

The paramyxovirus Hendra virus (HeV) causes severe respiratory illness and encephalitis in humans. To develop therapeutics for HeV and related viral infections, further studies are needed to understand the mechanisms underlying paramyxovirus fusion events. Knowledge gained in studies of the HeV fusion (F) protein may be applicable to a broad span of enveloped viruses. In this study, we demonstrate that disulfide bonds introduced between the HeV F transmembrane domains (TMDs) block fusion. Depending on the location of these disulfide bonds, HeV F can still fold properly and bind a prefusion conformation-specific antibody prior to cell disruption. These findings support our current model for HeV membrane fusion and expand our knowledge of the TMD and its role in HeV F stability and fusion promotion.
KEYWORDS: Hendra virus, fusion protein, membrane fusion, viral entry
ABSTRACT
Hendra virus (HeV) is a zoonotic paramyxovirus that utilizes a trimeric fusion (F) protein within its lipid bilayer to mediate membrane merger with a cell membrane for entry. Previous HeV F studies showed that transmembrane domain (TMD) interactions are important for stabilizing the prefusion conformation of the protein prior to triggering. Thus, the current model for HeV F fusion suggests that modulation of TMD interactions is critical for initiation and completion of conformational changes that drive membrane fusion. HeV F constructs (T483C/V484C, V484C/N485C, and N485C/P486C) were generated with double cysteine substitutions near the N-terminal region of the TMD to study the effect of altered flexibility in this region. Oligomeric analysis showed that the double cysteine substitutions successfully promoted intersubunit disulfide bond formation in HeV F. Subsequent fusion assays indicated that the introduction of disulfide bonds in the mutants prohibited fusion events. Further testing confirmed that T483C/V484C and V484C/N485C were expressed at the cell surface at levels that would allow for fusion. Attempts to restore fusion with a reducing agent were unsuccessful, suggesting that the introduced disulfide bonds were likely buried in the membrane. Conformational analysis showed that T483C/V484C and V484C/N485C were able to bind a prefusion conformation-specific antibody prior to cell disruption, indicating that the introduced disulfide bonds did not significantly affect protein folding. This study is the first to report that TMD dissociation is required for HeV F fusogenic activity and strengthens our model for HeV fusion.
IMPORTANCE The paramyxovirus Hendra virus (HeV) causes severe respiratory illness and encephalitis in humans. To develop therapeutics for HeV and related viral infections, further studies are needed to understand the mechanisms underlying paramyxovirus fusion events. Knowledge gained in studies of the HeV fusion (F) protein may be applicable to a broad span of enveloped viruses. In this study, we demonstrate that disulfide bonds introduced between the HeV F transmembrane domains (TMDs) block fusion. Depending on the location of these disulfide bonds, HeV F can still fold properly and bind a prefusion conformation-specific antibody prior to cell disruption. These findings support our current model for HeV membrane fusion and expand our knowledge of the TMD and its role in HeV F stability and fusion promotion.
INTRODUCTION
The Paramyxoviridae family consists of negative-sense single-stranded RNA viruses enclosed within lipid membranes. Hendra (HeV) and Nipah (NiV) viruses, members of the Henipavirus genus, are highly pathogenic zoonotic viruses within the Paramyxoviridae family (1). Due to the high mortality rates associated with HeV and NiV infections and the lack of a human vaccine or effective treatment, they have been designated biosafety level 4 pathogens (2). HeV and NiV were identified in Australia and Malaysia, respectively, in the 1990s following outbreaks of severe encephalitis and respiratory disease in humans (2–5). Further investigation revealed that fruit bats of the Pteropodidae family were the natural reservoir for the viruses, and transmission to other organisms, including pigs and horses, contributed to the zoonotic spread to humans (6–8). The potential for future outbreaks of henipavirus infections and for the emergence of similar zoonotic viruses warrants further research into the entry mechanisms of these pathogens.
Membrane fusion is an essential step in entry of enveloped viruses that relies on the coordination of specialized proteins at the viral membrane surface. HeV and NiV possess two surface glycoproteins: the attachment protein (G), which allows the virus to bind a target cell, and the fusion protein (F), which promotes merger of the viral membrane with the target membrane (9, 10). Both glycoproteins, F and G, are required for paramyxovirus membrane fusion, but it is still unclear how interactions between F and G and receptor binding promote triggering of F (11). The henipaviruses and other members of the Paramyxoviridae family use a trimeric class I F protein to drive membrane fusion (12–14). Before the F protein can participate in fusion events, the inactive precursor (F0) must be proteolytically cleaved within the host cell to form a fusion-active disulfide-linked heterodimer (F1+F2) (Fig. 1A). For HeV and NiV, the F protein traffics to the cell surface and is subsequently endocytosed to be cleaved by the protease cathepsin L before being recycled back to the surface (15–17). Following the cleavage event, the F protein is maintained at the surface in a metastable prefusion state until it is triggered to undergo the conformational changes needed to promote membrane fusion. These conformational changes from the prefusion to postfusion form involve an essentially irreversible rearrangement of the F protein ectodomain that results in formation of a stable six-helix bundle (Fig. 1B to F).
FIG 1.

HeV F protein schematic and fusion model. (A) Diagram of the fusion-active, disulfide (S-S)-linked F protein with the HeV F TMD sequence below. Domain structure includes the fusion peptide (FP), heptad repeat A (HRA), HRB, TMD, and the cytoplasmic tail (CT). In the fusion model, the TMDs of the metastable prefusion F interact as a trimer (B). Then triggering of F leads to dissociation of the TMDs and the HRB domains (C). Changes in TMD interactions promote extension of the HRA domains and insertion of the FP into the target membrane (D). (E and F) Further refolding of F leads to formation of the postfusion six-helix bundle conformation.
Studies of several viral fusion proteins have shown that the transmembrane domain (TMD) is critical for driving fusion events (18–28). For HeV, previous work has shown that TMD interactions within the F protein trimer help preserve the metastable prefusion conformation and play a role in fusion promotion (29–31). More specifically, these findings suggest that HeV F TMD interactions are needed to stabilize the heptad repeat B (HRB) domains that form the stalk of the protein prior to triggering fusion (Fig. 1B). Thus, the current model for HeV F fusion events suggests that dissociation of TMD trimeric interactions is required to initiate conformational changes that destabilize interactions between the HRB domains and eventually promote formation of the postfusion six-helix bundle to drive membrane fusion (29).
Based on this model, we hypothesized that fusion could be blocked by introducing disulfide bonds to covalently link the TMDs of HeV F. Studies using substituted cysteine residues to generate disulfide bonds have previously been conducted to examine conformational changes in paramyxovirus surface glycoproteins. For measles virus (MeV), residues in the attachment protein stalk were replaced with cysteines to promote disulfide bond formation to identify four conserved residues required for folding into a fusion-conducive conformation (32). In addition, studies of the attachment proteins from canine distemper virus and MeV showed that the disulfide bonds introduced in the central region of the stalk blocked fusion, but fusion activity was restored under reducing conditions (33). For the Newcastle disease virus attachment protein, disulfide bonds were generated across the dimer interface in the globular domain to show changes in receptor binding and fusion promotion (34).
