

HCV infection is a worldwide problem of public health and a major contributor to hepatocellular carcinoma. The single-stranded RNA virus with RNA-dependent RNA polymerase experiences a high error rate and develops strategies to escape the immune system and hepatocarcinogenesis. Studies have revealed the involvement of HCV proteins in the impairment of DNA repair. The present study aimed to further elucidate mechanisms by which the viral NS3 protein impairs the repair of DNA damage. Our results clearly indicate that HCV NS3/4A protease targets WRN for degradation, and, at the same time, diminishes the repair efficiency of nonhomologous end joining by interfering with the recruitment of Ku protein to the DNA double-strand break sites. The study describes a novel mechanism by which the NS3 protein influences DNA repair and provides new insight into the molecular mechanism of HCV pathogenesis.
KEYWORDS: hepatitis C virus, NS3 protease, RecQ helicase, Werner syndrome protein (WRN), Ku protein, DNA damage, nonhomologous end joining
ABSTRACT
Hepatitis C virus (HCV) NS3 protein possesses protease and helicase activities and is considered an oncoprotein in virus-derived hepatocellular carcinoma. The NS3-associated oncogenesis has been studied but not fully understood. In this study, we have identified novel interactions of the NS3 protein with DNA repair factors, Werner syndrome protein (WRN) and Ku70, in both an HCV subgenomic replicon system and Huh7 cells expressing NS3. HCV NS3 protein inhibits WRN-mediated DNA repair and reduces the repair efficiency of nonhomologous end joining. It interferes with Ku70 recruitment to the double-strand break sites and alters the nuclear distribution of WRN-Ku repair complex. In addition, WRN is a substrate of the NS3/4A protease; the level of WRN protein is regulated by both the proteasome degradation pathway and HCV NS3/4A protease activity. The dual role of HCV NS3 and NS3/4A proteins in regulating the function and expression level of the WRN protein intensifies the effect of impairment on DNA repair. This may lead to an accumulation of DNA mutations and genome instability and, eventually, tumor development.
IMPORTANCE HCV infection is a worldwide problem of public health and a major contributor to hepatocellular carcinoma. The single-stranded RNA virus with RNA-dependent RNA polymerase experiences a high error rate and develops strategies to escape the immune system and hepatocarcinogenesis. Studies have revealed the involvement of HCV proteins in the impairment of DNA repair. The present study aimed to further elucidate mechanisms by which the viral NS3 protein impairs the repair of DNA damage. Our results clearly indicate that HCV NS3/4A protease targets WRN for degradation, and, at the same time, diminishes the repair efficiency of nonhomologous end joining by interfering with the recruitment of Ku protein to the DNA double-strand break sites. The study describes a novel mechanism by which the NS3 protein influences DNA repair and provides new insight into the molecular mechanism of HCV pathogenesis.
INTRODUCTION
Hepatitis C virus (HCV) is classified in the Hepacivirus genus within the Flaviviridae family. The viral genome consists of a 9.6-kb single-stranded positive-sense RNA with 5′ and 3′ noncoding regions and a long open reading frame encoding a polyprotein precursor approximately 3,000 amino acids in length (1). Chronic liver infection with HCV affects more than 71 million people worldwide (http://www.who.int/news-room/fact-sheets/detail/hepatitis-c). The importance of HCV infection in hepatocellular carcinomas (HCC) (2) and non-Hodgkin’s B-cell lymphomas (3) has been well documented. However, the mechanism of its oncogenesis remains largely unknown. HCV polyprotein precursor is cleaved into 10 structural and nonstructural (NS) proteins through the action of cellular proteases and the virus-encoded proteases NS2 and NS3/4A. The NS4A protein that acts as a cofactor of the NS3 serine protease is required for trans cleavage at the NS4B/5A junction of the viral polyprotein and for internal NS3 cleavage (4). Although the oncogenesis driven by the viral NS3/4A protein is not fully understood, studies have indicated that NS3/4A impairs the efficiency of DNA repair and renders the cells more sensitive to DNA damage by causing cytoplasmic translocation of ATM and producing reactive oxygen species (ROS) (5–7). In addition, NS3 was found to enter into the cell nucleus and inhibit p53-dependent transcription through interacting with p53 (8). Furthermore, NS3 affects the functions of host cell proteins through its protease activity. With the cofactor NS4A, the NS3/4A protease cleaves mitochondrial antiviral signaling protein (MAVS) downstream of the retinoic acid-inducible gene I (RIG-I) (9) and TIR-domain-containing adapter-inducing interferon-β (TRIF) downstream of the Toll-like receptor 3 (TLR3) (10), resulting in the suppression of NF-κB activation and evasion of innate immunity. It was also demonstrated that NS3/4A protease cleaves T cell protein tyrosine phosphatase (TC-PTP), activates epidermal growth factor (EGF)-induced signal transduction, and increases Akt basal activity critical for the maintenance of HCV replication (11). It would be interesting to know whether HCV NS3/4A protease targets and disrupts the function of nuclear proteins involving in DNA repair.
HCV NS3 protein can also function as a helicase. It belongs to helicase superfamily 2 (SF2) and shares conserved domains with other family members (12, 13). In our previous study, we have demonstrated intermolecular interactions between the NS3 RNA-binding domain and ATPase domain (14). In this study, potential interactions between HCV NS3 protein and members of the RecQ family that also belong to the SF2 superfamily were examined. The RecQ helicases are functionally involved in homology-dependent recombination, replication initiation, replication restart or fork elongation, and DNA repair and are required for the maintenance of genomic stability (15). All five members in the human RecQ helicase family share a conserved helicase domain that possesses DNA-dependent ATPase and 3′-to-5′ helicase activities. One of the members, Werner syndrome protein (WRN), also possesses 3′-to-5′ exonuclease activity. In addition, purified human RecQ helicases were demonstrated to preferentially bind and unwind partially double-stranded DNA molecules, including model replication forks, D and T loops, or synthetic Holliday junctions, and highly structured DNAs such as G quadruplexes (16). WRN also interacts with topoisomerase I and regulates DNA topology during replication (17). Camptothecin (CPT) is a topoisomerase I inhibitor. It induces strand breaks at DNA sites where topoisomerase I is covalently linked (18) and causes cell cycle arrest at S phase (19). WRN helicase, recruited by RPA and Mre11, regulates the ATR-CHK1-induced S-phase checkpoint pathway and participates in DNA repair in response to CPT-induced DNA damage (20, 21).
In this study, we found that HCV NS3 protein interacts with WRN and Ku70 in HCV replicon cells. Ku70 is known to form a heterodimer with Ku80 and takes part as an initiation factor in nonhomologous end joining (NHEJ) repair (22, 23). As WRN participates in both NHEJ and homologous recombination (HR) repair (24), mechanisms by which the NS3 protein is involved in the WRN-mediated DNA repair were examined. We found that HCV NS3 protein impairs NHEJ repair. It disrupts binding of Ku70 protein to the double-strand break (DSB) sites and may thus alter the subcellular distribution of repair complexes. In addition, the level of WRN protein is regulated by both the proteasome degradation pathway and an HCV NS4A-dependent NS3 protease activity. The effects of HCV NS3 protein and NS3/4A protease activity on the functions and expression level of the WRN protein may result in an accumulation of DNA mutations, genome instability, and a higher sensitivity of host cells to DNA damage agents, leading to cell senescence as a critical part in HCC development.
RESULTS
HCV NS3 protein interacts with WRN, a member of the RecQ family.
To examine whether HCV NS3 protein interacts with members of the RecQ family, a pulldown assay was performed with glutathione S-transferase (GST)-NS3 and cell lysates prepared from Huh7 cells. This revealed an interaction of the NS3 protein with WRN protein but not with RecQL1, BLM, or RecQL5 and barely with RecQL4 (Fig. 1A). Consistently, immunoprecipitation (IP) assays with protein lysates derived from tetracycline/doxycycline (Dox)-inducible NS3-expressing cells (Tet-on repressor [TR]-NS3) detected an association of WRN with the NS3 protein (Fig. 1B). HCV NS3 protein also formed complexes with WRN in HCV replicon (HCVR) cells (Fig. 1C). Addition of ethidium bromide (EtBr), a DNA intercalator that distorts DNA structure, reduced the level of coprecipitated NS3 but maintained it at a certain level (Fig. 1C), indicating that the association between NS3 and WRN could be DNA dependent and independent. Cellular fractionation demonstrated codistribution of NS3 and WRN in the cytoplasm and chromatin-bound fractions of the HCVR cells (Fig. 1D).
FIG 1.

