

Type III interferons are potent antiviral cytokines important for regulation of viruses that infect at mucosal surfaces. Studies using mice lacking the Ifnlr1 gene encoding the type III interferon receptor have demonstrated that signaling through this receptor is critical for protection against influenza virus, norovirus, and reovirus. Using a genetic approach to disrupt murine type III interferon cytokine genes Ifnl2 and Ifnl3, we found that mice lacking these cytokines fully recapitulate the impaired control of viruses observed in mice lacking Ifnlr1. Our results support the idea of an exclusive role for known type III interferon cytokines in signaling via IFNLR to mediate antiviral effects at mucosal surfaces. These findings emphasize the importance of type III interferons in regulation of a variety of viral pathogens and provide important genetic evidence to support our understanding of the ligand-receptor interactions in this pathway.
KEYWORDS: cytokines, influenza, interferons, noroviruses, reovirus
ABSTRACT
Type III interferon (IFN), or IFN lambda (IFN-λ), is an essential component of the innate immune response to mucosal viral infections. In both the intestine and the lung, signaling via the IFN-λ receptor (IFNLR) controls clinically important viral pathogens, including influenza virus, norovirus, and rotavirus. While it is thought that IFN-λ cytokines are the exclusive ligands for signaling through IFNLR, it is not known whether genetic ablation of these cytokines phenotypically recapitulates disruption of the receptor. Here, we report the serendipitous establishment of Ifnl2−/− Ifnl3−/− mice, which lack all known functional murine IFN-λ cytokines. We demonstrate that, like Ifnlr1−/− mice lacking IFNLR signaling, these mice display defective control of murine norovirus, reovirus, and influenza virus and therefore genocopy Ifnlr1−/− mice. Thus, for regulation of viral infections at mucosal sites of both the intestine and lung, signaling via IFNLR can be fully explained by the activity of known cytokines IFN-λ2 and IFN-λ3. Our results confirm the current understanding of ligand-receptor interactions for type III IFN signaling and highlight the importance of this pathway in regulation of mucosal viral pathogens.
IMPORTANCE Type III interferons are potent antiviral cytokines important for regulation of viruses that infect at mucosal surfaces. Studies using mice lacking the Ifnlr1 gene encoding the type III interferon receptor have demonstrated that signaling through this receptor is critical for protection against influenza virus, norovirus, and reovirus. Using a genetic approach to disrupt murine type III interferon cytokine genes Ifnl2 and Ifnl3, we found that mice lacking these cytokines fully recapitulate the impaired control of viruses observed in mice lacking Ifnlr1. Our results support the idea of an exclusive role for known type III interferon cytokines in signaling via IFNLR to mediate antiviral effects at mucosal surfaces. These findings emphasize the importance of type III interferons in regulation of a variety of viral pathogens and provide important genetic evidence to support our understanding of the ligand-receptor interactions in this pathway.
INTRODUCTION
Interferons (IFNs) are a central part of the innate immune response to viral infections. Cells detect viruses through pattern recognition receptors, leading to the transcription of molecules that act via both autocrine signaling and paracrine signaling to induce an antiviral state. This response includes the production and release of IFNs, which bind cell surface receptors and drive gene expression through Janus kinase-signal transducer and activator of transcription (JAK-STAT) signaling pathways, resulting in the expression of hundreds of interferon-stimulated genes (ISGs) (1). Together, these signals activate innate immunity against viral infections and prime the adaptive immune response (1).
Humans have three classes of IFNs—types I, II, and III—that signal via distinct receptor complexes. Type I IFNs include 13 subtypes of alpha IFN (IFN-α) in addition to IFN-β, IFN-ε, IFN-κ, and IFN-ω, all of which bind the heterodimeric type I IFN receptor (IFNAR) composed of subunits IFNAR1 and IFNAR2 (2). In contrast to the diversity of type I IFNs, the type II IFN family consists only of IFN-γ, which binds the IFN-γ receptor complex (1). Type III IFNs consist of three to four functional IFN-λs in humans and signal through the heterodimeric type III IFN receptor (IFNLR). IFNLR is composed of the unique IFNLR1 subunit and the common interleukin-10Rβ (IL-10Rβ) subunit, the latter of which also pairs with distinct ligand-binding chains to form receptors for IL-10, IL-22, and IL-26 (3–6).
Although type I and type III IFNs bind distinct receptors, they both activate transcription factors STAT1, STAT2, and IRF9, leading to the upregulation of a largely overlapping set of ISGs (7, 8). Type III IFN signaling acts more slowly and leads to induction of ISGs that is somewhat weaker but more sustained than that seen with type I IFN signaling (9, 10). Additionally, the two have distinct effects in vivo based on the localization of their respective receptors. IFNAR is expressed on most nucleated cells, whereas IFNLR expression is restricted to distinct cell types, particularly epithelial cells (11–13). IFNLR expression at epithelial barriers explains the critical role for type III IFNs in regulating viral infections at sites such as the intestinal or respiratory epithelium (11, 14, 15).
Mouse models have been useful in dissecting IFN signaling pathways in response to viral infections. Human and murine IFN-λ loci share similar forms of organization, although Ifnl1 is a pseudogene and the region encoding Ifnl4 is absent in mice (16). Murine Ifnl2 and Ifnl3 are highly homologous to their human orthologs, and the murine Ifnlr1 receptor subunit likewise shares a high degree of sequence identity with the human subunit (17). As a result of these similarities, both human and murine IFN-λs display some cross-species reactivity (18).
To study the role of type III IFNs in vivo, Ifnlr1−/− mice have been tested for susceptibility to infection by a variety of pathogens (19) using animals that are deficient in type III IFN receptor signaling either constitutively (20) or conditionally (14). However, given the promiscuity of receptor subunits, it is not known whether disruption of IFNLR1 fully recapitulates an absence of IFN-λ cytokines. In this study, we used a new IFN-λ2/IFN-λ3 (Ifnl2−/− Ifnl3−/−) double-knockout mouse model to demonstrate that absence of IFN-λ expression genocopies absence of IFNLR for multiple viral infections.
