

PRRSV infection elicits a meager protective immune response in pigs. One of the possible reasons is that PRRSV antagonizes interferon induction and its downstream signaling. Interferons are key components in the innate immunity and play crucial roles against viral infection and in the activation of adaptive immune response via JAK/STAT signaling. STAT2 is indispensable in the JAK/STAT signaling since it is also involved in activation of antiviral activity in the absence of STAT1. Here, we discovered that PRRSV nsp11 downregulates STAT2. Interestingly, the N-terminal domain of nsp11 is responsible for inducing STAT2 degradation and directly interacts with STAT2 N-terminal domain. We also identified a crucial amino acid residue K59 in nsp11 since a mutation of it led to loss of the ability to downregulate STAT2. A mutant PRRSV with mutation of K59 had minimal effect on STAT2 reduction. Our data provide further insights into PRRSV interference with interferon signaling.
KEYWORDS: PRRSV, STAT2, nsp11, NTD, IFN signaling, immune response
ABSTRACT
Interferons (IFNs) play a crucial role in host antiviral response by activating the JAK/STAT (Janus kinase/signal transducer and activator of transcription) signaling pathway to induce the expression of myriad genes. STAT2 is a key player in the IFN-activated JAK/STAT signaling. Porcine reproductive and respiratory syndrome virus (PRRSV) is an important viral pathogen, causing huge losses to the swine industry. PRRSV infection elicits a meager protective immune response in pigs. The objective of this study was to investigate the effect of PRRSV on STAT2 signaling. Here, we demonstrated that PRRSV downregulated STAT2 to inhibit IFN-activated signaling. PRRSV strains of both PRRSV-1 and PRRSV-2 species reduced the STAT2 protein level, whereas the STAT2 transcript level had minimal change. PRRSV reduced the STAT2 level in a dose-dependent manner and shortened STAT2 half-life significantly from approximately 30 to 5 h. PRRSV-induced STAT2 degradation could be restored by treatment with the proteasome inhibitor MG132 and lactacystin. In addition, PRRSV nonstructural protein 11 (nsp11) was identified to interact with and reduce STAT2. The N-terminal domain (NTD) of nsp11 was responsible for STAT2 degradation and interacted with STAT2 NTD and the coiled-coil domain. Mutagenesis analysis showed that the amino acid residue K59 of nsp11 was indispensable for inducing STAT2 reduction. Mutant PRRSV with the K59A mutation generated by reverse genetics almost lost the ability to reduce STAT2. Together, these results demonstrate that PRRSV nsp11 antagonizes IFN signaling via mediating STAT2 degradation and provide further insights into the PRRSV interference of the innate immunity.
IMPORTANCE PRRSV infection elicits a meager protective immune response in pigs. One of the possible reasons is that PRRSV antagonizes interferon induction and its downstream signaling. Interferons are key components in the innate immunity and play crucial roles against viral infection and in the activation of adaptive immune response via JAK/STAT signaling. STAT2 is indispensable in the JAK/STAT signaling since it is also involved in activation of antiviral activity in the absence of STAT1. Here, we discovered that PRRSV nsp11 downregulates STAT2. Interestingly, the N-terminal domain of nsp11 is responsible for inducing STAT2 degradation and directly interacts with STAT2 N-terminal domain. We also identified a crucial amino acid residue K59 in nsp11 since a mutation of it led to loss of the ability to downregulate STAT2. A mutant PRRSV with mutation of K59 had minimal effect on STAT2 reduction. Our data provide further insights into PRRSV interference with interferon signaling.
INTRODUCTION
Interferons (IFNs) are an indispensable component of innate immunity. Three types of IFNs have been detected: types I, II, and III (1–3). Upon binding specific receptors, IFNs trigger a signaling cascade through Janus kinase (JAK) and signal transducer and activator of transcription (STAT) and induce an antiviral state by activating expression of a myriad of interferon-stimulated gene (ISG) products, such as ISG15, ISG56, protein kinase R, 2′,5′-oligoadenylate synthetase, and Mx GTPases (3, 4).
Both type I and type III IFNs can activate the same canonical JAK-STAT pathway, in which JAKs phosphorylate STAT1 and STAT2, followed by interaction with IFN regulatory factor 9 (IRF9) to form a heterotrimer termed IFN-stimulated gene factor 3 (ISGF3) (3, 5). The ISGF3 complex translocates into the nucleus to activate the expression of ISGs. On the other hand, there is a STAT1-independent pathway, also known as the noncanonical JAK/STAT signaling. In this pathway, STAT2 and IRF9 form a complex that has ISGF3-like functions to induce prolonged expression of ISGs, as well as STAT2/IRF9-specific ISGs, such as APOBEC3G (A3G), that have broad antiviral activity, retinoic acid-induced gene G (RIG-G), CCL8, and CX3CL1, in the absence of STAT1 (6). STAT2 is known to mediate innate immunity against dengue virus in the absence of STAT1 (7). Therefore, STAT2 is indispensable in both canonical and noncanonical IFN-activated signaling pathways (8).
Due to its essential role in the JAK-STAT pathway, many viruses target STAT2 to antagonize IFN signaling. For example, measles virus V protein binds to STAT2 (9). Yellow fever virus NS5 binds to STAT2 upon IFN stimulation (10). Dengue virus (DENV) NS5 protein recruits the E3 ligase UBR4 to induce STAT2 degradation (11). Similar to DENV, Zika virus NS5 also promotes STAT2 degradation (12).
Since the first emergence in the late 1980s, porcine reproductive and respiratory syndrome (PRRS) has remained a top challenge to the swine industry (13). PRRS virus (PRRSV), the causative agent, is a member of the genus Porartevirus, the family Arteriviridae, and the order Nidovirales (14–16). The conventional two genotypes, type 1 (European) and type 2 (North American) PRRSV, have been classified as two species, PRRSV-1 and PRRSV-2, respectively (17). PRRSV is a small enveloped virus containing a single-stranded, positive-sense RNA genome approximately 15 kb in length. The genome encodes 11 known open reading frames (ORFs) (13). The replicase-associated polyproteins pp1a and pp1ab are encoded by the ORF1a and ORF1b regions, which comprise 80% of the viral genome. The polyproteins are proteolytically processed into more than 14 nonstructural proteins (nsp1 to nsp12). ORF2, -2a, -3, -4, -5a, -5, -6, and -7 encode structural proteins, including the minor membrane-associated proteins GP2, E, GP3, GP4, and ORF5a, a major envelope glycoprotein (GP5), a membrane protein (M), and a nucleocapsid protein (N), respectively (13, 18). PRRSV has a very restricted tropism for cells of monocyte/macrophage lineage. In vivo, PRRSV preferentially targets porcine pulmonary alveolar macrophages (PAMs) (19). In vitro, PRRSV propagation in laboratories is generally conducted on MARC-145, an epithelial cell-derived cell line from African green monkey kidney (20).
PRRSV infection of pigs induces poor innate (21) and adaptive immune responses (22). It has been shown that PRRSV has several evasion strategies to interfere with the host innate immune response (23, 24). PRRSV appears to suppress the induction of type I IFNs by interfering with the RIG-I signaling pathway (25). PRRSV nsp1α, nsp1β, nsp2, and nsp11 modulate the induction of type I IFNs (26–30). Moreover, PRRSV interferes with the IFN downstream signaling by blocking STAT1/STAT2 nuclear translocation via nsp1β-mediated degradation of karyopherin-α1 (KPNA1), which is responsible for ISGF3 nuclear translocation (24, 31, 32). N protein could also inhibit ISGF3 nuclear translocation (33). PRRSV also antagonizes STAT3 signaling via nsp5 (34). PRRSV elevates miR-30c to downregulate JAK1 to inhibit IFN signaling (35). However, the effect of PRRSV infection on STAT2 signaling remains unknown.
In this study, we demonstrated that PRRSV induced STAT2 degradation to inhibit IFN-activated signaling. PRRSV strains from both PRRSV-1 and PRRSV-2 species reduced STAT2 protein level, whereas its transcript had minimal change. PRRSV infection shortened STAT2 half-life significantly. PRRSV nsp11 was demonstrated to reduce STAT2 protein level and interact with STAT2. Specifically, the nsp11 N-terminal domain (NTD) interacts with the STAT2 NTD and coiled-coil domain (CCD). The amino acid residue K59 of nsp11 is crucial for the reduction of STAT2. Together, these results demonstrate that PRRSV antagonizes STAT2 signaling via nsp11. The data improve our understanding of the mechanism of PRRSV interference with the IFN-activated JAK/STAT signaling.
RESULTS
PRRSV infection reduces STAT2 protein level protein level.
When we studied the effect of PRRSV infection on JAK/STAT signaling, we discovered that PRRSV reduced STAT2 and STAT3 (34) protein levels, while STAT1 remained stable. To confirm the effect of PRRSV infection on STAT2, we inoculated MARC-145 cells with PRRSV strain VR-2385 and harvested the cells at 36 h postinfection (hpi). Compared to mock-infected cells, PRRSV-infected cells had a lower level of STAT2 at 16% but a similar level of STAT1 (Fig. 1A). PRRSV nsp2 was determined to verify PRRSV infection. Since PRRSV targets PAMs during pig infection, we infected primary porcine PAM cells with VR-2385 to confirm the effect of PRRSV infection on STAT2. Due to the rapid replication of PRRSV in PAM cells, these cells were harvested at 16 hpi (34). As in MARC-145 cells, PRRSV infection reduced the STAT2 level in PAM cells to 16% compared to the mock-infected control, whereas STAT1 underwent minimal change (Fig. 1B).
FIG 1.

PRRSV infection reduces STAT2 protein level in MARC-145 and PAM cells. (A) PRRSV reduces STAT2 protein level but has minimal effect on STAT1. MARC-145 cells were infected with VR-2385 at an MOI of 1 and harvested 36 hpi for Western blotting (WB) with antibodies against STAT2, STAT1, PRRSV nsp2, and tubulin. The relative levels of STAT2 are shown below the images after normalization with housekeeping gene tubulin in densitometry analysis. (B) PRRSV reduces STAT2 in PAM cells but has a minimal effect on STAT1. The cells were infected with VR-2385 at an MOI of 1 and harvested for WB at 16 hpi. (C) PRRSV inhibits IFN-α-activated expression of ISG15 and ISG56 detected by reverse transcription quantitative PCR (RT-qPCR). MARC-145 cells were infected with VR-2385 at an MOI of 1 and, at 24 hpi, treated with IFN-α at a final concentration of 300 U/ml for another 24 h. VR, VR-2385. Error bars represent the standard errors of three repeated experiments. Significant differences between IFN and VR+IFN are denoted by asterisks (**, P < 0.01). (D) Reduction of STAT2 by different PRRSV strains in MARC-145 cells. The cells were infected with VR-2385, MLV (Ingelvac PRRS MLV), and LV (Lelystad virus) at an MOI of 1. The relative levels of STAT2 are shown below the images after normalization with tubulin. (E) PRRSV RNA levels of the PRRSV strains detected by RT-qPCR. Virus-infected MARC-145 cells were harvested for RNA isolation and RT-qPCR at 24 hpi. Error bars represent the standard errors of the results of three repeated experiments. (F) UV-inactivated PRRSV has minimal effect on the STAT2 level. The cells were inoculated with PRRSV VR-2385 at an MOI of 1 or UV-inactivated VR-2385 at MOIs of 5 and 10, followed by WB at 36 hpi.