HCV NS3 protein interacts with WRN. (A) GST pulldown assay. GST and GST-NS3 proteins expressed in E. coli were purified following incubation with glutathione agarose resin. The proteins were subjected to SDS-PAGE, followed by Coomassie blue staining (left) and Western blot analysis with antibodies specific to GST (middle). A GST pulldown assay was performed with Huh7 cell lysate, followed by Western blotting with antibodies as indicated (right). Input represents an aliquot of the Huh7 cell lysate run in parallel as a control. (B) Coimmunoprecipitation assay of NS3 with WRN. Expression of NS3 protein in the inducible cell line TR-NS3 upon doxycycline treatment is shown on the left. Coimmunoprecipitation was carried out with protein lysates prepared from the TR-NS3 cells and antibodies specific to WRN or control IgG, as indicated, followed by SDS-PAGE and Western blot analysis with antibodies against WRN, the V5 tag of the NS3-V5His protein, and the actin control. (C) DNA-dependent and -independent association of NS3 with WRN in HCVR cells. Protein lysates were prepared from HCVR and control Huh7 cells in the presence of EtBr and incubated for various time periods as indicated. Coimmunoprecipitation and Western blot analysis were performed with antibodies as indicated. (D) Fractionation of HCVR cell lysate. Cell lysates harvested from HCVR and control Huh7 cells were separated into cytoplasm (CP), nucleoplasm (NP), and chromatin-bound (CB) fractions. Western blot analysis was performed to detect the expression of WRN and NS3 protein. GAPDH, topoisomerase I (TOP1), and histone H3 served as markers and internal controls of the individual cellular compartments as indicated.
HCV NS3 and NS3/4A protein impair WRN-mediated DNA repair.
WRN is known to participate in multiple DNA repair pathways. To examine the effects of NS3 on DNA repair, NS3-expressing HCVR cells and TR-NS3 and TR-NS3/4A cells were exposed to CPT to induce DNA DSBs. Cells released from CPT treatment were then harvested at different time points and subjected to Western blotting to detect the level of the DNA damage marker, γH2AX, as an indication of the status of DNA repair (25). Meanwhile, the level of WRN protein was analyzed.
As shown in Fig. 2A, the level of γH2AX in the control Huh7 cells increased upon CPT treatment and declined at 1 h after release from CPT treatment but remained high in HCVR cells during the time period of 0 to 1 h after CPT release. A similar trend was observed in the NS3- and NS3/4A-expressing cells [Fig. 2B and C, Dox(+)]. The greater duration of a high γH2AX level implies a delay in DNA repair in the NS3-expressing HCVR cells and TR-NS3 and TR-NS3/4A cells. In addition, CPT treatment induced WRN expression in the control Huh7 and Dox(−) TR-NS3 and TR-NS3/4A cells; the level peaked at the 0.5- to 1-h time point after CPT release, temporally coincident with the increase of γH2AX (Fig. 2A to C). These results suggest that WRN is critical in the early phase of CPT-induced DNA damage repair in the control cells. The phenomenon of CPT-induced enhancement of WRN expression in the control cells was not observed in HCVR and Dox-pretreated NS3- and NS3/4A-expressing cells (Fig. 2A to C). This indicates that NS3 and NS3/4A proteins trigger impairment of the repair of CPT-induced DNA DSB, possibly associated with the function and protein level of WRN. A link between the impairment to the expression of HCV NS3 and NS3/4A proteins and its association with WRN was then examined by WRN knockdown and rescue experiments. As shown in Fig. 2D, 1 h after CPT release, both WRN-knockdown Huh7 and 293T cells showed a remarkably higher level of γH2AX than that of the control cells, suggesting increased damage of the WRN-knockdown cells and the critical role of WRN in the repair of CPT-induced DNA DSB. In addition, ectopic overexpression of WRN dose-dependently reduced the CPT-induced γH2AX level in both transfected NS3- and NS3/4A-expressing 293T cells and the HCVR cells (Fig. 2E). Taken together, these results indicate interference of HCV nonstructural proteins in WRN-mediated DNA repair, possibly through interaction of the NS3 protein with WRN. Alternatively, the viral proteins may enhance cellular sensitivity to DNA damage that requires the function of WRN for repair.
FIG 2.

HCV protein-induced impairment of DNA repair is associated with WRN. (A to C) The protein levels of γH2AX and WRN in NS3-expressing cells released from CPT treatment. HCVR and control Huh7 cells were left untreated (U) or treated with 1 μM CPT for 1 h and then released at different time points, as indicated (A). Similar studies were performed with TR-NS3 (B) and TR-NS3/4A cells (C) 48 h after doxycycline induction. Cell lysates were prepared for Western blot analysis. Relative intensities (RIs) of γH2AX and WRN are shown. (D) The γH2AX level in WRN knockdown cells. Huh7 and 293T cells were transduced with lentivirus carrying the gene of WRN shRNA and then treated with 1 μM CPT for 1 h. One hour after release from CPT treatment, the cells were harvested for Western blot analysis. Cells transduced with lentivirus carrying the gene of LacZ shRNA were analyzed in parallel as the control. (E) The γH2AX level in cells ectopically overexpressing WRN. 293T cells were transfected with plasmids encoding NS3-V5His, NS3/4A-V5His, and EGFP-WRN-mycHis or an empty vector control, as indicated. Meanwhile, HCVR cells were transfected with various doses of the EGFP-WRN-mycHis plasmid. Forty-eight hours posttransfection, the cells were kept untreated (U) or treated with CPT for 1 h and then harvested at 0 and 1 h after release from the treatment, as indicated. The cell lysates were subjected to Western blot analysis.
NS3 protein interacts with Ku70 and impairs NHEJ repair.
CPT treatment stimulates DNA DSBs, a severe form of damage hazardous to cells. Mechanisms participating in DSB repair in mammalian cells include NHEJ and HR (23, 26–31). As WRN participates in both NHEJ and HR repair (24), mechanisms by which HCV nonstructural proteins are involved in the WRN-mediated DNA repair were investigated. A pulldown assay was performed with GST-NS3 protein and Huh7 cell lysates to examine potential interactions of the NS3 protein with members participating in WRN-associated DNA repair pathways, including the catalytic subunit of DNA-PK (DNA-PKcs), Ku70/80, XRCC4, NBS1, Mre11, CtIP, Rad51, and PARP1. The results demonstrated that Ku70 was the major component that interacted with the NS3 protein (Fig. 3A). Coimmunoprecipitation assays also demonstrated the association of NS3 with Ku70 in both TR-NS3 and HCVR cells (Fig. 3B). It is known that Ku70 forms a heterodimer with Ku80 and takes part as the initiation factor in NHEJ repair (32). Following a subsequent recruitment of DNA-PKcs to phosphorylate a number of downstream targets, Artemis and WRN join the process of NHEJ (28, 33, 34).
FIG 3.
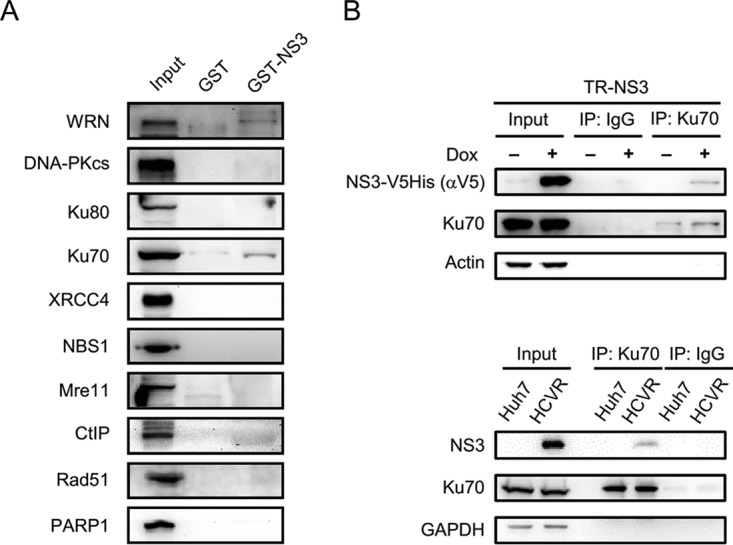
The interaction between NS3 protein and Ku70. (A) GST pulldown assay. A GST pulldown assay was performed with GST-NS3 protein and Huh7 cell lysates, followed by Western blotting with antibodies specific to proteins involved in the WRN-associated DNA repair system, as indicated. (B) Coimmunoprecipitation assay. An immunoprecipitation assay was performed with protein lysates prepared from TR-NS3, HCVR, and control Huh7 cells using polyclonal anti-Ku70 antibodies (XRCC6 A7330) or control IgG, followed by Western blotting, as indicated.