The introduction of disulfide bonds has also been an important tool for studying paramyxovirus F proteins. Single cysteine substitutions were made in the membrane-proximal region of the HRB domain of MeV F, generating disulfide-linked dimers, to study formation of the postfusion six-helix bundle. Results showed that the constructs were able to promote efficient viral entry even though two HRB domains were locked together by a disulfide bond (35). Other studies have used double cysteine substitutions to further restrict conformational changes in the paramyxovirus F protein. For MeV F, double cysteine substitutions were made to introduce disulfide bonds to link the globular head and stalk domains of different monomers within the F trimer blocked fusion activity, and fusion was partially restored by subsequently reducing the disulfide bridges. These findings suggested that fusion activity requires reversible interactions between the stalk and head domains of the F protein (36). For parainfluenza virus 5 (PIV5) F, double cysteine substitutions were used to introduce disulfide bonds in the membrane-proximal external region (MPER) which is N-terminal to the TMD. Results from this study showed that dissociation of the MPER within the PIV5 F trimer is necessary to promote the conformational changes that drive fusion events (37). Additionally, work on HeV F used double cysteine substitutions to block conformational changes in the ectodomain to analyze effects on fusion. This study showed that the disulfide bonds introduced in the ectodomain could inhibit fusion by stabilizing HeV F in the prefusion conformation (38).
Previous evidence and calculations for paramyxoviruses have shown that the TMDs of the F protein are potentially longer than a typical vertically inserted membrane-spanning helix (18). Due to the lack of structural data for membrane-spanning regions, the orientation of the paramyxovirus F protein TMDs within the membrane remains unclear. We selected residues near the N-terminal region of the predicted HeV F TMD for substitution with double cysteine residues to determine if TMD dissociation is essential to drive conformational changes required for fusion events (Fig. 1A). These TMD mutants were designed with the goal of introducing disulfide bonds that would link the three monomers of the F trimer in the TMD region to assess alterations in fusion activity, protein stability, and overall protein conformation.
Double cysteine substitutions in the HeV F TMD led to the formation of disulfide-linked trimers, and fusion was blocked for these mutants. Attempts to restore fusion for the mutants with a disulfide reducing agent were unsuccessful, suggesting that the introduced disulfide bonds were protected in the membrane. Further analysis showed that two of the mutants were expressed at the cell surface in the prefusion conformation at levels that would normally promote fusion. Our results suggest that these two mutants were properly folded and processed, supporting the conclusion that TMD dissociation is required for fusion promotion. This study is the first to show that HeV F fusogenic activity can be prohibited by blocking TMD dissociation. These findings provide important new information on paramyxovirus fusion and contribute to our current knowledge of HeV F TMD interactions in protein stability, conformation, and fusion promotion events.
RESULTS
Double cysteine substitutions in the HeV F TMD promote disulfide bond formation.
Our current model suggests that TMD dissociation is important for conformational changes in the ectodomain needed for fusion, so substitutions were made in the HeV F TMD to analyze the effects on protein folding, stability, and fusion promotion when the TMDs are locked together. HeV F associates as a homotrimer immediately following synthesis, so double cysteine substitutions were made to link the three monomers with disulfide bonds (T483C/V484C, V484C/N485C, and N485C/P486C). The mutation locations were selected based on the prediction that the residues were present in the N-terminal region of the HeV F TMD. HeV F is synthesized in the endoplasmic reticulum (ER), which has a thinner lipid bilayer than the plasma membrane (39). Our goal was to mutate residues in the TMD that could be exposed to the oxidizing environment of the ER to allow for disulfide bond formation before trafficking of the F protein to the cell surface.
The oligomeric state of the HeV F TMD mutants was analyzed to determine if disulfide bonds successfully linked the monomers of the HeV F trimer. Vero cells were transiently transfected with plasmids encoding wild-type (WT) HeV F or one of the TMD mutants. Then, cells were starved and metabolically labeled, and samples were immunoprecipitated using an anti-peptide antibody (Ab) that binds to the cytoplasmic tail of HeV F. Boiled samples were separated on 3.5% polyacrylamide gels under nonreducing conditions to allow for visualization of different oligomeric forms of HeV F. T483C/V484C, V484C/N485C, and N485C/P486C migrated primarily in the trimeric form, whereas WT HeV F migrated primarily as a monomer (Fig. 2A). This suggested that the double cysteine substitutions in HeV F resulted in disulfide bonds that covalently linked the monomers of the HeV F trimer. In contrast, WT HeV F migrated primarily in the monomeric form because the monomers of the trimer lack covalent interactions. To confirm that the trimeric form was a result of the introduced disulfide bonds, samples were alternatively treated under reducing conditions. Results showed that the HeV F TMD mutants, similar to WT HeV F, migrated in the monomeric form under reducing conditions (Fig. 2B). Taken together, these findings indicate that the monomers of T483C/V484C, V484C/N485C, and N485C/P486C were effectively cross-linked due to the introduction of disulfide bonds between the TMDs of the F trimer.
FIG 2.
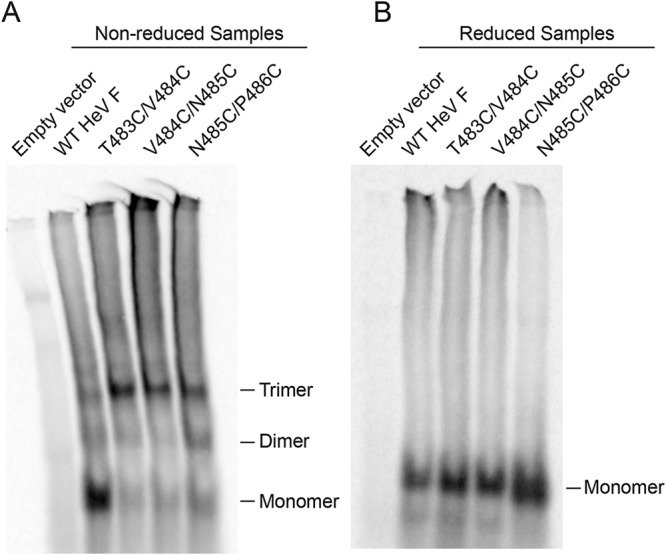
Double cysteine substitutions in the HeV F TMD promote disulfide bond formation. Vero cells were transfected with WT HeV F or one of the TMD mutants. At 24 h posttransfection, the cells were metabolically labeled for 3 h, and samples were immunoprecipitated. WT HeV F and the mutants were treated with nonreducing loading buffer (A) or reducing loading buffer (B) and separated using 3.5% SDS-PAGE.
Fusogenic activity is blocked for the HeV F TMD mutants.
Fusion assays were conducted to determine whether the mutants could promote fusion when TMD dissociation was inhibited. Vero cells were transiently transfected with plasmids encoding WT HeV G and WT HeV F or one of the TMD mutants. Cells transfected with WT HeV G alone or empty vector served as negative controls. At 48 h posttransfection, the cells were analyzed by microscopy for syncytium formation. As expected, cells transfected with a combination of WT HeV F and G showed the formation of small and large syncytia (Fig. 3A). However, samples transfected with WT HeV G and one of the TMD mutants showed no syncytium formation, suggesting that the introduced disulfide bonds blocked normal fusion promotion (Fig. 3A).
FIG 3.