STAT2 plays a critical role in the IFN-activated JAK/STAT signaling pathway. We conducted an IFN treatment to confirm that PRRSV infection interfered with type I IFN signaling (31, 32). MARC-145 cells were inoculated with VR-2385 and, at 24 hpi, treated with IFN-α for 24 h. The reverse transcription and quantitative PCR (RT-qPCR) results showed that the transcript levels of ISG15 and ISG56 in IFN-treated cells increased by 55- and 32-fold, respectively, compared to mock-treated cells (Fig. 1C). As expected, the transcript levels of ISG15 and ISG56 in PRRSV-infected cells with IFN treatment were at 9 and 11%, respectively, compared to the mock-infected cells (Fig. 1C), which is consistent with the previous observation (31).
We speculated that other PRRSV strains could also induce a reduction of STAT2. Therefore, Lelystad virus, the prototype of PRRSV-1, and Ingelvac PRRS MLV, a strain of PRRSV-2, were used in the experiment. Compared to the mock-infected control, VR-2385, MLV and Lelystad reduced STAT2 to 12, 10, and 33%, respectively, whereas the STAT1 level in the infected cells remained unchanged (Fig. 1D). The PRRSV RNA levels of these strains were determined by RT-qPCR (Fig. 1E). The Lelystad virus had the lowest RNA level, which is consistent with the least effect on the STAT2 level in the cells infected by this strain.
To determine whether PRRSV replication was indispensable for STAT2 reduction, we inactivated VR-2385 by UV light and inoculated MARC-145 cells with UV-inactivated VR-2385 at multiplicities of infection (MOIs) of 5 and 10. Western blotting (WB) result showed that only live PRRSV virions led to the STAT2 reduction, whereas the UV-inactivated PRRSV had a minimal effect (Fig. 1F), which suggests that PRRSV replication is needed to induce STAT2 reduction and that PRRSV structural proteins in the inactivated virions had no detectable effect.
PRRSV reduces STAT2 protein level in a dose- and time-dependent manner.
To further study the PRRSV-mediated reduction of STAT2, we infected MARC-145 cells with VR-2385 at MOIs of 0.1, 1, and 10. Along with the increased amount of VR-2385 inoculum, the STAT2 protein level decreased in a dose-dependent manner. Compared to the mock-infected cells, the cells inoculated at MOIs of 0.1, 1, and 10 had STAT2 levels at 29, 13, and 5%, respectively (Fig. 2A). The PRRSV RNA levels in the infected cells significantly increased, which was consistent with the incremental inoculum (Fig. 2B).
FIG 2.

PRRSV reduces STAT2 in a dose and time-dependent manner. (A) Dose-dependent reduction of STAT2 by PRRSV. MARC-145 cells were inoculated with incremental MOIs of VR-2385 and harvested for WB at 36 hpi. The relative levels of STAT2 are shown below the images after normalization with tubulin. (B) PRRSV RNA levels detected by RT-qPCR. Error bars represent the standard errors of the means of three repeated experiments. Significant differences in RNA level from an MOI of 0.1 are denoted by asterisks (**, P < 0.01; ***, P < 0.001). (C) Temporal kinetics of STAT2 levels in PRRSV-infected cells. MARC-145 cells were infected with VR-2385 at an MOI of 1. At different time points, the cells were harvested for WB. Mock-infected cells at corresponding time points were included as controls. The relative levels of STAT2 are shown below the images after normalization with tubulin at the corresponding time point. (D) PRRSV infection has minimal effect on STAT2 mRNA level. The cells were harvested at 24, 36, and 48 hpi for RNA isolation and RT-qPCR. The relative STAT2 mRNA levels are shown compared to the mock-infected cells at the corresponding time point. Error bars represent standard errors of the results of three repeated experiments.
The temporal kinetics of the STAT2 protein level in the infected cells were analyzed to determine whether the change of STAT2 level occurred during a specific period postinfection. MARC-145 cells were infected with PRRSV VR-2385 at an MOI of 1 and harvested at 24, 36, and 48 hpi. Compared to the mock-infected cells, the STAT2 levels in the virus-infected cells at 24, 36, and 48 hpi decreased to 60, 20, and 20%, respectively, while STAT1 levels remained stable (Fig. 2C). This result demonstrated that PRRSV infection reduced the STAT2 protein level in a time-dependent manner.
In mammalian cells, the protein reduction could be due to a decrease in transcription and/or translation or an increase in protein degradation. To investigate the reason for the PRRSV-induced STAT2 reduction, we performed RT-qPCR to determine the transcript level of STAT2 in PRRSV-infected cells at 24, 36, and 48 hpi. In comparison to mock-infected cells at each time point, PRRSV-infected cells at the corresponding time point had similar mRNA levels of endogenous STAT2 (Fig. 2D). The STAT2 transcript level at 36 h is the highest among the three time points tested. The results demonstrated that PRRSV-induced STAT2 reduction is not due to change at the transcript level.
PRRSV infection shortens the STAT2 half-life and mediates STAT2 reduction via the ubiquitin-proteasome degradation pathway.
Since PRRSV infection did not alter the transcript level of STAT2, we speculated that PRRSV might accelerate the rate of STAT2 degradation. We then wondered whether the half-life of STAT2 would be shortened by PRRSV. PRRSV-infected MARC-145 cells were treated with cycloheximide, a translation inhibitor, followed by harvesting and immunoblotting for densitometry analysis at the indicated time points. The average relative levels of STAT2 of multiple experiments for each time point were analyzed by using GraphPad Prism software (Fig. 3A). In the presence of PRRSV, the STAT2 half-life was shortened to ∼5 h compared to 30 h in mock-infected cells. The results suggest that the shortened STAT2 half-life may account for the STAT2 reduction in PRRSV-infected cells.
FIG 3.

PRRSV infection shortens STAT2 half-life and mediates STAT2 reduction via the ubiquitin-proteasome degradation pathway. (A) PRRSV shortens STAT2 half-life. MARC-145 cells were infected with VR-2385 at an MOI of 1. The cells were treated with cycloheximide at 24 hpi and harvested at the indicated time (h) for WB and densitometry analysis. The average relative levels of STAT2 of five experiments for each time point are shown. Error bars represent the standard errors of the repeated experiments. Significant differences between mock and VR-2385-infected cells are denoted by asterisks (***, P < 0.001). (B) MG132 treatment restores STAT2 level in PRRSV-infected cells. MARC-145 cells were infected with VR-2385 at an MOI of 1. At 24 hpi, the cells were treated with MG132 for 6 h and then harvested for WB. Nontreated and mock-infected cells were included as controls. WB with an antibody against PRRSV nsp1β was also done. The relative levels of STAT2 are shown below the images. (C) Lactacystin treatment restores STAT2 levels in PRRSV-infected cells. The cells were treated with lactacystin at 24 hpi and harvested 6 h later. (D) Lactacystin treatment has minimal effect on the viability of MARC-145 cells during the 6-h treatment. LAC, lactacystin. DMSO was included as a solvent control. (E) Lactacystin treatment has minimal effect on PRRSV RNA level during the 6-h treatment. Error bars represent the standard errors of the results of three repeated experiments.
Since PRRSV shortened the STAT2 half-life, we further determined the protein degradation pathway for PRRSV-induced STAT2 reduction. The ubiquitin-proteasome system accounts for the turnover of most short-lived cellular proteins. MG132, a proteasome inhibitor, was used to treat MARC-145 cells at 24 hpi. After MG132 treatment for 6 h (30 hpi), the cells were harvested for immunoblotting. The MG132-treated, PRRSV-infected cells displayed levels of STAT2 similar to those of mock-infected cells (Fig. 3B). As described previously, the short treatment with MG132 did not cause detectable cytotoxicity or inhibition of PRRSV replication (32, 34, 36). To further confirm this result, we treated MARC-145 cells with lactacystin, a more specific proteasome inhibitor, for 6 h. WB results showed that lactacystin could also block PRRSV-mediated STAT2 reduction (Fig. 3C). A cell viability assay and qPCR showed that lactacystin had a negligible effect on cell viability (Fig. 3D) and PRRSV replication (Fig. 3E), respectively, with a 6-h treatment. These results indicate that PRRSV induces STAT2 reduction through the ubiquitin-proteasome degradation pathway.
PRRSV nsp11 reduces STAT2 protein level.
Since Fig. 1F indicated that PRRSV replication was critical for STAT2 reduction, we speculated that certain nonstructural proteins (nsps) were responsible for the PRRSV-induced STAT2 reduction. HEK293 cells were transfected with plasmids encoding PRRSV VR-2385 nsps to assess their effects on STAT2 degradation. The WB results showed that nsp11 resulted in a lower level of STAT2 protein compared to that of empty vector (EV) and other PRRSV plasmids (Fig. 4A). Since nsp2, nsp3, and nsp5 induced slightly lower levels of STAT2, we further tested their effects on STAT2. The results confirmed that nsp11 induced much less STAT2 than did nsp2, nsp3, and nsp5 (Fig. 4B). Therefore, we further analyzed the role of nsp11 in STAT2 degradation.
FIG 4.

PRRSV nsp11 induces a reduction of STAT2. (A) nsp11 induces a lower STAT2 protein level than the empty vector (EV) and the other nsps. The HEK293 cells were transfected with individual HA-nsp plasmids and STAT2 plasmid. The relative levels of STAT2 protein are shown below the images. Molecular mass markers are added on left side of the nsp images. (B) nsp11 induces less STAT2 than do nsp2, nsp3, and nsp5 in HEK293 cells. (C) nsp11 reduces STAT2 in HeLa cells in a dose-dependent manner. HeLa cells were transfected with nsp11 plasmid in incremental amounts. (D) nsp11 has minimal effect on cell viability. HEK293 cells in one well of a 24-well plate were transfected with 0.5 μg of nsp11 plasmid. EV was included as a control. After 24 and 48 h, the cells were harvested for a cell viability assay. Error bars represent the standard errors of the means of three repeated experiments. (E) nsp11 inhibits IFN-α-activated expression of the ISRE reporter in HEK293 cells. The cells were cotransfected with ISRE luciferase reporter, Renilla luciferase plasmid, and HA-nsp11. The relative levels of firefly luciferase activity are shown as the fold changes compared to the EV control after normalization with the Renilla activity. A significant difference in firefly luciferase activity from the EV control is denoted by asterisks (***, P < 0.001). (F) nsp11 has a minimal effect on the STAT2 transcript level. HEK293 cells were transfected with nsp11 plasmid DNA for 24 h, followed by RNA isolation and real-time PCR.