To examine the effects of HCV NS3 protein on NHEJ repair, overall and precise NHEJ efficiencies were analyzed in HCVR cells and cells expressing the viral NS3 and NS3/4A proteins following cotransfection of reporter plasmids, as described in Materials and Methods. The results demonstrated that overall NHEJ efficiency decreased to 59% in HCVR cells compared to the level in the control Huh7 cells, whereas little difference was detected in precise NHEJ efficiency values (Fig. 4A). However, both NHEJ efficiencies were significantly reduced in NS3- and NS3/4A-expressing cells. The overall and precise NHEJ efficiencies were reduced to 40% and 62%, respectively, in Huh7 cells expressing the NS3 protein, and reduced to 46% and 64%, respectively, in NS3/4A-expressing cells compared to levels in the control Huh7 cells (Fig. 4B, top). Reduced efficiencies of overall and precise NHEJ repair were also detected in NS3- and NS3/4A-expressing 293T cells (Fig. 4B, bottom). These results indicate that NHEJ repair is impaired by the NS3 and NS3/4A proteins. It remains to be elucidated whether the viral NS4B/5A/5B proteins coexpressing with NS3/4A in the HCVR cells contribute to the precise NHEJ repair that recovers the efficiency inhibited by the NS3 and NS3/4A proteins. Alternatively, NS4B/5A/5B proteins may prevent NS3 and NS3/4A from interfering with the precise NHEJ repair. In addition, we also found that there was little difference in the efficiencies of HR repair between the control and the NS3/4A-expressing cells, but the efficiency was increased in the NS3-expressing cells (Fig. 4C). NS3 possesses helicase activity and can also function as a protease in the presence of NS4A cofactor (4, 35). This raised interesting questions of whether NS3 could interact with WRN via their helicase domains to regulate the functions of WRN and whether WRN could be a substrate of NS3 protease. In addition, NS3 and NS3/4A proteins may impair DNA repair by suppressing NHEJ through interaction with Ku70.
FIG 4.
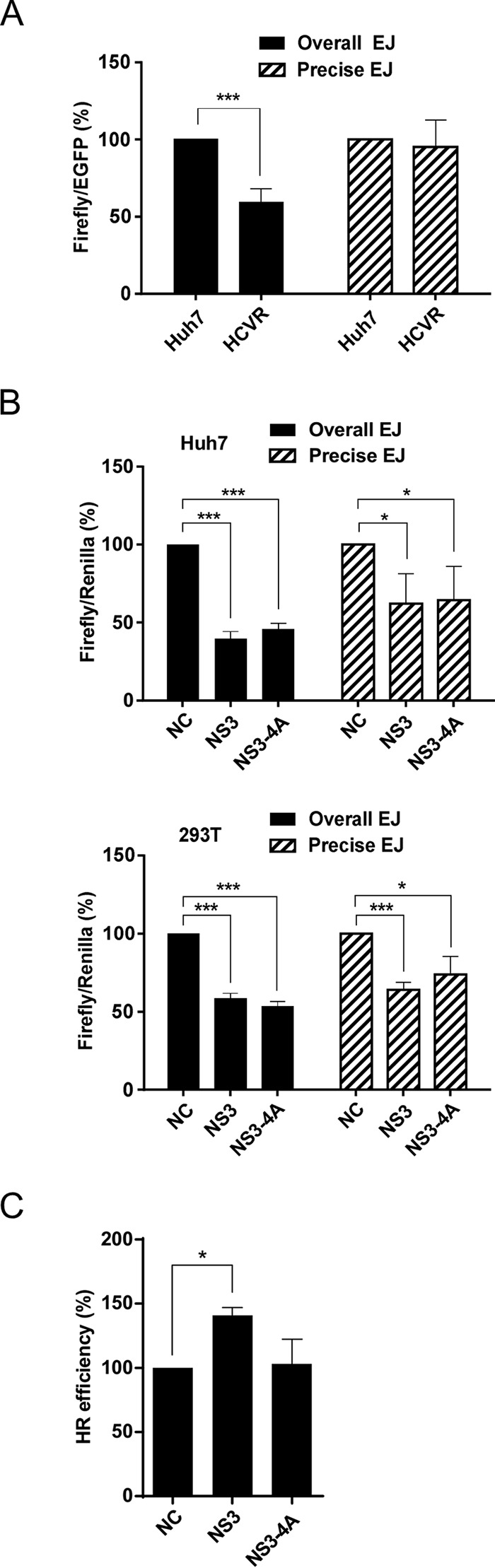
HCV NS3 and NS3/4A inhibit NHEJ repair. (A and B) NHEJ assay. The overall and precise NHEJ efficiencies in HCVR and in 293T or Huh7 cells expressing NS3 or NS3/4A protein were measured by luciferase assay. The luminescence value of firefly luciferase was normalized against the internal EGFP fluorescence control (A) or Renilla luminescence control (B). Statistical analysis is shown (n = 3). P values define significant differences between the NHEJ efficiencies of the Huh7 and HCVR cells or between those of the negative control (NC) and NS3- or NS3/4A-expressing cells. (C) HR assay. The HR efficiency in 293T cells expressing NS3 or NS3/4A protein was calculated by normalization of the percentage of EGFP-positive cells to the luminescence value of the firefly luciferase control. Statistical analysis is shown (n = 3). P values define significant differences between those of the negative control (NC) and NS3- or NS3/4A-expressing cells. *, P < 0.05; ***, P < 0.001.
HCV NS3 protein alters the nuclear distribution of the repair complex.
To elucidate the effects of NS3 and NS3/4A proteins on Ku70-initiated NHEJ, the distribution of WRN and Ku70 in CPT-treated NS3- and NS3/4A-expressing cells was analyzed by confocal microscopy. As shown in Fig. 5A, we found that WRN partially colocalized with Ku70 and γH2AX in the nuclei of the CPT-treated control Huh7 cells. The colocalization of WRN with Ku70 became remarkable in the NS3- and NS3/4A-expressing cells, but they did not localize to the γH2AX foci. These phenomena were also observed in HCVR cells (Fig. 5A). Statistical analysis showed a significant reduction in the percentage of cells with WRN and Ku70 localized to γH2AX foci in both NS3- and NS3/4A-expressing and HCVR cells (Fig. 5B). These results indicate that the association of WRN with Ku70 in the viral protein-expressing cells may not localize to the sites of CPT-induced DNA DSBs and explain the NS3- and NS3/4A-mediated impairment of NHEJ repair as a consequence of function loss of the WRN-Ku70 complex. In addition, the number of γH2AX foci in CPT-treated NS3- and NS3/4A-expressing cells was slightly lower than that in the CPT-treated control Huh7 cells, but the number in CPT-treated HCVR cells was higher than that in the control cells (Fig. 5C). The increase in γH2AX foci in HCVR cells may be partially explained by the effect of the viral NS5A protein on the impairment of HR repair through disruption of the RAD51/RAD51AP1/UAF1 complex (36).
FIG 5.

HCV nonstructural proteins alter the nuclear distribution of repair complex in CPT-treated cells. Doxycycline-treated TR-NS3 and TR-NS3/4A cells, the control Huh7 cells, and HCVR cells were subjected to CPT treatment for 1 h. Immunofluorescence staining was performed at 0.5 h after the cells were released from the CPT treatment (A). In addition, the percentages of cells in which WRN/Ku70 localized to the γH2AX foci were calculated (B) and numbers of γH2AX foci per cell were counted (C). Statistical analysis is shown (n = 3). At least 100 cells were analyzed for each experimental condition. P values define significant differences between two groups, as indicated. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
HCV NS3 protein interferes with Ku70 recruitment to the DSB sites.
To examine the hypothesis that HCV NS3 protein disrupts binding of the WRN-Ku70 complex to the DSB sites, immunoprecipitation and DNA pulldown assays were performed. Results demonstrated coimmunoprecipitation of Ku70 with WRN in both CPT-treated and untreated HCVR and NS3-expressing TR-NS3 cells (Fig. 6A). WRN-Ku70 complex was also detected, although at a much lower level, in the control Huh7 and Dox(−) TR-NS3 cells. In addition, consistent with the observation from confocal microscopy, an association of γH2AX with the WRN-Ku70 complex was detected only in the CPT-treated control Huh7 and Dox(−) TR-NS3 cells, not in the HCVR cells, and barely in the NS3-expressing TR-NS3 cells (Fig. 6A). The suppressive effect of NS3 on the interaction between WRN and γH2AX was also detected in ionizing radiation (IR)-exposed Dox(+) TR-NS3 cells (Fig. 6B). IR is known to induce DSBs that distribute randomly, and the repair of IR-induced DSBs in mammalian cells is mainly mediated by NHEJ (37). Further DNA pulldown assays demonstrated a reduction in the efficiency of Ku70 recruitment to the DNA DSBs in both NS3-expressing TR-NS3 and HCVR cells (Fig. 6C). Taken together, these data suggest that HCV NS3 protein interferes with the formation of WRN-Ku70 complex at the DSB sites, resulting in impairment of the Ku70-initiated NHEJ repair.
FIG 6.

