubMed] [Google Scholar]
- 16.Spiers H, Hannon E, Schalkwyk LC, Smith R, Wong CCY, O’Donovan MC, et al. Methylomic trajectories across human fetal brain development. Genome Res. 2015;25(5):338–352. doi: 10.1101/gr.180273.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schneider E, Dittrich M, Böck J, Nanda I, Müller T, Seidmann L, et al. CpG sites with continuously increasing or decreasing methylation from early to late human fetal brain development. Gene. 2016;592(1):110–118. doi: 10.1016/j.gene.2016.07.058. [DOI] [PubMed] [Google Scholar]
- 18.Ecker JR, Geschwind DH, Kriegstein AR, Ngai J, Osten P, Polioudakis D, et al. The BRAIN initiative cell census consortium: lessons learned toward generating a comprehensive BRAIN cell atlas. Neuron. 2017;96(3):542–557. doi: 10.1016/j.neuron.2017.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Florio E, Keller S, Coretti L, Affinito O, Scala G, Errico F, et al. Tracking the evolution of epialleles during neural differentiation and brain development: D-aspartate oxidase as a model gene. Epigenetics. 2017;12(1):41–54. doi: 10.1080/15592294.2016.1260211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Punzo D, Errico F, Cristino L, Sacchi S, Keller S, Belardo C, et al. Age-related changes in D-aspartate oxidase promoter methylation control extracellular D-aspartate levels and prevent precocious cell death during brain aging. J Neurosci. 2016;36(10):3064–3078. doi: 10.1523/JNEUROSCI.3881-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hashimoto A, Oka T. Free D-aspartate and D-serine in the mammalian brain and periphery. Prog Neurobiol. 1997;52(4):325–353. doi: 10.1016/S0301-0082(97)00019-1. [DOI] [PubMed] [Google Scholar]
- 22.Errico F, Napolitano F, Nisticò R, Usiello A. New insights on the role of free D-aspartate in the mammalian brain. Amino Acids. 2012;43(5):1861–1871. doi: 10.1007/s00726-012-1356-1. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki M, Imanishi N, Mita M, Hamase K, Aiso S, Sasabe J. Heterogeneity of D-serine distribution in the human central nervous system. ASN Neuro. 2017;9(3):1759091417713905. doi: 10.1177/1759091417713905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang LZ, Zhu XZ. Spatiotemporal relationships among D-serine, serine racemase, and D-amino acid oxidase during mouse postnatal development. Acta Pharmacol Sin. 2003;24(10):965–974. [PubMed] [Google Scholar]
- 25.Van Veldhoven PP, Brees C, Mannaerts GP. D-aspartate oxidase, a peroxisomal enzyme in liver of rat and man. BBA - Gen Subj. 1991;1073(1):203–208. doi: 10.1016/0304-4165(91)90203-S. [DOI] [PubMed] [Google Scholar]
- 26.Schell MJ, Molliver ME, Snyder SH. D-serine, an endogenous synaptic modulator: localization to astrocytes and glutamate-stimulated release. Proc Natl Acad Sci. 1995;92(9):3948–3952. doi: 10.1073/pnas.92.9.3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Katane M, Homma H. D-aspartate: an important bioactive substance in mammals: a review from an analytical and biological point of view. J Chromatogr B Anal Technol Biomed Life Sci. 2011;879(29):3108–3121. doi: 10.1016/j.jchromb.2011.03.062. [DOI] [PubMed] [Google Scholar]
- 28.Verrall L, Walker M, Rawlings N, Benzel I, Kew JNC, Harrison PJ, et al. D-amino acid oxidase and serine racemase in human brain: Normal distribution and altered expression in schizophrenia. Eur J Neurosci. 2007;26(6):1657–1669. doi: 10.1111/j.1460-9568.2007.05769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jagannath V, Marinova Z, Monoranu CM, Walitza S, Grünblatt E. Expression of D-amino acid oxidase (Dao/DAAO) and D-amino acid oxidase activator (DaoA/G72) during development and aging in the human post-mortem brain. Front Neuroanat. 2017;11:31. doi: 10.3389/fnana.2017.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Radzishevsky I, Sason H, Wolosker H. D-serine: physiology and pathology. Curr Opin Clin Nutr Metab Care. 2013;16(1):72–75. doi: 10.1097/MCO.0b013e32835a3466. [DOI] [PubMed] [Google Scholar]