HeLa cells were transfected with an nsp11 plasmid to confirm its effect on STAT2. Densitometry analysis showed that the levels of endogenous STAT2 in HeLa cells transfected with 0.25, 0.5, and 1.0 μg of nsp11 plasmid were reduced to 30, 20, and 10%, respectively, compared to the empty vector control (Fig. 4C). The nsp11 protein level increased along with the incremental DNA amount in the transfection. This indicated that nsp11 reduced STAT2 protein level in a dose-dependent manner. To exclude the possibility that nsp11-mediated STAT2 reduction was due to the cytotoxicity of nsp11 (37, 38), we conducted a cell viability assay. The results showed that nsp11 had minimal effect on cell viability at both 24 and 48 h posttransfection (Fig. 4D). In addition, the IFN-stimulated response element (ISRE) promoter luciferase assay confirmed that nsp11 significantly inhibited the signaling activated by type I IFN (Fig. 4E).
PRRSV nsp11 contains a nidovirus uridylate-specific endoribonuclease (NendoU) domain, which is indispensable for arterivirus replication (37, 39, 40). NendoU has been demonstrated to cleave 5′ uridine nucleotides of RNA substrates to generate a 2′-3′-cyclic phosphate end product, similar to XendoU, an endoribonuclease in eukaryotes (37, 41). The STAT2 mRNA level in PRRSV-infected cells remained the same as in mock-infected cells (Fig. 2D), suggesting that the NendoU does not contribute to the nsp11-mediated STAT2 reduction. To verify this, we transfected HEK293 cells with an nsp11 plasmid for 24 h, followed by RT-qPCR to determine the STAT2 mRNA level. The results showed that nsp11 had no significant effect on STAT2 transcript level compared to the empty vector (Fig. 4F).
The N-terminal domain of nsp11 is responsible for the reduction of STAT2 and interacts with it.
The crystal structure analysis of nsp11 shows that nsp11 has three different domains: an N-terminal domain (NTD), a linker domain, and a C-terminal domain (CTD) (40). To map the nsp11 domain that is responsible for the STAT2 degradation, we constructed four truncation plasmids of VR-2385 nsp11: an NTD from amino acids (aa) 1 to 90, an NTD with linker domain (NTDL) from aa 1 to 106, a linker and C-terminal domain (LCTD) from aa 91 to 223, and a CTD from aa 107 to 223 (Fig. 5A). HEK293 cells were transfected with the nsp11 truncation plasmids. WB results and densitometry analysis showed that the cells transfected with nsp11-NTD and nsp11-NTDL had lower STAT2 protein levels at 40 and 30%, respectively, than cells transfected with the empty vector (Fig. 5B). The expression of the nsp11 truncation constructs in HEK293 cells was confirmed with hemagglutinin (HA) antibody. The results indicate that the NTD of nsp11 is responsible for the PRRSV-induced degradation of STAT2.
FIG 5.

The N-terminal domain of PRRSV nsp11 is required for STAT2 reduction. (A) Schematic illustration of truncation plasmids of nsp11: NTD, NTDL, LCTD, and CTD. The numbers above the lines indicate amino acid positions in nsp11. NTD, N-terminal domain; CTD, C-terminal domain; L, linker domain. (B) The nsp11 NTD and NTDL lead to a STAT2 reduction, whereas LCTD and CTD have minimal effect. HEK293 cells were cotransfected with the HA-tagged nsp11 truncation plasmids and STAT2 plasmid for 36 h. EV was included as a control. (C) The nsp11 NTD induces the elevation of STAT2 polyubiquitination. The HEK293 cells were treated with MG132 for 6 h before harvesting to block STAT2 degradation. IP with cMyc antibody to pulldown STAT2 was done, followed by WB with the ubiquitin (Ub) antibody. WB of input was conducted. The relative levels of Ub after normalization with STAT2 are shown below the images. (D) IP of STAT2 coprecipitates nsp11-NTD, but not YFP. HEK293 cells were cotransfected with the YFP-tagged nsp11-NTD and STAT2 plasmids. IP with cMyc antibody to pulldown STAT2 was done, followed by WB with YFP and cMyc antibodies. Samples of input were included as controls. (E) IP of STAT2 coprecipitates full-length nsp11, but not YFP. HEK293 cells were cotransfected with the YFP-tagged full-length nsp11 and STAT2 plasmids. IP and WB were done as described above.
Since PRRSV mediates STAT2 reduction via the ubiquitin-proteasome degradation pathway (Fig. 3), we speculated that nsp11-NTD could increase the STAT2 polyubiquitination level. HEK293 cells transfected with YFP-nsp11-NTD, Myc-STAT2, and pRK5-HA-ubiquitin-K48 plasmids for 24 h were treated with MG132 for another 6 h. The cells were lysed for immunoprecipitation (IP) with the antibody against cMyc tag and subsequent WB with antibodies against ubiquitin and STAT2. The results showed that the polyubiquitination level of STAT2 in the presence of nsp11-NTD was 2.1-fold higher than that of empty vector, whereas there were similar levels of the total ubiquitination in the input with or without nsp11-NTD expression (Fig. 5C). The expression of YFP, YFP-nsp11-NTD, and Myc-STAT2 was confirmed in the input.
Furthermore, we evaluated whether nsp11-NTD had interaction with STAT2. HEK293 cells were transfected with Myc-STAT2 and YFP-nsp11-NTD plasmids. Indeed, IP of STAT2 coprecipitated nsp11-NTD (Fig. 5D). Since nsp11 exists as a homodimer and the NTD may not mimic the whole molecule, full-length nsp11 was used to verify the interaction with STAT2. Like NTD, full-length nsp11 was coprecipitated in the IP of STAT2 (Fig. 5E). The expression of Myc-STAT2, YFP-nsp11 or nsp11-NTD, and YFP was also confirmed in the input.
The N-terminal domain of STAT2 interacts with nsp11-NTD.
As a transcription factor, STAT2 has a conserved structure containing several domains, such as the coiled-coil domain (CCD), DNA-binding domain (DBD), Src homology 2 (SH2) domain, and transactivation domain (TAD) (42). To identify the interacting domain with nsp11-NTD, we constructed three truncates of STAT2: D1 (aa 1 to 315), D2 (aa 1 to 480), and D3 (aa 481 to 851) using Myc-tagged recombinant plasmids (Fig. 6A). HEK293 cells were transfected with YFP-nsp11-NTD, and one of the Myc-tagged STAT2 truncates, followed by IP with YFP antibody and WB with antibodies against Myc and YFP. The IP of nsp11-NTD coprecipitated STAT2, STAT2-D1, and STAT2-D2, whereas it failed to pull down STAT2-D3 (Fig. 6B), which suggests that nsp11-NTD interacts with STAT2-D1 and STAT2-D2. The expression of these plasmids was confirmed in HEK293 cell lysates. STAT2-D1 and STAT2-D2 have the same first 315 residues in the N-terminal and coiled-coil domains of STAT2, which suggests that the first 315 residues of STAT2 contain the nsp11 interaction site.
FIG 6.

The NTD of STAT2 interacts with nsp11. (A) Schematic illustration of truncation plasmids STAT2: D1, D2, and D3. The numbers above the lines indicate amino acid positions in STAT2. N, N-terminal domain; CCD, coiled-coil domain; DBD, DNA binding domain; L, linker domain; SH2,: Src homology 2 domain; TAD, transactivation domain. (B) IP of nsp11-NTD coprecipitates STAT2, STAT2-D1, and STAT2-D2, but not STAT2-D3. HEK293 cells were transfected with YFP-nsp11-NTD and Myc-STAT2 or STAT2 truncate plasmids for 36 h. IP with YFP antibody, followed by WB with cMyc and YFP antibodies, was done. EV was included as a control. WB of the input was done.
Three amino acids of nsp11 are crucial for the interaction with STAT2.
Structural analysis of the nsp11 showed several stretches of amino acids in nsp11-NTD with potential surface orientation, suggesting that some of these surface-oriented amino acids might interact with STAT2. Therefore, we constructed three truncates of nsp11-NTD: D1 (aa 1 to 51), D2 (aa 52 to 90), and D3 (aa 35 to 65) into pCDNA3-VenusC1 vector (Fig. 7A). HEK293 cells were transfected with nsp11, nsp11-NTD, and the three nsp11-NTD truncates to determine their effects on the STAT2 protein level. WB analyses showed that except for the empty vector and nsp11-NTD-D1, all the others induced STAT2 degradation (Fig. 7B). The nsp11-NTD-D2 induced the STAT2 reduction to 17% and nsp11-NTD-D3 to 46%. This indicates that aa 52 to 65 of nsp11 contain the key site for the induction of STAT2 decrease, as they are the same residues as those present in both D2 and D3. The expression of these proteins was confirmed by WB with YFP antibody.
FIG 7.

Mapping motifs in nsp11-NTD that are required for STAT2 reduction. (A) Schematic illustration of truncation plasmids nsp11-NTD: D1, D2, and D3. The numbers above the lines indicate amino acid positions in nsp11. (B) The NTD-D2 and NTD-D3 lead to a STAT2 reduction, whereas NTD-D1 has minimal effect. HEK293 cells were cotransfected with the YFP-tagged nsp11-NTD truncation plasmids and STAT2 plasmid. EV was included as a control. The cells were harvested at 36 h posttransfection for immunoblotting with antibodies against STAT2, YFP, and tubulin. The relative levels of STAT2 are shown below the images after normalization with tubulin. (C) Possible model of nsp11 protein constructed by SWISS-MODEL (https://swissmodel.expasy.org/). The loop of aa 57 to 62 is shown on the upper and right side of the nsp11 homodimer. (D) Alignment of nsp11 from PRRSV strains VR-2332 (GenBank accession no. U87392), MLV (GenBank accession no. AF066183), VR-2385 (GenBank accession no. JX044140), MN184 (GenBank accession no. EF488739), and Lelystad (GenBank accession no. M96262). Residues aa 40 to 65 are shown. The top row of residues denotes consensus amino acids. The numbers denote amino acid residues of nsp11. A period (“.”) denotes the same residues as the consensus; a dash (“-”) denotes deletion. Note that Lelystad nsp11 has an insertion of “R” as aa 61.
The region from aa 52 to 65 contains one stretch of amino acids, aa 57 to 62, with potential surface orientation. We then conducted SWISS-MODEL analysis (https://swissmodel.expasy.org/) to determine whether aa 57 to 62 (IHKYSR) of the nsp11 protein had a surface orientation. Indeed, aa 57 to 62 are located on one of the outside loops in the nsp11 NTD displayed in the structural model (Fig. 7C). PRRSV nsp11 is known to form a homodimer by the interaction between the NTD of one monomer and CTD of the other (40, 43). Accordingly, two loops of aa 57 to 62 are shown. Furthermore, the sequence alignment displayed that these stretch of amino acids are also highly conserved across different PRRSV-2 strains, including VR-2332, MLV, VR-2385, and MN184, whereas the corresponding residues of the Lelystad nsp11 are IDARYSK with an insertion of residue R as aa 60 (Fig. 7D).