RESULTS
Generation of Ifnl2−/− Ifnl3−/− mice.
Mice lack functional Ifnl1 and Ifnl4 genes, so Ifnl2 and Ifnl3 are responsible for all IFN-λ expression (16). The Ifnl2 and Ifnl3 genes are adjacent to each other and appear to have resulted from a head-to-head duplication event (21). These genes are 97.1% identical, with very similar promoters, and are thought to be functionally redundant. Our initial goal was to establish reporter mice for IFN-λ3 expression. We engineered a construct containing the coding sequence of tdTomato, an orange fluorescent protein (22), downstream of self-cleaving peptide T2A (Fig. 1A), and we used CRISPR/Cas9 to target this construct to exon 1 of Ifnl3. From 20 targeted pups, we obtained a single pup confirmed by PCR to have the transgene inserted in the Ifnl3 locus (Fig. 1B).
FIG 1.

Generation of Ifnl2−/− Ifnl3−/− mice. (A) Diagram depicting the guide RNA (bold purple arrow) used to target Ifnl3 for insertion of a construct with a left homology arm (HA), a single T2A sequence, a tdTomato-coding sequence with a polyadenylation signal, and a right HA (blue) by the use of CRISPR/Cas9. Locations of primers used to verify insertion are shown by small arrows over the 5′ and 3′ junctions. (B) PCR amplification of 5′ and 3′ insertion junctions using primers corresponding to the small arrows described in the panel A legend. (C) Schematic of Ifnl2 and Ifnl3 loci showing 7.2-kb deletion and insertion site of tdTomato with portions of genomic sequence (blue), nonspecific inserted sequences (black), and the construct sequence (red) at junctions.
However, while thoroughly analyzing the locus, we found that a 7.2-kb deletion had been inadvertently introduced 5′ of Ifnl3, resulting in removal of exon 1 of Ifnl2 (Fig. 1C) (17). The additional deletion within Ifnl2 most likely occurred due to the extremely high similarity of the two genes at this locus. Although the reporter was not functional in these mice, we had serendipitously introduced disruptions into both IFN-λ genes, establishing mice with linked Ifnl2 and Ifnl3 mutations. We bred this line to homozygosity, thereby generating Ifnl2−/− Ifnl3−/− mice that should lack all functional IFN-λ production.
The capacity of Ifnl2−/− Ifnl3−/− mice to produce IFN-λ was tested following treatment with the synthetic double-stranded RNA analogue poly(I·C), which induces Ifnl2/Ifnl3 expression (23). In contrast to wild-type mice, which had elevated Ifnl2/Ifnl3 expression in both epithelial and systemic tissues after intraperitoneal poly(I·C) injection, Ifnl2−/− Ifnl3−/− mice failed to upregulate IFN-λ expression (Fig. 2A to C). Additionally, IFN-λ was detected in the serum of wild-type mice (Fig. 2D) (24) but not Ifnl2−/− Ifnl3−/− mice (Fig. 2E) after poly(I·C) treatment. Thus, Ifnl2−/− Ifnl3−/− mice are incapable of producing type III IFN.
FIG 2.

Lack of type III IFN induction by poly(I·C) in Ifnl2−/− Ifnl3−/− mice. (A to C) Ileal (A), colonic (B), and splenic (C) Ifnl2/3 copies detected by quantitative PCR (qPCR) in wild-type (WT) and Ifnl2−/− Ifnl3−/− mice at 2 h after intraperitoneal injection of 100 μg of poly(I·C). (D) Serum IFN-λ protein levels in WT mice at 0, 2, 4, 6, 8, and 10 h after poly(I·C) treatment as quantified by ELISA. (E) IFN-λ protein levels in WT and Ifnl2−/− Ifnl3−/− mouse serum in naive mice and 4 h after poly(I·C) treatment as quantified by ELISA. Each symbol represents the value for an individual mouse. Data are combined from results of two separate experiments performed with 3 to 4 mice per group each (A to C) or from results of one experiment performed with 3 to 6 mice per group (D to E). Results were analyzed by Kruskal-Wallis test with Dunn’s multiple comparison.
Restriction of murine norovirus in Ifnl2−/− Ifnl3−/− mice is similar to that in Ifnlr1−/− mice.
To examine the response of Ifnl2−/− Ifnl3−/− mice to enteric viruses, we infected mice with the CR6 strain of murine norovirus (MNoV), which is sensitive to IFN-λ signaling (14, 25). CR6 replicates in the ileum and colon in specialized intestinal epithelial cells (IECs) called tuft cells (26, 27) and is shed at high levels in the stool during persistent infection (28). While the endogenous IFN-λ response to CR6 does not effectively clear infection, replication is significantly limited by intact IFNLR1 signaling, specifically in IECs (14, 25). CR6 shedding in the stool was significantly higher at day 7 postinfection in both Ifnl2−/− Ifnl3−/− mice and Ifnlr1−/− mice than in the wild-type controls (Fig. 3A). Numbers of CR6 genome copies in the colon were also significantly higher in Ifnl2−/− Ifnl3−/− mice than in the wild-type mice and were equivalent to the levels detected in Ifnlr1−/− mice (Fig. 3B). Thus, mice lacking IFN-λ cytokines fail to control persistent MNoV replication, genocopying mice lacking the IFN-λ receptor.
FIG 3.