HCV protein interferes with the formation of repair complex. (A and B) NS3 inhibits WRN-γH2AX interaction. NS3-expressing HCVR and Dox(+) TR-NS3 cells and their corresponding control cells were treated with CPT (A) or IR (B). Protein lysates were prepared from the cells 0.5 h after release from the treatment. Immunoprecipitation was performed with antibodies against WRN, followed by SDS-PAGE and Western blot analysis. Cells that remained untreated (U) were analyzed in parallel as controls. (C) DNA pulldown assay. Protein lysates prepared from HCVR and control Huh7 cells released from IR treatment were subjected to DNA pulldown assay by incubation with a biotinylated DSB DNA probe immobilized on streptavidin Sepharose beads. After washing, the reaction products were resolved by SDS-PAGE for Western blot analysis. Statistical analysis is shown (n = 3). P values define significant differences of Ku70 binding between two groups. **, P < 0.01.
The level of WRN protein is regulated by both proteasome and NS3/4A protease activity.
We have earlier shown a delay in the repair of CPT-induced DNA damage in both HCVR and Dox-inducible NS3- and NS3/4A-expressing cells (Fig. 2A to C). The impairment of DNA repair was linked to the level of WRN protein (Fig. 2D and E). To better understand the effects of NS3/4A protein on WRN-mediated DNA repair, we further elucidated whether NS3/4A protein can regulate the stability of WRN protein. The protein synthesis inhibitor cycloheximide (CHX) and proteasome inhibitor MG132 were applied. As expected, CHX treatment reduced the level of WRN protein in both the Dox(−) control and Dox(+) NS3/4A-expressing cells. The reduced WRN levels were alleviated when the cells were treated with MG132 alone or cotreated with CHX and MG132 (Fig. 7A). However, the protection from degradation in the NS3/4A-expressing cells did not reach the same extent as observed in the Dox(−) control cells. These results indicate involvement of both proteasome-dependent and -independent pathways in WRN protein degradation and suggest a potential role of the NS3/4A protease activity in downregulating the level of WRN protein. Mechanisms involved in the degradation of WRN protein were further examined by introducing TR-NS3pd/4A cells in which expression of an NS3/4A protein defective in protease activity, termed NS3pd/4A (4), can be induced by doxycycline. TR-NS3/4A and TR-NS3pd/4A cells were treated with CHX and harvested at different time points for Western blot analysis. As shown in Fig. 7B, WRN protein was nearly completely diminished at 6 h after CHX treatment in the NS3/4A-expressing cells but remained at a level of approximately 50% at the 6-h time point in TR-NS3pd/4A cells. The possibility that NS3/4A protein targets WRN for degradation was then examined.
FIG 7.

The level of WRN protein is regulated by proteasome and HCV NS3 protease activity. (A) WRN level is regulated by both proteasome-dependent and -independent degradation pathways. TR-NS3/4A cells induced by Dox for 48 h were treated with the proteasome inhibitor MG132 20 min before the CHX treatment or treated with CHX or MG132 alone, as indicated. The cells were harvested 8 h after the CHX treatment for Western blot analysis. Statistical analysis is shown (n = 3). *, P < 0.05; **, P < 0.01. (B) HCV NS3/4A reduces the stability of WRN. After Dox induction, TR-NS3/4A and TR-NS3pd/4A cells were treated with CHX and harvested at the indicated time points for Western blot analysis. Plot shows the statistical analysis (n = 3). ***, P < 0.001.
WRN is a substrate of NS3/4A protease.
To investigate whether WRN can be a target of NS3/4A protease, a cleavage assay was performed. Plasmid encoding an enhanced green fluorescent protein (EGFP)-WRN fusion protein with a molecular size of approximately 210 kDa with myc and His tags at the C terminus was cotransfected with NS3/4A-expressing plasmid into 293T cells. Cell lysates were harvested at 2 days posttransfection for Western blot analysis. As shown in Fig. 8A (left), in addition to the full-length EGFP-WRN-mycHis protein, an 80-kDa N-terminal product and a 130-kDa C-terminal product were generated, as detected by antibodies specific to EGFP and myc tag, respectively. These cleavage products were also detected in HCVR cells ectopically expressing the full-length EGFP-WRN-mycHis fusion protein (Fig. 8A, right) but not in either the NS3pd/4A-expressing cells or the control cells lacking the full-length EGFP-WRN-mycHis protein or the NS3/4A protein (Fig. 8A, left). These results demonstrated that WRN is a substrate of the NS3/4A protease. A search for the presence of the consensus sequence of the trans cleavage site of NS3/4A protease, (D/E)XXXX(T/C)↓(S/A) (38), in the WRN protein was conducted. This identified potential cleavage sites in the repeat sequences of the WRN acidic region, 424-HLSPNDNENDT↓SYVIESDEDLEMEMLK-450 and 451-HLSPNDNENDT↓SYVIESDEDLEMEMLK-477 (Fig. 8B). A mutant form of the full-length EGFP-WRN-mycHis protein, designated EGFP-WRN(ARm)-mycHis, in which both of the predicted consensus sequences DNENDTS have been mutated to DNENDAA, was expressed in the culture system for cleavage assay. However, both the 80-kDa and the 130-kDa cleavage products were detected in the cells coexpressing NS3/4A protein and EGFP-WRN(ARm)-mycHis (Fig. 8B). These results suggest the possibility that HCV NS3/4A protease targets the WRN mutant protein at cryptic sites near the predicted cleavage sequences. Although the cleavage site has not yet been defined, we tested the hypothesis that the protease activity of NS3/4A involved in WRN degradation would lead to the impairment of NHEJ repair. An NHEJ assay was performed in TR-NS3/4A and TR-NS3pd/4A cells. As shown in Fig. 8C, the overall and precise NHEJ efficiencies in NS3pd/4A-expressing cells were 2.2-fold and 1.7-fold, respectively, of those in the NS3/4A-expressing cells. Since NS3/4A protein reduced the NHEJ efficiency by half (Fig. 4), these results strongly suggest that WRN cleavage is critical for the NS3/4A-mediated impairment of DNA repair.
FIG 8.

WRN is a substrate of HCV NS3/4A protease. (A) An NS3/4A-dependent cleavage of WRN protein. A WRN cleavage assay was performed by cotransfecting 293T cells with plasmids encoding NS3/4A-V5His or NS3pd/4A-V5His and an EGFP-WRN fusion protein (approximately 210 kDa) with myc and His tags at the C terminus. Twenty-four hours posttransfection, the cells were treated with MG132 for 24 h and harvested for Western blot analysis. The cleavage assay in HCVR and control Huh7 cells was also analyzed 72 h after transfection of the plasmid encoding the EGFP-WRN fusion protein. (B) WRN cleavage in cells expressing NS3/4A protease and the WRN mutant protein EGFP-WRN(ARm)-mycHis. (C) NHEJ assay in TR-NS3/4A and TR-NS3pd/4A cells. The overall and precise NHEJ efficiencies in TR-NS3/4A and TR-NS3pd/4A cells were measured by luciferase assay. The luminescence value of firefly luciferase was normalized against the internal EGFP fluorescence control. Statistical analysis is shown (n = 3). P values define significant differences between the NHEJ efficiencies of TR-NS3/4A and TR-NS3pd/4A cells. **, P < 0.01; ***, P < 0.001.
DISCUSSION
HCV NS3 protein is considered a critical factor associated with cell oncogenesis in HCV-infected patients. ROS stimulation and impairment of DNA repair may provide clues to elucidate the development of oncogenesis with NS3 (6, 7), but the details remain unclear. In this study, we found that HCV NS3 protein interacts with WRN and plays a dual role in the suppression of WRN-mediated NHEJ repair. It interferes with Ku70 recruitment to the DSB sites in initiating DNA repair and at the same time participates in the degradation of WRN.
WRN protein possesses both helicase and 3′-to-5′ exonuclease activities and is known to participate in multiple DNA repair pathways (16, 39), whereas Ku is known to function as a guide to label the DSB sites and is required for NHEJ repair (40). In addition, Ku70 interacts with WRN and regulates its exonuclease activity (33). The interaction between WRN and Ku may play a key role in the stimulation of NHEJ repair (41). In this study, we identified the novel interactions of HCV NS3 protein with WRN and Ku70 (Fig. 1 and 3) and observed a delay in the repair of DNA damage induced by the topoisomerase I inhibitor CPT in both NS3-expressing TR-NS3 and TR-NS3/4A and HCVR cells (Fig. 2). In addition, an elevated WRN level temporally coincident with the duration of a high γH2AX level was detected in CPT-treated control cells but not in NS3-expressing cells. With Ku acting as the sensor of DNA DSB sites, recruiting DNA-PKcs to initialize NHEJ repair (42), it is likely that NS3 interferes with DNA repair through interaction with Ku and WRN. When an NHEJ assay was performed in NS3- and NS3/4A-expressing cells, reduced efficiencies were detected for levels of both overall and precise NHEJ repair (Fig. 4B). In addition, confocal microscopy and DNA pulldown assays demonstrated interference of the Ku-mediated recognition of DNA DSB ends in forming a Ku70/WRN repair complex in HCVR cells (Fig. 5 and 6). It remains to be elucidated whether the viral NS4B/5A/5B proteins coexpressing with NS3/4A are responsible for the recovered precise NHEJ repair efficiency in the HCVR cells (Fig. 4A). Recently, HCV NS5A protein was reported to contribute to increased cellular sensitivity to DNA damage agents (36, 43). An increased level of γH2AX foci was detected in NS5A-expressing cells. HCV NS5A protein impairs DNA damage HR repair by disrupting RAD51/RAD51AP1/UAF1 complex formation and by translocation of RAD51 to the cytoplasm (36). The effect of NS5A may partially explain the elevated level of γH2AX foci in the HCVR cells (Fig. 5C).