We speculated that residues aa 57 to 62 might correlate with STAT2 interaction or degradation. We constructed two mutants of nsp11-NTD-D2: D2M1 (I57A, H58A, and K59A) and D2M2 (Y60A, S61A, and R62A) each with three residues mutated to alanine (Fig. 8A). HEK293 cells were transfected with these mutants to test their effect on the STAT2 protein level. The WB results showed that nsp11 NTD-D2M1 failed to induce STAT2 reduction, whereas NTD-D2M2 had an effect similar to that of wild-type NTD-D2 (Fig. 8B). These results indicate that the mutations in NTD-D2M1 led to the loss of the capability to induce STAT2 reduction and that residues aa 57 to 59 (IHK) were crucial for the nsp11 induction of STAT2 degradation.
FIG 8.

The K59 residue in nsp11 is required for inducing STAT2 reduction. (A) Schematic illustration of mutant plasmids of nsp11-NTD-D2: D2M1 (I57A, H58A, and K59A) and D2M2 (Y60A, S61A, and R62A). (B) The NTD-D2M1 fails to reduce STAT2, whereas NTD-D2M2 induces STAT2 decrease similarly to wild-type NTD-D2. HEK293 cells were transfected with the nsp11-NTD-D2, mutant, and STAT2 plasmids. The cells were harvested at 36 h posttransfection for immunoblotting with antibodies against STAT2, YFP, and tubulin. The relative levels of STAT2 are shown below the images after normalization with tubulin. (C) IP of STAT2 coprecipitates nsp11 NTD-D2 and NTD-D2M2 but much less NTD-D2M1. IP of YFP-tagged nsp11 NTD-D2 and NTD-D2M2 coprecipitates STAT2, whereas much less STAT2 was coprecipitated in the NTD-D2M1 pulldown. (D) Schematic illustration of mutant plasmids of full-length nsp11: M1 (I57A, H58A, and K59A), M2 (Y60A, S61A, and R62A), and M3 (H129A, endoribonuclease inactive). (E) The nsp11-M1 has minimal effect on the STAT2 level, whereas nsp11-M2 and nsp11-M3 induce STAT2 reduction. (F) Schematic illustration of mutant plasmids of full-length nsp11: M4 (I57A), M5 (H58A), and M6 (K59A). (G) The nsp11-M6 has a minimal effect on the STAT2 level, whereas nsp11-M4 and nsp11-M5 induce STAT2 reduction.
We further conducted co-IP analyses to determine whether the mutation in NTD-D2M1 led to the loss of interaction with STAT2. IP of STAT2 coprecipitated nsp11 NTD-D2 and NTD-D2M2 in similar levels, but NTD-D2M1 at a reduced level (Fig. 8C). In contrast, cells transfected with EV and NTD-D2M1 had a higher level of STAT2 than those of NTD-D2 and NTD-D2M2, as shown in WB analyses of the input (Fig. 8C). Conversely, YFP IP of NTD-D2 and NTD-D2M2 coprecipitated STAT2, whereas less STAT2 could be coprecipitated by IP of NTD-D2M1, and no STAT2 could be coprecipitated by IP of YFP empty vector. These results demonstrated that the mutations of aa 57 to 59 in NTD-D2M1 affected nsp11 function in the STAT2 degradation via weakening its interaction with STAT2.
Amino acid residue K59 is critical for nsp11 induction of STAT2 reduction.
To further confirm the critical role of aa 57 to 59 in nsp11-mediated STAT2 reduction, we introduced mutations of I57A/H58A/K59A and Y60A/S61A/R62A into full-length nsp11, named nsp11-M1 and nsp11-M2, respectively (Fig. 8D). Like the nsp11-NTD mutants, nsp11-M1 failed to induce STAT2 reduction, whereas nsp11-M2 reduced STAT2 to a level similar to that of wild-type nsp11 (Fig. 8E). In addition, to strengthen our conclusion that the nsp11 NendoU has no role in the reduction of STAT2, we constructed nsp11-M3, a NendoU-inactive mutant nsp11 with a mutation of H129A (Fig. 8D) (23, 40, 44). WB results showed that nsp11-M3 also induced STAT2 reduction similarly to nsp11-M2 (Fig. 8E). These results demonstrated that amino acid residues 57 to 59 but not the NendoU activity of nsp11 are responsible for the STAT2 reduction.
To identify the most critical residue from aa 57 to 59 that is indispensable for the STAT2 reduction, we conducted single-residue mutations for nsp11-M4 (I57A), nsp11-M5 (H58A), and nsp11-M6 (K59A) mutants (Fig. 8F). Like wild-type nsp11, nsp11-M4 and nsp11-M5 reduced STAT2, whereas nsp11-M6 induced much less reduction (Fig. 8G). This result shows that K59 residue is the most crucial for nsp11 to induce STAT2 reduction.
nsp11 interacts with STAT2 in PRRSV-infected cells, and K59A mutant virus has minimal effect to reduce STAT2.
To further confirm that nsp11 interacts with STAT2 in PRRSV-infected cells, we conducted co-IP of STAT2 from the lysate of PRRSV-infected cells. WB results showed that nsp11 was present in the STAT2 precipitates, whereas PRRSV nsp1β was not (Fig. 9A). This indicates that nsp11 interacts with STAT2 in PRRSV-infected cells.
FIG 9.
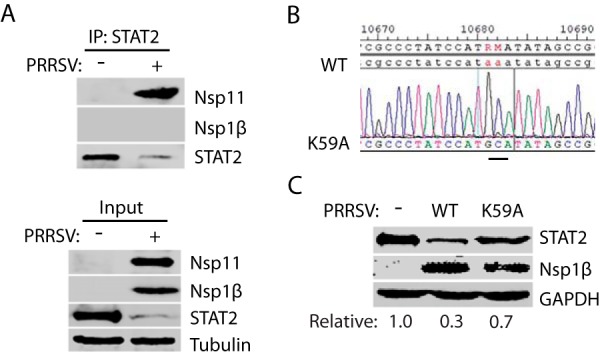
PRRSV nsp11 interacts with STAT2 in infected cells, and residue K59 is essential for STAT2 reduction. (A) STAT2 IP precipitates nsp11 in PRRSV-infected MARC-145 cells. The cells were infected with PRRSV VR-2385 at an MOI of 1 and harvested at 36 hpi for IP with STAT2 antibody, followed by WB with rabbit antibodies against PRRSV nsp11 and nsp1β. WB of input was included as s control. (B) Sequencing of the cDNA of mutant VR-2385 with nsp11 K59A confirms the presence of codon mutation in its genome. The numbers above the sequence indicate nucleotide positions in the cDNA of the VR-2385 genome. The bar below the chromatogram indicates the codon change from AAA(Lys) to GCA(Ala) in nsp11. (C) Mutant PRRSV VR-2385 with nsp11 K59A has much less effect on STAT2 than does the wild-type (WT) virus in MARC-145 cells. The cells were infected with the mutant virus and WT VR-2385 at an MOI of 1 and harvested at 36 hpi for WB.
Since K59 residue is critical for nsp11 induction of STAT2 reduction, we reasoned that the K59A mutation of the PRRSV infectious clone would abolish its capacity to reduce STAT2. K59A mutation was introduced into pIR-VR2385-CA, the cDNA infectious clone of VR-2385. Mutant virus was recovered, and DNA sequencing confirmed the presence of the mutant nucleotides GC for K59A mutation in the genome of the recovered virus (Fig. 9B). MARC-145 cells were infected with the mutant virus. The wild-type VR-2385 was included as a control. WB results showed that the VR-2385 K59A mutant virus had minimal effect on STAT2, whereas the wild-type virus reduced STAT2 (Fig. 9C). These data demonstrate that K59 residue of nsp11 is indispensable for PRRSV reduction of STAT2.
DISCUSSION
The canonical JAK-STAT pathway is activated by many cytokines, including IFNs, interleukins, and granulocyte-macrophage colony-stimulating factor. In mammalian cells, there are seven STAT members: STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6. Among these seven STATs, STAT2 is unique (42). STAT2 is only activated by type I and type III IFNs, whereas STAT1 can respond to all three types of IFNs, IL-4, and IL-6 (45). Except for STAT2, all other STAT proteins have at least two isoforms with specific functions. Moreover, STAT2 is the largest STAT, with the molecular mass of 113 kDa.
STAT2 is not only a key component in the canonical IFN-activated JAK-STAT signaling, but it is also involved in STAT1-independent IFN signaling (6). In the noncanonical IFN signaling pathway, the STAT2/IRF9 complex functions as an ISGF3-like response and provides an alternative antiviral activity in the deficiency of STAT1 (6). It was reported that STAT2 binds the ISREs of ISG15, ISG56, IFI27, MX1, OAS2, and IFIT3 genes in the absence of STAT1. Moreover, the study of STAT2/IRF9 identified a set of ISGF3-independent ISGs, including A3G, CCL8, and CX3CL1. In addition, during lymphocytic choriomeningitis virus infection, STAT2/IRF9 directs a proinflammatory immune response in the absence of STAT1, suggesting that the ISGF3-like complex has a novel role in inflammation (46). Therefore, STAT2 is indispensable in IFN-activated signaling.
In this study, we demonstrated that PRRSV induces STAT2 degradation to antagonize STAT2 signaling via nsp11. The observation was confirmed in multiple aspects. First, PRRSV induced STAT2 degradation in both MARC-145 and PAM cells but had no effect on the STAT1 protein level. Several strains from both PRRSV species reduced STAT2, without affecting STAT1, which indicates that the PRRSV-induced STAT2 reduction was specific and might be an inherent property of PRRSV infection. Second, the PRRSV-induced STAT2 reduction was dose and time dependent. Third, treatment with MG132 and lactacystin restored the STAT2 level in PRRSV-infected cells, which indicates that PRRSV-mediated STAT2 reduction was via the ubiquitin-proteasome protein degradation pathway. Consistent with this result, STAT2 half-life was shortened by PRRSV from approximately 30 to 5 h, and PRRSV infection had minimal effect on STAT2 mRNA level.
UV-inactivated PRRSV had no effect on the STAT2 protein level, which indicates that the structural proteins of PRRSV might have no detectable role in PRRSV-mediated STAT2 reduction. After screening of PRRSV nonstructural proteins, nsp2, nsp3, nsp5, and nsp11 were identified to be candidates responsible for the STAT2 reduction. Further experiments showed that nsp11 induced much less STAT2 than did nsp2, nsp3, and nsp5. However, nsp2, nsp3, and nsp5 might have a certain effect on STAT2, albeit much less than that of nsp11. Consequently, nsp11 was able to inhibit IFN-α-activated expression of ISRE reporter. nsp11 was previously shown to suppress MAVS and RIG-I expression through its endoribonuclease domain to antagonize type I IFN induction (44, 47).