Fusogenic activity is blocked for the HeV F TMD mutants. (A) Vero cells were transfected with the attachment protein WT HeV G and WT HeV F or one of the TMD mutants. Syncytium formation was analyzed at 48 h posttransfection. Images were taken with a Zeiss Axiovert 100 microscope. White arrowheads indicate syncytia. Images are representative. (B) Vero cells were transfected with luciferase, WT HeV G, and WT HeV F DNA or one of the TMD mutants. At 24 h posttransfection, the Vero cells were overlaid with BSR cells. After a 3-h incubation period, the cells were lysed and prepared for luminosity analysis to quantify fusion. Results were normalized to samples transfected with WT HeV F and G. All data are presented as the means ± standard deviations for three independent experiments. Statistical analysis was performed using a two-way analysis of variance with a Bonferroni correction. Asterisks indicate statistical significance compared to values for the WT HeV F (****, P < 0.0001).
A quantitative luciferase reporter gene assay was used to confirm the syncytium assay results. Vero cells were transiently transfected with plasmids encoding luciferase under the control of a T7 promoter, WT HeV G, and WT HeV F or one of the TMD mutants. At 24 h posttransfection, the Vero cells were overlaid with BSR cells containing the T7 polymerase. After a 3-h incubation, the cells were lysed and analyzed for luminescence as a measure of cell-cell fusion. Results showed that the TMD mutants did not promote fusion above the levels observed for the negative control (HeV G alone) (Fig. 3B). Together, these results indicate that the introduced disulfide bonds in the HeV F TMD prohibit fusion activity.
Cell surface expression is variably reduced for the HeV F TMD mutants.
Previous studies have shown that the increased cell surface density of WT HeV F correlates with increased fusion activity (40). Cell surface expression analysis was performed to determine if the TMD mutants were trafficked to the surface at levels that would normally allow for fusion promotion. Vero cells were transiently transfected with WT HeV F or one of the TMD mutants, starved, and metabolically labeled. Then, the samples were biotinylated prior to lysis and immunoprecipitation so that the cell surface protein population could be isolated and compared to total protein levels via SDS-PAGE analysis. Results for total protein expression showed no significant differences between the TMD mutants and the WT HeV F (Fig. 4A and C), whereas protein cleavage was significantly reduced for the mutants (Fig. 4D), indicating that they may be processed and trafficked less efficiently. Analysis of cell surface protein expression and cleavage showed significantly reduced levels for N485C/P486C, suggesting that this mutant may be misfolded or have severe trafficking defects (Fig. 4B and E). Similar to total protein cleavage results, surface protein cleavage levels for the TMD mutants were significantly reduced, further indicating that they are trafficked and processed less efficiently than WT HeV F (Fig. 4F). Although cleavage of T483C/V484C and V484C/N485C was reduced, the amount of fusion-active F1 on the surface was above the level previously shown to be needed for HeV F fusion (40). Based on these results, T483C/V484C and V484C/N485C are likely unable to promote fusion because TMD dissociation is an essential step for initiating conformational changes during fusion events.
FIG 4.

Cell surface expression is variably reduced for the HeV F TMD mutants. Total (A) and surface (B) expression levels were analyzed using a biotinylation method. Vero cells were transfected with WT HeV F or one of the TMD mutants. At 24 h posttransfection, the cells were metabolically labeled for 4 h, followed by biotinylation of the surface proteins and immunoprecipitation. The samples were analyzed by 15% SDS-PAGE. Total expression (F0+ F1) and cleavage [F1/(F0+F1)] (C and D), as well as surface expression and cleavage (E and F) were quantified by densitometry and normalized to levels for WT HeV F. All data are represented as the means ± standard deviations for three independent experiments. Statistical analyses were performed using a two-way analysis of variance with a Bonferroni correction. Asterisks indicate statistical significance compared to values for the WT HeV F (**, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
T483C/V484C and V484C/N485C are maintained over time at levels that normally allow for fusion.
Since the HeV F TMD mutants showed moderate differences in total expression levels and variable differences in surface expression levels compared to those of WT HeV F, a time course immunoprecipitation experiment was performed to monitor stability of the F protein over time (Fig. 5). Vero cells were transiently transfected with WT HeV F or one of the TMD mutants. Then, the cells were starved, metabolically labeled, and chased with regular medium for different amounts of time, as indicated in the figure. Finally, cells were lysed, and samples were immunoprecipitated for SDS-PAGE analysis. At early time points, WT HeV F and the TMD mutants were predominantly found in the F0 inactive form (Fig. 5A). Over time, levels of the F1 proteolytically active form increased, indicating that WT HeV F and the mutants were processed. Interestingly, quantification of expression levels at different time points showed that the mutants were highly expressed compared to expression of the WT HeV F at early time points (Fig. 5B). At later times, T483C/V484C and V484C/N485C showed reductions in protein levels compared to the level of WT HeV F, indicating that these mutants may have minor folding changes that target some of the protein for degradation. However, N485C/P486C showed a more severe reduction in protein level over time, suggesting that this mutant is likely targeted for degradation after synthesis due to improper folding. This result is consistent with the finding that N485C/P486C surface expression was significantly reduced (Fig. 4). Overall, these results show that the location of the introduced disulfide bonds in the HeV F TMD can variably affect protein folding and stability over time. These findings suggest that the minor changes observed for T483C/V484C and V484C/N485C protein stability are likely not the cause of fusion prohibition.
FIG 5.
T483C/V484C and V484C/N485C are present over time at levels that normally allow for fusion. (A) Vero cells were transfected with WT HeV F or one of the TMD mutants. At 24 h posttransfection, the cells were metabolically labeled for 30 min. Following different chase time points, the samples were immunoprecipitated and analyzed by 10% SDS-PAGE. (B) Expression was quantified by densitometry and normalized to the value for WT HeV F at each time point. All data are represented as the means ± standard deviations for three independent experiments.
Introduced disulfide bonds in the HeV F TMD mutants are poorly accessible to reducing agent.
Since fusion assays with the TMD mutants suggested that TMD dissociation is essential for fusion promotion, the mutants were analyzed to determine if fusion could be restored by reducing the introduced disulfide bonds. In theory, reduction of the disulfide bonds linking the TMDs would allow for dissociation of the TMDs to initiate the necessary conformational changes in the ectodomain for fusion activity. To test this, a luciferase reporter gene assay was conducted as described previously, except that the overlay medium contained the cell-impermeant reducing agent tris(2-carboxyethyl)phosphine (TCEP). Following the 3-h incubation with overlaid BSR cells in 6 mM TCEP-containing medium, the samples were analyzed for luminescence. Interestingly, fusion increased for WT HeV F treated with TCEP, suggesting that reduction of intramolecular disulfide bonds may impact overall protein stability and enhance the triggering of fusion (Fig. 6A). However, results for the TMD mutants showed no significant change in fusion levels between treated and untreated samples, indicating that fusion was not restored in the presence of TCEP (Fig. 6A).
FIG 6.

The introduced disulfide bonds in the HeV F TMD mutants are poorly accessible to reducing agent. (A) The samples were prepared as described in the legend of Fig. 3B, except the overlay medium consisted of BSRs in DMEM plus 10% FBS ± 6 mM TCEP. Results were normalized to levels of the samples transfected with WT HeV F and G (untreated). All data are presented as the means ± standard deviations for three independent experiments. Statistical analysis was performed using two-way analysis of variance with a Bonferroni correction. Asterisks indicate statistical significance compared to the level of the WT HeV F+G (untreated) (***, P < 0.005). (B) The samples were prepared as described in the legend of Fig. 2A, except that the samples were treated with DMEM plus 10% FBS ± 6 mM TCEP for 3 h following the metabolic label. Blue asterisks indicate trimer (F1), purple asterisk indicates dimer (F1), and orange asterisks indicate monomer (F1).
