

Abstract
Weight loss is an early manifestation of Alzheimer's disease that can precede the cognitive decline, raising the possibility that amyloid-β (Aβ) disrupts hypothalamic neurons critical for the regulation of body weight. We previously reported that, in young transgenic mice overexpressing mutated amyloid precursor protein (Tg2576), Aβ causes dysfunction in neuropeptide Y (NPY)-expressing hypothalamic arcuate neurons before plaque formation. In this study, we examined whether Aβ causes arcuate NPY neuronal dysfunction by disrupting intracellular Ca2+ homeostasis. Here, we found that the L-type Ca2+ channel blocker nimodipine could hyperpolarize the membrane potential, decrease the spontaneous activity, and reduce the intracellular Ca2+ levels in arcuate NPY neurons from Tg2576 brain slices. In these neurons, there was a shift from high to low voltage-threshold activated L-type Ca2+ currents, resulting in increased Ca2+ influx closer to the resting membrane potential, an effect recapitulated by Aβ1–42 and reversed by nimodipine. These low voltage-threshold activated L-type Ca2+ currents were dependent in part on calcium/calmodulin-dependent protein kinase II and IP3 pathways. Furthermore, the effects on intracellular Ca2+ signaling by both a positive (ghrelin) and negative (leptin) modulator were blunted in these neurons. Nimodipine pretreatment restored the response to ghrelin-mediated feeding in young (3–5 months), but not older (10 months), female Tg2576 mice, suggesting that intracellular Ca2+ dysregulation is only reversible early in Aβ pathology. Collectively, these findings provide evidence for a key role for low-threshold activated voltage gated L-type Ca2+ channels in Aβ-mediated neuronal dysfunction and in the regulation of body weight.
SIGNIFICANCE STATEMENT Weight loss is one of the earliest manifestations of Alzheimer's disease (AD), but the underlying cellular mechanisms remain unknown. Disruption of intracellular Ca2+ homeostasis by amyloid-β is hypothesized to be critical for the early neuronal dysfunction driving AD pathogenesis. Here, we demonstrate that amyloid-β causes a shift from high to low voltage-threshold activated L-type Ca2+ currents in arcuate neuropeptide Y neurons. This leads to increased Ca2+ influx closer to the resting membrane potential, resulting in intracellular Ca2+ dyshomeostasis and neuronal dysfunction, an effect reversible by the L-type Ca2+ channel blocker nimodipine early in amyloid-β pathology. These findings highlight a novel mechanism of amyloid-β-mediated neuronal dysfunction through L-type Ca2+ channels and the importance of these channels in the regulation of body weight.
Keywords: Alzheimer's disease, electrophysiology, ghrelin, hypothalamus, leptin, neuropeptide Y
Introduction
Weight loss is a common manifestation of Alzheimer's disease (AD) and since the 1984 consensus statement has been a criterion consistent with the diagnosis of AD (McKhann et al., 1984). In epidemiological studies, early weight loss in AD was associated with worsening disease progression and increased mortality, whereas weight gain was protective (White et al., 1998), suggesting that the factors involved in maintaining body weight are likely to be intrinsic to AD pathogenesis. Importantly, weight loss can precede the cognitive decline in AD, suggesting that systemic metabolic dysfunction occurs during the preclinical stage of AD, where neuropathological abnormalities, such as hyperphosphorylated tau and amyloid-β (Aβ), have started to accumulate but have not caused significant cognitive impairment (Johnson et al., 2006; Gao et al., 2011; Emmerzaal et al., 2015). Despite the evidence supporting the importance of early systemic metabolic dysfunction in AD, the underlying mechanisms have not been elucidated but could conceivably be due to disruption of brain circuits regulating body weight (Ishii and Iadecola, 2015).
