

Antiretroviral therapy with multiple drugs in combination can efficiently suppress HIV replication and dramatically reduce the morbidity and mortality associated with AIDS-related illness; however, antiretroviral therapy cannot eradiate the HIV reservoirs, and lifelong treatment is required, which often results in cumulative toxicities, drug resistance, and a multitude of complications, thus necessitating the development of sterilizing-cure or functional-cure strategies. Here, we report that genetically anchoring the short-peptide fusion inhibitor 2P23 to the cell membrane can fully prevent infections from divergent HIV-1, HIV-2, and SIV isolates as well as a panel of enfuvirtide-resistant mutants. Membrane-bound 2P23 also effectively blocks HIV-1 Env-mediated cell-cell fusion and cell-associated virion-mediated cell-cell transmission, renders CD4+ T cells nonpermissive to infection, and confers a robust survival advantage over unmodified cells. Thus, our studies verify a powerful strategy to generate resistant cells for gene therapy of both the HIV-1 and HIV-2 infections.
KEYWORDS: HIV-1, HIV-2, gene therapy, fusion inhibitor, glycosylphosphatidylinositol
ABSTRACT
Emerging studies demonstrate that the antiviral activity of viral fusion inhibitor peptides can be dramatically improved when being chemically or genetically anchored to the cell membrane, where viral entry occurs. We previously reported that the short-peptide fusion inhibitor 2P23 and its lipid derivative possess highly potent antiviral activities against human immunodeficiency virus type 1 (HIV-1), HIV-2, and simian immunodeficiency virus (SIV). To develop a sterilizing or functional-cure strategy, here we genetically linked 2P23 and two control peptides (HIV-1 fusion inhibitor C34 and hepatitis B virus [HBV] entry inhibitor 4B10) with a glycosylphosphatidylinositol (GPI) attachment signal. As expected, GPI-anchored inhibitors were efficiently expressed on the plasma membrane of transduced TZM-bl cells and primarily directed to the lipid raft site without interfering with the expression of CD4, CCR5, and CXCR4. GPI-anchored 2P23 (GPI-2P23) completely protected TZM-bl cells from infections of divergent HIV-1, HIV-2, and SIV isolates as well as a panel of enfuvirtide (T20)-resistant mutants. GPI-2P23 also rendered the cells resistant to viral envelope-mediated cell-cell fusion and cell-associated virion-mediated cell-cell transmission. Moreover, GPI-2P23-modified human CD4+ T cells (CEMss-CCR5) fully blocked both R5- and X4-tropic HIV-1 isolates and displayed a robust survival advantage over unmodified cells during HIV-1 infection. In contrast, it was found that GPI-anchored C34 was much less effective in inhibiting HIV-2, SIV, and T20-resistant HIV-1 mutants. Therefore, our studies have demonstrated that genetically anchoring a short-peptide fusion inhibitor to the target cell membrane is a viable strategy for gene therapy of both HIV-1 and HIV-2 infections.
IMPORTANCE Antiretroviral therapy with multiple drugs in combination can efficiently suppress HIV replication and dramatically reduce the morbidity and mortality associated with AIDS-related illness; however, antiretroviral therapy cannot eradiate the HIV reservoirs, and lifelong treatment is required, which often results in cumulative toxicities, drug resistance, and a multitude of complications, thus necessitating the development of sterilizing-cure or functional-cure strategies. Here, we report that genetically anchoring the short-peptide fusion inhibitor 2P23 to the cell membrane can fully prevent infections from divergent HIV-1, HIV-2, and SIV isolates as well as a panel of enfuvirtide-resistant mutants. Membrane-bound 2P23 also effectively blocks HIV-1 Env-mediated cell-cell fusion and cell-associated virion-mediated cell-cell transmission, renders CD4+ T cells nonpermissive to infection, and confers a robust survival advantage over unmodified cells. Thus, our studies verify a powerful strategy to generate resistant cells for gene therapy of both the HIV-1 and HIV-2 infections.
INTRODUCTION
More than 36 million people worldwide are currently living with human immunodeficiency virus (HIV), including about 1 million to 2 million HIV-2 infections (www.unaids.org). Despite tremendous efforts, the development of an effective preventive vaccine remains a daunting challenge. However, highly active antiretroviral therapy (HAART) with multiple drugs in combination can suppress HIV replication to below the limit of clinical detection, which dramatically reduces the morbidity and mortality associated with AIDS-related illness and the risk of viral transmission to contacts (treatment as prevention). But HAART cannot eradiate the HIV reservoirs, viral rebound within weeks of treatment interruption, while lifelong treatment often results in cumulative toxicities, drug resistance, and a multitude of complications (1, 2). Therefore, alternative therapies that can achieve a sterilizing or functional cure are urgently needed.
Gene therapy holds considerable promise for functional cure of HIV infection, and in this context, genetically engineered HIV-resistant cells represent a powerful strategy which has been proven by the cure of the “Berlin patient” and the “London patient”; these two patients were transplanted with allogeneic hematopoietic stem cells (HSCs) harboring a naturally occurring CCR5Δ32 mutation (3, 4). However, such a procedure is not easy to duplicate and thus is not practical for the treatment of a larger population; instead, disruption of CCR5 by gene editing has been extensively explored (5–7). While CCR5 editing confers resistance to CCR5-tropic (R5) viruses, this strategy would be ineffective for CXCR4-tropic (X4) and dual-tropic (R5X4) viruses. A shift of coreceptor usage to an X4-tropic virus was observed in a patient who received donated bone marrow carrying the CCR5Δ32 mutation (8). It was also found that individuals with the CCR5Δ32 mutation have increased susceptibility to infections from West Nile virus, tickborne encephalitis virus, and influenza virus (9–11), and the mutation in the homozygous state is associated with human death (12). Therefore, a safe and effective genetic approach that can render target cells resistant to both R5 and X4 viruses would be highly appreciated.
Entry of HIV into target cells is mediated by two envelope (Env) glycoprotein subunits: the receptor binding gp120 subunit and the fusogenic gp41 subunit. Refolding of the N-terminal heptad repeat (NHR) and the C-terminal heptad repeat (CHR) of gp41 into a six-helix bundle (6-HB) drives the viral and cellular membranes in close apposition, leading to a fusion reaction (13, 14). Peptides derived from the NHR and CHR sequences can competitively block viral 6-HB formation and inhibit Env-mediated membrane fusion and viral entry (15). The CHR-derived peptide drug T20 (enfuvirtide) was clinically approved in 2003 as the first viral membrane fusion inhibitor, which is used in combination therapy for HIV-1 infection (16, 17). In a pilot study (18), T20 was genetically engineered for cell surface expression through the membrane-spanning domain (MSD) of human low-affinity nerve growth factor receptor (LNGFR), leading to 100-fold increased anti-HIV activity independent of coreceptor usage. To develop a more efficient fusion inhibitor-based gene therapeutic strategy, the same authors applied the CHR peptide C46 (Fig. 1) to generate a membrane-anchoring fusion inhibitor, maC46, via the MSD of human tCD34 (19–23). In a nonhuman primate model, maC46-modified CD4+ T cells showed positive selection following simian-human immunodeficiency virus (SHIV) challenge, accounting for >90% of the total CD4+ T cell population in peripheral blood, the gastrointestinal tract, and lymph nodes (24). It was also found that maC46 macaques maintained high frequencies of SHIV-specific, gene-modified CD4+ T cells and enhanced cytotoxic T lymphocyte function and antibody responses (24). A phase I clinical trial showed that infusion of maC46-modified autologous T cells into HIV-infected patients with advanced disease and HAART failure was well tolerated, with a significant increase of CD4 counts (25). Moreover, maC46 has been explored for combination gene therapy with a short hairpin RNA (shRNA) or a high-affinity P2-CCL5 intrakine to CCR5 (6, 26–30). Compared to T20, the CHR-derived peptide C34 has more potent anti-HIV activity, and thus, it has been widely used as a template to develop new HIV-1 fusion inhibitors (31). C34 was also applied to create resistant cells by genetically fusing it with the amino terminus of CCR5 and CXCR4 or with a glycosylphosphatidylinositol (GPI) attachment signal (32, 33). In humanized mice, T cells expressing C34-CXCR4 were highly resistant to HIV-1 infection and exhibited a selective survival advantage (32).
FIG 1.

Diagram of HIV fusion protein gp41 and design strategy for membrane-anchored fusion inhibitors. (A) Functional domains of gp41 and fusion inhibitor peptides. The sequences of the M-T hook structure, pocket-binding domain (PBD), and tryptophan-rich motif (TRM) in the CHR or CHR-derived fusion inhibitor peptides are marked in green, red, and purple, respectively. PEG8 indicates a flexible linker of 8-unit polyethylene glycol; C16 in parentheses indicates palmitic acid. FP, fusion peptide; NHR, N-terminal heptad repeat; CHR, C-terminal heptad repeat; TM, transmembrane domain; CT, cytoplasmic tail; T20RS, T20-resistant site; DP, deep-pocket site. (B) Illustration of membrane-anchored fusion inhibitors. T20 is fused with the membrane-spanning domain (MSD) of low-affinity nerve growth factor receptor (LNGFR), C46 is fused with the MSD of human tCD34, C34 is fused with the coreceptor CXCR4 or attached to glycosylphosphatidylinositol (GPI), and 2P23 is attached to GPI. (C) Lentiviral vector expressing a GPI-anchored fusion inhibitor and its mechanism of action. The encoding sequence of a fusion inhibitor peptide was genetically linked with the sequences encoding the IgG3 hinge region, a His tag, and the GPI attachment signal of DAF. When 2P23 is expressed on the cell surface via a GPI anchor, it binds to the NHR target during the prehairpin state of gp41 and blocks 6-HB formation. LTR, long terminal repeat. RRE, Rev response element; cPPT, central polypurine track; WPRE, woodchuck hepatitits virus posttranscription regulatory element.
We previously developed short-peptide HIV fusion inhibitors that primarily target a conserved deep NHR pocket rather than the T20-resistant site (34–36). Of them, a helical peptide termed 2P23 exhibited highly potent activities in inhibiting divergent HIV-1, HIV-2, simian immunodeficiency virus (SIV), and T20-resistant mutants (34). By linking a lipid tail to the C terminus of 2P23, a lipopeptide derivative termed LP-19 with dramatically increased in vitro and in vivo anti-HIV activities and stability was generated (37). It is considered that lipopeptide-based fusion inhibitors can bind preferentially to the cell membranes where fusion occurs, thus elevating the local concentrations of the inhibitors (37–41). In this study, we focused on developing a 2P23-based gene therapeutic strategy by genetically linking it with the GPI attachment signal of decay-accelerating factor (DAF). As controls, C34 peptide and a hepatitis B virus (HBV) entry inhibitor peptide (4B10) were also engineered for cell surface expression. Our results demonstrate that genetically anchoring a short-peptide fusion inhibitor to the target cell membrane is a viable strategy for gene therapy of both HIV-1 and HIV-2 infections.