Our data ruled out that the endoribonuclease activity of nsp11 plays a role in STAT2 degradation. First, we found both PRRSV infection and nsp11 overexpression had no effect on STAT2 mRNA level. Second, the catalytic domain (C-terminal domain) of nsp11 has no effect on the STAT2 protein level, whereas the nsp11 NTD alone induced a STAT2 reduction similar to that of the full-length nsp11. Moreover, full-length nsp11 mutant H129A, which loses the endoribonuclease activity (40), had an effect on STAT2 reduction similar to that of the wild-type nsp11. These results confirmed that the nsp11 NTD but not the endoribonuclease activity was responsible for STAT2 degradation.
We further determined the mechanism of nsp11-mediated STAT2 reduction. The nsp11 NTD is formed by six β-strands and two α-helices and is connected to the catalytic CTD through a linker domain (40, 43). Mutations of S74A and F76A disrupt nsp11 dimerization. Our data showed that the nsp11-NTD (aa 1 to 90) led to a higher STAT2 polyubiquitination level to induce STAT2 degradation. Interestingly, the IP of STAT2 coprecipitated both nsp11-NTD and full-length nsp11, which suggests that nsp11 induces STAT2 degradation through its NTD interacting with STAT2. The interaction of nsp11 with STAT2 was also confirmed in the PRRSV-infected cells.
We also analyzed the interaction domain in STAT2. Like other STATs, STAT2 has six conserved domains. NTD is required for the tyrosine phosphorylation (48) and the interaction with the IFN receptor (49). CCD of STAT2 is the specific domain that IRF9 binds (50). Direct binding of ISGF3 to DNA is mediated by STAT1 and IRF9, not STAT2, so STAT2 DBD seems to bind the promoter of ISGF3-independent ISGs (51). Moreover, the STAT2 DBD contains a cluster of basic amino acids to function as a bipartite NLS when binding with the corresponding conserved region of STAT1 (52). The function of the linker domain remains unknown. The SH2 domain is responsible for STAT2 to bind to phosphorylated IFN receptor and interact with phosphorylated STAT1 to form a heterodimer (53). The TAD is indispensable for transcription factors to recruit other coactivators or transcriptional regulators (54). One nuclear export signal lies in the STAT2 TAD to mediate STAT2 exportation from the nucleus back to the cytoplasm (55). Our IP data showed that the NTD and CCD of STAT2 interacted with nsp11.
In addition, we identified the critical amino acids in nsp11 that correlate with the STAT2 reduction. Our results showed that the nsp11 NTD-D2 and NTD-D3 led to the STAT2 reduction, whereas NTD-D1 had minimal effect. We conducted mutagenesis of NTD-D2 based on sequence and computational structural analyses and generated two mutants. Mutation at aa 57 to 59 (IHK) in NTD-D2M1 led to the loss of the nsp11 capability in inducing STAT2 degradation. This suggests that amino acid residues 57 to 59 are crucial to the nsp11-induced STAT2 reduction. Co-IP results showed that NTD-D2M1 had much weaker interaction with STAT2 than NTD-D2M2 and wild-type NTD-D2. The NTD-D2M2 could coprecipitate STAT2 at a level similar to that for NTD-D2. These results indicate that aa 57 to 59 are indispensable for nsp11 to interact with and downregulate STAT2.
To exclude the possibility that truncated and mutated nsp11 could not fold properly, we introduced mutations I57A/H58A/K59A (nsp11-M1) and Y60A/S61A/R62A (nsp11-M2) into full-length nsp11 to detect their effect on STAT2. As expected, the full-length mutants are similar to the corresponding nsp11-NTD mutants, which confirms that aa 57 to 59 of nsp11 are critical for STAT2 reduction. Furthermore, we determined that K59 of nsp11 is the critical residue for the STAT2 reduction since the mutant nsp11-K59A loses the ability to reduce STAT2, whereas the other two mutants nsp11-I57A and nsp11-H58A reduced STAT2 like wild-type nsp11.
Moreover, we constructed a mutant full-length PRRSV cDNA clone, pIR-VR2385-K59A, with K59A mutation in nsp11. Our results showed that the mutant virus had much less effect on STAT2 reduction than did the wild-type virus, which further confirms that K59 residue of nsp11 is indispensable for PRRSV reduction of STAT2. Consistent with this observation, sequence alignment shows that K59 is highly conserved in different PRRSV-2 strains, whereas there is an arginine residue inserted at aa 60 in nsp11 of Lelystad virus, a prototype strain of PRRSV-1. We constructed mutant nsp11 K59R of VR-2385 and noticed an effect on STAT2 reduction similar to that of wild-type nsp11 (data not shown), which suggests that the arginine residue is anticipated to be indispensable for Lelystad reduction of STAT2. It is not known whether other members of the family Arteriviridae can reduce STAT2 like PRRSV. There is a lysine residue at aa 60 (K60) in nsp11 of equine arteritis virus (EAV). SWISS-MODEL analysis indicates that the K60 is surface oriented on the homodimer of EAV nsp11 (data not shown). Sequencing alignment indicates that there are about 50% identical amino acids between nsp11 of EAV and PRRSV. Further study is needed to determine whether EAV can also induce STAT2 degradation and whether the K60 residue is indispensable for the effect.
As the first line of defense against viral infections, IFN is indispensable in innate immunity. To antagonize IFNs, PRRSV suppresses the production of type I IFNs by inhibiting IRF3 phosphorylation (26), interfering with the polyubiquitination of IκBα (29), cleaving MAVS (56), and suppressing RIG-I and MAVS expression (47) and also blocks IFN-activated downstream signaling by degrading KPNA1 to block STAT1/STAT2 nuclear translocation (32) and inducing STAT3 degradation (34). In this study, we further demonstrated that PRRSV also targets STAT2 to antagonize IFN signaling. However, the exact mechanism of PRRSV nsp11-mediated STAT2 degradation is not known. We speculate that the nsp11 interaction with STAT2 might recruit an E3 ubiquitin ligase to promote STAT2 turnover as nsp11 increases STAT2 polyubiquitination. It is known that dengue virus NS5 interacts with E3 ubiquitin ligase UBR4 to degrade STAT2 (11), whereas Zika virus NS5 does not need UBR4 to induce STAT2 degradation (12). Respiratory syncytial virus NS1 protein uses the elongin-cullin E3 ligase to degrade STAT2 (58). DCST1 E3 ligase interacts and promotes STAT2 degradation (59). Our attempt to determine the E3 ligase for nsp11-induced STAT2 degradation was not fruitful. Considering the complexity of STAT2 turnover by multiple E3 ligases, further work is warranted to determine whether PRRSV nsp11 recruits a specific E3 ligase.
In conclusion, our results demonstrated that PRRSV infection induced STAT2 degradation via the ubiquitin-proteasome pathway. Specifically, PRRSV induces STAT2 degradation by the interaction between nsp11 and STAT2. The K59 residue of nsp11 is indispensable for PRRSV-mediated STAT2 turnover. This provides further insights into PRRSV interference with IFN signaling, in addition to inhibiting STAT1/2 translocation from the cytoplasm into the nucleus. This study further elucidates PRRSV interference with innate immunity and the consequent immune response.
MATERIALS AND METHODS
Cells, viruses, and chemicals.
MARC-145 (20), HEK293 (ATCC CRL-1573), HeLa (ATCC CCL-2), and BHK-21 (ATCC CCL-10) cells were cultured in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum (FBS). Primary porcine PAM cells were revived from cryopreserved stock and cultured in RPMI 1640 medium with 10% FBS as previously described (31, 60).
PRRSV strains VR-2385 (GenBank accession number JX044140) (61), Ingelvac PRRS MLV (GenBank accession AF066183) (62), and Lelystad (GenBank accession M96262) (63) were used to inoculate MARC-145 cells at an MOI of 1 or using the amounts indicated in figure legends or results. The median tissue culture infectious dose of PRRSV was determined in MARC-145 cells (64).
For virus inactivation, the virus inoculum was treated in a UV cross-linker (Spectrolinker XL-1500; Agilent Technologies, Santa Clara, CA) at 1,200 mJ/cm2 for two 10-min pulses with a 1-min interval. Virus inactivation was confirmed by lack of PRRSV-positive cells in inoculated MARC-145 cells at 72 hpi (31).
Cycloheximide (Sigma-Aldrich, St. Louis, MO), a protein translation inhibitor, was added to PRRSV-infected and mock-infected cells at a final concentration of 50 μg/ml to determine the half-life of STAT2. MG132 (Sigma), a proteasome inhibitor, was used to treat cells at a final concentration of 10 μM for 6 h prior to harvesting. Lactacystin (Sigma), a specific proteasome inhibitor, was utilized to treat cells for 6 h at a final concentration of 5 μM. Human IFN-α (GenScript) was used to activate the JAK-STAT signaling pathway at a final concentration of 300 U/ml.
Plasmids.
The nsp11 of PRRSV VR-2385 was cloned into a pCAGEN-HA vector and pCDNA3-VenusC1 vector (34) with the primers 32nsp11F1 and 85nsp11R1 (Table 1). The nsp11 deletion mutants were cloned into pCAGEN-HA vector using the following primer pairs: 32nsp11F1 and 85nsp11-NTD-R for nsp11-NTD, 32nsp11F1 and 85nsp11-NL-R for nsp11-NTDL, 85nsp11-LC-F and 85nsp11R1 for nsp11-LCTD, and 85nsp11-CTD-F and 85nsp11R1 for nsp11-CTD (Table 1). nsp11-NTD was also cloned into a pCDNA3-VenusC1 vector with the same primers. The nsp11-NTD fragments were cloned into pCDNA3-VenusC1 by using the primer pairs 32nsp11F1 and 85nsp11-NTD-1R for nsp11-NTD-D1, 85nsp11-NTD-2F and 85nsp11-NTD-R for nsp11-NTD-D2, and 85nsp11-NTD-3F and 85nsp11-NTD-3R for nsp11-NTD-D3 (Table 1). STAT2 was cloned into a pCAGEN-Myc vector and pCDNA3-VenusC1 vector with the primers STAT2F1 and STAT2R1. The STAT2 deletion mutants were cloned into the pCAGEN-Myc vector by using the following primer pairs: STAT2F1 and STAT2R2 for STAT2-D1, STAT2F1 and STAT2R3 for STAT2-D2, and STAT2F2 and STAT2R1 for STAT2-D3. Mutants of nsp11-NTD-D2 were cloned into the pCDNA3-VenusC1 vector using the following primers: (i) 85nsp11N-D2-m1-F1 and 85nsp11-NTD-R for D2M1 and (ii) VenusC1F-HindIII, 85nsp11N-D2-m2-F1, 85nsp11N-D2-m2-R1, and 85nsp11-NTD-R for D2M2 with overlapping PCR (Table 1). The resulting recombinant plasmids were confirmed by restriction enzyme digestion and DNA sequencing.
TABLE 1.