Neurons that coexpress neuropeptide Y (NPY) and agouti-related peptide in the arcuate nucleus of the hypothalamus are major orexigenic or positive regulators of body weight (Loh et al., 2015). Leptin is an adipocyte-derived hormone that maintains body weight homeostasis by acting as the negative afferent signal to the brain, where it can inhibit arcuate NPY neurons (McGuire and Ishii, 2016). Conversely, ghrelin is a stomach-derived peptide that can increase feeding and alter systemic metabolism by stimulating arcuate NPY neurons (Yanagi et al., 2018). Therefore, leptin and ghrelin regulate body weight in large part by modulating arcuate NPY neuronal function. We previously found that a transgenic mouse model of Aβ accumulation (Tg2576 mice), at a young age (3 months) before developing amyloid plaques, exhibited early body weight deficits and low plasma leptin levels that were due to increased energy expenditure and not from changes in feeding behavior (Ishii et al., 2014). Furthermore, the systemic metabolic deficits in this mouse model were associated with depolarized membrane potentials in arcuate NPY neurons and a blunted response to both leptin and ghrelin (Ishii et al., 2014). Collectively, these results are consistent with Aβ causing arcuate NPY neuronal dysfunction, leading to early systemic metabolic deficits; however, how Aβ causes dysfunction in these neurons is not known.
Since intracellular Ca2+ signaling mechanisms play a key role in the regulation of arcuate NPY neurons by leptin and ghrelin (Kohno et al., 2003; Wang et al., 2008), intracellular Ca2+ dyshomeostasis may underlie the Aβ-mediated dysfunction of these neurons. Decades of accumulating evidence have led to the hypothesis that early disruption of intracellular Ca2+ signaling is critical for the neuronal dysfunction and driving AD pathogenesis (Alzheimer's Association Calcium Hypothesis Workgroup, 2017). This disruption of intracellular Ca2+ homeostasis by Aβ is thought to begin at the plasma membrane (Goodison et al., 2012). Therefore, in this study, we tested the hypothesis that Aβ pathology can cause arcuate NPY neuronal dysfunction by disrupting intracellular Ca2+ homeostasis through the modulation of voltage-gated Ca2+ channels. Using brain slices or dissociated neurons from young Tg2576 mice, we found that arcuate NPY neurons have aberrant hyperactivity and increased intracellular Ca2+ levels, an effect recapitulated in WT arcuate NPY neurons treated with exogenous Aβ1–42 and reversed by the dihydropyridine L-type Ca2+ channel blocker nimodipine. Whole-cell recordings demonstrated that these arcuate NPY neurons switched from high to low voltage-threshold activated L-type Ca2+ currents, resulting in increased Ca2+ influx and neuronal dysfunction. Furthermore, nimodipine rescued ghrelin-mediated feeding in young (3–5 months), but not older (10 months), Tg2576 mice, suggesting that Aβ-mediated Ca2+ neurotoxicity is reversible only during the early stages of Aβ pathology. These findings show that Aβ disrupts intracellular Ca2+ homeostasis and neuronal function through low voltage-threshold activated L-type Ca2+ currents and that L-type Ca2+ channels in arcuate NPY neurons play an important role in the regulation of body weight.
Materials and Methods
Animals.
All procedures were approved by the Institutional Animal Care and Use Committee of Weill Cornell Medicine. Experiments were performed in the Tg2576 transgenic line, which overexpresses the Swedish mutant (K670N, M671L) human amyloid precursor protein gene (APPswe) driven by the hamster prion protein promoter (RRID:MGI:3710766) (Hsiao et al., 1996). For NPY-GFP mice, we used the previously well-characterized BAC transgenic NPY-hrGFP mouse line (B6.FVB-Tg(NPY-hrGFP)1Lowl/J, The Jackson Laboratory, catalog #006417, RRID:IMSR_JAX:006417) (van den Pol et al., 2009; Ishii et al., 2014). To generate Tg2576 mice with GFP labeling in NPY neurons, we crossed male Tg2576 mice with female NPY-GFP mice to produce hemizygous NPY-GFP mice with or without a copy of the APPswe transgene. WT littermates lacking the APPswe transgene were used as controls. All mice were derived from an in-house colony and housed in climate-controlled 12 h light-dark cycle rooms with free access to water and standard rodent chow (LabDiet, catalog #5053).