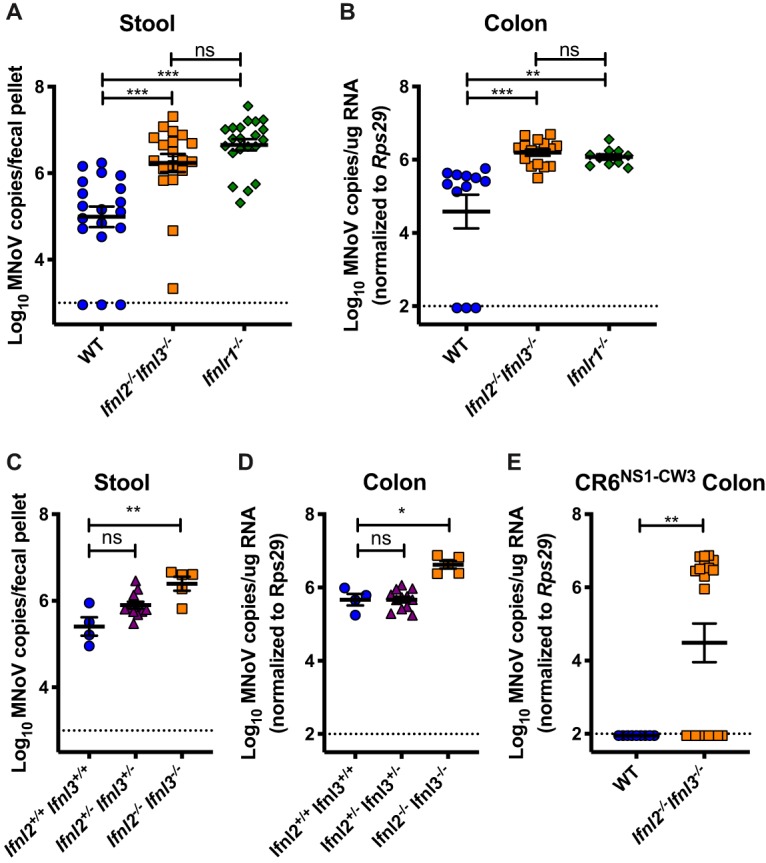
IFN-λ cytokines limit MNoV infection. MNoV.CR6 genome copies were detected by RT-qPCR in stool at day 7 postinfection (A) and in colon at day 14 postinfection (B) in WT, Ifnl2−/− Ifnl3−/−, and Ifnlr1−/− mice after peroral inoculation with 106 PFU. (C and D) MNoV.CR6 viral copies in stool (C) and colon (D) of littermate WT, Ifnl2+/− Ifnl3+/−, and Ifnl2−/− Ifnl3−/− mice 14 days postinfection with 106 PFU. (E) MNoV copies detected in WT and Ifnl2−/− Ifnl3−/− mice 14 days postinoculation with 106 PFU MNoV.CR6NS1−CW3. Data were combined from results from at least two experiments performed with 3 to 14 mice per group per experiment. MNoV.CR6 and MNoV.CR6NS1−CW3 infection data were analyzed by Kruskal-Wallis test with Dunn’s multiple-comparison test and Mann-Whitney test, respectively.
Ifnl2−/− Ifnl3−/− mice were initially bred as homozygotes independently from the wild-type mice used for controls, leading to the possibility of differences in gut microbiota composition between the strains. Since MNoV infection is influenced by commensal bacteria in the gut (29), we sought to exclude the possibility of microbiota effects in our experiments. Ifnl2+/− Ifnl3+/− mice were interbred, and the resultant Ifnl2+/+ Ifnl3+/+, Ifnl2+/− Ifnl3+/−, and Ifnl2−/− Ifnl3−/− progeny were infected with CR6. Ifnl2−/− Ifnl3−/− mice displayed significantly higher numbers of CR6 genome copies in stool and colon than the heterozygote and wild-type littermate controls (Fig. 3C and D). Viral loads in Ifnl2+/− Ifnl3+/− mice were similar to those in wild-type mice, indicating that one functional copy each of Ifnl2 and Ifnl3 is sufficient to control MNoV infection.
The viral nonstructural protein 1 (NS1) serves an important function in tropism of CR6 for IECs and in suppression of IFN-λ signaling (26, 28, 30). The chimeric virus CR6NS1−CW3, in which the CR6 NS1 has been replaced with the NS1 of acute MNoV strain CW3, fails to replicate in colonic IECs in wild-type mice because it lacks an IFN-λ-evading NS1 (26, 30). Intestinal infection with this virus is rescued in Ifnlr1−/− mice (26). Therefore, we infected Ifnl2−/− Ifnl3−/− mice with CR6NS1−CW3 and observed that, in contrast to wild-type controls, the virus replicated in a significant number of mice lacking IFN-λ cytokines (Fig. 3E), similarly to our prior findings with mice lacking the IFN-λ receptor (26). Together, our data indicate that Ifnl2−/− Ifnl3−/− and Ifnlr1−/− mice are similarly susceptible to MNoV infection.
Reovirus replicates similarly in Ifnl2−/− Ifnl3−/− mice and Ifnlr1−/− mice.
IFN-λ is important in the control of multiple enteric viral pathogens (19). To test the effects of loss of Ifnl2/Ifnl3 expression on a virus distinct from MNoV but also sensitive to IFN-λ signaling, we used reovirus, which is controlled by IFNLR1 expression on IECs (14). Mice were inoculated perorally with reovirus strain type 1 Lang (T1L), and viral titers in stool, jejunum, and ileum were assessed 4 days postinfection. Reovirus titers in the stool were elevated in Ifnl2−/− Ifnl3−/− and Ifnlr1−/− mice relative to those in wild-type mice, indicating increased levels of virus shedding (Fig. 4A). Increased viral loads were also observed in jejunal and ileal tissues (Fig. 4B and C). Viral loads in Ifnl2−/− Ifnl3−/− and Ifnlr1−/− mice were similar in stool and ileal tissue, while jejunal tissues from Ifnlr1−/− mice had moderately higher viral titers than those from Ifnl2−/− Ifnl3−/− mice. Thus, like the IFN-λ receptor, IFN-λ cytokines are required for control of multiple enteric viruses.
FIG 4.

Increased reovirus titers in mice lacking IFN-λ cytokines. Titers of reovirus strain T1L in stool (A), ileum (B), and jejunum (C) were assessed by plaque assay at 4 days after peroral infection with 108 PFU. Data are combined from results of two experiments performed with a total of 9 or 10 mice per group. Results were analyzed by one-way ANOVA with Tukey’s multiple-comparison test.