RESULTS
Expression of antiviral peptides in the lipid raft of the plasma membrane through a GPI anchor.
To generate GPI-anchored antiviral peptides, the sequence encoding 2P23, C34, or 4B10 was genetically linked with sequences encoding the IgG3 hinge region, a His tag, and the GPI attachment signal of DAF. Three fusion genes, designated 2P23/Hinge/His/DAF, C34/Hinge/His/DAF, and 4B10/Hinge/His/DAF, were respectively inserted into a self-inactivating lentiviral vector, pRRLsin-18.PPT.hPGK.WPRE (Fig. 1C). The recombinant viruses were then packaged and applied to transduce target cells. To determine whether fusion genes were expressed on the cell surface through a GPI anchor, the transduced TZM-bl cells were treated with or without phosphatidylinositol-specific phospholipase C (PI-PLC) and then stained with an anti-His tag antibody, followed by fluorescence-activated cell sorter (FACS) analysis. As shown in Fig. 2A, three transgenes were highly expressed on the surface of the transduced cells, and their expression was substantially reduced after PI-PLC treatment, verifying that each peptide inhibitor was tethered to the cell surface through a GPI anchor. Here, we refer to the three transgenes as GPI-2P23, GPI-C34, and GPI-4B10, respectively.
FIG 2.
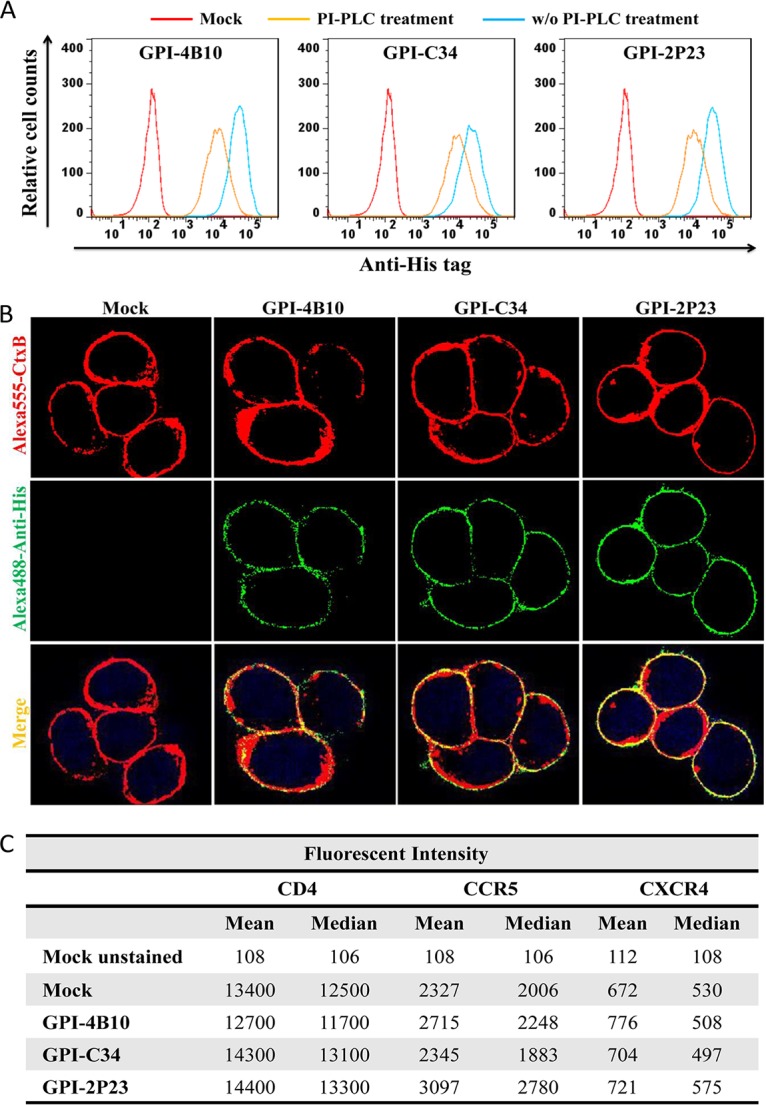
Expression of GPI-anchored peptides in transduced TZM-bl cells and their effects on CD4, CCR5, and CXCR4. (A) FACS analysis of cell surface expression of GPI-anchored peptides in transduced TZM-bl cells with or without PI-PLC treatment detected by an anti-His tag antibody. (B) Confocal analysis of GPI-anchored peptides in transduced TZM-bl cells. Alexa555-CtxB, cells stained with the Alexa 555-conjugated cholera toxin B subunit; Alexa488-Anti-His, cells stained with mouse anti-His tag antibody followed by Alexa 488-conjugated goat anti-mouse IgG antibody. (C) Expression levels of CD4, CCR5, and CXCR4 on the surface of TZM-bl cells transduced with GPI-anchored peptides as judged by the fluorescence intensity.
In essence, the GPI anchor is a posttranslation modification, and many GPI-anchored proteins are directed to the lipid raft sites, the specialized dynamic microdomains of the plasma membrane that serve as gateways for HIV entry and budding (42, 43). To determine if GPI-anchored peptides were located in the lipid rafts, mock-, GPI-2P23-, GPI-C34-, or GPI-4B10-transduced TZM-bl cells were costained with (i) mouse anti-His tag antibody, followed by Alexa 488-conjugated goat anti-mouse IgG antibody; (ii) Alexa 555-conjugated cholera toxin subunit B (CtxB); and (iii) 4′,6-diamidino-2-phenylindole (DAPI). As visualized in Fig. 2B, CtxB bound to the lipid raft marker GM1 efficiently; GPI-2P23, GPI-C34, and GPI-4B10 were colocalized with GM1 on the cell surface, hence suggesting that they were located in the lipid rafts of the plasma membrane.
It is also critical to know whether peptide anchoring affected the expression of the HIV receptor CD4 and the coreceptors CCR5 and CXCR4 on the cell surface. We thus stained mock-, GPI-2P23-, GPI-C34-, or GPI-4B10-transduced TZM-bl cells with phycoerythrin (PE)-conjugated anti-human CD4, CCR5, or CXCR4 antibody, followed by FACS analysis. As judged by the fluorescence intensity (Fig. 2C), transduction of GPI-anchored peptides in TZM-bl cells had virtually no effect on the expression levels of the receptor or coreceptors compared to those in the parental TZM-bl cells. Additionally, we did not observe that the expression of the transgenes impacted the viability and growth of the transduced cells.
GPI-2P23-modified TZM-bl cells are highly resistant to divergent HIV-1, HIV-2, and SIV isolates.
We next focused on characterizing whether 2P23 maintained its potent and broad antiviral activity when attached to the cell membrane via a GPI anchor. First, we compared the antiviral activities of GPI-2P23, GPI-C34, and GPI-4B10 in transduced TZM-bl cells with a panel of six representative viruses. As shown in Fig. 3, mock- and GPI-4B10-transduced TZM-bl cells were susceptible to infections from all the tested viruses, including two HIV-1 (X4-tropic NL4-3 and R5-tropic JRFL), two HIV-2 (R5X4-tropic ROD and R5-tropic ST), and two SIV (R5-tropic mac239 and smmPBj) isolates. In contrast, GPI-2P23-transduced TZM-bl cells were highly resistant to all the HIV-1, HIV-2, and SIV isolates; however, GPI-C34-transduced TZM-bl cells resisted HIV-1 but not the HIV-2 and SIV isolates. Serving as a virus control, none of the transduced cells opposed infection by vesicular stomatitis virus (VSV).
FIG 3.

Inhibitory activity of GPI-anchored peptides in transduced TZM-bl cells against HIV-1, HIV-2, and SIV isolates. The inhibition of GPI-2P23, GPI-C34, and GPI-4B10 on two HIV-1 pseudoviruses (NL4-3 and JRCSF), two replicative HIV-2 isolates (ROD and ST), two SIV pseudoviruses (mac239 and smmPBj), and VSV-G was determined. The transduced cells were sorted so as to express the cognate GPI-anchored peptide in close to 100% of the cells. Error bars represent the means ± standard errors of the means (SEM) from three independent experiments with triplicate samples. Mock, parental TZM-bl cells; GPI-2P23, GPI-2P23-transduced TZM-bl cells; GPI-C34, GPI-C34-transduced TZM-bl cells; GPI-4B10, GPI-4B10-transduced TZM-bl cells.
To validate the potency and breadth of GPI-2P23-modified cells, a panel of eight replication-competent HIV-1 isolates was further used to assess the antiviral activity of GPI-2P23 in TZM-bl cells. As shown in Table 1, GPI-2P23 and GPI-C34, but not GPI-4B10, efficiently protected transduced cells from infections by all replication-competent HIV-1 isolates. Moreover, a “global panel” of 12 HIV-1 pseudoviruses that represent diverse subtypes from the global AIDS epidemic was prepared and applied to verify the antiviral activity of GPI-2P23 in TZM-bl cells by a single-cycle infection assay. Consistently, GPI-2P23 and GPI-C34 rendered the transduced cells fully resistant to the pseudoviruses.
TABLE 1.
Inhibitory activity of GPI-2P23 against divergent HIV-1 and T20-resistant mutantsa
| HIV-1 isolate | Subtype | Tropism | Mean % infection ± SEM |
||
|---|---|---|---|---|---|
| GPI-4B10 | GPI-C34 | GPI-2P23 | |||
| Replication-competent viruses | |||||
| JR-CSF | B | CCR5 | 92 ± 3 | 0 | 0 |
| THRO.c/2626 | B | CCR5 | 91 ± 3 | 0 | 0 |
| CH077.t/2627 | B | CCR5 | 83 ± 3 | 0 | 0 |
| NL4-3 | B | CXCR4 | 97 ± 3 | 0 | 0 |
| LAI.2 | B | CXCR4 | 84 ± 3 | 0 | 0 |
| SG3.1 | B | CXCR4 | 92 ± 3 | 0 | 0 |
| 89.6 | B | R5X4 | 91 ± 3 | 0 | 0 |
| Mj4 | C | CCR5 | 94 ± 3 | 0 | 0 |
| “Global panel” pseudovirus | |||||
| 398-F1_F6_20 | A | CCR5 | 91 ± 3 | 0 | 0 |
| TRO.11 | B | CCR5 | 89 ± 3 | 0 | 0 |
| X2278_C2_B6 | B | CCR5 | 98 ± 3 | 0 | 0 |
| CE1176_A3 | C | CCR5 | 87 ± 3 | 0 | 0 |
| CE703010217_B6 | C | CCR5 | 86 ± 2 | 0 | 0 |
| HIV_25710-2.43 | C | CCR5 | 84 ± 2 | 0 | 0 |
| X1632-S2-B10 | G | CCR5 | 99 ± 4 | 0 | 0 |
| 246_F3_C10_2 | A/C | CCR5 | 91 ± 4 | 0 | 0 |
| CNE8 | A/E | CCR5 | 89 ± 3 | 0 | 0 |
| CNE55 | A/E | CCR5 | 91 ± 2 | 0 | 0 |
| CH119.10 | B/C | CCR5 | 97 ± 5 | 0 | 0 |
| BJOX002000.03 | B/C | CCR5 | 99 ± 4 | 0 | 0 |
| T20-resistant isolates | |||||
| NL4-3WT | B | CXCR4 | 98 ± 3 | 0 | 0 |
| NL4-3I37T | B | CXCR4 | 96 ± 2 | 0 | 0 |
| NL4-3V38A | B | CXCR4 | 93 ± 2 | 0 | 0 |
| NL4-3V38M | B | CXCR4 | 97 ± 2 | 0 | 0 |
| NL4-3Q40H | B | CXCR4 | 97 ± 2 | 0 | 0 |
| NL4-3N43K | B | CXCR4 | 98 ± 3 | 0 | 0 |
| NL4-3D36S/V38M | B | CXCR4 | 98 ± 3 | 0 | 0 |
| NL4-3I37T/N43K | B | CXCR4 | 101 ± 5 | 1 | 0 |
| NL4-3V38A/N42T | B | CXCR4 | 103 ± 3 | 1 | 0 |
The assay was performed in triplicate and repeated 3 times. The data for all 3 assays are expressed as means ± standard errors of means. NL4-3WT, wild-type NL4-3.