Preparation of hypothalamic slices and whole-cell patch-clamp studies.
Three- to 4-month-old young male and female NPY-GFP hemizygous mice with or without a copy of the APPswe transgene were used in all electrophysiology experiments. The mice were anesthetized with 2% isoflurane, and their brains were rapidly removed and immersed into ice-cold sucrose (s)-ACSF composed of the following (in mm): 248 sucrose, 26 NaHCO3, 1 NaH2PO4, 5 KCl, 5 MgSO4, 0.5 CaCl2, and 10 glucose, gassed with 95% O2/5% CO2, pH 7.3. Coronal slices (200 μm thick) containing the hypothalamic arcuate nuclei were obtained using a VT1000s Vibratome (Leica Microsystems) and stored in a self-designed chamber containing lactic acid (l)-ACSF composed of the following (in mm): 124 NaCl, 26 NaHCO3, 5 KCl, 1 NaH2PO4, 2 MgSO4, 2 CaCl2, 10 glucose, and 4.5 lactic acid, gassed with 95% O2/5% CO2 and pH 7.4, at 32°C for 1 h, and then the hypothalamic slice was transferred to a glass-bottom recording chamber (P-27; Warner Instrument) mounted on an E600 epifluorescence microscope (Nikon) stage. The slices were continuously perfused with the oxygenated l-ACSF at 2 ml/min. Under the microscope equipped with 40× water-immersion lens and the FITC filter (Chroma Technology), the arcuate nuclei were identified in the ventromedial portion near the base of the third ventricle with GFP-labeled NPY neurons in slices consistently displaying intense green fluorescence and the visualized whole-cell recordings were conducted only on GFP-labeled neurons (Ishii et al., 2014). The patch glass electrode was pulled using borosilicate capillaries (OD 1.5 mm/ID 0.86 mm; World Precision Instruments) and P-80 micropipette puller (Sutter Instruments).
Current-clamp mode was used to record the membrane potential and spontaneous discharges in arcuate NPY neurons in slices.
The resistance of the pipette was 5–7 mΩ when filled with an intracellular solution containing the following (in mm): 130 K-gluconate, 10 NaCl, 1.6 MgCl2, 0.1 EGTA, 10 HEPES, and 2 Mg-ATP, adjusted to pH 7.3. The GFP-positive arcuate neurons were current-clamped using an Axopatch 200A amplifier (filtered at 2 kHz, digitized at 10 kHz) and Digidata 1320A (Molecular Devices). After a Giga Ω seal formation, brief negative pressure was further applied to obtain the whole-cell configuration. The recording began with the membrane test for monitoring the access resistance, which was continuously monitored through the recording. Only those cells in which access resistance was stable (change <10%) were included in data analysis. The voltage and current signals were filtered at 2 kHz, digitalized online at a sampling rate of 10 kHz, and stored for offline analysis using pClamp 10 software (Molecular Devices). Stable baseline recordings were achieved before local application of vehicle (PBS), ghrelin (100 nm), ω-Cgtx-GVIA (1 μm), ω-AgaIVA (1 μm), oligomeric Aβ1–42 (100 nM) or nimodipine (2 μm) to the bath. The concentration of ghrelin was based on previously published studies showing good physiological responses in arcuate NPY neurons (Cowley et al., 2003).
Voltage-clamp mode was used to record L-type voltage-gated Ca2+ currents in arcuate NPY neurons in slices.