Influenza virus infection is controlled by IFN-λ cytokines.
Antiviral effects of IFN-λ occur at multiple epithelial barriers (11). To test whether type III IFN cytokines protect against viral infection at an epithelial barrier other than the intestine, we compared wild-type and Ifnl2−/− Ifnl3−/− mice for susceptibility to influenza A virus (IAV) infection. Type III IFNs couple with type I IFNs for antiviral effects against IAV infection and can also preempt the detrimental effects of a strong type I IFN response in the lung (31, 32). We inoculated mice intranasally with mouse-adapted IAV PR8 and monitored weight over a 14-day course of infection. While all mouse strains had recovered from weight loss by day 14, Ifnl2−/− Ifnl3−/− mice displayed significantly more weight loss than the wild-type mice (Fig. 5A). We also quantified lung viral loads in these mice 3 days postinfection and found significantly elevated levels of PR8 virus in Ifnl2−/− Ifnl3−/− mice that were comparable to the levels observed in Ifnlr1−/− mice (Fig. 5B). These results demonstrate that Ifnl2−/− Ifnl3−/− mice display defects in antiviral responses at multiple epithelial sites.
FIG 5.

Ifnl2−/− Ifnl3−/− mice have impaired control of nonlethal IAV infection. Male mice that were between 53 and 59 days old were intranasally inoculated with 100 TCID50 of strain PR8. (A) Mice were monitored for weight loss for 14 days after inoculation. Data represent results of one experiment performed with 9 to 12 mice per group. Results were analyzed by two-way ANOVA with Tukey’s multiple-comparison test. (B) Viral loads in the lungs 3 days after inoculation as quantified by hemagglutination assay. Data are from three experiments performed with 3 to 5 mice per group each. Results were analyzed by Kruskal-Wallis test with Dunn’s multiple-comparison test.
DISCUSSION
Studies using mice lacking the IFNLR1 subunit of the type III IFN receptor have demonstrated an essential function for IFN-λ in innate antiviral responses. In this study, we used a new Ifnl2−/− Ifnl3−/− mouse model to show that abrogation of type III IFN cytokine production leads to deficiencies in antiviral responses against both enteric and respiratory viral infections that are comparable to those seen with mice lacking the IFN-λ receptor. We demonstrated that Ifnl2−/− Ifnl3−/− mice did not express IFN-λ in response to Toll-like receptor 3 (TLR3) agonist poly(I·C). We also demonstrated that lack of type III IFN expression was associated with increased titers of MNoV and reovirus in the intestine and increased shedding in the stool that were comparable to the levels seen in Ifnlr1−/− mice. We used littermate controls to verify that this effect was not due to microbiota differences (33, 34), as phenotypic differences unrelated to genotype can arise in independently bred mouse lines (35). Using low-dose influenza virus infection of the upper respiratory tract, an infection model dependent on intact signaling through IFNLR (32, 36), we found that type III IFN cytokines also function in control of a respiratory viral infection. We conclude that in nearly all contexts tested, viral control mediated by the type III IFN receptor is fully explained by IFN-λs acting as IFNLR ligands.
While Ifnl2−/− Ifnl3−/− mice genocopy Ifnlr1−/− mice in terms of viral loads in the ileum and viral shedding in stool, we cannot fully exclude the possibility of a contribution from another signal in the jejunum during reovirus infection, as Ifnlr1−/− mice exhibit an approximately 3-fold increase in viral titers compared with Ifnl2−/− Ifnl3−/− mice. However, we observed greater variation in reovirus titers among Ifnlr1−/− mice, suggesting the possibility of cage-to-cage variation.
Ifnl2 and Ifnl3 are adjacent to each other in an inverted orientation in the mouse genome, with the genes and flanking genomic regions sharing high sequence identity (37). Due to this sequence similarity, although our guide RNAs (gRNAs) successfully disrupted the Ifnl3 locus and permitted insertion of a reporter gene, we also unintentionally caused a large deletion in Ifnl2, leading to disruption of both Ifnl2 and Ifnl3 genes and promoter activity. While off-target effects may occur with CRISPR/Cas9-mediated genomic engineering (38, 39), here we highlight the importance of this consideration in targeting members of highly homologous gene families. Validating that engineered mouse lines do not harbor additional mutations that may affect the phenotypes observed is of critical importance. Although we did not alter the coding sequence of any additional genes in establishing the Ifnl2−/− Ifnl3−/− line, as is the case with any new mouse line, we cannot exclude the possibility that the large deletions in our mouse line might affect regulatory elements of distant genes.
Fortuitously, an Ifnl2Egfp reporter mouse has been recently developed, providing a useful model for studying IFN-λ in vivo (32). This reporter line accomplishes our original goal by allowing the visualization and isolation of IFN-λ-expressing cells while maintaining normal production of IFN-λ in response to TLR stimulation.
Studies of the role of type III IFNs have largely relied on mice lacking the IFNLR1 subunit of the type III IFN receptor. However, it remained possible that this receptor subunit could respond to ligands other than IFN-λs, as is the case for the type I IFN receptor. Using our newly established double-knockout mouse line, we found that the absence of known type III IFNs genocopied IFNLR1 deficiency. Thus, we confirm that in the context of MNoV, reovirus, and IAV infection, all phenotypic effects observed by disruption of Ifnlr1 can be explained by interactions of ligands IFN-λ2 and IFN-λ3 with this receptor.
MATERIALS AND METHODS
Ethics statement.
All mice were used according to regulations stipulated by Washington University Institutional Animal Care and Use Committee and to protocols approved by the Washington University Animal Studies Committee.
Animals and mouse line generation.
C57BL/6J mice were originally purchased from Jackson Laboratories (stock 000664; Jackson Laboratories, Bar Harbor, ME) and were bred and housed in animal facilities in Washington University in Saint Louis under specific-pathogen-free (including murine norovirus-free) conditions. Generation of Ifnlr1−/− mice was previously described (14); briefly, these mice were established by interbreeding Ifnlr1tm1a(EUCOMM)Wtsi mice and Deleter-Cre mice, followed by backcrossing by speed congenics onto a C57BL/6J background.