Primers used in this study
| Primera | Sequence (5′–3′)b | Target gene/vector |
|---|---|---|
| 32nsp11F1 | CGGAATTCGGGTCGAGCTCTCCGCTCC | nsp11 |
| 32nsp11R1 | CCGCTCGAGTTATTCAAGTTGGAAATAGGC | nsp11 |
| 85nsp11-NTD-R | CCGCTCGAGTTAAAATTTTGTGAGGTAGTA | nsp11-NTD |
| 85nsp11-NL-R | CCGCTCGAGTTAGCCGGTGCTGAAGATCGT | nsp11-NTDL |
| 85nsp11-LC-F | CGGAATTCGTTAAGGGCGAGGCTCAA | nsp11-LCTD |
| 85nsp11-CTD-F | CGGAATTCCGAATTGAGGTAGATTGC | nsp11-CTD |
| 85nsp11-NTD-1R | CCGCTCGAGTTAAACCAGCCGATCTGGCCA | nsp11-NTD-D1 |
| 85nsp11-NTD-2F | CGGAATTCACCAGCCTTCGCCCTATC | nsp11-NTD-D2 |
| 85nsp11-NTD-3F | CGGAATTCTGGCCCGTGGTGACAACT | nsp11-NTD-D3 |
| 85nsp11-NTD-3R | CCGCTCGAGTTAAATGCACGCGCGGCTATA | nsp11-NTD-D3 |
| STAT2F1 | GCGAATTCGCGCAGTGGGAAATGCTGCA | STAT2 |
| STAT2R1 | GACTCGAGCTAGAAGTCAGAAGGCATCA | STAT2 |
| STAT2R2 | CCGCTCGAGTTAGGCTCTGTGGAGCAGACG | STAT2D1 |
| STAT2R3 | CCGCTCGAGTTACTGAAGGTTTGGGCTGAG | STAT2D2 |
| STAT2F2 | GCGAATTCAACCAGCAGTTCTTCTCC | STAT2D3 |
| R-STAT2-F1 | AACCGTACACGAAGGAGGTG | STAT2 |
| R-STAT2-R1 | GATTCGGGGATAGAGGAAGC | STAT2 |
| 85nsp11N-D2-m1-F1 | GGAATTCACCAGCCTTCGCCCTGCAGCAGCATATAGCCGCGCGTGCAT | nsp11-NTD-D2M1 |
| 85nsp11N-D2-m1-R1 | ACGCGCGGCTATATGCTGCTGCAGGGCGAAGGCTGGTAACCAGC | nsp11-M1 |
| 85nsp11N-D2-m1-F2 | GCAGCAGCATATAGCCGCGCGTGCATTGGTGCCGGCTATATGG | nsp11-M1 |
| 85nsp11N-D2-m2-F1 | CATAAAGCAGCAGCAGCGTGCATTGGTGCCGGCTATATGGTGG | nsp11-NTD-D2M2 |
| 85nsp11N-D2-m2-R1 | CAATGCACGCTGCTGCTGCTTTATGGATAGGGCGAAGGCTGGT | nsp11-NTD-D2M2 |
| 85nsp11-H129A-F1 | CGTCCCTCCCAGCAGCCTTCATTGGTGACGTCAAAGGCAC | nsp11-M3 |
| 85nsp11-H129A-R1 | CCAATGAAGGCTGCTGGGAGGGACGCAGCAACTTCTCGCT | nsp11-M3 |
| 85nsp11D2M5-F1 | CTTCGCCCTGCACATAAATATAGCCGCGCGTGCATTGGTG | nsp11-M4 |
| 85nsp11D2M5-R1 | TATATTTATGTGCAGGGCGAAGGCTGGTAACCAGCCGATC | nsp11-M4 |
| 85nsp11D2M6-F1 | CGCCCTATCGCAAAATATAGCCGCGCGTGCATTGGTGCCG | nsp11-M5 |
| 85nsp11D2M6-R1 | GGCTATATTTTGCGATAGGGCGAAGGCTGGTAACCAGCCG | nsp11-M5 |
| 85nsp11D2M7-F1 | CCTATCCATGCATATAGCCGCGCGTGCATTGGTGCCGGCT | nsp11-M6 |
| 85nsp11D2M7-R1 | CGCGGCTATATGCATGGATAGGGCGAAGGCTGGTAACCAG | nsp11-M6 |
| VenusC1F-HindIII | CGAAGCTTCGCCACCATGGTGAGCAAG | pCDNA3-VenucsC1 |
F, forward primer; R, reverse primer. A “32” or “85” prefix in a primer designation indicates that the primer is based on sequences of PRRSV VR-2332 (GenBank accession no. U87392) or VR-2385 (GenBank accession no. JX044140), respectively. An “R-” prefix primer name indicates the primer was designed for real-time PCR.
Italicized letters indicate restriction enzyme cleavage sites for cloning.
The construction of the other PRRSV nsp plasmids was described previously (33). The pCDNA3-STAT2 was a gift from Curt M. Horvath (65). The pGL-ISRE used for the IFN-STAT1/2 reporter assay was described previously (33). The pRK5-HA-ubiquitin-K48 plasmid was a gift from Ted Dawson (Addgene, plasmid 17605) (66).
The construction of mutant nsp11 for nsp11-M1 to nsp11-M6 were done with overlapping PCR with the primers listed in Table 1 as described previously (32). For example, primers 32nsp11F1, 85nsp11N-D2m1-R1, 85nsp11N-D2m1-F2, and 32nsp11R1 were used in overlapping PCR for cloning nsp11-M1; 32nsp11F1, 85nsp11N-D2m2-R1, 85nsp11N-D2m2-F1, and 32nsp11R1 were used for nsp11-M2. The mutagenesis of the cDNA infectious clone of PRRSV VR-2385 for nsp11 K59A was done similarly to the construction of nsp11-M6 and ligated into pIR-VR2385-CA as described previously (67, 68). The resulting mutant plasmids were confirmed by DNA sequencing. For virus recovery, the mutant plasmid was transfected into BHK-21 cells, with the wild-type plasmid included as a control. The cell culture supernatant at 72 h posttransfection was collected and used to inoculate fresh MARC-145 cells. RNA was isolated from the recovered mutant virus for cDNA synthesis and DNA sequencing to confirm the presence of the mutant nucleotides.
RNA isolation and real-time PCR.
Total RNA was isolated from MARC-145 cells with TRIzol reagent (Thermo Fisher Scientific, Waltham, MA) in accordance with the manufacturer’s instructions. Reverse transcription and real-time PCR (RT-qPCR; Thermo Fisher Scientific) was conducted as described previously (60). Transcripts of ribosomal protein L32 (RPL32), the reference gene, were also detected. The relative transcript levels were shown as the fold changes compared to the control cells after RPL32 normalization. The real-time PCR primers used for STAT2 were R-STAT2-F1 and R-STAT2-R1 (Table 1). The primers used for PRRSV, ISG15, ISG56, and RPL32 were as described previously (31). All experiments were repeated at least three times, with each conducted in triplicate.
Western blot analysis.
Cell lysates in Laemmli sample buffer were subjected to SDS-PAGE and WB as described previously (69). The primary antibodies used in this study were against STAT2 (Santa Cruz Biotechnology, Inc., Dallas, TX), STAT1 (Santa Cruz), PRRSV nsp2 protein (70, 71), nsp11 (47), β-tubulin (Sigma), GAPDH (glyceraldehyde-3-phosphate dehydrogenase; Santa Cruz), ubiquitin (Santa Cruz), HA tag (Thermo Fisher Scientific), cMyc tag (Thermo Fisher Scientific), and green fluorescent protein (BioLegend, Inc., San Diego, CA). Affinity-purified rabbit antibody against PRRSV nsp1β was prepared by GenScript (Piscataway, NJ) with the synthetic peptide NGPIVVQYFSVKES (aa 142 to 155 of nsp1β of VR-2385; GenBank accession no. JX044140). The secondary antibodies used in this study were goat anti-mouse or goat anti-rabbit IgG conjugated with horseradish peroxidase (Rockland Immunochemicals, Inc., Gilbertsville, PA). Chemiluminescence signal acquisition was conducted by using the Quantity One program (v4.6) and a Chemi-Doc XRS imaging system (Bio-Rad Laboratories, Hercules, CA). All experiments were repeated at least three times to warrant the reliability of the results. To monitor the STAT2 half-life, WB was conducted, and the band intensity was quantified with Quantity One. This experiment was repeated five times. The data were processed with GraphPad Prism (v7) to show the half-life curve.
Cell viability assay.
To test the cytotoxicity of lactacystin, lactacystin was added into MARC-145 cells infected with PRRSV for 24 h, and dimethyl sulfoxide (DMSO)-treated and mock-infected cells were included as controls. After 6 h of treatment, a cell viability assay was conducted with CellTiter-Glo (Promega, Madison, WI) according to the manufacturer’s instructions. To test the toxicity of PRRSV nsp11, the same amounts of empty vector and nsp11 plasmids were transfected into the same number of cells in different wells. After 24 and 48 h, the cell viability was detected by using CellTiter-Glo. All experiments were repeated at least three times, with each performed in triplicate.
Immunoprecipitation.
IP was conducted as previously described (34, 36). The clarified cell lysates were incubated with specific antibodies indicated in Results or the figure legends, followed by incubation with protein A/G-magnetic beads (Bimake.com, Houston, TX). The IP products were subjected to WB for detection of target proteins with corresponding antibodies indicated. To determine ubiquitinated STAT2, ubiquitin aldehyde (Boston Biochem, Inc., Cambridge, MA), a specific inhibitor of ubiquitin C-terminal hydrolases, was added to the lysis buffer at a final concentration of 2.53 μM.
ISRE reporter assay.
HEK293 cells were transfected with the ISRE reporter plasmid pGL3-ISRE and nsp11 plasmids. Renilla luciferase vector pRL-TK (Promega), expressing Renilla luciferase under the control of a constitutively active promoter, was also transfected for normalization. At 24 h posttransfection, the cells were treated with 300 U/ml IFN-α and, 24 h later, harvested for luciferase activity assay of firefly and Renilla luciferases according to the manufacturer’s instructions (Promega). The relative levels of firefly luciferase activity are shown as fold changes compared to the EV control after normalization with the Renilla activity, as described previously (33, 34).
Statistical analysis.
Differences in indicators between the treatment group and control were assessed by using a Student t test. A two-tailed P value of 0.05 was considered significant.
ACKNOWLEDGMENTS
We thank Jie Peng of the Department of Mechanical Engineering, University of Maryland, for helping us illustrate the nsp11 protein structure. We thank Ying Fang at Kansas State University for monoclonal antibodies against PRRSV nsp2 and nsp1β proteins and Dongwan Yoo at the University of Illinois at Urbana—Champaign for rabbit antibody against PRRSV nsp11 protein.
J.H., X.Z., S.L., and Z.M. were partially sponsored by the China Scholarship Council. This study was partially funded by a seed grant from the University of Maryland.