The resistance of the pipette was 3–5 mΩ when filled with an intracellular solution containing the following (in mm): 100 CsCl, 30 TEA-Cl, 1 MgCl2, 10 EGTA, 4 NaCl, 10 HEPES, 3 Na2-ATP, adjusted to pH 7.3. Voltage-gated Ca2+ currents were recorded from GFP-labeled arcuate NPY neurons using an Axopatch 200A amplifier (filtered at 2 kHz, digitized at 10 kHz), Digidata 1320A (Molecular Devices), and pClamp 10 software (Molecular Devices). Following a GigaΩ seal formation with a further brief negative pressure applied to obtain the whole-cell configuration. Using 5 mm Ba2+ as the charge carrier, the voltage-gated Ca2+ channel currents were elicited from the holding potential of −60 mV to stepping potentials ranging from −50 to 20 mV. The recording began with the membrane test for monitoring the access resistance, which was continuously monitored throughout the recording. Stable baseline recordings were achieved before local application of vehicle (PBS), leptin (100 nm, Sigma-Aldrich), or oligomeric Aβ1–42 (100 nm) to the bath. The concentrations of leptin and oligomeric Aβ1–42 were chosen based on previously published studies showing good physiological responses in arcuate NPY neurons (Berman et al., 2008; Baver et al., 2014; Ishii et al., 2014). Only those cells in which access resistance was stable (change < 5%) were included for data analysis. The Ca2+ channel current amplitudes were expressed as mean ± SEM, with N as the number of neurons tested from a minimum of 3 mice per group. The L-type Ca2+ current was measured as the amplitude of Ca2+ currents at the end of the 500 ms depolarizing pulse, whereas the amplitude of the transient fast inactivating Ca2+ current was obtained by subtracting the amplitude of the L-type Ca2+ current from the peak transient Ca2+ currents measured at 30 ms after initiation of depolarization pulse. The current-voltage relationship (I–V) curves were plotted for each recording. The cell area was determined using the cell membrane capacitance (pF), and the density of the Ca2+ channel currents (pA) was expressed as pA/pF.
Assessment of cytoplasmic Ca2+ levels in arcuate NPY-GFP neurons.
Three- to 4-month-old young male or female NPY-GFP hemizygous mice with or without a copy of the APPswe transgene were used. Cytoplasmic-free Ca2+ was measured using fura-2 AM as the indicator (Invitrogen), as previously described (Koizumi et al., 2018). The isolated arcuate NPY-GFP neurons were obtained using enzymatic digestion with 0.05% thermolysin plus 0.05% pronase (Sigma-Aldrich). Fura-2 AM (25 μm) was loaded to the cells for 45 min at 37°C and then transferred to polyornithine-coated glass-bottom Petri dish (Warner Instruments). Images were acquired on a Nikon 300 inverted microscope by using a fluorite oil-immersion lens (Nikon CF UV-F X40; NA, 1.3). Fura-2 AM was alternately excited through narrow bandpass filters (340 and 380 nm). An intensified CCD camera (Retiga, ExI) recorded the fluorescence emitted by the indicator (510 ± 40 nm). Fluorescent images were digitized, and backgrounds were subtracted. GFP-labeled cells were simultaneously recorded in a randomly selected field. Fluorescence measurements were obtained from cell body, and ratios were calculated for each pixel by using a standard formula. Results were expressed as 340/380 nm ratio. Leptin (100 nm), ghrelin (100 nm), Aβ1–42 (100 nm), and nimodipine (2 μm) were perfused for 20 min, respectively.
Preparation of oligomeric Aβ1-42.
Soluble oligomeric Aβ1–42 was freshly prepared from lyophilized solid human synthetic Aβ1–42 (rPeptide, catalog #A-1002) as previously described (Ishii et al., 2014). Briefly, lyophilized Aβ1–42 was suspended in 1,1,1, 3,3,3 hexafluoro-2-propanol (Sigma-Aldrich) to 1 mm using a gas-tight syringe. The peptide solution was aliquoted and lyophilized using a Speed-Vac. Dried peptide was stored at −80°C until additional processing. Immediately before the electrophysiology or calcium imaging experiments, dried peptide was resuspended in anhydrous DMSO (Sigma-Aldrich), bath-sonicated for 10 min, and diluted to 100 mm in PBS. The Aβ1–42 was then allowed to oligomerize by incubation at 4°C for 24 h. After incubation, the peptide solution was centrifuged to remove insoluble aggregates. The supernatant containing the soluble oligomeric Aβ1–42 was removed and used for the electrophysiology and calcium imaging experiments as described.