Ifnl2−/− Ifnl3−/− mice were developed with assistance from the Genome Engineering and iPSC Center (GEiC) at Washington University in Saint Louis, where gRNAs were designed to target the Ifnl3 locus for insertion of a construct that included the T2A self-cleaving peptide sequence and the tdTomato coding sequence followed by the bovine growth hormone polyadenylation signal sequence. The sequence of the selected gRNA for CRISPR/Cas9-mediated cleavage was 5′-ACTGGGAGCCTGGTGGCCCTNGG-3′. C57BL/6J (Jackson Laboratories, Bar Harbor, ME) fertilized zygotes were injected with Cas9 mRNA and gRNA. Founder mice were genotyped for insertion of the tdTomato locus by PCR amplification of a 919-bp region of the 5′ junction with primers 5′-AGGCACTCACCTACAATGGC-3′ and 5′-GGGATTCTCCTCCACGTCAC-3′. The 3′ junction was also verified with PCR amplification of a 1,002-bp region with primers 5′-TCGCATTGTCTGAGTAGGTGT-3′ and 5′-GGAGGGAGAACCCGTGAGT-3′. Identification of the 7.2-kb deletion in Ifnl2 was confirmed by Sanger sequencing using primers 5′-CACACTGTGGACAGGCCAT-3′ and 5′-GACGGCGGCAATGTATTAAC-3′. Additional generations were genotyped by Transnetyx (Cordova, TN) from tail biopsy specimens using real-time PCR with mutation-specific probes.
Equal ratios of male and female mice, aged 5 to 10 weeks, were used in all experiments for all strains unless otherwise noted. Experimental mice were cohoused with up to 5 mice of the same sex per cage, with autoclaved standard chow pellets and water provided ad libitum.
Generation of viral stocks.
Stocks of MNoV strains CR6 and CR6NS1−CW3 were generated from molecular clones as previously described (26, 40). Plasmids encoding the viral genomes were transfected into 293T cells to produce infectious virus, which was subsequently passaged on BV2 cells. After two passages, BV2 cultures were frozen and then thawed to liberate virus. Cultures then were cleared of cellular debris, and virus was concentrated by ultracentrifugation through a 30% sucrose cushion. Titers of virus stocks were determined by plaque assay on BV2 cells (41).
For propagation of reovirus stocks, spinner-adapted murine L929 (L) cells were grown in either suspension cultures or monolayer cultures in Joklik’s modified Eagle’s minimal essential medium (SMEM; Lonza) supplemented with 5% fetal bovine serum (Gibco); 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (Gibco); and 25 ng/ml amphotericin B (Sigma). Strain type 1 Lang (T1L) was recovered using plasmid-based reverse genetics (42). Purified reovirus was prepared from second-passage or third-passage L-cell lysate stocks (43). Viral particles were extracted from infected cell lysates by the use of Vertrel XF (Dupont), layered onto 1.2-g/cm3 to 1.4-g/cm3 CsCl gradients, and centrifuged at 62,000 × g for 16 h. Bands corresponding to virions (1.36 g/cm3) were collected and dialyzed in virion storage buffer (150 mM NaCl, 15 mM MgCl2, and 10 mM Tris-HCl [pH 7.4]) (44). Viral titer was determined by plaque assay using L cells (45).
A/Puerto Rico/8/1934 influenza A virus was prepared in 10-day-old embryonated chicken eggs. The viral titer was determined by virus titration assay in MDCK cells.
Mouse treatments and infections.
Treatment of mice with polyinosine-poly(C) [poly(I·C)] was done via intraperitoneal injection with 100 μg of high-molecular-weight poly(I·C) (InvivoGen) diluted in phosphate-buffered saline (PBS). Tissues were collected at 2 h, and serum was collected at 2 to 10 h after challenge.
For MNoV infections, mice were inoculated with 106 PFU of the indicated strains perorally in a volume of 25 μl. Stool and tissues were collected at 7 or 14 days postinfection. All stool and tissues were harvested into 2-ml tubes (Sarstedt, Germany) with 1-mm-diameter zirconia/silica beads (Biospec, Bartlesville, OK). Tissues were flash frozen in a bath of ethanol and dry ice and either processed on the same day or stored at −80°C.
For reovirus infections, mice were orally gavaged with 108 PFU of strain T1L virus in a volume of 100 μl. Fecal samples and 10-cm-long sections of small intestine were harvested into 1.0 ml of CaCl2+ and MgCl2+ plus PBS (PBS+/+) and flash frozen for determination of viral titers.
Mice used for IAV experiments were intranasally inoculated with 100 50% tissue culture infective doses (TCID50) of A/Puerto Rico/8/1934 (PR8) strain virus in a 15-μl volume. Weight was monitored every 2 days. For IAV infection experiments, only male mice were used. Lung tissue was harvested and washed in cold PBS before being placed in 2-ml homogenizer tubes (Qiagen) with 1 ml of minimal essential medium (MEM) and 2.0-mm-diameter stainless steel beads and stored on ice until further processing was performed.
RNA extraction, quantitative reverse transcription-PCR (RT-qPCR), and enzyme-linked immunosorbent assay (ELISA).
RNA was isolated from stool using a ZR-96 viral RNA kit (Zymo Research, Irvine, CA) (14). RNA from tissues or cells was isolated using TRI reagent with a Direct-zol-96 RNA kit (Zymo Research, Irvine, CA) according to the manufacturer’s protocol. A 5-μl volume of RNA was used for cDNA synthesis with an ImPromII reverse transcriptase system (Promega, Madison, WI).