- 31.Mothet JP, Le Bail M, Billard JM. Time and space profiling of NMDA receptor co-agonist functions. J Neurochem. 2015;135(2):210–225. doi: 10.1111/jnc.13204. [DOI] [PubMed] [Google Scholar]
- 32.Rulands S, Lee HJ, Clark SJ, Angermueller C, Smallwood SA, Krueger F, et al. Genome-scale oscillations in DNA methylation during exit from pluripotency. Cell Syst. 2018;7(1):63–76. doi: 10.1016/j.cels.2018.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Szyf M. The elusive role of 5′-hydroxymethylcytosine. Epigenomics. 2016;8(11):1539–1551. doi: 10.2217/epi-2016-0076. [DOI] [PubMed] [Google Scholar]
- 34.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Booth MJ, Ost TW, Beraldi D, Bell NM, Branco MR, Reik W, Balasubramanian S. Oxidative bisulfite sequencing of 5-methylcytosine and 5-hydroxymethylcytosine. Nat Protoc. 2013;8(10):1841–1851. doi: 10.1038/nprot.2013.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sasabe J, Miyoshi Y, Suzuki M, Mita M, Konno R, Matsuoka M, et al. D-amino acid oxidase controls motoneuron degeneration through D-serine. Proc Natl Acad Sci. 2012;109(2):627–632. doi: 10.1073/pnas.1114639109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shibuya N, Koike S, Tanaka M, Ishigami-Yuasa M, Kimura Y, Ogasawara Y, et al. A novel pathway for the production of hydrogen sulfide from D-cysteine in mammalian cells. Nat Commun. 2013;4:1366. doi: 10.1038/ncomms2371. [DOI] [PubMed] [Google Scholar]
- 38.Farhy-Tselnicker I, Allen NJ. Astrocytes, neurons, synapses: a tripartite view on cortical circuit development. Neural Dev. 2018;13(1):7. doi: 10.1186/s13064-018-0104-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Affinito O, Scala G, Palumbo D, Florio E, Monticelli A, Miele G, et al. Modeling DNA methylation by analyzing the individual configurations of single molecules. Epigenetics. 2016;11(12):881–888. doi: 10.1080/15592294.2016.1246108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Keller S, Punzo D, Cuomo M, Affinito O, Coretti L, Sacchi S, et al. DNA methylation landscape of the genes regulating D-serine and D-aspartate metabolism in post-mortem brain from controls and subjects with schizophrenia. Sci Rep. 2018;8(1):10163. doi: 10.1038/s41598-018-28332-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scala G, Affinito O, Palumbo D, Florio E, Monticelli A, Miele G, et al. Amplimeth profiler: a pipeline for the analysis of CpG methylation profiles of targeted deep bisulfite sequenced amplicons. BMC Bioinformatics. 2016;17(1):484. doi: 10.1186/s12859-016-1380-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wolosker H, Blackshaw S, Snyder SH. Serine racemase: a glial enzyme synthesizing D-serine to regulate glutamate-N-methyl-D-aspartate neurotransmission. Proc Natl Acad Sci U S A. 1999;96(23):13409–13414. doi: 10.1073/pnas.96.23.13409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pollegioni L, Sacchi S. Metabolism of the neuromodulator D-serine. Cell Mol Life Sci. 2010;67(14):2387–2404. doi: 10.1007/s00018-010-0307-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wolosker H, Mori H. Serine racemase: an unconventional enzyme for an unconventional transmitter. Amino Acids. 2012;43(5):1895–1904. doi: 10.1007/s00726-012-1370-3. [DOI] [PubMed] [Google Scholar]
- 45.Billard JM. D-amino acids in brain neurotransmission and synaptic plasticity. Amino Acids. 2012;43(5):1851–1860. doi: 10.1007/s00726-012-1346-3. [DOI] [PubMed] [Google Scholar]
- 46.Van Horn MR, Sild M, Ruthazer ES. D-serine as a gliotransmitter and its roles in brain development and disease. Front Cell Neurosci. 2013;7:39. doi: 10.3389/fncel.2013.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Papouin T, Ladépêche L, Ruel J, Sacchi S, Labasque M, Hanini M, et al. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell. 2012;150(3):633–646. doi: 10.1016/j.cell.2012.06.029. [DOI] [PubMed] [Google Scholar]