GPI-2P23-modified TZM-bl cells are fully resistant to T20-resistant HIV-1 mutants.
T20 remains the only membrane fusion inhibitor available for the treatment of viral infection, but it easily induces drug resistance both in vitro and in vivo, with the resistant mutations largely mapped to amino acids 36 to 45 in the NHR target site of gp41 (44). Our previous studies demonstrated that 2P23 sustained its potent activity in inhibiting a panel of T20-resistant mutants (34). Here, we tested whether TZM-bl cells expressing GPI-2P23 were resistant to infections with the panel of T20-resistant pseudoviruses by a single-cycle infection assay. As shown in Table 1, TZM-bl cells carrying GPI-4B10 were still susceptible to infections by diverse T20-resitant viruses; in contrast, the cells with GPI-2P23 and GPI-C34 were not permissive to the infections. Collectively, these results demonstrate that membrane-anchored 2P23 can efficiently protect target cells from infections by HIV-1, HIV-2, SIV, and T20-resistant mutants.
GPI-2P23 efficiently blocks HIV-1 Env-mediated cell-cell fusion.
We previously adapted a dual-split-protein (DSP)-based cell-cell fusion assay to evaluate the inhibitory activities of various HIV fusion inhibitors (45), in which 293FT cells expressing CCR5/CXCR4/DSP8–11 were used as a target (referred to as 293FTTarget). To characterize the inhibitory activities of the membrane-anchored inhibitors against HIV Env-mediated cell-cell fusion, here we transduced 293FTTarget cells with lentiviral vectors encoding GPI-2P23, GPI-C34, or GPI-4B10. First, the cell surface expression of the transgenes as well as their potential effects on the expression of CD4, CCR5, and CXCR4 were analyzed by FACS analysis. Similar to that in transduced TZM-bl cells, three transgenes were efficiently expressed on the surface of 293FTTarget cells and had no appreciable effect on the expression levels of the receptor and coreceptors (Fig. 4A and B). Next, the inhibitory activities of three GPI-anchored peptides on Env-mediated cell-cell fusion were determined by the DSP-based fusion assay. As shown in Table 2, GPI-2P23 could efficiently block cell-cell fusion mediated by divergent subtypes of primary HIV-1 Envs and the panel of T20-resistant mutant Envs. In contrast, GPI-C34 was effective against HIV-1 Envs, but its inhibitory activity against the T20-resistant mutants was obviously reduced, especially against the two mutant viruses carrying the I37T/N43K or V38A/N42T double mutation. As expected, GPI-4B10 could not significantly affect Env’s fusion capabilities.
FIG 4.

Expression of GPI-anchored peptides in transduced 293FT target cells expressing CCR5/CXCR4/DSP8–11 and their effects on CD4, CCR5, and CXCR4. (A) FACS analysis of the expression of GPI-anchored peptides on the surface of transduced 293FT cells that express CCR5/CXCR4/DSP8–11. (B) Expression levels of CD4, CCR5, and CXCR4 on the surface of transduced cells as judged by the fluorescence intensity.
TABLE 2.
Inhibitory activity of GPI-2P23 against HIV-1 Env-mediated cell-cell fusiona
| HIV-1 Env | Subtype | Tropism | Mean % cell-cell fusion ± SEM |
||
|---|---|---|---|---|---|
| GPI-4B10 | GPI-C34 | GPI-2P23 | |||
| Primary Envs | |||||
| 92RW020 | A | CCR5 | 112 ± 5 | 0 | 0 |
| JRFL | B | CCR5 | 107 ± 4 | 0 | 0 |
| R3A | B | R5/X4 | 99 ± 2 | 0 | 0 |
| CNE11 | B′ | R5/X4 | 92 ± 8 | 0 | 0 |
| CAP210.2.00.E8 | C | CCR5 | 100 ± 5 | 0 | 0 |
| CNE8 | A/E | CCR5 | 89 ± 6 | 0 | 0 |
| CH70.1 | B/C | R5/X4 | 102 ± 1 | 0 | 0 |
| Envs of T20-resistant mutants | |||||
| NL4-3WT | B | CXCR4 | 92 ± 3 | 0 | 0 |
| NL4-3I37T | B | CXCR4 | 98 ± 3 | 5 ± 1 | 0 |
| NL4-3V38A | B | CXCR4 | 99 ± 3 | 5 ± 1 | 0 |
| NL4-3V38M | B | CXCR4 | 93 ± 3 | 4 ± 1 | 0 |
| NL4-3Q40H | B | CXCR4 | 89 ± 3 | 8 ± 1 | 0 |
| NL4-3N43K | B | CXCR4 | 92 ± 2 | 3 ± 1 | 0 |
| NL4-3D36S/V38M | B | CXCR4 | 101 ± 6 | 0 | 0 |
| NL4-3I37T/N43K | B | CXCR4 | 99 ± 2 | 26 ± 3 | 0 |
| NL4-3V38A/N42T | B | CXCR4 | 98 ± 3 | 16 ± 3 | 0 |
The assay was performed in triplicate and repeated 3 times. The data for all 3 assays are expressed as means ± standard errors of means.
GPI-2P23 efficiently blocks cell-cell HIV-1 transmission.
Previous studies reported that CCR5-using HIV-infected cells cocultured with TZM-bl target cells can result in rapid and efficient infection by the virus through a cell-cell pathway, which can be monitored by induction of the reporter luciferase (46, 47). Here, we adopted a similar experimental protocol to determine whether GPI-2P23 in transduced TZM-bl cells blocked HIV-1 transmission through cell-cell contact. First, three replication-competent R5-tropic HIV-1 isolates, including two subtype B transmitted/founder viruses (RHPA.c/2635 and THRO.c/2626) and one subtype C virus (MJ4), were selected to infect CEMss-CCR5 cells as a donor, based on results showing that the infectivity of cell-free viruses in TZM-bl cells is dramatically reduced in the absence of a polycationic DEAE-dextran supplement (Fig. 5A), whereas the cell-cell transmission of the viruses was independent of DEAE-dextran (Fig. 5B). We accordingly assessed the inhibitory activities of GPI-anchored peptides against cell-cell HIV-1 transmission using the CEMss-CCR5HIV+/TZM-bl system without the addition of DEAE-dextran. As shown in Fig. 5C, both GPI-2P23 and GPI-C34 efficiently blocked the transmission of the viruses MJ4, RHPA.c/2635, and THRO.c/2626 from the donor CEMss-CCR5 cells to the transduced TZM-bl target cells, while GPI-4B10 could not block the viruses as anticipated.
FIG 5.

Inhibitory activity of GPI-anchored peptides during cell-cell transmission of replication-competent HIV-1 isolates. (A) Replicative infections of the R5-tropic HIV-1 isolates MJ4, THRO.c/2626, and RHPA.c/2635 in TZM-bl cells depend on the presence of DEAE-dextran as cell-free viruses. RLU, relative luciferase units; T/F, transmitted/founder. (B) Infections by cell-associated MJ4, THRO.c/2626, and RHPA.c/2635 viruses in TZM-bl cells are independent of DEAE-dextran during cell-cell transmission. (C) Inhibition of GPI-anchored peptides during cell-cell HIV-1 transmission. TZM-bl cells expressing GPI-anchored peptides were used as a target and cocultured for 36 h with donor CEMss-CCR5 cells that were preinfected with one of the three R5-tropic viruses. Percent infection of TZM-bl cells was monitored by quantifying the production of the reporter luciferase. Error bars represent the means ± SEM from 3 independent experiments with triplicate samples.
We also evaluated the inhibitory activities of GPI-anchored peptides in transduced TZM-bl cells against the cell-cell transmission of HIV-1 pseudoviruses that restrict our assessment to single-round transmission events. To this end, a panel of six R5-tropic pseudoviruses with different subtypes (398-F1_F6_20, X2278_C2_B6, RHPA4259.7, HIV_25710-2.43, CNE8, and CH119.10) was selected. Similar to that with the replicative viruses, the infectivity of cell-free pseudoviruses in TZM-bl cells was markedly reduced in the absence of DEAE-dextran (Fig. 6A), whereas cell-cell virus transmission between pseudovirus-infected HEK293T cells (donor) and TZM-bl cells (target) occurred irrespective of DEAE-dextran (Fig. 6B). When GPI-2P23 and GPI-C34 were transduced into TZM-bl cells, they efficiently blocked all the pseudoviruses from spreading between the donor cells and target cells (Fig. 6C). GPI-4B10 had no significant activities in inhibiting the panel of pseudoviruses, verifying the antiviral specificities of two GPI-anchored HIV fusion inhibitor peptides (2P23 and C34).
FIG 6.

Inhibitory activity of GPI-anchored peptides during cell-cell transmission of HIV-1 pseudoviruses. (A) Single-cycle infections of TZM-bl cells by a panel of cell-free HIV-1 pseudoviruses are dependent on DEAE-dextran. (B) Single-cycle infections of TZM-bl cells by the cell-associated HIV-1 pseudoviruses are independent of DEAE-dextran. (C) Inhibition of GPI-anchored peptides during cell-cell transmission mediated by the HIV-1 pseudoviruses. Transduced TZM-bl cells were used as a target and cocultured for 36 h with donor HEK293T cells that were preinfected by pseudoviruses, and percent infection of TZM-bl cells was monitored by quantifying the production of the reporter luciferase. Error bars represent the means ± SEM from three independent experiments with triplicate samples.
GPI-2P23-transduced human CD4+ T cells fully resist HIV infection.