MNoV TaqMan assays were conducted using a standard curve for determination of absolute numbers of viral genome copies, as previously described (46). A TaqMan assay for Ifnl2/Ifnl3 (Mm04204156_gH) (Thermo Fisher Scientific) was conducted with AmpliTaq gold DNA polymerase (Applied Biosystems) as previously described (14). Quantitative PCR was also performed for housekeeping gene Rps29 as previously described (14), and the results were used to normalize absolute values of Ifnl2/Ifnl3 and MNoV per microgram of RNA.
For IFN-λ ELISA, serum was collected at indicated time points and IFN-λ2/IFN-λ3 levels were analyzed by ELISA (R&D Systems).
Viral titration assays.
Reovirus titers in organs and stool homogenates were determined by plaque assay using L cells (45). Samples were homogenized for 8 min using TissueLyser LT (Qiagen), frozen at –80°C, and homogenized again immediately prior to titer determination. Titers are expressed as PFU per milliliter of tissue or stool homogenate.
For determinations of influenza A virus titers, lungs were homogenized in a TissueLyser II system (Qiagen). Sample homogenate was cleared via centrifugation at 1,000 rpm for 5 min before aliquoting and cold storage. After freezing, serial dilutions of homogenates were incubated on MDCK cells for 1 h at 37°C followed by a PBS wash and addition of 200 μl of infection media (MEM with 1% antibiotics, 1% l-glutamine, 1% vitamins, 0.1% bovine serum albumin [BSA], 1 μg/ml tosylsulfonyl phenylalanyl chloromethyl ketone [TPCK]-treated trypsin). After 3 days of incubation at 37°C, 50 μl of culture supernatant was tested for the presence of influenza virus by hemagglutination assay using 0.5% turkey red blood cells. End point dilution of hemagglutination was scored, and TCID50 titers were calculated as previously described (47).
Statistical analysis.
Data were analyzed with Prism 8 software (GraphPad Software, San Diego, CA). In all graphs, three asterisks indicate a P value of <0.001, two asterisks indicate a P value of <0.01, one asterisk indicates a P value of <0.05, and “ns” indicates that the result(s) was not significant (P > 0.05) as determined by Mann-Whitney test, one-way analysis of variance (ANOVA), or Kruskal-Wallis test with Dunn’s multiple-comparison test or by two-way ANOVA with Tukey’s multiple-comparison test, as specified in the relevant figure legends.
ACKNOWLEDGMENTS
We thank the staff members of the Genome Engineering and iPSC Center (GEiC) at Washington University in St. Louis for designing and validating gRNAs, J. Michael White and the Transgenic, Knockout and Micro-Injection Core for embryonic stem (ES) cell microinjection services, and D. Kreamalmeyer for animal care and breeding. We acknowledge all members of the Baldridge laboratory for helpful review and discussions.
This work was supported by funds from NIH grants K22 AI127846, R01 AI127552, R01 AI139314, and R01 AI141478 (M.T.B.); R01 AI038296 (T.S.D.); and R01 AI11893801 and R21 AI137450 (A.C.M.B.) and by funds from the Pew Biomedical Scholars Program (M.T.B.) and by the Global Probiotics Council’s Young Investigator Grant for Probiotics Research (M.T.B.) and the NSF Graduate Research Fellowship (DGE-1745038 [to E.A.K.]).
REFERENCES
- 1.Schneider WM, Chevillotte MD, Rice CM. 2014. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol 32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ingle H, Peterson ST, Baldridge MT. 2018. Distinct effects of type I and III interferons on enteric viruses. Viruses 10:46. doi: 10.3390/v10010046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sheppard P, Kindsvogel W, Xu W, Henderson K, Schlutsmeyer S, Whitmore TE, Kuestner R, Garrigues U, Birks C, Roraback J, Ostrander C, Dong D, Shin J, Presnell S, Fox B, Haldeman B, Cooper E, Taft D, Gilbert T, Grant FJ, Tackett M, Krivan W, McKnight G, Clegg C, Foster D, Klucher KM. 2003. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol 4:63–68. doi: 10.1038/ni873. [DOI] [PubMed] [Google Scholar]
- 4.Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, Langer JA, Sheikh F, Dickensheets H, Donnelly RP. 2003. IFN-λs mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol 4:69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- 5.Prokunina-Olsson L, Muchmore B, Tang W, Pfeiffer RM, Park H, Dickensheets H, Hergott D, Porter-Gill P, Mumy A, Kohaar I, Chen S, Brand N, Tarway M, Liu L, Sheikh F, Astemborski J, Bonkovsky HL, Edlin BR, Howell CD, Morgan TR, Thomas DL, Rehermann B, Donnelly RP, O'Brien TR. 2013. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet 45:164–171. doi: 10.1038/ng.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoon S-I, Jones BC, Logsdon NJ, Harris BD, Deshpande A, Radaeva S, Halloran BA, Gao B, Walter MR. 2010. Structure and mechanism of receptor sharing by the IL-10R2 common chain. Structure 18:638–648. doi: 10.1016/j.str.2010.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doyle SE, Schreckhise H, Khuu-Duong K, Henderson K, Rosler R, Storey H, Yao L, Liu H, Barahmand-Pour F, Sivakumar P, Chan C, Birks C, Foster D, Clegg CH, Wietzke-Braun P, Mihm S, Klucher KM. 2006. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology 44:896–906. doi: 10.1002/hep.21312. [DOI] [PubMed] [Google Scholar]