Ghrelin-induced feeding experiments.
A randomized controlled crossover design was used for the feeding experiments. Three- to 5-month-old female Tg2576 or WT littermates were randomly assigned to receive either vehicle (PBS) or ghrelin (0.5 mg per kg body weight in PBS, Phoenix Pharmaceuticals). Ghrelin dosage was determined based on previously published experiments in mice (Wang et al., 2002; Ueno et al., 2004). Each mouse was lightly restrained and given a single intraperitoneal injection of PBS or ghrelin 3 h into the light cycle, when mice are in a satiated state. Immediately after the injection, each mouse was individually housed in a standard cage with free access to water and a premeasured standard rodent chow (LabDiet, catalog #5053). One hour after the injection, food intake was measured with the cage carefully inspected to ensure there were no missing or spilled pellets. After the experiment, mice were group housed with the same littermates as before. One week later, mice that had been given previously PBS received ghrelin, and those that were given ghrelin received PBS injections. Food intake was measured as before. For the nimodipine pretreatment experiment, 3- to 5-month-old mice were first administered a single intraperitoneal injection of nimodipine (10 mg per kg body weight in 2% DMSO and PBS, Sigma-Aldrich). Thirty minutes after the nimodipine injection, mice were randomly given an intraperitoneal bolus injection of either ghrelin (0.5 mg per kg body weight in PBS) or PBS alone. Food intake was measured as before. The following week, all mice were pretreated as before with nimodipine, those that received PBS the prior week received ghrelin, and those that received ghrelin received PBS. Experiments were repeated in 10-month-old Tg2576 and WT littermate mice. Since adult male Tg2576 mice have to be single-housed at all times due to aggressive behavior, female mice were used in all feeding experiments to minimize any confounding effects caused by stress due to prolonged social isolation. The food intake measured was normalized to the body weight (per 100 g) of each mouse to minimize any variations due to differences in body weight as Tg2576 mice as a group have lower body weight compared with WT littermates throughout adulthood (Ishii et al., 2014).
Experimental design and statistical analyses.
Experiments were designed to determine changes associated with experimental manipulations (e.g., vehicle and drug) and/or genotype (i.e., Tg2576 mice compared with WT littermate mice). Unless otherwise indicated, all data are presented as mean ± SEM and analyzed using Prism version 8 (GraphPad Software). Two-group comparisons were analyzed by the two-tailed unpaired Student's t test. Two-tailed paired Student's t test was used for comparisons between two different treatments (i.e., vehicle and drug) used in the same cell or mouse. Multiple three or more group comparisons were evaluated by one-way or two-way ANOVA followed by post hoc Tukey or Sidak's test for multiple comparisons. Differences were considered statistically significant for p values < 0.05.
Results
Arcuate NPY neurons from Tg2576 mice exhibit nimodipine-sensitive changes in membrane potential and spike frequency
Using whole-cell current-clamp recordings, we investigated the electrophysiological properties of arcuate NPY neurons in brain slices of Tg2576 mice expressing GFP in NPY neurons (Ishii et al., 2014). Since voltage-gated Ca2+ channels are involved in Aβ-mediated intracellular Ca2+ dysregulation in other systems (Mattson, 2007; Alzheimer's Association Calcium Hypothesis Workgroup, 2017; Frere and Slutsky, 2018), we tested whether these channels could be responsible for the aberrant electrophysiological properties of arcuate NPY neurons in Tg2576 brain slices. The L-type Ca2+ channel antagonist nimodipine (2 μm) significantly hyperpolarized the membrane potential and lowered the spike frequency in arcuate NPY neurons from Tg2576 slices (Fig. 1A–C); however, antagonists against other types of voltage-gated Ca2+ channels, such as ω-Cgtx-GVIA (1 μm) and ω-AgaIVA (1 μm), which respectively block N-type and P/Q-type Ca2+ channels, had no effect (Fig. 1D,E).