Having demonstrated that GPI-2P23 and GPI-C34 in transduced TZM-bl cells blocked both cell-free and cell-associated HIV-1 with exceptional potency and breadth, we were intrigued to know whether GPI-2P23 and GPI-C34 could protect human CD4+ T cells from HIV-1 infection. To facilitate monitoring of positive transduced cells, we constructed fusion genes encoding three GPI-anchored peptide inhibitors, which were genetically linked with a green fluorescent protein (GFP)-encoding sequence via an internal 2A protein splicing signal in the lentiviral transfer vector pRRLsin-18.PPT.EF1α.WPRE (Fig. 7A). The recombinant viruses were then produced to transduce human CEMss-CCR5 cells. As anticipated, three transgenes (GPI-2P23/GFP, GPI-C34/GFP, and GPI-4B10/GFP) were well expressed on the cell surface of CEMss-CCR5 cells as detected by GFP and anti-His antibody (Fig. 7B). Similarly, no significant difference was observed for the expression of CD4, CCR5, and CXCR4 in transduced cells (Fig. 7C).
FIG 7.

Expression of GPI-anchored peptides in transduced CD4+ T cells and their effects on CD4, CCR5, and CXCR4. (A) Schematic view of the lentiviral vector expressing a GPI-anchored fusion inhibitor peptide linked to GFP-encoding sequences via a 2A signal. (B) Expression of GPI-anchored peptides along with GFP in transduced CEMss-CCR5 cells analyzed by FACS analysis. (C) Expression of CD4, CCR5, and CXCR4 on the surface of transduced CEMss-CCR5 cells as judged by the fluorescence intensity.
The transduced CEMss-CCR5 cells were then infected with the X4-tropic virus NL4-3 or the R5-tropic virus RHPA.c/2635 and cultured in complete Dulbecco’s modified Eagle’s medium (DMEM) for 11 and 17 days, respectively. As monitored for intracellular HIV-1 P24-Gag and GFP expression by flow cytometry, 0.55%, 24.6%, and 80.5% P24-Gag- and GFP-double-positive cells were observed in GPI-4B10/GFP-transduced CEMss-CCR5 cells after NL4-3 inoculation for 5, 8, and 11 days, respectively (Fig. 8, left); in sharp contrast, only <0.3% P24-Gag- and GFP-double-positive cells were seen in CEMss-CCR5 cells transduced with GPI-2P23/GFP (Fig. 8, right) or GPI-C34/GFP (middle) during the 11-day observation. Results for RHPA.c/2635 infection are shown in Fig. 9. Similarly, GPI-4B10/GFP-transduced CEMss-CCR5 cells were effectively infected over time, with the percentages of P24-Gag- and GFP-double-positive cells being 1.1%, 3.56%, 28.5%, and 64.9% after viral inoculation for 8, 11, 14, and 17 days, respectively (Fig. 9, left); however, the cells transduced with GPI-2P23/GFP (right) or GPI-C34/GFP (middle) were highly resistant to infection by RHPA.c/2635, having <0.3% P24-Gag- and GFP-double-positive cells over time.
FIG 8.

Inhibitory activity of GPI-anchored peptides in transduced human T cells against an X4-tropic HIV-1 isolate. CEMss-CCR5 cells transduced with GPI-2P23/GFP, GPI-C34/GFP, or GPI-4B10/GFP were infected with 1,000 TCID50 of the X4-tropic NL4-3 isolate and monitored by intracellular HIV-1 P24 Gag and GFP expression by flow cytometry. Data from a representative experiment of three independent experiments are shown.
FIG 9.

Inhibitory activity of GPI-anchored peptides in transduced human T cells against an R5-tropic HIV-1 isolate. CEMss-CCR5 cells transduced with GPI-2P23/GFP, GPI-C34/GFP, or GPI-4B10/GFP were infected with 1,000 TCID50 of the R5-tropic transmitted/founder virus RHPA.c/2635 and monitored by intracellular HIV-1 P24 Gag and GFP expression by flow cytometry. Data from a representative experiment of three independent experiments are shown.
Selective survival and expansion of GPI-2P23-transduced CEMss-CCR5 cells during HIV-1 infection.
As a potential gene therapy approach, it is very important to know whether CD4+ T cells expressing GPI-anchored 2P23 or C34 can confer a selective survival and expansion advantage. Thus, we mixed approximately 15 to 20% GPI-2P23/GFP- or GPI-C34/GFP-expressing CEMss-CCR5 cells with untransduced cells prior to HIV-1 infection. As shown in Fig. 10 and 11, following viral infection, the proportion of GFP-positive cells (GFP+) cells gradually increased over time, and by day 22, >99% of cells in the NL4-3-infected culture and >95% in the RHPA.c/2635-infected culture expressed GPI-2P23/GFP or GPI-C34/GFP. In the absence of HIV-1 infection, the percentage of GFP+ cells in the mixed populations was relatively stable (data not shown). These data indicate that both GPI-2P23 and GPI-C34 can confer an effective selective survival advantage to CEMss-CCR5 cells following HIV-1 infection. Here, it was noticed that obvious subsets of GFP+ GPI-C34 and GPI-2P23 populations were p24 positive (p24+) on days 11 and 15 after NL4-3 challenge (Fig. 10A) and on days 14 and 17 after RHPA.c/2635 challenge (Fig. 11A), which were never seen in purified CEMss-CCR5 cells bearing GPI-C34 or GPI-2P23 (Fig. 8 and 9). We speculated that GPI-C34- and GPI-2P23-expressing cells might trap HIV-1 virions at the cell surface during the peak time of viral replication that occurred in unmodified cells.
FIG 10.

Selective survival and expansion of CEMss-CCR5 cells expressing GPI-anchored fusion inhibitors during X4-tropic HIV-1 infection. (A) CEMss-CCR5 T cells were transduced with GPI-2P23/GFP or GPI-C34/GFP and mixed with untransduced cells at a ratio of approximately 20% GFP-positive cells. The mixed population was inoculated with 1,000 TCID50 of the X4-tropic virus NL4-3, and the proportion of transgene-expressing cells was monitored over time by flow cytometry. (B) Survival curve of transduced CEMss-CCR5 T cells. Data from a representative experiment of three independent experiments are shown.
FIG 11.

Selective survival and expansion of CEMss-CCR5 cells expressing GPI-anchored fusion inhibitors during R5-tropic HIV-1 infection. (A) CEMss-CCR5 T cells were transduced with GPI-2P23/GFP or GPI-C34/GFP and mixed with untransduced cells at a ratio of approximately 15% GFP-positive cells. The mixed population was inoculated with 1,000 TCID50 of the X5-tropic virus RHPA.c/2635, and the proportions of transgene-expressing cells was monitored over time by flow cytometry. (B) Survival curve of transduced CEMss-CCR5 T cells. Data from a representative experiment of three independent experiments are shown.
DISCUSSION
The major obstacle to curing HIV/AIDS is latently infected CD4+ T cells that are quiescent and insensitive to antiretroviral drugs in HIV-1-infected patients (48–51). Upon withdrawal of antiretroviral therapy, dormant viruses transforming into an active state are able to rapidly replicate. A variety of gene therapy strategies have been explored for cure or functional cure of HIV-1 infection, including RNA-based and protein-based approaches or their combinations (6, 7, 52, 53). In an advanced stage, a group of approaches is currently being investigated in clinical settings (6). Gene therapy establishing a subset of HIV-resistant target cells has shown promising effectiveness and has the potential to mimic the successful cases known as the “Berlin patient” and the “London patient” (3–5). Notably, the viral fusion inhibitor peptide C46 has been comprehensively characterized for HIV-1 gene therapy in a membrane-anchoring format, which aims to generate resistant cells capable of blocking the gateway of virus entry (19–30, 54–58). The fusion inhibitor peptide C34 has also been applied in a membrane-bound approach (32, 33), and CXCR4-conjugated C34 is currently being evaluated in a clinical trial (6).
In the past decade, we have dedicated our efforts to developing HIV fusion inhibitors. We highlight several structural features critical for gp41-dependent fusion that have been identified, including salt bridges (59, 60), the “QIWNNMT” motif (61), the M-T hook structure (62, 63), the pocket-2 conformation (64), and the Asn145 motif (65); a group of novel fusion inhibitor peptides was thereby designed, including sifuvirtide (66), CP32M (44), MT-SC22EK (36), HP23 (35), 2P23 (34), LP-11 (39), LP-19 (37), LP-46 (67), LP-50 and LP-51 (68), LP-52 (69), LP-80 (70), and LP-83 (45). Our series of data provide important information for understanding the mechanisms of HIV Env-mediated cell fusion and its inhibition, and especially, the results have testified that a lipopeptide fusion inhibitor can interact with the cell membrane to enhance its antiviral activity by acting locally at the viral entry site. In the present study, we focused on developing a fusion inhibitor-based gene therapy strategy that can render modified target cells with potent and broad HIV resistance. The rationale that we used to select a helical short peptide is based on the following: first, 2P23 was specifically designed to target the highly conserved deep NHR pocket rather than the T20-resistant site, and thus, it showed very potent antiviral activity against divergent HIV-1 and T20-resistant mutants (34); second, 2P23 was also highly active in binding to and inhibiting HIV-2 and SIV isolates (34); third, its membrane-anchoring lipopeptide derivative (LP-19) exhibited dramatically increased antiviral activity and high therapeutic efficacy in a nonhuman primate model (37); fourth, we have repeatedly failed to select HIV-1 mutants escaping the 2P23 and LP-19 inhibitors, highlighting their high genetic barriers to induction of resistance; and fifth, compared to other gp41-derived fusion inhibitor peptides, 2P23-based inhibitors might be less immunogenic and cytotoxic in vivo owing to an artificial, minimal amino acid sequence. We therefore constructed glycosylphosphatidylinositol (GPI)-anchored 2P23 along with C34 and 4B10 control peptides through a third-generation lentiviral vector and transduced them into different target cells for characterization. As shown, three transgenes were efficiently expressed on the cell surface and targeted to the lipid rafts of the plasma membranes without interfering with the expression of the cell surface markers CD4, CCR5, and CXCR4 as well as the viability and growth of the transduced cells. Promisingly, GPI-2P23-transduced TZM-bl cells were highly resistant to infections by divergent HIV-1, HIV-2, SIV, and T20-resistant mutants; in contrast, GPI-C34-transduced TZM-bl cells were resistant to HIV-1 but not to the HIV-2 and SIV isolates. GPI-2P23 in transduced cells also effectively blocked both wild-type and T20-resistant Env-mediated cell-cell fusion, whereas GPI-C34 displayed a markedly reduced potency in inhibiting cell fusion by T20-resistant Env mutants. Moreover, we demonstrate that both GPI-2P23 and GPI-C34 could efficiently inhibit cell-cell HIV-1 transmission by R5- and X4-tropic HIV-1 isolates and that the modified human CD4+ T cells (CEMss-CCR5) were also nonpermissive to infections and had a robust survival advantage over unmodified cells following HIV-1 challenge. Taken together, the present data demonstrate that genetically expressing the short-peptide fusion inhibitor 2P23 on the target cell membrane is a more efficient strategy for gene therapy of both HIV-1 and HIV-2 infections. Considering that cell-cell fusion and cell-cell transmission might be dominant pathways of HIV spread in infected individuals and that such viruses may easily evade inhibition by many neutralizing antibodies (46, 47, 71), fusion inhibitor peptide-based genetic approaches should be highly appreciated for developing HIV-resistant cells.