REFERENCES
- 1.Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, Stark GR. 2007. Interferons at age 50: past, current, and future impact on biomedicine. Nat Rev Drug Discov 6:975–990. doi: 10.1038/nrd2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nan Y, Nan G, Zhang YJ. 2014. Interferon induction by RNA viruses and antagonism by viral pathogens. Viruses 6:4999–5027. doi: 10.3390/v6124999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nan Y, Wu C, Zhang YJ. 2017. Interplay between Janus kinase/signal transducer and activator of transcription signaling activated by type I interferons and viral antagonism. Front Immunol 8:1758. doi: 10.3389/fimmu.2017.01758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rusinova I, Forster S, Yu S, Kannan A, Masse M, Cumming H, Chapman R, Hertzog PJ. 2013. Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res 41:D1040–D1046. doi: 10.1093/nar/gks1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schindler C, Levy DE, Decker T. 2007. JAK-STAT signaling: from interferons to cytokines. J Biol Chem 282:20059–20063. doi: 10.1074/jbc.R700016200. [DOI] [PubMed] [Google Scholar]
- 6.Blaszczyk K, Olejnik A, Nowicka H, Ozgyin L, Chen YL, Chmielewski S, Kostyrko K, Wesoly J, Balint BL, Lee CK, Bluyssen HA. 2015. STAT2/IRF9 directs a prolonged ISGF3-like transcriptional response and antiviral activity in the absence of STAT1. Biochem J 466:511–524. doi: 10.1042/BJ20140644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perry ST, Buck MD, Lada SM, Schindler C, Shresta S. 2011. STAT2 mediates innate immunity to dengue virus in the absence of STAT1 via the type I interferon receptor. PLoS Pathog 7:e1001297. doi: 10.1371/journal.ppat.1001297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chowdhury FZ, Farrar JD. 2013. STAT2: a shape-shifting antiviral super STAT. JAKSTAT 2:e23633. doi: 10.4161/jkst.23633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramachandran A, Parisien JP, Horvath CM. 2008. STAT2 is a primary target for measles virus V protein-mediated alpha/beta interferon signaling inhibition. J Virol 82:8330–8338. doi: 10.1128/JVI.00831-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laurent-Rolle M, Morrison J, Rajsbaum R, Macleod JML, Pisanelli G, Pham A, Ayllon J, Miorin L, Martinez C, tenOever BR, García-Sastre A. 2014. The interferon signaling antagonist function of yellow fever virus NS5 protein is activated by type I interferon. Cell Host Microbe 16:314–327. doi: 10.1016/j.chom.2014.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morrison J, Laurent-Rolle M, Maestre AM, Rajsbaum R, Pisanelli G, Simon V, Mulder LCF, Fernandez-Sesma A, García-Sastre A. 2013. Dengue virus co-opts UBR4 to degrade STAT2 and antagonize type I interferon signaling. PLoS Pathog 9:e1003265. doi: 10.1371/journal.ppat.1003265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grant A, Ponia SS, Tripathi S, Balasubramaniam V, Miorin L, Sourisseau M, Schwarz MC, Sánchez-Seco MP, Evans MJ, Best SM, García-Sastre A. 2016. Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe 19:882–890. doi: 10.1016/j.chom.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lunney JK, Fang Y, Ladinig A, Chen N, Li Y, Rowland B, Renukaradhya GJ. 2016. Porcine reproductive and respiratory syndrome virus (PRRSV): pathogenesis and interaction with the immune system. Annu Rev Anim Biosci 4:129–154. doi: 10.1146/annurev-animal-022114-111025. [DOI] [PubMed] [Google Scholar]
- 14.Rossow KD. 1998. Porcine reproductive and respiratory syndrome. Vet Pathol 35:1–20. doi: 10.1177/030098589803500101. [DOI] [PubMed] [Google Scholar]
- 15.Bilodeau R, Archambault D, Vezina SA, Sauvageau R, Fournier M, Dea S. 1994. Persistence of porcine reproductive and respiratory syndrome virus infection in a swine operation. Can J Vet Res 58:291–298. [PMC free article] [PubMed] [Google Scholar]
- 16.Adams MJ, Lefkowitz EJ, King AMQ, Harrach B, Harrison RL, Knowles NJ, Kropinski AM, Krupovic M, Kuhn JH, Mushegian AR, Nibert M, Sabanadzovic S, Sanfacon H, Siddell SG, Simmonds P, Varsani A, Zerbini FM, Gorbalenya AE, Davison AJ. 2017. Changes to taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2017). Arch Virol 162:2505–2538. doi: 10.1007/s00705-017-3358-5. [DOI] [PubMed] [Google Scholar]
- 17.Kuhn JH, Lauck M, Bailey AL, Shchetinin AM, Vishnevskaya TV, Bào Y, Ng TFF, LeBreton M, Schneider BS, Gillis A, Tamoufe U, Diffo JLD, Takuo JM, Kondov NO, Coffey LL, Wolfe ND, Delwart E, Clawson AN, Postnikova E, Bollinger L, Lackemeyer MG, Radoshitzky SR, Palacios G, Wada J, Shevtsova ZV, Jahrling PB, Lapin BA, Deriabin PG, Dunowska M, Alkhovsky SV, Rogers J, Friedrich TC, O’Connor DH, Goldberg TL. 2016. Reorganization and expansion of the nidoviral family Arteriviridae. Arch Virol 161:755–768. doi: 10.1007/s00705-015-2672-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mardassi H, Mounir S, Dea S. 1995. Molecular analysis of the ORFs 3 to 7 of porcine reproductive and respiratory syndrome virus, Quebec reference strain. Arch Virol 140:1405–1418. doi: 10.1007/bf01322667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rossow KD, Collins JE, Goyal SM, Nelson EA, Christopher-Hennings J, Benfield DA. 1995. Pathogenesis of porcine reproductive and respiratory syndrome virus infection in gnotobiotic pigs. Vet Pathol 32:361–373. doi: 10.1177/030098589503200404. [DOI] [PubMed] [Google Scholar]
- 20.Kim HS, Kwang J, Yoon IJ, Joo HS, Frey ML. 1993. Enhanced replication of porcine reproductive and respiratory syndrome (PRRS) virus in a homogeneous subpopulation of MA-104 cell line. Arch Virol 133:477–483. doi: 10.1007/BF01313785. [DOI] [PubMed] [Google Scholar]
- 21.Buddaert W, Van Reeth K, Pensaert M. 1998. In vivo and in vitro interferon (IFN) studies with the porcine reproductive and respiratory syndrome virus (PRRSV). Adv Exp Med Biol 440:461–467. doi: 10.1007/978-1-4615-5331-1_59. [DOI] [PubMed] [Google Scholar]
- 22.Labarque GG, Nauwynck HJ, Van Reeth K, Pensaert MB. 2000. Effect of cellular changes and onset of humoral immunity on the replication of porcine reproductive and respiratory syndrome virus in the lungs of pigs. J Gen Virol 81:1327–1334. doi: 10.1099/0022-1317-81-5-1327. [DOI] [PubMed] [Google Scholar]
- 23.Wang R, Zhang YJ. 2014. Antagonizing interferon-mediated immune response by porcine reproductive and respiratory syndrome virus. Biomed Res Int 2014:315470. doi: 10.1155/2014/315470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang L, Zhang YJ. 2017. Antagonizing cytokine-mediated JAK-STAT signaling by porcine reproductive and respiratory syndrome virus. Vet Microbiol 209:57–65. doi: 10.1016/j.vetmic.2016.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo R, Xiao S, Jiang Y, Jin H, Wang D, Liu M, Chen H, Fang L. 2008. Porcine reproductive and respiratory syndrome virus (PRRSV) suppresses interferon-beta production by interfering with the RIG-I signaling pathway. Mol Immunol 45:2839–2846. doi: 10.1016/j.molimm.2008.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beura LK, Sarkar SN, Kwon B, Subramaniam S, Jones C, Pattnaik AK, Osorio FA. 2010. Porcine reproductive and respiratory syndrome virus nonstructural protein 1beta modulates host innate immune response by antagonizing IRF3 activation. J Virol 84:1574–1584. doi: 10.1128/JVI.01326-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim O, Sun Y, Lai FW, Song C, Yoo D. 2010. Modulation of type I interferon induction by porcine reproductive and respiratory syndrome virus and degradation of CREB-binding protein by nonstructural protein 1 in MARC-145 and HeLa cells. Virology 402:315–326. doi: 10.1016/j.virol.2010.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Z, Lawson S, Sun Z, Zhou X, Guan X, Christopher-Hennings J, Nelson EA, Fang Y. 2010. Identification of two auto-cleavage products of nonstructural protein 1 (nsp1) in porcine reproductive and respiratory syndrome virus-infected cells: nsp1 function as interferon antagonist. Virology 398:87–97. doi: 10.1016/j.virol.2009.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun Z, Chen Z, Lawson SR, Fang Y. 2010. The cysteine protease domain of porcine reproductive and respiratory syndrome virus nonstructural protein 2 possesses deubiquitinating and interferon antagonism functions. J Virol 84:7832–7846. doi: 10.1128/JVI.00217-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Kasteren PB, Bailey-Elkin BA, James TW, Ninaber DK, Beugeling C, Khajehpour M, Snijder EJ, Mark BL, Kikkert M. 2013. Deubiquitinase function of arterivirus papain-like protease 2 suppresses the innate immune response in infected host cells. Proc Natl Acad Sci U S A 110:E838–E847. doi: 10.1073/pnas.1218464110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patel D, Nan Y, Shen M, Ritthipichai K, Zhu X, Zhang YJ. 2010. Porcine reproductive and respiratory syndrome virus inhibits type I interferon signaling by blocking STAT1/STAT2 nuclear translocation. J Virol 84:11045–11055. doi: 10.1128/JVI.00655-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang R, Nan Y, Yu Y, Zhang YJ. 2013. Porcine reproductive and respiratory syndrome virus Nsp1β inhibits interferon-activated JAK/STAT signal transduction by inducing karyopherin-α1 degradation. J Virol 87:5219–5228. doi: 10.1128/JVI.02643-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang R, Nan Y, Yu Y, Yang Z, Zhang YJ. 2013. Variable interference with interferon signal transduction by different strains of porcine reproductive and respiratory syndrome virus. Vet Microbiol 166:493–503. doi: 10.1016/j.vetmic.2013.07.022. [DOI] [PubMed] [Google Scholar]
- 34.Yang L, Wang R, Ma Z, Xiao Y, Nan Y, Wang Y, Lin S, Zhang YJ. 2017. Porcine reproductive and respiratory syndrome virus antagonizes JAK/STAT3 signaling via nsp5, which induces STAT3 degradation. J Virol 91:e02087-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Q, Huang C, Yang Q, Gao L, Liu HC, Tang J, Feng WH. 2016. MicroRNA-30c modulates type I IFN responses to facilitate porcine reproductive and respiratory syndrome virus infection by targeting JAK1. J Immunol 196:2272–2282. doi: 10.4049/jimmunol.1502006. [DOI] [PubMed] [Google Scholar]