- 48.Errico F, Nuzzo T, Carella M, Bertolino A, Usiello A. The emerging role of altered d-aspartate metabolism in schizophrenia: new insights from preclinical models and human studies. Front Psychiatry. 2018;9:559. doi: 10.3389/fpsyt.2018.00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sacchi S, Novellis V, Paolone G, Nuzzo T, Iannotta M, Belardo C, et al. Olanzapine, but not clozapine, increases glutamate release in the prefrontal cortex of freely moving mice by inhibiting D-aspartate oxidase activity. Sci Rep. 2017;7:46288. doi: 10.1038/srep46288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Errico F, Mothet JP, Usiello A. D-aspartate: an endogenous NMDA receptor agonist enriched in the developing brain with potential involvement in schizophrenia. J Pharm Biomed Anal. 2015;116:7–17. doi: 10.1016/j.jpba.2015.03.024. [DOI] [PubMed] [Google Scholar]
- 51.Szilágyi B, Ferenczy GG, Keserű GM. Drug discovery strategies and the preclinical development of D-amino-acid oxidase inhibitors as antipsychotic therapies. Expert Opin Drug Discov. 2018;13(10):973–982. doi: 10.1080/17460441.2018.1524459. [DOI] [PubMed] [Google Scholar]
- 52.Ma T, Wu Y, Chen B, Zhang W, Jin L, Shen C, et al. D-serine contributes to seizure development via ERK signaling. Front Neurosci. 2019;13:254. doi: 10.3389/fnins.2019.00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kölker S. Metabolism of amino acid neurotransmitters: the synaptic disorder underlying inherited metabolic diseases. J Inherit Metab Dis. 2018;41(6):1055–1063. doi: 10.1007/s10545-018-0201-4. [DOI] [PubMed] [Google Scholar]
- 54.Feng J, Fan G. The role of DNA methylation in the central nervous system and neuropsychiatric disorders. Int Rev Neurobiol. 2009;89:67–84. doi: 10.1016/S0074-7742(09)89004-1. [DOI] [PubMed] [Google Scholar]
- 55.Cholewa-Waclaw J, Bird A, von Schimmelmann M, Schaefer A, Yu H, Song H, et al. The role of epigenetic mechanisms in the regulation of gene expression in the nervous system. J Neurosci. 2016;36(45):11427–11434. doi: 10.1523/JNEUROSCI.2492-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weaver ICG, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, et al. (2004) Epigenetic programming by maternal behavior. Nat Neurosci 2004;7(8):847–854. [DOI] [PubMed]
- 57.Yamanaka M, Miyoshi Y, Ohide H, Hamase K, Konno R. D-amino acids in the brain and mutant rodents lacking D-amino-acid oxidase activity. Amino Acids. 2012;43(5):1811–1821. doi: 10.1007/s00726-012-1384-x. [DOI] [PubMed] [Google Scholar]
- 58.Guo JU, Ma DK, Mo H, Ball MP, Jang MH, Bonaguidi MA, et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat Neurosci. 2011;14(10):1345–1351. doi: 10.1038/nn.2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guo JU, Su Y, Zhong C, Ming GL, Song H. Emerging roles of TET proteins and 5-hydroxymethylcytosines in active DNA demethylation and beyond. Cell Cycle. 2011;10(16):2662–2668. doi: 10.4161/cc.10.16.17093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hashimoto A, Kumashiro S, Nishikawa T, Oka T, Takahashi K, Mito T, et al. Embryonic development and postnatal changes in free d-aspartate and d-serine in the human prefrontal cortex. J Neurochem. 1993;61(1):348–351. doi: 10.1111/j.1471-4159.1993.tb03575.x. [DOI] [PubMed] [Google Scholar]
- 61.Sakai K, Homma H, Lee JA, Fukushima T, Santa T, Tashiro K, et al. Emergence of D-aspartic acid in the differentiating neurons of the rat central nervous system. Brain Res. 1998;808(1):65–71. doi: 10.1016/S0006-8993(98)00599-X. [DOI] [PubMed] [Google Scholar]
- 62.Katane M, Homma H. D-aspartate oxidase: the sole catabolic enzyme acting on free D-aspartate in mammals. Chem Biodivers. 2010;7(6):1435–1449. doi: 10.1002/cbdv.200900250. [DOI] [PubMed] [Google Scholar]