Apart from the inhibitor peptide 2P23, our selection of GPI as a membrane-anchoring platform is also based on multifaceted considerations. Because many cell surface proteins are naturally expressed on the plasma membrane by covalently attaching a GPI anchor (72, 73), we think that a GPI scaffold might be a safer one than other membrane-anchored scaffolds, especially for linking a small peptide; the GPI attachment signal of decay-accelerating factor (DAF) efficiently anchors the antiviral peptides in the lipid rafts of the plasma membrane, which can result in a locally high peptide concentration to maximize antiviral activity (42, 43). By using the GPI scaffold, Zhou and coworkers successfully anchored HIV-1-neutralizing antibodies on the lipid rafts of the transduced target cells, conferring high-level resistance to HIV-1 infection (33, 74–77). It was found that GPI-anchored scFv X5 efficiently protected CD4+ T cells from HIV-1 infection and deletion in humanized mice (76). As mentioned above, maC46 used the membrane-spanning domain (MSD) of human tCD34 as an anchoring scaffold, while C34-CXCR4 used the coreceptor CXCR4 as an anchoring scaffold. There are concerns over the effect of the overexpression of these scaffolds on the normal functions of cellular proteins per se; in other words, the extent to which the expression of tCD34msd and CXCR4 would affect the natural function of the modified cells remains elusive.
Here, we emphasize another prominent inherent advantage of GPI-2P23. The first is its potential low immunogenicity when used in vivo. Previous studies found that when autologous CD4+ T cells expressing maC46 were infused into HIV-positive (HIV+) patients who had drug-resistant virus and HAART failure, no evidence of positive selection was observed; conversely, maC46-modified cells quickly declined within a week after infusion (25). Actually, maC46 inherits a full-length native CHR sequence of the viral gp41 protein, which could have induced immune responses to eliminate the maC46-expressing cells. In that study, it was also found that the HIV+ patients had preexisting antibodies to maC46, which might mediate antibody-dependent cell-mediated cytotoxicity (ADCC) or other immune responses responsible for the observed decline (6, 25). Different from the viral native sequence-derived T20, C34, and C46 peptides, 2P23 is composed of a largely artificial amino acid sequence (Fig. 1) and has low immunogenicity in mice and rabbits. Accordingly, preexisting antibodies specific for 2P23 in HIV+ individuals are not expected. Regardless, we hope to test the efficacy and immunogenicity of the GPI-2P23 strategy in humanized mouse and/or nonhuman primate models in our future studies.
Moreover, we also address the problem associated with HIV-2 infection. Currently, HIV-2 has infected about 1 million to 2 million people worldwide, mostly in West Africa, but the virus is increasingly spreading to other regions of the world through immigration, causing concerns over its potential epidemics (78). However, all available antiretroviral drugs are specifically developed against HIV-1 replication, and consequently, some clinical drugs have limited or no activity in inhibiting HIV-2, including all nonnucleoside reverse transcriptase inhibitors, some protease inhibitors, and the fusion inhibitor T20 (79–81). Similarly, emerging gene therapeutic strategies are primarily focused on HIV-1, and little attention has been paid to curing or functionally curing HIV-2 infection. In this study, we show that fusion inhibitor C34-based resistant cells were still permissive to HIV-2 and SIV isolates. Therefore, the newly developed GPI-2P23 strategy holds great potential as a genetic intervention approach for the treatment of HIV-2 infection.
MATERIALS AND METHODS
Cells and plasmids.
HEK293T cells were purchased from the American Type Culture Collection (ATCC) (Rockville, MD). TZM-bl cells, plasmids encoding the “global panel” HIV-1 Envs (subtypes A, B, C, G, A/C, A/E, and B/C), molecular clones of HIV-1 (NL4-3, SG3.1, LAI.2, JR-CSF, MJ4, 89.6, THRO.c/2626, CH077.t/2627, and RHPA.c/2635), and molecular clones of HIV-2 (ROD and ST) were obtained through the AIDS Reagent Program, Division of AIDS, NIAID, NIH. Two plasmids encoding SIV Env (mac239 and smmPBj) were kindly gifted by Jianqing Xu at the Shanghai Public Health Clinical Center, Fudan University, China. The plasmids encoding DSP1–7 and 293FT cells stably expressing CXCR4/CCR5/DSP8 − 11 were a kind gift from Zene Matsuda at the Institute of Medical Science of the University of Tokyo (Tokyo, Japan). The human CD4+ T cell line CEM-SS expressing CCR5 (CEMss-CCR5) was a kind gift from Paul Zhou at the Institute Pasteur of Shanghai, Chinese Academy of Sciences, China.
Generation of recombinant lentiviruses.
Fusion genes encoding the 2P23, C34, or 4B10 peptide; an IgG3 hinge; and a His tag, with a GPI attachment signal (C-terminal 34 amino acids of DAF) at the C terminus, were synthesized (Genewiz, China) and subsequently ligated between the BamHI and SalI sites of a third-generation lentiviral transfer vector, pRRLsin.PPT.hPGK.WPRE (hPGK promoter). Fusion genes encoding GFP and GPI-2P23, -C34, or -4B10 linked via an internal 2A peptide signal were synthesized (Genewiz, China) and subsequently ligated between the BamHI and SalI sites of a third-generation lentiviral transfer vector, pRRLsin-18.PPT.hEF1α.2A.GFP.WPRE (hEF1α promoter).
Recombinant lentiviruses expressing fusion genes were generated as described previously (75). Briefly, 1.5 × 107 HEK293T cells were seeded onto a P-150 dish in 25 ml complete DMEM. After culturing overnight, cells were cotransfected with 50 μg of the lentiviral transfer vector encoding a GPI-anchored peptide inhibitor, 18.75 μg of the packaging construct delta8.9 encoding Gag/Pol/Rev, and 7.5 μg of a plasmid encoding the vesicular stomatitis virus G (VSV-G) envelope, using a linear polyethyleneimine (PEI) transfection reagent. Cell culture supernatants were removed at 20 h posttransfection and replaced with fresh complete DMEM plus 10% fetal bovine serum (FBS). After 24 h, the supernatants were harvested and centrifuged at 4,000 rpm for 15 min. The harvested supernatants were filtered through a 0.45-mm filter and subsequently concentrated by ultracentrifugation. The lentiviral vector pellets were resuspended in RPMI 1640 with 25 mM HEPES and stored in aliquots in a freezer at −80°C. Lentivirus titers were determined on HEK293T cells by monitoring His tag or GFP expression by flow cytometry (FACSCanto II; Becton, Dickinson, Mountain View, CA), and the titers were expressed as transducing units (TU) per milliliter.
Generation of stable cell lines expressing GPI-anchored peptides.
To generate target cells stably expressing GPI-anchored peptides, TZM-bl, 293FT, or CEMss-CCR5 cells were seeded onto a 24-well plate (5 × 104 cells/well). On the next day, 2 × 106 TU of recombinant lentiviruses were added to the cells in the presence of 8 μg/ml of Polybrene (Sigma). After 24 h, the transduced cells were extensively washed and cultured in complete DMEM. TZM-bl and 293FT cells expressing the transgenes were sorted and collected using mouse anti-His tag antibody (Invitrogen Life Technologies) and phycoerythrin (PE)-conjugated goat anti-mouse IgG antibody (eBioscience), and CEMss-CCR5 cells expressing the transgenes were purified by GFP expression.
Flow cytometry analysis.
To detect the cell surface expression of GPI-anchored peptides, the transduced TZM-bl, 293FT, and CEMss-CCR5 cells were sequentially stained with a mouse anti-His tag antibody and a PE-conjugated goat anti-mouse IgG antibody or Alexa Fluor 647-conjugated goat anti-mouse IgG antibody (Invitrogen Life Technologies) for 60 min at 4°C. Next, the stained cells were washed twice with FACS buffer (phosphate-buffered saline [PBS] solution with 0.5% bovine serum albumin [BSA] and 2 mM EDTA) and resuspended in 0.2 ml of FACS buffer containing 4% formaldehyde. FACS analysis was performed with a FACSCanto II instrument (Becton, Dickinson, Mountain View, CA). To determine whether the expression of transgenes was truly targeted through a GPI anchor, the transduced cells were first incubated with 5 U/ml phosphatidylinositol-specific phospholipase C (PI-PLC) (Invitrogen Life Technologies) in 0.5 ml 1× PBS and rocked at 4°C for 30 min, followed by two washes to remove the remaining PI-PLC. The cells were then stained by antibodies and analyzed by FACS analysis as described above.
To analyze whether GPI-anchored peptides affected the expression of CD4, CCR5, and CXCR4, the transduced cells were stained with PE-conjugated anti-human CD4, CD195, or CD184 or allophycocyanin (APC)-conjugated anti-human CD4, CD195, or CD184 (BD Science) antibodies for 30 min at 4°C. The cells were then washed twice with FACS buffer and fixed with 4% formaldehyde, followed by FACS analysis.
Confocal analysis.
The transduced TZM-bl cells were seeded (8,000 cells/well) onto a 35-mm glass dish with a 14-mm bottom well (Cellvis) and incubated at 37°C in 5% CO2 for 2 days. Cells were washed twice with 300 μl PBS, fixed for 15 min with 4% formaldehyde in PBS containing 1% BSA, and blocked for 1 h with blocking buffer (5% goat serum in PBS plus 1% BSA). Next, the cells were sequentially stained with mouse anti-His tag antibody, Alexa 488-conjugated goat anti-mouse IgG antibody, and Alexa 555-conjugated cholera toxin subunit B (CtxB) (Invitrogen Life Technologies). After three washes with PBS, cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) in permeabilization buffer (blocking buffer plus 0.5% saponin) for 7 min. Images were captured with a laser confocal microscope (Leica Microsystems, Wetzlar, Germany).
Inhibition of GPI-anchored peptides on replication-competent HIV-1/2 isolates.
A panel of infectious HIV-1 (NL4-3, SG3.1, LAI.2, JRCSF, MJ4, 89.6, THRO.c/2626, CH077.t/2627, and RHPA.c/2635) and two HIV-2 (ROD and ST) isolates was generated by transfection of HEK293T cells (6 × 106 cells in a P-100 dish) with a plasmid (24 μg) of the molecular clones using a linear PEI transfection reagent. The virus-containing supernatants were harvested at 48 h posttransfection and stored in aliquots in a freezer at −80°C. Titers of the viruses were determined by a 50% tissue culture infectious dose (TCID50) assay in TZM-bl cells.