- 36.Yang L, Wang R, Yang S, Ma Z, Lin S, Nan Y, Li Q, Tang Q, Zhang YJ. 2018. Karyopherin α6 is required for the replication of porcine reproductive and respiratory syndrome virus and Zika virus. J Virol 92:e00072-18. doi: 10.1128/JVI.00072-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nedialkova DD, Ulferts R, van den Born E, Lauber C, Gorbalenya AE, Ziebuhr J, Snijder EJ. 2009. Biochemical characterization of arterivirus nonstructural protein 11 reveals the nidovirus-wide conservation of a replicative endoribonuclease. J Virol 83:5671–5682. doi: 10.1128/JVI.00261-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun Y, Li D, Giri S, Prasanth SG, Yoo D. 2014. Differential host cell gene expression and regulation of cell cycle progression by nonstructural protein 11 of porcine reproductive and respiratory syndrome virus. Biomed Res Int 2014:430508. doi: 10.1155/2014/430508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Posthuma CC, Nedialkova DD, Zevenhoven-Dobbe JC, Blokhuis JH, Gorbalenya AE, Snijder EJ. 2006. Site-directed mutagenesis of the nidovirus replicative endoribonuclease NendoU exerts pleiotropic effects on the arterivirus life cycle. J Virol 80:1653–1661. doi: 10.1128/JVI.80.4.1653-1661.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi Y, Li Y, Lei Y, Ye G, Shen Z, Sun L, Luo R, Wang D, Fu ZF, Xiao S, Peng G. 2016. A dimerization-dependent mechanism drives the endoribonuclease function of porcine reproductive and respiratory syndrome virus nsp11. J Virol 90:4579–4592. doi: 10.1128/JVI.03065-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laneve P, Altieri F, Fiori ME, Scaloni A, Bozzoni I, Caffarelli E. 2003. Purification, cloning, and characterization of XendoU, a novel endoribonuclease involved in processing of intron-encoded small nucleolar RNAs in Xenopus laevis. J Biol Chem 278:13026–13032. doi: 10.1074/jbc.M211937200. [DOI] [PubMed] [Google Scholar]
- 42.Blaszczyk K, Nowicka H, Kostyrko K, Antonczyk A, Wesoly J, Bluyssen HA. 2016. The unique role of STAT2 in constitutive and IFN-induced transcription and antiviral responses. Cytokine Growth Factor Rev 29:71–81. doi: 10.1016/j.cytogfr.2016.02.010. [DOI] [PubMed] [Google Scholar]
- 43.Zhang M, Li X, Deng Z, Chen Z, Liu Y, Gao Y, Wu W, Chen Z. 2017. Structural biology of the arterivirus nsp11 endoribonucleases. J Virol 91:e01309-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shi X, Wang L, Li X, Zhang G, Guo J, Zhao D, Chai S, Deng R. 2011. Endoribonuclease activities of porcine reproductive and respiratory syndrome virus nsp11 was essential for nsp11 to inhibit IFN-beta induction. Mol Immunol 48:1568–1572. doi: 10.1016/j.molimm.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ramana CV, Chatterjee-Kishore M, Nguyen H, Stark GR. 2000. Complex roles of Stat1 in regulating gene expression. Oncogene 19:2619–2627. doi: 10.1038/sj.onc.1203525. [DOI] [PubMed] [Google Scholar]
- 46.Hofer MJ, Li W, Manders P, Terry R, Lim SL, King NJ, Campbell IL. 2012. Mice deficient in STAT1 but not STAT2 or IRF9 develop a lethal CD4+ T-cell-mediated disease following infection with lymphocytic choriomeningitis virus. J Virol 86:6932–6946. doi: 10.1128/JVI.07147-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun Y, Ke H, Han M, Chen N, Fang W, Yoo D. 2016. Nonstructural protein 11 of porcine reproductive and respiratory syndrome virus suppresses both MAVS and RIG-I expression as one of the mechanisms to antagonize type I interferon production. PLoS One 11:e0168314. doi: 10.1371/journal.pone.0168314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qureshi SA, Leung S, Kerr IM, Stark GR, Darnell JE Jr.. 1996. Function of Stat2 protein in transcriptional activation by alpha interferon. Mol Cell Biol 16:288–293. doi: 10.1128/MCB.16.1.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li X, Leung S, Kerr IM, Stark GR. 1997. Functional subdomains of STAT2 required for preassociation with the alpha interferon receptor and for signaling. Mol Cell Biol 17:2048–2056. doi: 10.1128/mcb.17.4.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martinez-Moczygemba M, Gutch MJ, French DL, Reich NC. 1997. Distinct STAT structure promotes interaction of STAT2 with the p48 subunit of the interferon-alpha-stimulated transcription factor ISGF3. J Biol Chem 272:20070–20076. doi: 10.1074/jbc.272.32.20070. [DOI] [PubMed] [Google Scholar]
- 51.Brierley MM, Fish EN. 2005. Stats: multifaceted regulators of transcription. J Interferon Cytokine Res 25:733–744. doi: 10.1089/jir.2005.25.733. [DOI] [PubMed] [Google Scholar]
- 52.Melen K, Kinnunen L, Julkunen I. 2001. Arginine/lysine-rich structural element is involved in interferon-induced nuclear import of STATs. J Biol Chem 276:16447–16455. doi: 10.1074/jbc.M008821200. [DOI] [PubMed] [Google Scholar]
- 53.Gupta S, Yan H, Wong LH, Ralph S, Krolewski J, Schindler C. 1996. The SH2 domains of Stat1 and Stat2 mediate multiple interactions in the transduction of IFN-alpha signals. EMBO J 15:1075–1084. doi: 10.1002/j.1460-2075.1996.tb00445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wojciak JM, Martinez-Yamout MA, Dyson HJ, Wright PE. 2009. Structural basis for recruitment of CBP/p300 coactivators by STAT1 and STAT2 transactivation domains. EMBO J 28:948–958. doi: 10.1038/emboj.2009.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Banninger G, Reich NC. 2004. STAT2 nuclear trafficking. J Biol Chem 279:39199–39206. doi: 10.1074/jbc.M400815200. [DOI] [PubMed] [Google Scholar]
- 56.Dong J, Xu S, Wang J, Luo R, Wang D, Xiao S, Fang L, Chen H, Jiang Y. 2015. Porcine reproductive and respiratory syndrome virus 3C protease cleaves the mitochondrial antiviral signaling complex to antagonize IFN-β expression. J Gen Virol 96:3049–3058. doi: 10.1099/jgv.0.000257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reference deleted.
- 58.Elliott J, Lynch OT, Suessmuth Y, Qian P, Boyd CR, Burrows JF, Buick R, Stevenson NJ, Touzelet O, Gadina M, Power UF, Johnston JA. 2007. Respiratory syncytial virus NS1 protein degrades STAT2 by using the Elongin-Cullin E3 ligase. J Virol 81:3428–3436. doi: 10.1128/JVI.02303-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nair S, Bist P, Dikshit N, Krishnan MN. 2016. Global functional profiling of human ubiquitome identifies E3 ubiquitin ligase DCST1 as a novel negative regulator of type I interferon signaling. Sci Rep 6:36179. doi: 10.1038/srep36179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Patel D, Opriessnig T, Stein DA, Halbur PG, Meng XJ, Iversen PL, Zhang YJ. 2008. Peptide-conjugated morpholino oligomers inhibit porcine reproductive and respiratory syndrome virus replication. Antiviral Res 77:95–107. doi: 10.1016/j.antiviral.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Meng XJ, Paul PS, Halbur PG. 1994. Molecular cloning and nucleotide sequencing of the 3′-terminal genomic RNA of the porcine reproductive and respiratory syndrome virus. J Gen Virol 75:1795–1801. doi: 10.1099/0022-1317-75-7-1795. [DOI] [PubMed] [Google Scholar]
- 62.Yuan S, Nelsen CJ, Murtaugh MP, Schmitt BJ, Faaberg KS. 1999. Recombination between North American strains of porcine reproductive and respiratory syndrome virus. Virus Res 61:87–98. doi: 10.1016/S0168-1702(99)00029-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Conzelmann KK, Visser N, Van Woensel P, Thiel HJ. 1993. Molecular characterization of porcine reproductive and respiratory syndrome virus, a member of the arterivirus group. Virology 193:329–339. doi: 10.1006/viro.1993.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang YJ, Stein DA, Fan SM, Wang KY, Kroeker AD, Meng XJ, Iversen PL, Matson DO. 2006. Suppression of porcine reproductive and respiratory syndrome virus replication by morpholino antisense oligomers. Vet Microbiol 117:117–129. doi: 10.1016/j.vetmic.2006.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Parisien JP, Lau JF, Rodriguez JJ, Ulane CM, Horvath CM. 2002. Selective STAT protein degradation induced by paramyxoviruses requires both STAT1 and STAT2 but is independent of alpha/beta interferon signal transduction. J Virol 76:4190–4198. doi: 10.1128/jvi.76.9.4190-4198.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lim KL, Chew KC, Tan JM, Wang C, Chung KK, Zhang Y, Tanaka Y, Smith W, Engelender S, Ross CA, Dawson VL, Dawson TM. 2005. Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation. J Neurosci 25:2002–2009. doi: 10.1523/JNEUROSCI.4474-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ma Z, Yu Y, Xiao Y, Opriessnig T, Wang R, Yang L, Nan Y, Samal SK, Halbur PG, Zhang YJ. 2017. The middle half genome of interferon-inducing porcine reproductive and respiratory syndrome virus strain A2MC2 is essential for interferon induction. J Gen Virol 98:1720–1729. doi: 10.1099/jgv.0.000819. [DOI] [PubMed] [Google Scholar]
- 68.Ni YY, Huang YW, Cao D, Opriessnig T, Meng XJ. 2011. Establishment of a DNA-launched infectious clone for a highly pneumovirulent strain of type 2 porcine reproductive and respiratory syndrome virus: identification and in vitro and in vivo characterization of a large spontaneous deletion in the nsp2 region. Virus Res 160:264–273. doi: 10.1016/j.virusres.2011.06.027. [DOI] [PubMed] [Google Scholar]
- 69.Zhang YJ, Wang KY, Stein DA, Patel D, Watkins R, Moulton HM, Iversen PL, Matson DO. 2007. Inhibition of replication and transcription activator and latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus by morpholino oligomers. Antiviral Res 73:12–23. doi: 10.1016/j.antiviral.2006.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li Y, Treffers EE, Napthine S, Tas A, Zhu L, Sun Z, Bell S, Mark BL, van Veelen PA, van Hemert MJ, Firth AE, Brierley I, Snijder EJ, Fang Y. 2014. Transactivation of programmed ribosomal frameshifting by a viral protein. Proc Natl Acad Sci U S A 111:E2172–E2181. doi: 10.1073/pnas.1321930111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guo R, Katz BB, Tomich JM, Gallagher T, Fang Y. 2016. Porcine reproductive and respiratory syndrome virus utilizes nanotubes for intercellular spread. J Virol 90:5163–5175. doi: 10.1128/JVI.00036-16. [DOI] [PMC free article] [PubMed] [Google Scholar]