HCV NS3 protein possesses protease activity. With the cofactor NS4A, the NS3 protease cleaves the viral nonstructural proteins at the consensus sequences (D/E)XXXX(C/T)↓(S/A). The NS3/4A protease also cleaves host proteins MAVS, TRIF, and TC-PTP at 503-EXXXPC↓H-509, 367-PXXXXC↓S-373, and 211-PXXXXC↓S-217, respectively (9–11). In this study, a WRN cleavage assay identified NS3/4A-dependent production of the proteolysed WRN fragments (Fig. 8A). Further analysis demonstrated that both overall and precise NHEJ efficiencies in the TR-NS3pd/4A cells were approximately twice the levels in the TR-NS3/4A cells (Fig. 8C), indicating that NS3 protease activity plays a key role in the suppression of WRN-mediated NHEJ repair.
The absence of WRN protein in individuals has been associated with Werner syndrome, an inherited disorder with the characteristics of premature aging and a higher risk of cancer (44–46). It is not clear whether HCV infection would generate genome instability associated with WRN syndrome. However, our results clearly indicate that HCV would target WRN for degradation while diminishing NHEJ repair by interfering with the recruitment of the Ku-WRN complex to the DNA DSB sites.
MATERIALS AND METHODS
Plasmids.
Plasmids pGST-NS3, pcDNA-NS3-V5HisTopo, pcDNA-NS(3/4A)-V5HisTopo, pcDNA-NS(3pd/4A)-V5HisTopo, pLenti-Flag-NS3-V5His, pLenti-Flag-NS(3/4A)-V5His, and pLenti-Flag-NS(3pd/4A)-V5His were constructed as follows. Plasmid pGST-NS3 encodes a GST-NS3 fusion protein and was generated by inserting the NS3 cDNA fragment into pGEX6p-1. Plasmids pcDNA-NS3-V5HisTopo (47), pcDNA-NS(3/4A)-V5HisTopo (4), and pcDNA-NS(3pd/4A)-V5HisTopo (4) have been described previously. For construction of plasmids pLenti-Flag-NS3-V5His, pLenti-Flag-NS3/4A-V5His, and pLenti-Flag-NS3pd/4A-V5His, DNA fragments of NS3-V5His, NS3/4A-V5His, and NS3pd/4A-V5His with RsrII sites created on both ends were obtained by PCR amplification from pcDNA-NS3-V5HisTopo, pcDNA-NS(3/4A)-V5HisTopo, and pcDNA-NS(3pd/4A)-V5HisTopo, respectively, followed by digestion with RsrII restriction endonuclease and independently cloned into the RsrII site of pLenti4-Flag-CPO (48). These constructs, under the transcriptional control of a reverse tetracycline-controlled transactivator-dependent promoter, allow expression of the respective viral proteins with a Flag tag at the N terminus and V5 and His tags at the C terminus upon doxycycline induction.
Plasmids pWRN-mycHis, pEGFP-WRN-mycHis, and pEGFP-WRN(ARm)-mycHis were constructed as follows. For construction of plasmid pWRN-mycHis, a DNA fragment of WRN with KpnI and XhoI sites at the ends was obtained by PCR amplification from pLXSN-WRN (49) and digestion with restriction endonucleases. The DNA fragment was then cloned into pcDNA4-mycHisA (Invitrogen) to generate pWRN-mycHis. For construction of plasmid pEGFP-WRN-mycHis, an EGFP cDNA fragment with KpnI sites created on both ends was obtained by PCR amplification from pAdTrack-CMV (where CMV is cytomegalovirus) and digestion with KpnI. The DNA fragment was then cloned into the KpnI site of pWRN-mycHis to yield plasmid pEGFP-WRN-mycHis. Plasmid pEGFP-WRN-mycHis can express EGFP-WRN fusion protein with myc and His tags at the C terminus when introduced into cultured cells. In addition, plasmid pEGFP-WRN(ARm)-mycHis encodes a GFP-WRN-mycHis protein with T434A/S435A and T461A/S462A mutations in the acidic region, designated GFP-WRN(ARm)-mycHis. For construction of plasmid pEGFP-WRN(ARm)-mycHis, two DNA fragments with sizes of 0.91-kb and 2.9-kb, designated WRN(148–450)ARm1 and WRN(446–1432)ARm2, which represent WRN fragments from amino acid residues 148 to 450 with T434A/S435A mutations and amino acids 446 to 1432 with T461A/S462A mutations, respectively, were generated. These two DNA fragments were then used to replace the 3.8-kb EcoRV-XhoI fragment of pEGFP-WRN-mycHis to generate plasmid pEGFP-WRN(ARm)-mycHis by using an In-Fusion cloning kit (Clontech).
Cell lines. (i) 293T and Huh7 cells.
Human embryonic kidney 293T and hepatocellular carcinoma Huh7 cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% heat-inactivated fetal bovine serum and 1% 100× nonessential amino acids plus 100 units of penicillin and 100 μg of streptomycin per ml in a humidified 5% CO2 atmosphere at 37°C.
(ii) H7-HCVR cells and inducible cell lines TR-NS3, TR-NS3/4A, and TR-NS3pd/4A.
H7-HCVR cells were derived from Huh7 cells by inclusion of an HCV 1b subgenomic replicon as previously described (50). For establishment of inducible cell lines (TR-NS3, TR-NS3/4A, and TR-NS3pd/4A), Huh7-TR cells which express an inducible Tet-on repressor (TR) was first established by transduction of lentivirus carrying the TR gene (TR virion) into Huh7 cells, followed by selection under blasticidin. Huh7-TR cells were then transduced with lentivirus expressing Flag-NS3-V5His (NS3 virion), Flag-NS3/4A-V5His (NS3/4A virion), and Flag-NS3pd/4A-V5His (NS3pd/4A virion) and selected under zeocin (50 μg/ml) for the establishment of, respectively, inducible TR-NS3, TR-NS3/4A, and TR-NS3pd/4A cell lines. Expression of the viral proteins were induced by treatment of the cells with doxycycline for 48 h.
Production of lentivirus particles.
For production of TR, NS3, NS3/4A, and NS3pd/4A virions, 293T cells in 10-cm plates (at 2 × 106 cells/plate) were transfected with the packaging vector pCMVdeltaR8.91, the vesicular stomatitis virus G (VSV-G) envelope glycoprotein-expressing vector pMD.G, and the corresponding expression vectors, pLenti6/TR, pLenti-Flag-NS3-V5His, pLenti-Flag-NS3/4A-V5His, and pLenti-Flag-NS3pd/4A-V5His, respectively, with T-Pro NTR II transfection reagent (T-Pro Biotechnology). The cultured medium was collected at 48 h posttransfection and subjected to centrifugation and filtration to remove cell debris. The lentivirus particle suspensions were stored at −80°C.
Antibodies.
Monoclonal anti-V5 antibody was purchased from Invitrogen. Monoclonal antibodies against RecQL1 (A-9), BLM (B-4), RecQ5 (C-9), XRCC4 (C-4), Rad51 (3C10), PARP1 (B-10), CtIP (D-4), NBS1 (B-5), Ku70 (E-5; for Western bot analysis and immunofluorescence staining), Ku86 (B-1), DNA-PKcs (G-12), RPA, and c-myc (9E10) and polyclonal antibodies against WRN (H300) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were from Santa Cruz. Polyclonal anti-RecQL4 (A6846) and anti-Ku70 (XRCC6 A7330; for immunoprecipitation and immunofluorescence staining) antibodies were from ABclonal. Polyclonal anti-histone H3 (ab1791), anti-Mre11, and anti-TOP1 (A302-589A) antibodies were from Abcam, Calbiochem, and Bethyl Laboratories, respectively. Monoclonal anti-NS3 antibody (8G-2) was from Abcam, anti-γ-H2AX antibody (JBW301) was from Millipore, and anti-β-actin antibody was from Sigma-Aldrich. Polyclonal antibodies against α-tubulin (GTX628802) and GST (GTX114099) were from GeneTex. Rabbit polyclonal antibodies against EGFP were generously provided by Shin-Lian Doong (National Taiwan University, Taiwan). Horseradish peroxidase-conjugated secondary antibodies were purchased from Jackson Laboratory. Alexa Fluor 488- and Alexa Fluor 594-conjugated secondary antibodies were from Invitrogen.
Expression of a GST fusion protein and GST pulldown assay.