Oligomeric analysis was performed to further understand the effects of TCEP on the introduced disulfide bonds in the TMD mutants. The samples were prepared as described in the legend of Fig. 2, except that a 3-h incubation with 6 mM TCEP or untreated medium was included after the metabolic label. Then, samples were immunoprecipitated and analyzed by SDS-PAGE. Interestingly, results of the analysis showed that TCEP was capable of reducing disulfide bonds within the WT F protein, as indicated by the shift in bands for the trimer, dimer, and monomer in treated samples (Fig. 6B). This shift is consistent with loss of the extracellular F2 subunit when F1+F2 is reduced in the presence of TCEP. The TMD mutants also showed shifts consistent with loss of the F2 subunit (Fig. 6B, asterisks). T483C/V484C showed some reduction from the trimeric to monomeric form, but a portion of the trimeric form was still present in the TCEP-treated samples. The trimeric form of V484C/N485C partially shifted to a trimer lacking F2 in the TCEP-treated samples, but there was little change in the amount of monomer. The oligomeric forms of N485C/P486C remained relatively unchanged following TCEP treatment. This indicates that TCEP was poorly able to access the introduced disulfide bonds linking the TMDs of the mutant HeV F proteins, and accessibility decreased as the mutations went further into the TMD region. Altogether, these data suggest that the disulfide bonds introduced in the mutants are likely buried in cell membrane, making them, in some cases, inaccessible to the reducing agent.
T483C/V484C and V484C/N485C bind a prefusion conformation-specific antibody prior to cell disruption.
Since T483C/V484C and V484C/N485C were present at the cell surface at levels that would allow for fusion, further studies were conducted to analyze the conformation of the mutant protein structures. A HeV F prefusion conformation-specific antibody, monoclonal antibody (MAb) 5B3, was used to compare WT HeV F to the TMD mutants (38, 41). Vero cells were transiently transfected with WT HeV F or one of the mutants. At 24 h posttransfection, the cells were metabolically labeled overnight and treated with the prefusion antibody prior to cell lysis and immunoprecipitation. Control samples were treated with the HeV F anti-peptide antibody after cell lysis. Results showed that T483C/V484C and V484C/N485C were able to bind the prefusion conformation-specific antibody at moderately reduced levels compared to the level of WT HeV F (Fig. 7A and B). This result is consistent with the cell surface protein expression levels observed for T483C/V484C and V484C/N485C, suggesting that the TMD mutants trafficked to the surface are present in the prefusion form (Fig. 4).
FIG 7.

T483C/V484C and V484C/N485C bind a prefusion conformation-specific antibody prior to cell disruption. (A) Vero cells were transfected with WT HeV F or one of the TMD mutants. At 24 h posttransfection, the cells were metabolically labeled for 24 h. Then, the cells were treated with HeV F MAb 5B3 antibody for 1 h, followed by lysis and pulldown. Control samples were lysed after the overnight label and treated with HeV F anti-peptide Ab for immunoprecipitation. The samples were analyzed by 10% SDS-PAGE. (B) 5B3 binding was quantified by densitometry and normalized to the level of WT HeV F. All data are represented as the means ± standard deviations for three independent experiments. (C) Vero cells were transfected with WT HeV F or one of the TMD mutants. At 24 h posttransfection, the cells were metabolically labeled for 3 h. Then, the samples were immunoprecipitated with anti-peptide Ab or MAb 5B3 and analyzed by 10% SDS-PAGE.
Prior work with the prefusion conformation-specific antibody has shown that WT HeV F is unable to bind the antibody when it is applied following cell lysis, likely due to disruption of the metastable prefusion conformation of HeV F following lysis buffer treatment. Since the TMDs of T483C/V484C and V484C/N485C are locked together by disulfide bonds, these mutants were tested to determine if the introduced disulfide bonds permanently lock the prefusion conformation of the ectodomain. The HeV F TMD mutants were tested with MAb 5B3 after cell lysis to analyze prefusion conformation-specific antibody binding, and results showed that treating the mutants with MAb 5B3 following cell disruption dramatically reduced levels of binding (Fig. 7C). Together, these results suggest that T483C/V484C and V484C/N485C are synthesized in a prefusion conformation, but locking the TMDs together with disulfide bonds does not completely prevent unfolding of the ectodomain.
DISCUSSION
The model for HeV fusion suggests that dissociation of the F protein TMDs is an essential step for initiating and completing conformational changes in the ectodomain required for membrane fusion (29). We tested this model by designing HeV F TMD mutants to introduce disulfide bonds that would link the TMDs and prevent trimeric dissociation. Results showed that the mutants were successfully synthesized as disulfide-linked trimers, but fusion was prohibited for the mutants, suggesting that TMD dissociation is critical for the conformational changes in HeV F needed for fusion. Whereas surface expression and stability of T483C/V484C and V484C/N485C were maintained at levels that would allow for fusion, our results showed that N485C/P486C surface expression was significantly reduced, suggesting that the position of these introduced disulfide bonds interfered with proper protein folding. Attempts to restore fusion for the TMD mutants were unsuccessful, indicating that the introduced disulfide bonds were poorly accessible to the reducing agent due to their position in the membrane. Additional analysis of the TMD mutants showed that T483C/V484C and V484C/N485C were maintained in a prefusion conformation prior to cell disruption. Together, these findings support the hypothesis that TMD dissociation is required for HeV fusogenic activity and that TMD interactions play a crucial role in F protein folding and stability.
Cleavage of the HeV F TMD mutants was significantly reduced compared to that of the WT HeV F, which could contribute to the lack of fusion. Previous work from our lab showed that decreased WT HeV F expression leads to decreased fusion activity, but fusion was still detectable when normal WT HeV F surface expression was reduced by 80% (40). The amount of active F1 protein at the surface for the mutants T483C/V484C and V484C/N485C was above that needed for fusion for the WT protein. However, our reporter gene assay results showed no fusion above background levels for the HeV F TMD mutants, supporting our conclusion that fusion is blocked.
Previous work with other paramyxoviruses has utilized introduced disulfide bonds to probe the effects of limiting mobility within the F protein. For PIV5, the introduced disulfide bonds that linked the monomers within the MPER, N-terminal to the predicted TMD, blocked fusion activity (37). This was consistent with the inhibition of fusion observed for the HeV F TMD mutants. Treatment of the PIV5 F MPER mutant with TCEP restored fusion activity. However, similar treatment of the HeV F TMD mutants with TCEP did not restore fusion. This suggests that the disulfide bonds of the HeV F TMD mutants in this study were protected in the membrane, whereas the MPER disulfide bonds in the PIV5 F mutant were exposed at the cell surface (37).
Additional work related to PIV5 F has addressed the role of the TMD in fusion. Investigations showed that single cysteine substitutions near the N terminus of the TMD led to disulfide bond formation in the absence of an oxidative cross-linker, similar to the disulfide bond formation we observed in the HeV F TMD mutants (18). Further analysis of this region of PIV5 F using alanine-scanning mutagenesis indicated that two residues, L486 and I488, were required for efficient fusion activity. Different amino acids were substituted at these sites to test the effects of amino acid side chains on fusion activity. Interestingly, substitution with cysteine led to a minor reduction in fusion activity compared to the severe reductions observed with other substitutions. This finding is consistent with the idea that disulfide bond formation, rather than the presence of substituted cysteine residues, drove the prohibition of fusion activity for the HeV F TMD mutants. In addition, an alanine substitution was previously made at residue 486 in HeV F, resulting in minimal changes in fusion activity compared to that of the WT, further suggesting that disulfide bond formation, rather than the cysteine substitutions, played a key role in blocking fusion activity for the HeV F TMD mutants (C. T. Barrett, A. Popa, and R. E. Dutch, unpublished data).