- 8.Zhou Z, Hamming OJ, Ank N, Paludan SR, Nielsen AL, Hartmann R. 2007. Type III interferon (IFN) induces a type I IFN-like response in a restricted subset of cells through signaling pathways involving both the Jak-STAT pathway and the mitogen-activated protein kinases. J Virol 81:7749–7758. doi: 10.1128/JVI.02438-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pervolaraki K, Rastgou Talemi S, Albrecht D, Bormann F, Bamford C, Mendoza JL, Garcia KC, McLauchlan J, Höfer T, Stanifer ML, Boulant S. 2018. Differential induction of interferon stimulated genes between type I and type III interferons is independent of interferon receptor abundance. PLoS Pathog 14:e1007420. doi: 10.1371/journal.ppat.1007420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jilg N, Lin W, Hong J, Schaefer EA, Wolski D, Meixong J, Goto K, Brisac C, Chusri P, Fusco DN, Chevaliez S, Luther J, Kumthip K, Urban TJ, Peng LF, Lauer GM, Chung RT. 2014. Kinetic differences in the induction of interferon stimulated genes by interferon-α and interleukin 28B are altered by infection with hepatitis C virus. Hepatology 59:1250–1261. doi: 10.1002/hep.26653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sommereyns C, Paul S, Staeheli P, Michiels T. 2008. IFN-lambda (IFN-λ) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog 4:e1000017. doi: 10.1371/journal.ppat.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pervolaraki K, Stanifer ML, Münchau S, Renn LA, Albrecht D, Kurzhals S, Senís E, Grimm D, Schröder-Braunstein J, Rabin RL, Boulant S. 21 April 2017, posting date Type I and type III interferons display different dependency on mitogen-activated protein kinases to mount an antiviral state in the human gut. Front Immunol doi: 10.3389/fimmu.2017.00459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Selvakumar TA, Bhushal S, Kalinke U, Wirth D, Hauser H, Köster M, Hornef MW. 16 October 2017, posting date Identification of a predominantly interferon-λ-induced transcriptional profile in murine intestinal epithelial cells. Front Immunol doi: 10.3389/fimmu.2017.01302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baldridge MT, Lee S, Brown JJ, McAllister N, Urbanek K, Dermody TS, Nice TJ, Virgin HW. 2017. Expression of Ifnlr1 on intestinal epithelial cells is critical to the antiviral effects of interferon lambda against norovirus and reovirus. J Virol 91:e02079-16. doi: 10.1128/JVI.02079-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mordstein M, Neugebauer E, Ditt V, Jessen B, Rieger T, Falcone V, Sorgeloos F, Ehl S, Mayer D, Kochs G, Schwemmle M, Günther S, Drosten C, Michiels T, Staeheli P. 2010. Lambda interferon renders epithelial cells of the respiratory and gastrointestinal tracts resistant to viral infections. J Virol 84:5670–5677. doi: 10.1128/JVI.00272-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lazear HM, Nice TJ, Diamond MS. 2015. Interferon-λ: immune functions at barrier surfaces and beyond. Immunity 43:15–28. doi: 10.1016/j.immuni.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lasfar A, Lewis-Antes A, Smirnov SV, Anantha S, Abushahba W, Tian B, Reuhl K, Dickensheets H, Sheikh F, Donnelly RP, Raveche E, Kotenko SV. 2006. Characterization of the mouse IFN-lambda ligand-receptor system: IFN-lambdas exhibit antitumor activity against B16 melanoma. Cancer Res 66:4468–4477. doi: 10.1158/0008-5472.CAN-05-3653. [DOI] [PubMed] [Google Scholar]
- 18.Jacobs S, Wavreil F, Schepens B, Gad HH, Hartmann R, Rocha-Pereira J, Neyts J, Saelens X, Michiels T. 2018. Species specificity of type III interferon activity and development of a sensitive luciferase-based bioassay for quantitation of mouse interferon-λ. J Interferon Cytokine Res 38:469–479. doi: 10.1089/jir.2018.0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee S, Baldridge MT. 30 June 2017, posting date Interferon-lambda: a potent regulator of intestinal viral infections. Front Immunol doi: 10.3389/fimmu.2017.00749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ank N, Iversen MB, Bartholdy C, Staeheli P, Hartmann R, Jensen UB, Dagnaes-Hansen F, Thomsen AR, Chen Z, Haugen H, Klucher K, Paludan SR. 2008. An important role for type III interferon (IFN-λ/IL-28) in TLR-induced antiviral activity. J Immunol 180:2474–2485. doi: 10.4049/jimmunol.180.4.2474. [DOI] [PubMed] [Google Scholar]
- 21.Donnelly RP, Kotenko SV. 2010. Interferon-lambda: a new addition to an old family. J Interferon Cytokine Res 30:555–564. doi: 10.1089/jir.2010.0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shaner NC, Campbell RE, Steinbach PA, Giepmans BNG, Palmer AE, Tsien RY. 2004. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol 22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 23.Mahlakõiv T, Hernandez P, Gronke K, Diefenbach A, Staeheli P. 2015. Leukocyte-derived IFN-α/β and epithelial IFN-λ constitute a compartmentalized mucosal defense system that restricts enteric virus infections. PLoS Pathog 11:e1004782. doi: 10.1371/journal.ppat.1004782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lauterbach H, Bathke B, Gilles S, Traidl-Hoffmann C, Luber CA, Fejer G, Freudenberg MA, Davey GM, Vremec D, Kallies A, Wu L, Shortman K, Chaplin P, Suter M, O’Keeffe M, Hochrein H. 2010. Mouse CD8α+ DCs and human BDCA3+ DCs are major producers of IFN-λ in response to poly IC. J Exp Med 207:2703–2717. doi: 10.1084/jem.20092720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nice TJ, Baldridge MT, McCune BT, Norman JM, Lazear HM, Artyomov M, Diamond MS, Virgin HW. 2015. Interferon-λ cures persistent murine norovirus infection in the absence of adaptive immunity. Science 347:269–273. doi: 10.1126/science.1258100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee S, Wilen CB, Orvedahl A, McCune BT, Kim K-W, Orchard RC, Peterson ST, Nice TJ, Baldridge MT, Virgin HW. 2017. Norovirus cell tropism is determined by combinatorial action of a viral non-structural protein and host cytokine. Cell Host Microbe 22:449–459. doi: 10.1016/j.chom.2017.