GST fusion plasmids were transformed into Escherichia coli BL21. Expression of GST fusion proteins was induced following treatment of the bacterial cultures with isopropyl-1-thio-β-d-galactopyranoside (IPTG) at 25°C for 18 h. For preparation of bacterial lysates, the bacterial cells were lysed in NETN buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.1% Nonidet P-40, 1 mM phenylmethylsulfonyl fluoride [PMSF], 0.5 mM NaF, 0.5 mM Na3VO4, and 1% complete EDTA-free protease inhibitor cocktail [Roche]) with sonication. Supernatants of the bacterial lysates were collected and incubated with glutathione agarose resin at 4°C for 18 h for partial purification of the GST fusion proteins. Following centrifugation, GST fusion proteins immobilized on the glutathione agarose resin were washed and resuspended in NETN buffer for GST pulldown assay. To perform the GST pulldown assay, Huh7 cell lysates in NETN buffer were incubated with GST fusion protein-immobilized glutathione resin at 4°C for 18 h. After an extensive wash with NETN buffer, GST fusion proteins and their interacting proteins were separated by SDS-PAGE, followed by Coomassie blue staining or Western blot analysis.
Immunoprecipitation and Western blot analysis.
Cell lysates prepared from cultured cells treated with immunoprecipitation-radioimmunoprecipitation assay (IP-RIPA) buffer (10 mM Tris-HCl, pH 8.0, 150 mM NaCl, 5 mM EDTA, 0.1% Nonidet P-40, and 1% complete EDTA-free protease inhibitor cocktail [Roche]) were incubated with primary antibody overnight at 4°C. Protein A beads (GE Healthcare) were then added, and the mixtures were incubated for an additional 4 h at 4°C to capture the antibody complex. For analysis of DNA-dependent protein association, EtBr (50 μg/ml) was added to the cell lysates as described previously (51) and incubated for various time periods. The immunoprecipitated proteins were run on an SDS-polyacrylamide gel and subjected to Western blot analysis.
To perform Western blot analysis, proteins resolved by SDS-PAGE were electrotransferred onto an Immobilon-P membrane (Millipore). The membranes were incubated with primary antibodies and then reacted with horseradish peroxidase-conjugated secondary antibodies. Immunoreactivity was visualized by an ECL LumiFast Chemiluminescent Substrate kit (T-Pro Biotechnology).
Cellular fractionation.
A 2-step centrifugation was carried out for separation of cytoplasmic, nucleoplasmic, and chromatin-bound proteins. In brief, cell lysate harvested after a treatment of cultured cells with NP-40 lysis buffer (50 mM Tris-HCl, pH 8.0, 10 mM NaCl, 5 mM MgCl2, and 0.5% Nonidet P-40) was subjected to centrifugation at 15,000 × g for 15 min. Supernatant representing the cytoplasmic fraction was collected, and the nuclear pellet was resuspended in high-salt buffer (20 mM HEPES, pH 7.9, 0.5 M NaCl, 1 mM EDTA, and 1 mM dithiothreitol [DTT]). Following a second centrifugation at 15,000 × g for 15 min, supernatant was collected as the nucleoplasm fraction. Chromatin-bound proteins were obtained by resuspending the pellet in Western blot sample buffer and heating at 95°C for 5 min. GAPDH, topoisomerase I, and histone H3 were used as markers of the cytoplasm, nucleoplasm, and chromatin-bound fractions, respectively, in Western blot analysis.
Immunofluorescence staining.
Cells cultured on glass slides were washed three times with phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde for 10 min, and then permeabilized in 0.5% Triton X-100 for 10 min. Primary antibodies were diluted in 4% bovine serum albumin (BSA) and incubated with cells for 1 h at room temperature. After three washes in PBS, the cells were stained with fluorescein- and/or rhodamine-conjugated secondary antibodies for 1 h at room temperature, washed in PBS, and mounted in 4′,6′-diamidino-2-phenylindole (DAPI) Fluoromount-G (SouthernBiotech). Photographs of the cells were taken with a broadband confocal microscope (Leica TCS SP5). Image analysis was performed using the Leica Application Suite provided with the microscope.
Knockdown of WRN.
For knockdown of WRN, cultured cells were transduced with lentivirus carrying the gene of a WRN short hairpin RNA (shRNA). After 72 h, the cells were selected by puromycin (2 μg/ml) for 2 to 3 days. Lentiviruses expressing WRN shRNA and LacZ shRNA were generated as described above (see “Production of lentivirus particles”). Lentiviral vectors pLKO.1-shRNAWRN and pLKO.1-shRNALacZ were purchased from the National RNAi core Facility, Academia Sinica, Taiwan. The shRNA sequence targeting WRN is 5′-CCTGTTTATGTAGGCAAGATT-3′ (TRCN0000004902). A lacZ shRNA construct was used as a nontargeting control.
NHEJ assay.
An NHEJ assay was performed as previously described (52) with modifications. In brief, H7-HCVR cells were transiently transfected with a linearized pGL3-promoter reporter plasmid harboring a firefly luciferase gene and the transfection efficiency internal control plasmid pAdTrack-CMV carrying an EGFP gene. For the overall NHEJ assay, the pGL3-promoter reporter plasmid was linearized with HindIII located at the linker region between the promoter and the coding sequences of the luciferase gene, and for the precise NHEJ assay, the plasmid was linearized with SphI located in the coding region of the luciferase gene. Two days posttransfection, cells were harvested and subjected to luciferase assay using a Dual-Glo luciferase assay kit (Promega). NHEJ assay was also performed in NS3-, NS3/4A-, and NS3pd/4A-expressing cells following transfection of Huh7 or 293T cells with the linearized pGL3-promoter reporter plasmid, the internal control plasmid pAdTrack-CMV carrying an EGFP gene, or pRL-TK carrying a Renilla luciferase gene and the effector plasmid pcDNA-NS3-V5HisTopo, pcDNA-NS(3/4A)-V5HisTopo, pcDNA-NS(3pd/4A)-V5HisTopo, or control vector pcDNA-ctrl-V5HisTopo (53). NHEJ efficiency was determined by normalization of the luminescence value of firefly luciferase with that of Renilla luciferase or with the fluorescence value of EGFP. Detection of luminescence and fluorescence was performed using an Orion II microplate luminometer (Berthold Detection Systems GmbH) and SpectraMax M5 microplate reader (Molecular Devices LLC), respectively.
HR assay.
The HR assay was performed by cotransfecting 293T cells with the HR reporter plasmid pDR-GFP (54), the I-SceI endonuclease-expressing plasmid pCβA-I-SceI, the internal control plasmid pGL3-promoter, and the effector plasmid pcDNA-NS3-V5HisTopo, pcDNA-NS(3/4A)-V5HisTopo, or control vector pcDNA-ctrl-V5HisTopo. Plasmid pDR-GFP carries two GFP mutant genes oriented as direct repeats (DR), and expression of the plasmid pCβA-I-SceI will produce a DSB in one of the GFP genes. The DSB can be repaired by HR between the two GFP mutant genes, resulting in the restoration of a functional GFP gene. Two days posttransfection, cells were pelleted, washed with PBS, and resuspended in PBS for flow cytometer analysis to detect GFP-positive cells. Meanwhile, a luciferase assay was performed for measuring the luminescence value as a control for transfection efficiency. HR efficiency was determined by normalization of the percentage of GFP-positive cells to the luminescence value of firefly luciferase.
DNA pulldown assay.
To perform a DNA pulldown assay, DSB probe was generated by annealing the single strand DNA, 5′-GGCTGGGGGGCGCGTACCAGTGACGTGAGTTGCGGAGGAG-3′ [pGRP78(−210 ∼ −171)] (55), with biotinylated antisense DNA, 5′-CTCCTCCGCAACTCACGTCACTGGTACGCGCCCCCCAGCC-3′ [pGRP78(−210 ∼ −171) complement]. The biotinylated DNA probe was bound to streptavidin Sepharose and blocked in ITB buffer (20 mM HEPES, pH 7.9, 0.2 mM EDTA, 100 mM KCl, 6.25 mM MgCl2, 20% glycerol, 0.01% Triton X-100, 1 mM PMSF, 0.5 mM NaF, 0.5 mM Na3VO4, and 1% complete EDTA-free protease inhibitor cocktail [Roche]) with 5% BSA at 4°C for 1 h. The DNA probe-bound Sepharose was then cleared and incubated with TR-NS3 cell lysate with rocking for 2 h at 4°C. After samples were washed in ITB buffer, proteins bound on the DNA-Sepharose were subjected to SDS-PAGE and Western blot analysis.
WRN cleavage assay.