Single cysteine substitutions were also used in a study to examine conformational changes in MeV F. They generated disulfide-linked mutants that were predicted to be unable to fully close the six-helix bundle (35). The single substitutions were made near the membrane-spanning region in the HRB domain. Despite this restriction on F protein flexibility, the mutants were able to efficiently open and stabilize fusion pores for viral entry, suggesting that these fusion events occur independently of complete six-helix bundle assembly (35). This work indicates that these single cysteine substitutions allow for greater flexibility than double cysteine substitutions in the membrane-proximal region of the F protein and provides evidence to support the idea that flexibility of interactions in the HRB and TMD are required for efficient fusion activity.
When TCEP was used in an attempt to restore fusion activity in our study, results showed that fusion was enhanced for WT HeV F. This increase may be due to the reduction of other disulfide bonds within the ectodomain of HeV F that are required for prefusion stability. Work on Newcastle disease virus identified free thiols in the surface-expressed F protein, a result of thiol/disulfide exchange, and blocking these groups with a thiol-specific biotin inhibited fusion (42). Thiol/disulfide exchange also plays a role in the entry of other enveloped viruses such as human immunodeficiency virus type I (43). These findings suggest that reduction of specific disulfide bonds in the F protein of paramyxoviruses may affect the efficiency of fusion activity. For HeV F, reducing disulfide bonds in the ectodomain by adding TCEP may cause the protein to trigger and promote fusion more readily than untreated HeV F. The HeV F TMD mutants showed small increases in fusion following TCEP treatment. However, these changes in fusion activity were not significant compared to the activity of the untreated mutants.
The paired cysteine substitutions we made in HeV F were located near the N terminus of the TMD, but our results suggested that the introduced disulfide bonds were buried within the plasma membrane. The F protein is synthesized in the ER, which consists of a slightly thinner lipid bilayer than the plasma membrane (39). This difference in membrane thickness may be important for exposing the N-terminal region of the HeV F TMD to the oxidizing environment of the ER to allow for disulfide bond formation immediately following synthesis. Once the mutant HeV F proteins are trafficked through the secretory pathway to the thicker plasma membrane, the disulfide bonds in the TMD may be shielded by the lipid environment from extracellular factors that could be introduced to disrupt the bonds.
Membrane thickness is largely determined by lipid composition, and a number of membrane lipids have been identified as important players in viral infectivity (44–50). Additionally, several studies of nonviral proteins have shown that cholesterol and sphingolipids play a role in promoting TMD helix interactions (51–53). Work with the paramyxovirus MeV has shown that the F protein is enriched in lipid rafts, and this partitioning is important for MeV assembly at the plasma membrane (49). Studies of NiV have shown evidence of F protein clustering in the plasma membrane, suggesting that membrane domains may be needed for proper surface glycoprotein organization for assembly and fusion (54). For Newcastle disease virus, lipid rafts have been shown to participate in forming and maintaining F protein and attachment protein complexes in the plasma membrane (55). Beyond paramyxoviruses, lipid raft domains have been implicated in the assembly and spread of filoviruses, retroviruses, and orthomyxoviruses (56). It is possible that lipid rafts also play a role in HeV assembly and TMD helix interactions. Additionally, partitioning of the F protein into rafts could increase the number of TMD residues accommodated by the membrane. Therefore, HeV F localization to lipid rafts could explain why the introduced disulfide bonds are initially formed at the ER but are protected from reducing agents at the plasma membrane. Future studies will analyze the importance of lipid composition for stability of the prefusion conformation and promotion of fusion events.
Here, we showed that disulfide bonds can be introduced to covalently link the TMDs of the HeV F trimer. Blocking TMD dissociation with introduced disulfide bonds prohibits fusion events, but further studies are needed to address the conformational changes that can occur in HeV F when the TMDs are locked. Our studies suggest that mutant HeV F proteins with linked TMDs are in some cases capable of maintaining a prefusion conformation. If cells expressing these constructs are disrupted, then the mutant HeV F proteins refold into a conformation that is no longer recognizable by a prefusion-specific antibody. Additional analysis is needed to understand if the disulfide-linked HeV F mutants are capable of refolding into protein intermediates that are suggested to occur after an initial triggering event and prior to formation of the six-helix bundle. Our findings reported here and future studies will contribute to understanding the HeV F dynamics required for fusion events and mechanisms of enveloped virus entry.
MATERIALS AND METHODS
Plasmids.
Plasmids containing HeV F and G were kindly provided by Lin-Fa Wang from the Australian Animal Health Laboratory. The HeV F TMD mutants were generated in pGEM using a QuikChange site-directed mutagenesis kit from Stratagene and subcloned into pCAGGS using the forward primer 5′-GCG ATT GAA TTC TAA GCA ATG GCT ACA CAA GAG-3′ and reverse primer 5′-CG GCG GCC ATG CAT ATT TTA TGT TCC AAT ATA ATA-3′ for PCR amplification. The constructs were verified by sequencing.
Antibodies.
Anti-peptide antibody to the HeV F cytoplasmic tail residues 527 to 539 (15) was used to pull down WT HeV F or the TMD mutant constructs. The prefusion conformation-specific antibody, MAb 5B3, generously provided by Christopher Broder (Uniformed Services University of the Health Sciences) was also used to detect HeV F (41).
Cell lines.
Vero cells (ATCC) and BSR cells (provided by Karl-Klaus Conzelman, Pettenkofer Institut) were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum (FBS; Sigma). For the BSR cells, 0.5 mg/ml Geneticin (Gibco) was added to the medium with every third passage to select for T7 polymerase-expressing cells.
Oligomeric analysis.
Vero cells in six-well plates were transiently transfected using Lipofectamine and Plus (Invitrogen) per the manufacturer’s protocol with pCAGGS-HeV F or one of the TMD mutants. At 24 h posttransfection, the cells were washed two times with phosphate-buffered saline (PBS) and starved with DMEM lacking cysteine and methionine (Cys–/Met–) for 45 min. Then, the cells were labeled for 3 h with Cys–/Met– DMEM containing Tran-35S label (100 μCi/ml; MP Biomedicals). The cells were washed three times with PBS and lysed with 500 μl of radioimmunoprecipitation assay (RIPA) lysis buffer (100 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 1% deoxycholic acid, 1 mM phenylmethylsulfonyl fluoride [Sigma], 25 mM iodoacetamide [Sigma], 1:100 aprotinin [Calbiochem]). The sample lysate was centrifuged at 136,500 × g for 15 min at 4°C, and the supernatant was incubated for 3 h with 4 μl of anti-peptide antibody. Then, the sample was incubated with 30 μl of protein A-Sepharose beads (ThermoFisher) for 30 min at 4°C with rocking and washed two times with RIPA buffer plus 0.30 M NaCl, two times with RIPA buffer plus 0.15 M NaCl, and two times with SDS wash II (150 mM NaCl, 50 mM Tris-HCl, pH 7.4, 2.5 mM EDTA). Depending on the experiment, the samples were resuspended with loading buffer lacking or containing dithiothreitol (DTT; Goldbio) for nonreducing or reducing conditions, respectively. Then, the samples were boiled and separated using 3.5% polyacrylamide gels for SDS-PAGE and visualized using a Typhoon imaging system (GE Healthcare). For the tris(2-carboxyethyl) (TCEP) (Calbiochem) treatment experiments, 1 ml of 6 mM TCEP in DMEM supplemented with 10% FBS was added to the cells following the metabolic label. The cells were incubated with the TCEP solution for 3 h at 37°C before two washes with PBS and addition of RIPA lysis buffer. Results were visualized using a Typhoon imaging system.