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilen CB, Lee S, Hsieh LL, Orchard RC, Desai C, Hykes BL, McAllaster MR, Balce DR, Feehley T, Brestoff JR, Hickey CA, Yokoyama CC, Wang Y-T, MacDuff DA, Kreamalmayer D, Howitt MR, Neil JA, Cadwell K, Allen PM, Handley SA, van Lookeren Campagne M, Baldridge MT, Virgin HW. 2018. Tropism for tuft cells determines immune promotion of norovirus pathogenesis. Science 360:204–208. doi: 10.1126/science.aar3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nice TJ, Strong DW, McCune BT, Pohl CS, Virgin HW. 2013. A single-amino-acid change in murine norovirus NS1/2 is sufficient for colonic tropism and persistence. J Virol 87:327–334. doi: 10.1128/JVI.01864-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baldridge MT, Nice TJ, McCune BT, Yokoyama CC, Kambal A, Wheadon M, Diamond MS, Ivanova Y, Artyomov M, Virgin HW. 2015. Commensal microbes and interferon-λ determine persistence of enteric murine norovirus infection. Science 347:266–269. doi: 10.1126/science.1258025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee S, Liu H, Wilen CB, Sychev ZE, Desai C, Hykes BL, Orchard RC, McCune BT, Kim K-W, Nice TJ, Handley SA, Baldridge MT, Amarasinghe GK, Virgin HW. 2019. A secreted viral nonstructural protein determines intestinal norovirus pathogenesis. Cell Host Microbe 25:845–857. doi: 10.1016/j.chom.2019.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mordstein M, Kochs G, Dumoutier L, Renauld J-C, Paludan SR, Klucher K, Staeheli P. 2008. Interferon-lambda contributes to innate immunity of mice against influenza A virus but not against hepatotropic viruses. PLoS Pathog 4:e1000151. doi: 10.1371/journal.ppat.1000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Galani IE, Triantafyllia V, Eleminiadou EE, Koltsida O, Stavropoulos A, Manioudaki M, Thanos D, Doyle SE, Kotenko SV, Thanopoulou K, Andreakos E. 2017. Interferon-λ mediates non-redundant front-line antiviral protection against influenza virus infection without compromising host fitness. Immunity 46:875–890. doi: 10.1016/j.immuni.2017.04.025. [DOI] [PubMed] [Google Scholar]
- 33.Walker FC, Baldridge MT. 2019. Interactions between noroviruses, the host, and the microbiota. Curr Opin Virol 37:1–9. doi: 10.1016/j.coviro.2019.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sullender ME, Baldridge MT. 2018. Norovirus interactions with the commensal microbiota. PLoS Pathog 14:e1007183. doi: 10.1371/journal.ppat.1007183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moon C, Baldridge MT, Wallace MA, Burnham CAD, Virgin HW, Stappenbeck TS. 2015. Vertically transmitted faecal IgA levels determine extra-chromosomal phenotypic variation. Nature 521:90–93. doi: 10.1038/nature14139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klinkhammer J, Schnepf D, Ye L, Schwaderlapp M, Gad HH, Hartmann R, Garcin D, Mahlakõiv T, Staeheli P. 2018. IFN-λ prevents influenza virus spread from the upper airways to the lungs and limits virus transmission. Elife 7:e33354. doi: 10.7554/eLife.33354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fox BA, Sheppard PO, O'Hara PJ. 2009. The role of genomic data in the discovery, annotation and evolutionary interpretation of the interferon-lambda family. PLoS One 4:e4933. doi: 10.1371/journal.pone.0004933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aryal NK, Wasylishen AR, Lozano G. 27 October 2018, posting date CRISPR/Cas9 can mediate high-efficiency off-target mutations in mice in vivo. Cell Death Dis doi: 10.1038/s41419-018-1146-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anderson KR, Haeussler M, Watanabe C, Janakiraman V, Lund J, Modrusan Z, Stinson J, Bei Q, Buechler A, Yu C, Thamminana SR, Tam L, Sowick MA, Alcantar T, O’Neil N, Li J, Ta L, Lima L, Roose-Girma M, Rairdan X, Durinck S, Warming S. 2018. CRISPR off-target analysis in genetically engineered rats and mice. Nat Methods 15:512–514. doi: 10.1038/s41592-018-0011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Strong DW, Thackray LB, Smith TJ, Virgin HW. 2012. Protruding domain of capsid protein is necessary and sufficient to determine murine norovirus replication and pathogenesis in vivo. J Virol 86:2950–2958. doi: 10.1128/JVI.07038-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Orchard RC, Wilen CB, Doench JG, Baldridge MT, McCune BT, Lee Y-C, Lee S, Pruett-Miller SM, Nelson CA, Fremont DH, Virgin HW. 2016. Discovery of a proteinaceous cellular receptor for a norovirus. Science 353:933–936. doi: 10.1126/science.aaf1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kobayashi T, Ooms LS, Ikizler M, Chappell JD, Dermody TS. 2010. An improved reverse genetics system for mammalian orthoreoviruses. Virology 398:194–200. doi: 10.1016/j.virol.2009.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Furlong DB, Nibert ML, Fields BN. 1988. Sigma 1 protein of mammalian reoviruses extends from the surfaces of viral particles. J Virol 62:246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith RE, Zweerink HJ, Joklik WK. 1969. Polypeptide components of virions, top component and cores of reovirus type 3. Virology 39:791–810. doi: 10.1016/0042-6822(69)90017-8. [DOI] [PubMed] [Google Scholar]
- 45.Virgin HW, Bassel-Duby R, Fields BN, Tyler KL. 1988. Antibody protects against lethal infection with the neurally spreading reovirus type 3 (Dearing). J Virol 62:4594–4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baert L, Wobus CE, Van Coillie E, Thackray LB, Debevere J, Uyttendaele M. 2008. Detection of murine norovirus 1 by using plaque assay, transfection assay, and real-time reverse transcription-PCR before and after heat exposure. Appl Environ Microbiol 74:543–546. doi: 10.1128/AEM.01039-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reed LJ, Muench H. 1938. A simple method of estimating fifty per cent endpoints. Am J Hyg 27:493–497. doi: 10.1093/oxfordjournals.aje.a118408. [DOI] [Google Scholar]