A WRN cleavage assay was performed by cotransfecting 293T cells with plasmids encoding NS3/4A-V5His or NS3pd/4A-V5His and the wild-type EGFP-WRN-mycHis fusion protein or its mutant form EGFP-WRN(ARm)-mycHis. Twenty-four hours posttransfection, the cells were treated with MG132 to avoid potential degradation of short-half-life products derived from WRN. After an additional 24 h, the cells were lysed with RIPA buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 0.5% sodium deoxycholate, 1% NP-40, 0.1% SDS, 1 mM PMSF, 0.5 mM NaF, 0.5 mM Na3VO4, and 1% complete EDTA-free protease inhibitor cocktail [Roche]) for Western blot analysis. For the analysis of WRN cleavage in H7-HCVR cells, transfection was performed with plasmid pEGFP-WRN-mycHis, and MG132 was added at 48 h posttransfection. The WRN cleavage products were detected by using anti-EGFP and anti-myc antibodies.
Statistical analysis.
Each experiment was repeated at least three times with comparable results. Results are shown as means ± standard deviations (SD). Analysis was performed using Excel 2016 (Microsoft). The P values were obtained using two-tailed Student’s t tests to evaluate differences between two groups, with a P value of < 0.05 considered statistically significant.
ACKNOWLEDGMENTS
We thank Junko Oshima (University of Washington School of Medicine, Seattle) for providing pLXSN-WRN, Maria Jasin (Memorial Sloan Kettering Cancer Center, New York) for providing plasmids pDR-GFP and pCβA-I-SceI, and Shin-Lian Doong (National Taiwan University College of Medicine, Taiwan) for providing rabbit polyclonal antibodies against EGFP. We also thank the National RNAi Core Facility (Academia Sinica, Taiwan) for providing plasmids pCMVdeltaR8.91 and pMD.G and the BioMed Resource Core of the 1st Core Facility Lab, National Taiwan University College of Medicine, for assistance in constructing the pEGFP-WRN(ARm)-mycHis plasmid. We are grateful to Shu-Chun Teng (National Taiwan University College of Medicine), Woei-Horng Fang (National Taiwan University College of Medicine), and Su-Fang Lin (National Health Research Institutes, Taiwan) for their helpful comments on the manuscript.
This work was supported by Research Grants 97-2320-B-002-013-MY3 and 102-2320-B-002-025 from the Ministry of Science and Technology, Republic of China.
We have no conflicts of interest to declare.
T.-I.C. conceived the study and wrote the first draft of the manuscript. T.-I.C., Y.-K.H., C.-Y.C., and Y.-H.C. conducted the experiments and analyzed the results. S.-T.H., Y.-S.L., and Y.-C.D. participated in plasmid construction and provided technical assistance on the expression of recombinant proteins. M.-F.C. and S.C.C. conceived and coordinated the study, interpreted the data, and revised the manuscript. All authors reviewed and approved the final version of the manuscript. The study was supervised by S.C.C.
REFERENCES
- 1.Choo QL, Richman K, Han JH, Berger K, Lee C, Dong C, Gallegos C, Coit D, Medina-Selby A, Barr PJ, Weiner AJ, Bradley DW, Kuo G, Houghton M. 1991. Genetic organization and diversity of the hepatitis C virus. Proc Natl Acad Sci U S A 88:2451–2455. doi: 10.1073/pnas.88.6.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lemon SM, McGivern DR. 2012. Is hepatitis C virus carcinogenic? Gastroenterology 142:1274–1278. doi: 10.1053/j.gastro.2012.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferri C, Caracciolo F, Zignego AL, Civita LL, Monti M, Longombardo G, Lombardini F, Greco F, Capochiani E, Mazzoni A, Mazzaro C, Pasero G. 1994. Hepatitis C virus infection in patients with non-Hodgkin’s lymphoma. Br J Haematol 88:392–394. doi: 10.1111/j.1365-2141.1994.tb05036.x. [DOI] [PubMed] [Google Scholar]
- 4.Kou YH, Chang MF, Wang YM, Hung TM, Chang SC. 2007. Differential requirements of NS4A for internal NS3 cleavage and polyprotein processing of hepatitis C virus. J Virol 81:7999–8008. doi: 10.1128/JVI.00348-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lai CK, Jeng KS, Machida K, Cheng YS, Lai MM. 2008. Hepatitis C virus NS3/4A protein interacts with ATM, impairs DNA repair and enhances sensitivity to ionizing radiation. Virology 370:295–309. doi: 10.1016/j.virol.2007.08.037. [DOI] [PubMed] [Google Scholar]
- 6.Machida K, Cheng KT, Lai CK, Jeng KS, Sung VM, Lai MM. 2006. Hepatitis C virus triggers mitochondrial permeability transition with production of reactive oxygen species, leading to DNA damage and STAT3 activation. J Virol 80:7199–7207. doi: 10.1128/JVI.00321-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Machida K, McNamara G, Cheng KT, Huang J, Wang CH, Comai L, Ou JH, Lai MM. 2010. Hepatitis C virus inhibits DNA damage repair through reactive oxygen and nitrogen species and by interfering with the ATM-NBS1/Mre11/Rad50 DNA repair pathway in monocytes and hepatocytes. J Immunol 185:6985–6998. doi: 10.4049/jimmunol.1000618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deng L, Nagano-Fujii M, Tanaka M, Nomura-Takigawa Y, Ikeda M, Kato N, Sada K, Hotta H. 2006. NS3 protein of hepatitis C virus associates with the tumour suppressor p53 and inhibits its function in an NS3 sequence-dependent manner. J Gen Virol 87:1703–1713. doi: 10.1099/vir.0.81735-0. [DOI] [PubMed] [Google Scholar]
- 9.Li XD, Sun L, Seth RB, Pineda G, Chen ZJ. 2005. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci U S A 102:17717–17722. doi: 10.1073/pnas.0508531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li K, Foy E, Ferreon JC, Nakamura M, Ferreon AC, Ikeda M, Ray SC, Gale M Jr, Lemon SM. 2005. Immune evasion by hepatitis C virus NS3/4A cleavages of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci U S A 102:2992–2997. doi: 10.1073/pnas.0408824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brenndörfer ED, Karthe J, Frelin L, Cebula P, Erhardt A, Schulte Am Esch J, Hengel H, Bartenschlager R, Sällberg M, Häussinger D, Bode JG. 2009. Nonstructural 3/4A protease of hepatitis C virus activates epithelial growth factor-induced signal transduction by cleavage of the T-cell protein tyrosine phosphatase. Hepatology 49:1810–1820. doi: 10.1002/hep.22857. [DOI] [PubMed] [Google Scholar]
- 12.Kadaré G, Haenni AL. 1997. Virus-encoded RNA helicases. J Virol 71:2583–2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fairman-Williams ME, Guenther UP, Jankowsky E. 2010. SF1 and SF2 helicases: family matters. Curr Opin Struct Biol 20:313–324. doi: 10.1016/j.sbi.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pan RY, Hung TM, Kou YH, Chan NL, Chang MF, Chang SC. 2010. In trans interaction of hepatitis C virus helicase domains mediates protease activity critical for internal NS3 cleavage and cell transformation. FEBS Lett 584:482–486. doi: 10.1016/j.febslet.2009.11.090. [DOI] [PubMed] [Google Scholar]
- 15.Bernstein KA, Gangloff S, Rothstein R. 2010. The RecQ DNA helicases in DNA repair. Annu Rev Genet 44:393–417. doi: 10.1146/annurev-genet-102209-163602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Opresko PL. 2003. Werner syndrome and the function of the Werner protein; what they can teach us about the molecular aging process. Carcinogenesis 24:791–802. doi: 10.1093/carcin/bgg034. [DOI] [PubMed] [Google Scholar]
- 17.Lebel M, Spillare EA, Harris CC, Leder P. 1999. The Werner syndrome gene product co-purifies with the DNA replication complex and interaction with PCNA and topoisomerase I. J Biol Chem 274:37795–37799. doi: 10.1074/jbc.274.53.37795. [DOI] [PubMed] [Google Scholar]
- 18.Hsiang YH, Hertzberg R, Hecht S, Liu LF. 1985. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J Biol Chem 260:14873–14878. [PubMed] [Google Scholar]
- 19.Cliby WA, Lewis KA, Lilly KK, Kaufmann SH. 2002. S phase and G2 arrests induced by topoisomerase I poisons are dependent on ATR kinase function. J Biol Chem 277:1599–1606. doi: 10.1074/jbc.M106287200. [DOI] [PubMed] [Google Scholar]
- 20.Hyun M, Choi S, Stevnsner T, Ahn B. 2016. The caenorhabditis elegans Werner syndrome protein participates in DNA damage checkpoint and DNA repair in response to CPT-induced double-strand breaks. Cell Signal 28:214–223. doi: 10.1016/j.cellsig.2015.12.006. [DOI] [PubMed] [Google Scholar]