Syncytium assay.
Vero cells in six-well plates were transiently transfected using Lipofectamine 3000 (Invitrogen) per the manufacturer’s protocol with pCAGGS-HeV F and pCAGGS-HeV G at a 1:3 ratio. At 48 h posttransfection, the cells were imaged using a Zeiss Axiovert 100 microscope with a 10× objective.
Reporter gene assay.
Vero cells in six-well plates were transiently transfected using Lipofectamine and Plus per the manufacturer’s protocol with 0.8 μg of luciferase under the control of the T7 promoter, 0.9 μg pCAGGS-HeV G, and 0.3 μg pCAGGS-HeV F or one of the TMD mutants. At 24 h posttransfection, the Vero cells were washed once with PBS and overlaid with BSR cells, previously lifted with trypsin (Invitrogen) and diluted in DMEM supplemented with 10% FBS, that stably express the T7 polymerase for 3 h at 37°C. For specific experiments, 6 mM TCEP was added to the overlay medium for the 3-h incubation. Then, the cells were lysed with reporter lysis buffer (Promega) and analyzed for luciferase activity using the luciferase assay system (Promega) per the manufacturer’s instructions. An Lmax luminometer (Molecular Devices; Sunnyvale, CA) was used with a 2-s delay and a 5-s integration time. Results were normalized to samples expressing WT HeV F and G.
Cell surface biotinylation.
Vero cells in 60-mm dishes were transiently transfected using Lipofectamine and Plus reagent or Lipofectamine 3000 per the manufacturer’s protocol with 4 μg of pCAGGS-HeV F or one of the TMD mutants. At 24 h posttransfection, the cells were washed two times with PBS and starved with Cys–/Met– DMEM for 45 min. Then, the cells were radiolabeled for 4 h in Cys–/Met– DMEM containing Tran-35S label. The cells were washed two times with ice-cold PBS (pH 8.0) and incubated with 1 ml of 1 mg/ml EZ-Link Sulfo-NHS-Biotin (sulfo-N-hydroxysuccinimide-biotin; Pierce) in PBS (pH 8.0) for 35 min at 4°C with rocking, followed by 15 min at room temperature. Next, the cells were washed three times with ice-cold PBS (pH 8.0), and 500 μl of RIPA lysis buffer was added. The sample lysate was centrifuged at 136,500 × g for 15 min at 4°C. The supernatant was transferred to 1.5-ml tubes and incubated with 8 μl of the anti-peptide HeV F antibody for 3 h at 4°C with rocking. Next, each sample was incubated with 30 μl of protein A-Sepharose beads for 30 min at 4°C with rocking. The samples were washed two times with RIPA buffer plus 0.30 M NaCl, two times with RIPA buffer plus 0.15 M NaCl, and two times with SDS wash II. Following the washes, 60 μl of 10% SDS was added, and the samples were boiled for 10 min, transferred to a new tube, and repeated with 40 μl of 10% SDS to give a total of 100 μl. Ten microliters of each sample was separated and resuspended in 2× SDS loading buffer containing DTT for total protein expression analysis. The remaining 90 μl of sample was treated with 400 μl of biotinylation dilution buffer (20 mM Tris [pH 8.0], 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 0.2% bovine serum albumin [US Biological Life Sciences]) and 30 μl of streptavidin beads for 1 h at 4°C with rocking. The washes described previously were repeated, and the samples were resuspended in 2× SDS loading buffer containing DTT. After the samples were boiled, analysis of HeV F was conducted using 15% SDS-PAGE and visualized using a Typhoon imaging system. Quantifications from band densitometry using ImageQuant, version 5.2, were reported as relative expression (percent) compared to WT HeV F. The quantification is the sum of F0 and F1 for each sample.
Time course immunoprecipitation.
Vero cells in six-well plates were transiently transfected using Lipofectamine and Plus per the manufacturer’s protocol with pCAGGS-HeV F or one of the TMD mutants. At 24 h posttransfection, the cells were washed two times with PBS, starved for 45 min, and metabolically labeled for 3 h (described previously). Then, the cells were washed three times with PBS and chased with DMEM plus 10% FBS. At different time points, the cells were washed two times with PBS and lysed with RIPA lysis buffer. Immunoprecipitation was conducted as described for oligomeric analysis, and samples were resuspended in 2× SDS loading buffer containing DTT. Samples were boiled and analyzed by 10% SDS-PAGE, and band densitometry quantifications were performed as described for surface biotinylation.
Immunoprecipitation with prefusion conformation-specific antibody.
To analyze MAb 5B3 binding prior to cell disruption, Vero cells in 60-mm dishes were transiently transfected using Lipofectamine and Plus per the manufacturer’s protocol with pCAGGS-HeV F or one of the TMD mutants. At 24 h posttransfection, the cells were washed two times with PBS, starved for 45 min, and metabolically labeled in 2 ml of overnight label medium (85% Cys–/Met– DMEM, 10% DMEM plus 10% FBS, 5% FBS) containing Tran-35S label for 24 h. Then, the cells were washed three times with PBS and treated with MAb 5B3 at 1 μg/ml in 1× PBS plus 1% bovine serum albumin for 1 h at 4°C with rocking. Following the antibody incubation, the cells were lysed with RIPA lysis buffer and centrifuged at 136,500 × g for 15 min at 4°C. Control samples were treated with 8 μl of anti-peptide antibody for 1.5 h after being lysed with RIPA lysis buffer and centrifuged. All samples were treated with 30 μl of protein A-Sepharose beads for 1 h. Then, the samples were washed as described previously. After the samples were boiled, they were separated by 10% SDS-PAGE, and band densitometry quantifications were performed as described for surface biotinylation.
To analyze MAb 5B3 binding following cell disruption, Vero cells in six-well plates were transiently transfected using Lipofectamine and Plus per the manufacturer’s protocol with pCAGGS-HeV F or one of the TMD mutants. At 24 h posttransfection, the cells were starved, labeled, lysed, and centrifuged as described for oligomeric analysis. Then, the cells were treated with 1 μg/ml MAb 5B3 or 4 μl of anti-peptide Ab for 3 h. The rest of the immunoprecipitation was performed as described for oligomeric analysis. Then, the samples were boiled and separated using 10% SDS-PAGE, and band densitometry quantifications were performed as described for surface biotinylation.
ACKNOWLEDGMENTS
We thank Chelsea Barrett for the initial design of the fusion model and for carefully reading the manuscript.
This work was supported by NIAID grant R01AI051517 and NIH grant 2P20 RR02017 to R.E.D.