To determine the inhibitory activity of GPI-anchored peptides in transduced TZM-bl cells, 200 TCID50 of the viruses in a 50-μl volume were added to the cells (1 × 104/well) and incubated at 37°C with 5% CO2 for 2 days. The cells were then lysed with lysis buffer, and luciferase activity was measured by a BrightGlo luciferase assay with a luminescence counter (Promega). To determine the inhibitory activity of GPI-anchored peptides in transduced CEMss-CCR5 cells, 1,000 TCID50 of the viruses were added to the cells (1 × 106/well) at 37°C with 5% CO2 and incubated at 37°C with 5% CO2 overnight. The infected cells were extensively washed with Hanks’ balanced salt solution (HBSS) and then cultured in complete DMEM for observation over time. Intracellular HIV-1 P24 expression in the infected cells was detected using PE-conjugated anti-P24 Gag antibody (clone KC57; Beckman Coulter, Brea, CA) and analyzed along with GFP expression by FACS analysis.
Inhibition of GPI-anchored peptides on HIV-1 and SIV pseudoviruses.
A single-cycle infection assay was performed as described previously (34). The Env-pseudotyped viruses for the global panel of HIV-1 isolates, two SIV isolates (mac239 and smmPBj), and VSV-G were generated by cotransfecting HEK293T cells with a pSG3Δenv backbone plasmid and an Env-expressing plasmid using a linear PEI transfection reagent. The pseudovirus-containing supernatants were harvested at 48 h posttransfection and stored in aliquots in a freezer at −80°C. Titers of pseudotyped virions were determined by a TCID50 assay in TZM-bl cells. To determine the inhibitory activity of GPI-anchored peptides in transduced TZM-bl cells, 200 TCID50 of the viruses were added to the cells (1 × 104/well) and incubated for 48 h at 37°C. The cells were lysed for determining the luciferase activity as described above.
Inhibition of GPI-anchored peptides during Env-mediated cell-cell fusion.
A dual-split-protein (DSP)-based cell-cell fusion assay was performed as described previously (45). To determine the inhibitory activities of GPI-anchored peptides, 293FT target cells stably expressing CCR5/CXCR4/DSP8–11 (referred to as 293FTTarget) were first transduced with the lentiviral vector encoding GPI-2P23, GPI-C34, or GPI-4B10. Effector HEK293T cells were seeded on a 96-well plate (1.5 × 104 cells/well) and incubated at 37°C with 5% CO2 overnight, and the cells were then cotransfected with an Env-expressing plasmid and a DSP1–7-expressing plasmid and cultured at 37°C for 24 h. Mock-, GPI-2P23-, GPI-C34-, or GPI-4B10-transduced 293FTTarget cells were resuspended in prewarmed culture medium and then added to the EnduRen live-cell substrate (Promega), followed by incubation at 37°C for 30 min. Next, the target cells (2.5 × 104/well) were added to effector cells and spun down to maximize cell-cell contact. After coculturing for 6 h, the fusion activity between the target and effector cells was monitored by quantifying the production of the reporter luciferase as described above.
Inhibition of GPI-anchored peptides during cell-cell HIV-1 transmission.
A cell-cell HIV-1 transmission assay was performed according to protocols described previously (46, 47), based on the fact that infection by many R5-tropic viruses in TZM-bl cells depends on polycationic supplements as cell-free virions but not during cell-cell transmission. To assess cell-cell transmission and the inhibition thereof, the dependence of HIV-1 infection on DEAE-dextran was initially characterized for both replication-competent and pseudotyped viruses, and TZM-bl cells were transduced with mock, GPI-2P23, GPI-C34, or GPI-4B10 as a target. For the inhibitory activities of GPI-anchored peptides during the cell-cell transmission of replication-competent HIV-1 isolates, CEMss-CCR5 cells were infected with the viruses as a donor. To determine the inhibitory activities of GPI-anchored peptides during the single-cycle cell-cell transmission of HIV-1 Env-pseudotyped viruses, HEK293T cells (104/well) were seeded on a 96-well plate, cultured for 24 h, and then cotransfected with a pSG3Δenv backbone plasmid and an Env-expressing plasmid. After culturing for 12 h, the transfected cells were washed twice with prewarmed DMEM and added to the transduced TZM-bl cells (104/well) in the absence of DEAE-dextran. After coculturing for 36 h, infection of TZM-bl cells was monitored by quantifying the production of the reporter luciferase as described above.
ACKNOWLEDGMENTS
We thank Zene Matsuda at the Institute of Medical Science, University of Tokyo, for providing plasmids and cells for the DSP-based cell-cell fusion assay and Jianqing Xu at the Shanghai Public Health Clinical Center and Institutes of Biomedical Sciences of Fudan University for providing the plasmids encoding SIV Env.
This work was supported by grants from the CAMS Innovation Fund for Medical Sciences (2017-I2M-1-014), the National Science and Technology Major Project of China (2018ZX10301103), and the National Natural Science Foundation of China (81630061 and 81673484).
REFERENCES
- 1.Katlama C, Deeks SG, Autran B, Martinez-Picado J, van Lunzen J, Rouzioux C, Miller M, Vella S, Schmitz JE, Ahlers J, Richman DD, Sekaly RP. 2013. Barriers to a cure for HIV: new ways to target and eradicate HIV-1 reservoirs. Lancet 381:2109–2117. doi: 10.1016/S0140-6736(13)60104-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sengupta S, Siliciano RF. 2018. Targeting the latent reservoir for HIV-1. Immunity 48:872–895. doi: 10.1016/j.immuni.2018.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hutter G, Nowak D, Mossner M, Ganepola S, Mussig A, Allers K, Schneider T, Hofmann J, Kucherer C, Blau O, Blau IW, Hofmann WK, Thiel E. 2009. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med 360:692–698. doi: 10.1056/NEJMoa0802905. [DOI] [PubMed] [Google Scholar]
- 4.Gupta RK, Abdul-Jawad S, McCoy LE, Mok HP, Peppa D, Salgado M, Martinez-Picado J, Nijhuis M, Wensing AMJ, Lee H, Grant P, Nastouli E, Lambert J, Pace M, Salasc F, Monit C, Innes AJ, Muir L, Waters L, Frater J, Lever AML, Edwards SG, Gabriel IH, Olavarria E. 2019. HIV-1 remission following CCR5Delta32/Delta32 haematopoietic stem-cell transplantation. Nature 568:244–248. doi: 10.1038/s41586-019-1027-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tebas P, Stein D, Tang WW, Frank I, Wang SQ, Lee G, Spratt SK, Surosky RT, Giedlin MA, Nichol G, Holmes MC, Gregory PD, Ando DG, Kalos M, Collman RG, Binder-Scholl G, Plesa G, Hwang WT, Levine BL, June CH. 2014. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med 370:901–910. doi: 10.1056/NEJMoa1300662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Falkenhagen A, Joshi S. 2018. Genetic strategies for HIV treatment and prevention. Mol Ther Nucleic Acids 13:514–533. doi: 10.1016/j.omtn.2018.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Mendoza C, Barreiro P, Benitez L, Soriano V. 2015. Gene therapy for HIV infection. Expert Opin Biol Ther 15:319–327. doi: 10.1517/14712598.2015.967208. [DOI] [PubMed] [Google Scholar]
- 8.Kordelas L, Verheyen J, Beelen DW, Horn PA, Heinold A, Kaiser R, Trenschel R, Schadendorf D, Dittmer U, Esser S, Essen HIV AlloSCT Group. 2014. Shift of HIV tropism in stem-cell transplantation with CCR5 Delta32 mutation. N Engl J Med 371:880–882. doi: 10.1056/NEJMc1405805. [DOI] [PubMed] [Google Scholar]
- 9.Glass WG, McDermott DH, Lim JK, Lekhong S, Yu SF, Frank WA, Pape J, Cheshier RC, Murphy PM. 2006. CCR5 deficiency increases risk of symptomatic West Nile virus infection. J Exp Med 203:35–40. doi: 10.1084/jem.20051970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kindberg E, Mickiene A, Ax C, Akerlind B, Vene S, Lindquist L, Lundkvist A, Svensson L. 2008. A deletion in the chemokine receptor 5 (CCR5) gene is associated with tickborne encephalitis. J Infect Dis 197:266–269. doi: 10.1086/524709. [DOI] [PubMed] [Google Scholar]
- 11.Hayashi T, MacDonald LA, Takimoto T. 2015. Influenza A virus protein PA-X contributes to viral growth and suppression of the host antiviral and immune responses. J Virol 89:6442–6452. doi: 10.1128/JVI.00319-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wei X, Nielsen R. 2019. CCR5-32 is deleterious in the homozygous state in humans. Nat Med 25:909–910. doi: 10.1038/s41591-019-0459-6. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. 1997. Atomic structure of the ectodomain from HIV-1 gp41. Nature 387:426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 14.Chan DC, Fass D, Berger JM, Kim PS. 1997. Core structure of gp41 from the HIV envelope glycoprotein. Cell 89:263–273. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 15.Chan DC, Kim PS. 1998. HIV entry and its inhibition. Cell 93:681–684. doi: 10.1016/s0092-8674(00)81430-0. [DOI] [PubMed] [Google Scholar]
- 16.Lalezari JP, Henry K, O’Hearn M, Montaner JS, Piliero PJ, Trottier B, Walmsley S, Cohen C, Kuritzkes DR, Eron JJ Jr, Chung J, DeMasi R, Donatacci L, Drobnes C, Delehanty J, Salgo M, TORO 1 Study Group. 2003. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in North and South America. N Engl J Med 348:2175–2185. doi: 10.1056/NEJMoa035026. [DOI] [PubMed] [Google Scholar]
- 17.Kilby JM, Hopkins S, Venetta TM, DiMassimo B, Cloud GA, Lee JY, Alldredge L, Hunter E, Lambert D, Bolognesi D, Matthews T, Johnson MR, Nowak MA, Shaw GM, Saag MS. 1998. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat Med 4:1302–1307. doi: 10.1038/3293. [DOI] [PubMed] [Google Scholar]