- 63.Balu DT, Coyle JT. Neuronal d-serine regulates dendritic architecture in the somatosensory cortex. Neurosci Lett. 2012;517(2):77–81. doi: 10.1016/j.neulet.2012.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ivanov AD, Mothet JP. The plastic d-serine signaling pathway: sliding from neurons to glia and vice-versa. Neurosci Lett. 2018;689:21–25. doi: 10.1016/j.neulet.2018.05.039. [DOI] [PubMed] [Google Scholar]
- 65.Hashimoto K, Malchow B, Falkai P, Schmitt A. Glutamate modulators as potential therapeutic drugs in schizophrenia and affective disorders. Eur Arch Psychiatry Clin Neurosci. 2013;263(5):367–377. doi: 10.1007/s00406-013-0399-y. [DOI] [PubMed] [Google Scholar]
- 66.Sacchi S, Rosini E, Pollegioni L, Molla G. D-amino acid oxidase inhibitors as a novel class of drugs for schizophrenia therapy. Curr Pharm Des. 2013;19(14):2499–2511. doi: 10.2174/1381612811319140002. [DOI] [PubMed] [Google Scholar]
- 67.Chang HJ, Lane HY, Tsai GE. NMDA pathology and treatment of schizophrenia. Curr Pharm Des. 2014;20(32):5118–5126. doi: 10.2174/1381612819666140110121908. [DOI] [PubMed] [Google Scholar]
- 68.Coyle JT, Balu DT. The role of serine racemase in the pathophysiology of brain disorders. Adv Pharmacol. 2017;82:35–56. doi: 10.1016/bs.apha.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Topo E, Fisher G, Sorricelli A, Errico F, Usiello A, D’Aniell A. Thyroid hormones and D-aspartic acid, D-aspartate oxidase, D-aspartate racemase, H2O2, and ROS in rats and mice. Chem Biodivers. 2010;7(6):1467–1478. doi: 10.1002/cbdv.200900360. [DOI] [PubMed] [Google Scholar]
- 70.Nuzzo T, Sacchi S, Errico F, Keller S, Palumbo O, Florio E, et al. Decreased free d -aspartate levels are linked to enhanced d-aspartate oxidase activity in the dorsolateral prefrontal cortex of schizophrenia patients. NPJ Schizophr. 2017;3:16. doi: 10.1038/s41537-017-0015-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Boscia F, Esposito CL, Casamassa A, de Franciscis V, Annunziato L, Cerchia L. The isolectin IB4 binds RET receptor tyrosine kinase in microglia. J Neurochem. 2013;126(4):428–436. doi: 10.1111/jnc.12209. [DOI] [PubMed] [Google Scholar]
- 72.Boscia F, Pannaccione A, Ciccone R, Casamassa A, Franco C, Piccialli I, et al. The expression and activity of KV3.4 channel subunits are precociously upregulated in astrocytes exposed to Aβ oligomers and in astrocytes of Alzheimer’s disease Tg2576 mice. Neurobiol Aging. 2017;54:187–198. doi: 10.1016/j.neurobiolaging.2017.03.008. [DOI] [PubMed] [Google Scholar]
- 73.Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: a fast and accurate Illumina paired-end reAd mergeR. Bioinformatics. 2014;30(5):614–620. doi: 10.1093/bioinformatics/btt593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27(6):863–864. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Ddo epiallelic distribution analysis in all analyzed brain regions during ontogenesis. Hierarchical cluster based on the epiallelic composition of each developmental stage is presented for HIPP, CX and CB. Developmental stages are indicated with different colors (P1 = red; P15 = blue; P30 = yellow; P60 = green). (JPG 87 kb)
Figure S2. Regression analysis associating the degree of methylation with mRNA expression levels of the Ddo gene. Regression analysis associating DNA methylation and mRNA expression of Ddo gene is shown in each analyzed brain area during development. Ddo mRNA expression is normalized to the mean values for two housekeeping genes and expressed as 2–∆Ct values. The three mice for each time point (P1 = red; P15 = blue; P30 = yellow; P60 = green) are indicated. Statistical analyses were performed using Pearson correlation. * p ≤ 0.05 (PDF 86 kb)
Data Availability Statement
The datasets supporting the results of this article are included within the article. All raw data have been deposited in a public database (ENA) under accession number: PRJEB28662 and no restrictions will be applied.