REFERENCES
- 1.Lee B, Ataman ZA. 2011. Modes of paramyxovirus fusion: a Henipavirus perspective. Trends Microbiol 19:389–399. doi: 10.1016/j.tim.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eaton BT, Broder CC, Middleton D, Wang LF. 2006. Hendra and Nipah viruses: different and dangerous. Nat Rev Microbiol 4:23–35. doi: 10.1038/nrmicro1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harcourt BH, Tamin A, Ksiazek TG, Rollin PE, Anderson LJ, Bellini WJ, Rota PA. 2000. Molecular characterization of Nipah virus, a newly emergent paramyxovirus. Virology 271:334–349. doi: 10.1006/viro.2000.0340. [DOI] [PubMed] [Google Scholar]
- 4.O’Sullivan JD, Allworth AM, Paterson DL, Snow TM, Boots R, Gleeson LJ, Gould AR, Hyatt AD, Bradfield J. 1997. Fatal encephalitis due to novel paramyxovirus transmitted from horses. Lancet 349:93–95. doi: 10.1016/S0140-6736(96)06162-4. [DOI] [PubMed] [Google Scholar]
- 5.Murray K, Selleck P, Hooper P, Hyatt A, Gould A, Gleeson L, Westbury H, Hiley L, Selvey L, Rodwell B. 1995. A morbillivirus that caused fatal disease in horses and humans. Science 268:94–97. doi: 10.1126/science.7701348. [DOI] [PubMed] [Google Scholar]
- 6.Marsh GA, Wang LF. 2012. Hendra and Nipah viruses: why are they so deadly? Curr Opin Virol 2:242–247. doi: 10.1016/j.coviro.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 7.Chua KB, Bellini WJ, Rota PA, Harcourt BH, Tamin A, Lam SK, Ksiazek TG, Rollin PE, Zaki SR, Shieh W, Goldsmith CS, Gubler DJ, Roehrig JT, Eaton B, Gould AR, Olson J, Field H, Daniels P, Ling AE, Peters CJ, Anderson LJ, Mahy BW. 2000. Nipah virus: a recently emergent deadly paramyxovirus. Science 288:1432–1435. doi: 10.1126/science.288.5470.1432. [DOI] [PubMed] [Google Scholar]
- 8.Halpin K, Young PL, Field HE, Mackenzie JS. 2000. Isolation of Hendra virus from pteropid bats: a natural reservoir of Hendra virus. J Gen Virol 81:1927–1932. doi: 10.1099/0022-1317-81-8-1927. [DOI] [PubMed] [Google Scholar]
- 9.Bossart KN, Wang LF, Eaton BT, Broder CC. 2001. Functional expression and membrane fusion tropism of the envelope glycoproteins of Hendra virus. Virology 290:121–135. doi: 10.1006/viro.2001.1158. [DOI] [PubMed] [Google Scholar]
- 10.Bossart KN, Wang LF, Flora MN, Chua KB, Lam SK, Eaton BT, Broder CC. 2002. Membrane fusion tropism and heterotypic functional activities of the Nipah virus and Hendra virus envelope glycoproteins. J Virol 76:11186–11198. doi: 10.1128/jvi.76.22.11186-11198.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jardetzky TS, Lamb RA. 2014. Activation of paramyxovirus membrane fusion and virus entry. Curr Opin Virol 5:24–33. doi: 10.1016/j.coviro.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dutch RE, Jardetzky TS, Lamb RA. 2000. Virus membrane fusion proteins: biological machines that undergo a metamorphosis. Biosci Rep 20:597–612. doi: 10.1023/a:1010467106305. [DOI] [PubMed] [Google Scholar]
- 13.Smith EC, Popa A, Chang A, Masante C, Dutch RE. 2009. Viral entry mechanisms: the increasing diversity of paramyxovirus entry. FEBS J 276:7217–7227. doi: 10.1111/j.1742-4658.2009.07401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harrison SC. 2008. Viral membrane fusion. Nat Struct Mol Biol 15:690–698. doi: 10.1038/nsmb.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pager CT, Wurth MA, Dutch RE. 2004. Subcellular localization and calcium and pH requirements for proteolytic processing of the Hendra virus fusion protein. J Virol 78:9154–9163. doi: 10.1128/JVI.78.17.9154-9163.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pager CT, Dutch RE. 2005. Cathepsin L is involved in proteolytic processing of the Hendra virus fusion protein. J Virol 79:12714. doi: 10.1128/JVI.79.20.12714-12720.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meulendyke KA, Wurth MA, McCann RO, Dutch RE. 2005. Endocytosis plays a critical role in proteolytic processing of the Hendra virus fusion protein. J Virol 79:12643–12649. doi: 10.1128/JVI.79.20.12643-12649.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bissonnette ML, Donald JE, DeGrado WF, Jardetzky TS, Lamb RA. 2009. Functional analysis of the transmembrane domain in paramyxovirus F protein-mediated membrane fusion. J Mol Biol 386:14–36. doi: 10.1016/j.jmb.2008.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kemble GW, Danieli T, White JM. 1994. Lipid-anchored influenza hemagglutinin promotes hemifusion, not complete fusion. Cell 76:383–391. doi: 10.1016/0092-8674(94)90344-1. [DOI] [PubMed] [Google Scholar]
- 20.Armstrong RT, Kushnir AS, White JM. 2000. The transmembrane domain of influenza hemagglutinin exhibits a stringent length requirement to support the hemifusion to fusion transition. J Cell Biol 151:425–437. doi: 10.1083/jcb.151.2.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miyauchi K, Komano J, Yokomaku Y, Sugiura W, Yamamoto N, Matsuda Z. 2005. Role of the specific amino acid sequence of the membrane-spanning domain of human immunodeficiency virus type 1 in membrane fusion. J Virol 79:4720–4729. doi: 10.1128/JVI.79.8.4720-4729.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Owens RJ, Burke C, Rose JK. 1994. Mutations in the membrane-spanning domain of the human immunodeficiency virus envelope glycoprotein that affect fusion activity. J Virol 68:570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salzwedel K, Johnston PB, Roberts SJ, Dubay JW, Hunter E. 1993. Expression and characterization of glycophospholipid-anchored human immunodeficiency virus type 1 envelope glycoproteins. J Virol 67:5279–5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shang L, Hunter E. 2010. Residues in the membrane-spanning domain core modulate conformation and fusogenicity of the HIV-1 envelope glycoprotein. Virology 404:158–167. doi: 10.1016/j.virol.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilk T, Pfeiffer T, Bukovsky A, Moldenhauer G, Bosch V. 1996. Glycoprotein incorporation and HIV-1 infectivity despite exchange of the gp160 membrane-spanning domain. Virology 218:269–274. doi: 10.1006/viro.1996.0190. [DOI] [PubMed] [Google Scholar]
- 26.Weiss CD, White JM. 1993. Characterization of stable Chinese hamster ovary cells expressing wild-type, secreted, and glycosylphosphatidylinositol-anchored human immunodeficiency virus type 1 envelope glycoprotein. J Virol 67:7060–7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shang L, Yue L, Hunter E. 2008. Role of the membrane-spanning domain of human immunodeficiency virus type 1 envelope glycoprotein in cell-cell fusion and virus infection. J Virol 82:5417–5428. doi: 10.1128/JVI.02666-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fritz R, Blazevic J, Taucher C, Pangerl K, Heinz FX, Stiasny K. 2011. The unique transmembrane hairpin of flavivirus fusion protein E is essential for membrane fusion. J Virol 85:4377–4385. doi: 10.1128/JVI.02458-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith EC, Culler MR, Hellman LM, Fried MG, Creamer TP, Dutch RE. 2012. Beyond anchoring: the expanding role of the Hendra virus fusion protein transmembrane domain in protein folding, stability, and function. J Virol 86:3003–3013. doi: 10.1128/JVI.05762-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Webb S, Nagy T, Moseley H, Fried M, Dutch R. 