- 18.Hildinger M, Dittmar MT, Schult-Dietrich P, Fehse B, Schnierle BS, Thaler S, Stiegler G, Welker R, von Laer D. 2001. Membrane-anchored peptide inhibits human immunodeficiency virus entry. J Virol 75:3038–3042. doi: 10.1128/JVI.75.6.3038-3042.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Egelhofer M, Brandenburg G, Martinius H, Schult-Dietrich P, Melikyan G, Kunert R, Baum C, Choi I, Alexandrov A, von Laer D. 2004. Inhibition of human immunodeficiency virus type 1 entry in cells expressing gp41-derived peptides. J Virol 78:568–575. doi: 10.1128/JVI.78.2.568-575.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Melikyan GB, Egelhofer M, von Laer D. 2006. Membrane-anchored inhibitory peptides capture human immunodeficiency virus type 1 gp41 conformations that engage the target membrane prior to fusion. J Virol 80:3249–3258. doi: 10.1128/JVI.80.7.3249-3258.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hermann FG, Martinius H, Egelhofer M, Giroglou T, Tonn T, Roth SD, Zahn R, Schult-Dietrich P, Alexandrov A, Dietrich U, Baum C, von Laer D. 2009. Protein scaffold and expression level determine antiviral activity of membrane-anchored antiviral peptides. Hum Gene Ther 20:325–336. doi: 10.1089/hum.2006.158. [DOI] [PubMed] [Google Scholar]
- 22.Perez EE, Riley JL, Carroll RG, von Laer D, June CH. 2005. Suppression of HIV-1 infection in primary CD4 T cells transduced with a self-inactivating lentiviral vector encoding a membrane expressed gp41-derived fusion inhibitor. Clin Immunol 115:26–32. doi: 10.1016/j.clim.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 23.Hermann FG, Egerer L, Brauer F, Gerum C, Schwalbe H, Dietrich U, von Laer D. 2009. Mutations in gp120 contribute to the resistance of human immunodeficiency virus type 1 to membrane-anchored C-peptide maC46. J Virol 83:4844–4853. doi: 10.1128/JVI.00666-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Younan PM, Polacino P, Kowalski JP, Peterson CW, Maurice NJ, Williams NP, Ho O, Trobridge GD, Von Laer D, Prlic M, Beard BC, DeRosa S, Hu SL, Kiem HP. 2013. Positive selection of mC46-expressing CD4+ T cells and maintenance of virus specific immunity in a primate AIDS model. Blood 122:179–187. doi: 10.1182/blood-2013-01-482224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lunzen JV, Glaunsinger T, Stahmer I, Baehr VV, Baum C, Schilz A, Kuehlcke K, Naundorf S, Martinius H, Hermann F, Giroglou T, Newrzela S, Muller I, Brauer F, Brandenburg G, Alexandrov A, von Laer D. 2007. Transfer of autologous gene-modified T cells in HIV-infected patients with advanced immunodeficiency and drug-resistant virus. Mol Ther 15:1024–1033. doi: 10.1038/mt.sj.6300124. [DOI] [PubMed] [Google Scholar]
- 26.Wolstein O, Boyd M, Millington M, Impey H, Boyer J, Howe A, Delebecque F, Cornetta K, Rothe M, Baum C, Nicolson T, Koldej R, Zhang J, Keech N, Camba Colon J, Breton L, Bartlett J, An DS, Chen IS, Burke B, Symonds GP. 2014. Preclinical safety and efficacy of an anti-HIV-1 lentiviral vector containing a short hairpin RNA to CCR5 and the C46 fusion inhibitor. Mol Ther Methods Clin Dev 1:11. doi: 10.1038/mtm.2013.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burke BP, Levin BR, Zhang J, Sahakyan A, Boyer J, Carroll MV, Colon JC, Keech N, Rezek V, Bristol G, Eggers E, Cortado R, Boyd MP, Impey H, Shimizu S, Lowe EL, Ringpis GE, Kim SG, Vatakis DN, Breton LR, Bartlett JS, Chen IS, Kitchen SG, An DS, Symonds GP. 2015. Engineering cellular resistance to HIV-1 infection in vivo using a dual therapeutic lentiviral vector. Mol Ther Nucleic Acids 4:e236. doi: 10.1038/mtna.2015.10. [DOI] [PubMed] [Google Scholar]
- 28.Symonds G, Bartlett JS, Kiem HP, Tsie M, Breton L. 2016. Cell-delivered entry inhibitors for HIV-1: CCR5 downregulation and blocking virus/membrane fusion in defending the host cell population. AIDS Patient Care STDS 30:545–550. doi: 10.1089/apc.2016.0245. [DOI] [PubMed] [Google Scholar]
- 29.Petit NY, Baillou C, Burlion A, Dorgham K, Levacher B, Amiel C, Schneider V, Lemoine FM, Gorochov G, Marodon G. 2016. Gene transfer of two entry inhibitors protects CD4(+) T cell from HIV-1 infection in humanized mice. Gene Ther 23:144–150. doi: 10.1038/gt.2015.101. [DOI] [PubMed] [Google Scholar]
- 30.Delville M, Touzot F, Couzin C, Hmitou I, Djerroudi L, Ouedrani A, Lefrère F, Tuchman-Durand C, Mollet C, Fabreguettes J-R, Ferry N, Laganier L, Magnani A, Magrin E, Jolaine V, Saez-Cirion A, Wolstein O, Symonds G, Frange P, Moins-Teisserenc H, Chaix-Baudier M-L, Toubert A, Larghero J, Parquet N, Brignier AC, Barré-Sinoussi F, Oksenhendler E, Cavazzana M. 2019. Safety of CD34(+) hematopoietic stem cells and CD4(+) T lymphocytes transduced with LVsh5/C46 in HIV-1 infected patients with high-risk lymphoma. Mol Ther Methods Clin Dev 13:303–309. doi: 10.1016/j.omtm.2019.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He Y. 2013. Synthesized peptide inhibitors of HIV-1 gp41-dependent membrane fusion. Curr Pharm Des 19:1800–1809. doi: 10.2174/1381612811319100004. [DOI] [PubMed] [Google Scholar]
- 32.Leslie GJ, Wang J, Richardson MW, Haggarty BS, Hua KL, Duong J, Secreto AJ, Jordon AP, Romano J, Kumar KE, DeClercq JJ, Gregory PD, June CH, Root MJ, Riley JL, Holmes MC, Hoxie JA. 2016. Potent and broad inhibition of HIV-1 by a peptide from the gp41 heptad repeat-2 domain conjugated to the CXCR4 amino terminus. PLoS Pathog 12:e1005983. doi: 10.1371/journal.ppat.1005983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu L, Wen M, Zhu Q, Kimata JT, Zhou P. 2016. Glycosyl phosphatidylinositol-anchored C34 peptide derived from human immunodeficiency virus type 1 Gp41 is a potent entry inhibitor. J Neuroimmune Pharmacol 11:601–610. doi: 10.1007/s11481-016-9681-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiong S, Borrego P, Ding X, Zhu Y, Martins A, Chong H, Taveira N, He Y. 2017. A helical short-peptide fusion inhibitor with highly potent activity against human immunodeficiency virus type 1 (HIV-1), HIV-2, and simian immunodeficiency virus. J Virol 91:e01839-16. doi: 10.1128/JVI.01839-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chong H, Qiu Z, Su Y, Yang L, He Y. 2015. Design of a highly potent HIV-1 fusion inhibitor targeting the gp41 pocket. AIDS 29:13–21. doi: 10.1097/QAD.0000000000000498. [DOI] [PubMed] [Google Scholar]
- 36.Chong H, Yao X, Qiu Z, Sun J, Zhang M, Waltersperger S, Wang M, Liu SL, Cui S, He Y. 2013. Short-peptide fusion inhibitors with high potency against wild-type and enfuvirtide-resistant HIV-1. FASEB J 27:1203–1213. doi: 10.1096/fj.12-222547. [DOI] [PubMed] [Google Scholar]
- 37.Chong H, Xue J, Xiong S, Cong Z, Ding X, Zhu Y, Liu Z, Chen T, Feng Y, He L, Guo Y, Wei Q, Zhou Y, Qin C, He Y. 2017. A lipopeptide HIV-1/2 fusion inhibitor with highly potent in vitro, ex vivo, and in vivo antiviral activity. J Virol 91:e00288-17. doi: 10.1128/JVI.00288-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ingallinella P, Bianchi E, Ladwa NA, Wang YJ, Hrin R, Veneziano M, Bonelli F, Ketas TJ, Moore JP, Miller MD, Pessi A. 2009. Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc Natl Acad Sci U S A 106:5801–5806. doi: 10.1073/pnas.0901007106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chong H, Wu X, Su Y, He Y. 2016. Development of potent and long-acting HIV-1 fusion inhibitors. AIDS 30:1187–1196. doi: 10.1097/QAD.0000000000001073. [DOI] [PubMed] [Google Scholar]
- 40.Wexler-Cohen Y, Shai Y. 2009. Membrane-anchored HIV-1 N-heptad repeat peptides are highly potent cell fusion inhibitors via an altered mode of action. PLoS Pathog 5:e1000509. doi: 10.1371/journal.ppat.1000509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wexler-Cohen Y, Ashkenazi A, Viard M, Blumenthal R, Shai Y. 2010. Virus-cell and cell-cell fusion mediated by the HIV-1 envelope glycoprotein is inhibited by short gp41 N-terminal membrane-anchored peptides lacking the critical pocket domain. FASEB J 24:4196–4202. doi: 10.1096/fj.09-151704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Waheed AA, Freed EO. 2010. The role of lipids in retrovirus replication. Viruses 2:1146–1180. doi: 10.3390/v2051146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leung K, Kim JO, Ganesh L, Kabat J, Schwartz O, Nabel GJ. 2008. HIV-1 assembly: viral glycoproteins segregate quantally to lipid rafts that associate individually with HIV-1 capsids and virions. Cell Host Microbe 3:285–292. doi: 10.1016/j.chom.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.He Y, Cheng J, Lu H, Li J, Hu J, Qi Z, Liu Z, Jiang S, Dai Q. 2008. Potent HIV fusion inhibitors against enfuvirtide-resistant HIV-1 strains. Proc Natl Acad Sci U S A 105:16332–16337. doi: 10.1073/pnas.0807335105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu Y, Chong H, Yu D, Guo Y, Zhou Y, He Y. 2019. Design and characterization of cholesterylated peptide HIV-1/2 fusion inhibitors with extremely potent and long-lasting antiviral activity. J Virol 93:e02312-18. doi: 10.1128/JVI.02312-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abela IA, Berlinger L, Schanz M, Reynell L, Gunthard HF, Rusert P, Trkola A. 2012. Cell-cell transmission enables HIV-1 to evade inhibition by potent CD4bs directed antibodies. PLoS Pathog 8:e1002634. doi: 10.1371/journal.ppat.1002634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gombos RB, Kolodkin-Gal D, Eslamizar L, Owuor JO, Mazzola E, Gonzalez AM, Korioth-Schmitz B, Gelman RS, Montefiori DC, Haynes BF, Schmitz JE. 2015. Inhibitory effect of individual or combinations of broadly neutralizing antibodies and antiviral reagents against cell-free and cell-to-cell HIV-1 transmission. J Virol 89:7813–7828. doi: 10.1128/JVI.00783-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JA, Baseler M, Lloyd AL, Nowak MA, Fauci AS. 1997. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci U S A 94:13193–13197. doi: 10.1073/pnas.94.24.13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davey RT Jr, Bhat N, Yoder C, Chun TW, Metcalf JA, Dewar R, Natarajan V, Lempicki RA, Adelsberger JW, Miller KD, Kovacs JA, Polis MA, Walker RE, Falloon J, Masur H, Gee D, Baseler M, Dimitrov DS, Fauci AS, Lane HC. 1999. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc Natl Acad Sci U S A 96:15109–15114. doi: 10.1073/pnas.96.26.15109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pierson T, McArthur J, Siliciano RF. 2000. Reservoirs for HIV-1: mechanisms for viral persistence in the presence of antiviral immune responses and antiretroviral therapy. Annu Rev Immunol 18:665–708. doi: 10.1146/annurev.immunol.18.1.665. [DOI] [PubMed] [Google Scholar]