- 21.Patro BS, Frohlich R, Bohr VA, Stevnsner T. 2011. WRN helicase regulates the ATR-CHK1-induced S-phase checkpoint pathway in response to topoisomerase-I-DNA covalent complexes. J Cell Sci 124:3967–3979. doi: 10.1242/jcs.081372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gell D, Jackson SP. 1999. Mapping of protein-protein interactions within the DNA-dependent protein kinase complex. Nucleic Acids Res 27:3494–3502. doi: 10.1093/nar/27.17.3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jackson SP, Boulton SJ. 1996. Saccharomyces cerevisiae Ku70 potentiates illegitimate DNA double-strand break repair and serves as a barrier to error-prone DNA repair pathways. EMBO J 15:5093–5103. doi: 10.1002/j.1460-2075.1996.tb00890.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shamanna RA, Croteau DL, Lee JH, Bohr VA. 2017. Recent advances in understanding Werner syndrome. F1000Res 6:1779. doi: 10.12688/f1000research.12110.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Turinetto V, Giachino C. 2015. Multiple facets of histone variant H2AX: a DNA double-strand-break marker with several biological functions. Nucleic Acids Res 43:2489–2498. doi: 10.1093/nar/gkv061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liang L, Deng L, Chen Y, Li GC, Shao C, Tischfield JA. 2005. Modulation of DNA end joining by nuclear proteins. J Biol Chem 280:31442–31449. doi: 10.1074/jbc.M503776200. [DOI] [PubMed] [Google Scholar]
- 27.Mimitou EP, Symington LS. 2009. Nucleases and helicases take center stage in homologous recombination. Trends Biochem Sci 34:264–272. doi: 10.1016/j.tibs.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 28.Chang HHY, Pannunzio NR, Adachi N, Lieber MR. 2017. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat Rev Mol Cell Biol 18:495–506. doi: 10.1038/nrm.2017.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jung D, Alt FW. 2004. Unraveling V(D)J recombination insight into gene regelation. Cell 116:299–311. doi: 10.1016/S0092-8674(04)00039-X. [DOI] [PubMed] [Google Scholar]
- 30.Shrivastav M, De Haro LP, Nickoloff JA. 2008. Regulation of DNA double-strand break repair pathway choice. Cell Res 18:134–147. doi: 10.1038/cr.2007.111. [DOI] [PubMed] [Google Scholar]
- 31.Lorenzini A, Johnson FB, Oliver A, Tresini M, Smith JS, Hdeib M, Sell C, Cristofalo VJ, Stamato TD. 2009. Significant correlation of species longevity with DNA double strand break recognition but not with telomere length. Mech Ageing Dev 130:784–792. doi: 10.1016/j.mad.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rathmell WK, Chu G. 1994. Involvement of the Ku autoantigen in the cellular response to DNA double-strand breaks. Proc Natl Acad Sci U S A 91:7623–7627. doi: 10.1073/pnas.91.16.7623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karmakar P, Snowden CM, Ramsden DA, Bohr VA. 2002. Ku heterodimer binds to both ends of the Werner protein and functional interaction occurs at the Werner N-terminus. Nucleic Acids Res 30:3583–3591. doi: 10.1093/nar/gkf482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li B, Comai L. 2000. Functional interaction between Ku and the Werner syndrome protein in DNA end processing. J Biol Chem 275:28349–28352. doi: 10.1074/jbc.C000289200. [DOI] [PubMed] [Google Scholar]
- 35.Chang SC, Cheng JC, Kou YH, Kao CH, Chiu CH, Wu HY, Chang MF. 2000. Roles of the AX4GKS and arginine-rich motifs of hepatitis C virus RNA helicase in ATP- and viral RNA-binding activity. J Virol 74:9732–9737. doi: 10.1128/JVI.74.20.9732-9737.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nguyen TTT, Park EM, Lim YS, Hwang SB. 2018. Nonstructural protein 5A impairs DNA damage repair: implications for hepatitis C virus-mediated hepatocarcinogenesis. J Virol 92:e00178-18. doi: 10.1128/JVI.00178-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mahaney BL, Meek K, Lees-Miller SP. 2009. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem J 417:639–650. doi: 10.1042/BJ20080413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grakoui A, McCourt DW, Wychowski C, Feinstone SM, Rice CM. 1993. Characterization of the hepatitis C virus-encoded serine proteinase: determination of proteinase-dependent polyprotein cleavage sites. J Virol 67:2832–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Gent DC, Hoeijmakers JH, Kanaar R. 2001. Chromosomal stability and the DNA double-stranded break connection. Nat Rev Genet 2:196–206. doi: 10.1038/35056049. [DOI] [PubMed] [Google Scholar]
- 40.Walker JR, Corpina RA, Goldberg J. 2001. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 412:607–614. doi: 10.1038/35088000. [DOI] [PubMed] [Google Scholar]
- 41.Grundy GJ, Rulten SL, Arribas-Bosacoma R, Davidson K, Kozik Z, Oliver AW, Pearl LH, Caldecott KW. 2016. The Ku-binding motif is a conserved module for recruitment and stimulation of non-homologous end-joining proteins. Nat Commun 7:11242. doi: 10.1038/ncomms11242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uematsu N, Weterings E, Yano K, Morotomi-Yano K, Jakob B, Taucher-Scholz G, Mari PO, van Gent DC, Chen BP, Chen DJ. 2007. Autophosphorylation of DNA-PKcs regulates its dynamics at DNA double-strand breaks. J Cell Biol 177:219–229. doi: 10.1083/jcb.200608077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pham HT, Nguyen TTT, Nguyen LP, Han SS, Lim YS, Hwang SB. 2019. Hepatitis C virus downregulates ubiquitin-conjugating enzyme E2S expression to prevent proteasomal degradation of NS5A, leading to host cells more sensitive to DNA damage. J Virol 93:e01240-18. doi: 10.1128/JVI.01240-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gray MD, Shen JC, Kamath-Loeb AS, Blank A, Sopher BL, Martin GM, Oshima J, Loeb LA. 1997. The Werner syndrome protein is a DNA helicase. Nat Genet 17:100–103. doi: 10.1038/ng0997-100. [DOI] [PubMed] [Google Scholar]
- 45.Hasty P, Campisi J, Hoeijmakers J, van Steeg H, Vijg J. 2003. Aging and genome maintenance: lessons from the mouse? Science 299:1355–1359. doi: 10.1126/science.1079161. [DOI] [PubMed] [Google Scholar]
- 46.Navarro CL, Cau P, Levy N. 2006. Molecular bases of progeroid syndromes. Hum Mol Genet 15:R151–R161. doi: 10.1093/hmg/ddl214. [DOI] [PubMed] [Google Scholar]
- 47.Kou YH, Chou SM, Wang YM, Chang YT, Huang SY, Jung MY, Huang YH, Chen MR, Chang MF, Chang SC. 2006. Hepatitis C virus NS4A inhibits cap-dependent and the viral IRES-mediated translation through interacting with eukaryotic elongation factor 1A. J Biomed Sci 13:861–874. doi: 10.1007/s11373-006-9104-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ko YC, Tsai WH, Wang PW, Wu IL, Lin SY, Chen YL, Chen JY, Lin SF. 2012. Suppressive regulation of KSHV RTA with O-GlcNAcylation. J Biomed Sci 19:12. doi: 10.1186/1423-0127-19-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen L, Huang S, Lee L, Davalos A, Schiest RH, Campisi J, Oshima J. 2003. WRN, the protein deficient in Werner syndrome, plays a critical structural role in optimizing DNA repair. Aging Cell 2:191–199. doi: 10.1046/j.1474-9728.2003.00052.x. [DOI] [PubMed] [Google Scholar]
- 50.Wu SC, Chang SC, Wu HY, Liao PJ, Chang MF. 2008. Hepatitis C virus NS5A protein down-regulates the expression of spindle gene Aspm through PKR-p38 signaling pathway. J Biol Chem 283:29396–29404. doi: 10.1074/jbc.M802821200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lai JS, Herr W. 1992. Ethidium bromide provides a simple tool for identifying genuine DNA-independent protein associations. Proc Natl Acad Sci U S A 89:6958–6962. doi: 10.1073/pnas.89.15.6958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bau DT, Mau YC, Ding SL, Wu PE, Shen CY. 2007. DNA double-strand break repair capacity and risk of breast cancer. Carcinogenesis 28:1726–1730. doi: 10.1093/carcin/bgm109. [DOI] [PubMed] [Google Scholar]
- 53.Hsin WC, Chang CH, Chang CY, Peng WH, Chien CL, Chang MF, Chang SC. 2018. Nucleocapsid protein-dependent assembly of the RNA packaging signal of Middle East respiratory syndrome coronavirus. J Biomed Sci 25:47. doi: 10.1186/s12929-018-0449-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pierce AJ, Johnson RD, Thompson LH, Jasin M. 1999. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev 13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alexandre S, Nakaki T, Vanhamme L, Lee AS. 1991. A binding site for the cyclic adenosine 3’, 5’-monophosphate response element-binding protein as a regulatory element in the grp78 promoter. Mol Endocrinol 5:1862–1872. doi: 10.1210/mend-5-12-1862. [DOI] [PubMed] [Google Scholar]