2017. Hendra virus fusion protein transmembrane domain contributes to pre-fusion protein stability. J Biol Chem 292:5685–5694. doi: 10.1074/jbc.M117.777235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Webb SR, Smith SE, Fried MG, Dutch RE. 2018. Transmembrane domains of highly pathogenic viral fusion proteins exhibit trimeric association in vitro. mSphere 3:e00047-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Navaratnarajah CK, Kumar S, Generous A, Apte-Sengupta S, Mateo M, Cattaneo R. 2014. The measles virus hemagglutinin stalk: structures and functions of the central fusion activation and membrane-proximal segments. J Virol 88:6158–6167. doi: 10.1128/JVI.02846-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ader N, Brindley MA, Avila M, Origgi FC, Langedijk JP, Orvell C, Vandevelde M, Zurbriggen A, Plemper RK, Plattet P. 2012. Structural rearrangements of the central region of the morbillivirus attachment protein stalk domain trigger F protein refolding for membrane fusion. J Biol Chem 287:16324–16334. doi: 10.1074/jbc.M112.342493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mahon PJ, Mirza AM, Musich TA, Iorio RM. 2008. Engineered intermonomeric disulfide bonds in the globular domain of Newcastle disease virus hemagglutinin-neuraminidase protein: implications for the mechanism of fusion promotion. J Virol 82:10386–10396. doi: 10.1128/JVI.00581-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brindley MA, Plattet P, Plemper RK. 2014. Efficient replication of a paramyxovirus independent of full zippering of the fusion protein six-helix bundle domain. Proc Natl Acad Sci U S A 111:E3795–E3804. doi: 10.1073/pnas.1403609111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee JK, Prussia A, Snyder JP, Plemper RK. 2007. Reversible inhibition of the fusion activity of measles virus F protein by an engineered intersubunit disulfide bridge. J Virol 81:8821–8826. doi: 10.1128/JVI.00754-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zokarkar A, Connolly SA, Jardetzky TS, Lamb RA. 2012. Reversible inhibition of fusion activity of a paramyxovirus fusion protein by an engineered disulfide bond in the membrane-proximal external region. J Virol 86:12397–12401. doi: 10.1128/JVI.02006-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wong JJ, Paterson RG, Lamb RA, Jardetzky TS. 2016. Structure and stabilization of the Hendra virus F glycoprotein in its prefusion form. Proc Natl Acad Sci U S A 113:1056–1061. doi: 10.1073/pnas.1523303113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamamoto T. 1963. On the thickness of the unit membrane. J Cell Biol 17:413–421. doi: 10.1083/jcb.17.2.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith EC, Dutch RE. 2010. Side chain packing below the fusion peptide strongly modulates triggering of the Hendra virus F protein. J Virol 84:10928–10932. doi: 10.1128/JVI.01108-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chan YP, Lu M, Dutta S, Yan L, Barr J, Flora M, Feng YR, Xu K, Nikolov DB, Wang LF, Skiniotis G, Broder CC. 2012. Biochemical, conformational, and immunogenic analysis of soluble trimeric forms of henipavirus fusion glycoproteins. J Virol 86:11457–11471. doi: 10.1128/JVI.01318-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jain S, McGinnes LW, Morrison TG. 2007. Thiol/disulfide exchange is required for membrane fusion directed by the Newcastle disease virus fusion protein. J Virol 81:2328–2339. doi: 10.1128/JVI.01940-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stantchev TS, Paciga M, Lankford CR, Schwartzkopff F, Broder CC, Clouse KA. 2012. Cell-type specific requirements for thiol/disulfide exchange during HIV-1 entry and infection. Retrovirology 9:97. doi: 10.1186/1742-4690-9-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aizaki H, Morikawa K, Fukasawa M, Hara H, Inoue Y, Tani H, Saito K, Nishijima M, Hanada K, Matsuura Y, Lai MM, Miyamura T, Wakita T, Suzuki T. 2008. Critical role of virion-associated cholesterol and sphingolipid in hepatitis C virus infection. J Virol 82:5715–5724. doi: 10.1128/JVI.02530-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang H, Li Y, Sadaoka T, Tang H, Yamamoto T, Yamanishi K, Mori Y. 2006. Human herpesvirus 6 envelope cholesterol is required for virus entry. J Gen Virol 87:277–285. doi: 10.1099/vir.0.81551-0. [DOI] [PubMed] [Google Scholar]
- 46.Miller ME, Adhikary S, Kolokoltsov AA, Davey RA. 2012. Ebolavirus requires acid sphingomyelinase activity and plasma membrane sphingomyelin for infection. J Virol 86:7473–7483. doi: 10.1128/JVI.00136-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun X, Whittaker GR. 2003. Role for influenza virus envelope cholesterol in virus entry and infection. J Virol 77:12543–12551. doi: 10.1128/jvi.77.23.12543-12551.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Til NP, Heutinck KM, van der Rijt R, Paulusma CC, van Wijland M, Markusic DM, Elferink RP, Seppen J. 2008. Alteration of viral lipid composition by expression of the phospholipid floppase ABCB4 reduces HIV vector infectivity. Retrovirology 5:14. doi: 10.1186/1742-4690-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vincent S, Gerlier D, Manie SN. 2000. Measles virus assembly within membrane rafts. J Virol 74:9911–9915. doi: 10.1128/jvi.74.21.9911-9915.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adu-Gyamfi E, Johnson KA, Fraser ME, Scott JL, Soni SP, Jones KR, Digman MA, Gratton E, Tessier CR, Stahelin RV. 2015. Host cell plasma membrane phosphatidylserine regulates the assembly and budding of Ebola virus. J Virol 89:9440–9453. doi: 10.1128/JVI.01087-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cristian L, Lear JD, DeGrado WF. 2003. Use of thiol-disulfide equilibria to measure the energetics of assembly of transmembrane helices in phospholipid bilayers. Proc Natl Acad Sci U S A 100:14772–14777. doi: 10.1073/pnas.2536751100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.de Almeida RF, Loura LM, Prieto M, Watts A, Fedorov A, Barrantes FJ. 2004. Cholesterol modulates the organization of the γM4 transmembrane domain of the muscle nicotinic acetylcholine receptor. Biophys J 86:2261–2272. doi: 10.1016/S0006-3495(04)74284-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sparr E, Ash WL, Nazarov PV, Rijkers DT, Hemminga MA, Tieleman DP, Killian JA. 2005. Self-association of transmembrane alpha-helices in model membranes: importance of helix orientation and role of hydrophobic mismatch. J Biol Chem 280:39324–39331. doi: 10.1074/jbc.M502810200. [DOI] [PubMed] [Google Scholar]
- 54.Liu Q, Chen L, Aguilar HC, Chou KC. 2018. A stochastic assembly model for Nipah virus revealed by super-resolution microscopy. Nat Commun 9:3050. doi: 10.1038/s41467-018-05480-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Laliberte JP, McGinnes LW, Morrison TG. 2007. Incorporation of functional HN-F glycoprotein-containing complexes into Newcastle disease virus is dependent on cholesterol and membrane lipid raft integrity. J Virol 81:10636–10648. doi: 10.1128/JVI.01119-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ono A, Freed EO. 2005. Role of lipid rafts in virus replication. Adv Virus Res 64:311–358. doi: 10.1016/S0065-3527(05)64010-9. [DOI] [PubMed] [Google Scholar]