- 51.Churchill MJ, Deeks SG, Margolis DM, Siliciano RF, Swanstrom R. 2016. HIV reservoirs: what, where and how to target them. Nat Rev Microbiol 14:55–60. doi: 10.1038/nrmicro.2015.5. [DOI] [PubMed] [Google Scholar]
- 52.Herrera-Carrillo E, Berkhout B. 2015. Potential mechanisms for cell-based gene therapy to treat HIV/AIDS. Expert Opin Ther Targets 19:245–263. doi: 10.1517/14728222.2014.980236. [DOI] [PubMed] [Google Scholar]
- 53.Herrera-Carrillo E, Berkhout B. 2015. Bone marrow gene therapy for HIV/AIDS. Viruses 7:3910–3936. doi: 10.3390/v7072804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zahn RC, Hermann FG, Kim EY, Rett MD, Wolinsky SM, Johnson RP, Villinger F, von Laer D, Schmitz JE. 2008. Efficient entry inhibition of human and nonhuman primate immunodeficiency virus by cell surface-expressed gp41-derived peptides. Gene Ther 15:1210–1222. doi: 10.1038/gt.2008.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kimpel J, Braun SE, Qiu G, Wong FE, Conolle M, Schmitz JE, Brendel C, Humeau LM, Dropulic B, Rossi JJ, Berger A, von Laer D, Johnson RP. 2010. Survival of the fittest: positive selection of CD4+ T cells expressing a membrane-bound fusion inhibitor following HIV-1 infection. PLoS One 5:e12357. doi: 10.1371/journal.pone.0012357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brauer F, Schmidt K, Zahn RC, Richter C, Radeke HH, Schmitz JE, von Laer D, Egerer L. 2013. A rationally engineered anti-HIV peptide fusion inhibitor with greatly reduced immunogenicity. Antimicrob Agents Chemother 57:679–688. doi: 10.1128/AAC.01152-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Savkovic B, Nichols J, Birkett D, Applegate T, Ledger S, Symonds G, Murray JM. 2014. A quantitative comparison of anti-HIV gene therapy delivered to hematopoietic stem cells versus CD4+ T cells. PLoS Comput Biol 10:e1003681. doi: 10.1371/journal.pcbi.1003681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sather BD, Romano Ibarra GS, Sommer K, Curinga G, Hale M, Khan IF, Singh S, Song Y, Gwiazda K, Sahni J, Jarjour J, Astrakhan A, Wagner TA, Scharenberg AM, Rawlings DJ. 2015. Efficient modification of CCR5 in primary human hematopoietic cells using a megaTAL nuclease and AAV donor template. Sci Transl Med 7:307ra156. doi: 10.1126/scitranslmed.aac5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.He Y, Liu S, Li J, Lu H, Qi Z, Liu Z, Debnath AK, Jiang S. 2008. Conserved salt bridge between the N- and C-terminal heptad repeat regions of the human immunodeficiency virus type 1 gp41 core structure is critical for virus entry and inhibition. J Virol 82:11129–11139. doi: 10.1128/JVI.01060-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.He Y, Liu S, Jing W, Lu H, Cai D, Chin DJ, Debnath AK, Kirchhoff F, Jiang S. 2007. Conserved residue Lys574 in the cavity of HIV-1 Gp41 coiled-coil domain is critical for six-helix bundle stability and virus entry. J Biol Chem 282:25631–25639. doi: 10.1074/jbc.M703781200. [DOI] [PubMed] [Google Scholar]
- 61.He Y, Cheng J, Li J, Qi Z, Lu H, Dong M, Jiang S, Dai Q. 2008. Identification of a critical motif for the human immunodeficiency virus type 1 (HIV-1) gp41 core structure: implications for designing novel anti-HIV fusion inhibitors. J Virol 82:6349–6358. doi: 10.1128/JVI.00319-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chong H, Yao X, Sun J, Qiu Z, Zhang M, Waltersperger S, Wang M, Cui S, He Y. 2012. The M-T hook structure is critical for design of HIV-1 fusion inhibitors. J Biol Chem 287:34558–34568. doi: 10.1074/jbc.M112.390393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chong H, Yao X, Qiu Z, Qin B, Han R, Waltersperger S, Wang M, Cui S, He Y. 2012. Discovery of critical residues for viral entry and inhibition through structural insight of HIV-1 fusion inhibitor CP621-652. J Biol Chem 287:20281–20289. doi: 10.1074/jbc.M112.354126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Qiu Z, Chong H, Yao X, Su Y, Cui S, He Y. 2015. Identification and characterization of a subpocket on the N-trimer of HIV-1 Gp41: implication for viral entry and drug target. AIDS 29:1015–1024. doi: 10.1097/QAD.0000000000000683. [DOI] [PubMed] [Google Scholar]
- 65.Geng X, Liu Z, Yu D, Qin B, Zhu Y, Cui S, Chong H, He Y. 2019. Conserved residue Asn-145 in the C-terminal heptad repeat region of HIV-1 gp41 is critical for viral fusion and regulates the antiviral activity of fusion inhibitors. Viruses 11:609. doi: 10.3390/v11070609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.He Y, Xiao Y, Song H, Liang Q, Ju D, Chen X, Lu H, Jing W, Jiang S, Zhang L. 2008. Design and evaluation of sifuvirtide, a novel HIV-1 fusion inhibitor. J Biol Chem 283:11126–11134. doi: 10.1074/jbc.M800200200. [DOI] [PubMed] [Google Scholar]
- 67.Zhu Y, Zhang X, Ding X, Chong H, Cui S, He J, Wang X, He Y. 2018. Exceptional potency and structural basis of a T1249-derived lipopeptide fusion inhibitor against HIV-1, HIV-2, and simian immunodeficiency virus. J Biol Chem 293:5323–5334. doi: 10.1074/jbc.RA118.001729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chong H, Xue J, Zhu Y, Cong Z, Chen T, Guo Y, Wei Q, Zhou Y, Qin C, He Y. 2018. Design of novel HIV-1/2 fusion inhibitors with high therapeutic efficacy in rhesus monkey models. J Virol 92:e00775-18. doi: 10.1128/JVI.00775-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chong H, Zhu Y, Yu D, He Y. 2018. Structural and functional characterization of membrane fusion inhibitors with extremely potent activity against human immunodeficiency virus type 1 (HIV-1), HIV-2, and simian immunodeficiency virus. J Virol 92:e01088-18. doi: 10.1128/JVI.01088-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chong H, Xue J, Zhu Y, Cong Z, Chen T, Wei Q, Qin C, He Y. 2019. Monotherapy with a low-dose lipopeptide HIV fusion inhibitor maintains long-term viral suppression in rhesus macaques. PLoS Pathog 15:e1007552. doi: 10.1371/journal.ppat.1007552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reh L, Magnus C, Schanz M, Weber J, Uhr T, Rusert P, Trkola A. 2015. Capacity of broadly neutralizing antibodies to inhibit HIV-1 cell-cell transmission is strain- and epitope-dependent. PLoS Pathog 11:e1004966. doi: 10.1371/journal.ppat.1004966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lu L, Gao J, Guo Z. 2015. Labeling cell surface GPIs and GPI-anchored proteins through metabolic engineering with artificial inositol derivatives. Angew Chem Int Ed Engl 54:9679–9682. doi: 10.1002/anie.201503814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Medof ME, Nagarajan S, Tykocinski ML. 1996. Cell-surface engineering with GPI-anchored proteins. FASEB J 10:574–586. doi: 10.1096/fasebj.10.5.8621057. [DOI] [PubMed] [Google Scholar]
- 74.Liu L, Wen M, Wang W, Wang S, Yang L, Liu Y, Qian M, Zhang L, Shao Y, Kimata JT, Zhou P. 2011. Potent and broad anti-HIV-1 activity exhibited by a glycosyl-phosphatidylinositol-anchored peptide derived from the CDR H3 of broadly neutralizing antibody PG16. J Virol 85:8467–8476. doi: 10.1128/JVI.00520-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu L, Wang W, Matz J, Ye C, Bracq L, Delon J, Kimata JT, Chen Z, Benichou S, Zhou P. 2016. The glycosylphosphatidylinositol-anchored variable region of llama heavy chain-only antibody JM4 efficiently blocks both cell-free and T cell-T cell transmission of human immunodeficiency virus type 1. J Virol 90:10642–10659. doi: 10.1128/JVI.01559-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ye C, Wang W, Cheng L, Li G, Wen M, Wang Q, Zhang Q, Li D, Zhou P, Su L. 2017. Glycosylphosphatidylinositol-anchored anti-HIV scFv efficiently protects CD4 T cells from HIV-1 infection and deletion in hu-PBL mice. J Virol 91:e01389-16. doi: 10.1128/JVI.01389-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wen M, Arora R, Wang H, Liu L, Kimata JT, Zhou P. 2010. GPI-anchored single chain Fv—an effective way to capture transiently-exposed neutralization epitopes on HIV-1 envelope spike. Retrovirology 7:79. doi: 10.1186/1742-4690-7-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Visseaux B, Damond F, Matheron S, Descamps D, Charpentier C. 2016. HIV-2 molecular epidemiology. Infect Genet Evol 46:233–240. doi: 10.1016/j.meegid.2016.08.010. [DOI] [PubMed] [Google Scholar]
- 79.Borrego P, Calado R, Marcelino JM, Bartolo I, Rocha C, Cavaco-Silva P, Doroana M, Antunes F, Maltez F, Caixas U, Barroso H, Taveira N. 2012. Baseline susceptibility of primary HIV-2 to entry inhibitors. Antivir Ther 17:565–570. doi: 10.3851/IMP1996. [DOI] [PubMed] [Google Scholar]
- 80.Ntemgwa ML, d’Aquin Toni T, Brenner BG, Camacho RJ, Wainberg MA. 2009. Antiretroviral drug resistance in human immunodeficiency virus type 2. Antimicrob Agents Chemother 53:3611–3619. doi: 10.1128/AAC.00154-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Witvrouw M, Pannecouque C, Switzer WM, Folks TM, De Clercq E, Heneine W. 2004. Susceptibility of HIV-2, SIV and SHIV to various anti-HIV-1 compounds: implications for treatment and postexposure prophylaxis. Antivir Ther 9:57–65. [PubMed] [Google Scholar]