Figure 1.
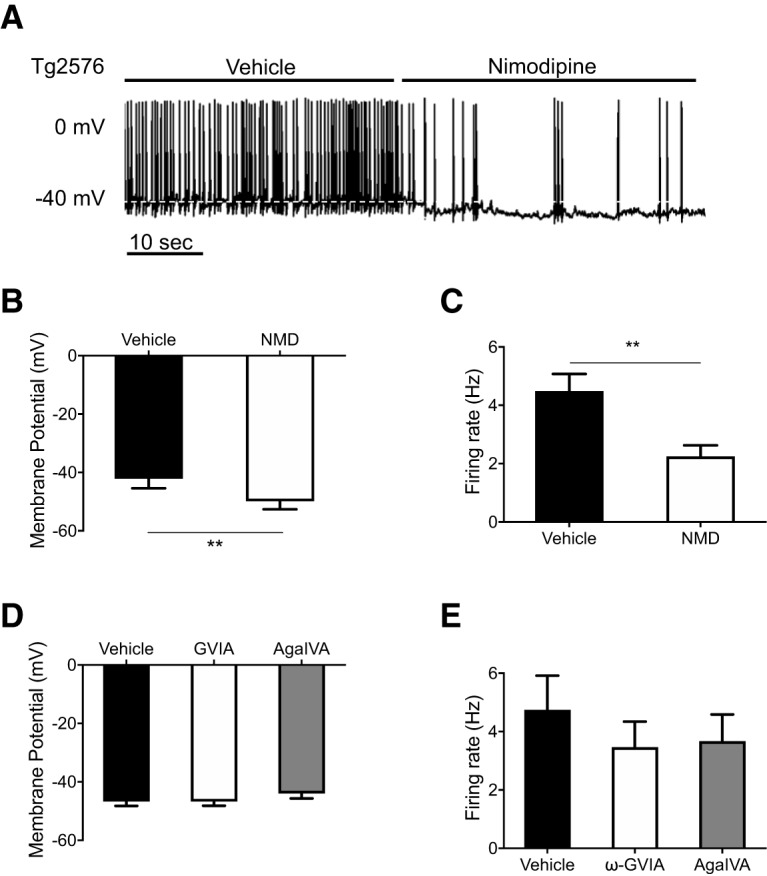
The membrane potential and spike frequency in arcuate NPY neurons from Tg2576 mice are sensitive to the L-type Ca2+ channel blocker nimodipine (NMD). A, Brain slices containing the hypothalamic arcuate nucleus from young (3- to 4-month-old) NPY-GFP mice with APPswe transgene (Tg2576) were used for whole-cell patch-clamp recordings. Representative traces are shown before and after nimodipine (2 μm). B, Application of the L-type voltage-gated Ca2+ channel antagonist nimodipine (2 μm) hyperpolarized the membrane potential of arcuate NPY neurons in Tg2576 brain slices. n = 8 cells from 6 mice per group. Statistical analysis: two-tailed paired Student's t test (t = 4.508, df = 7, p = 0.0028). C, Application of the L-type voltage-gated Ca2+ channel antagonist nimodipine (2 μm) reduced the spike frequency of arcuate NPY neurons in Tg2576 brain slices. n = 8 cells from 6 mice per group. Statistical analysis: two-tailed paired Student's t test (t = 4.548, df = 7, p = 0.0026). D, Application of the N-type (ω-Cgtx-GVIA,1 μm) or the P/Q-type Ca2+ channel (ω-AgaIVA, 1 μm) antagonists had no effect on membrane potential of arcuate NPY neurons in Tg2576 brain slices. n = 4 cells from 3 mice per group. Statistical analysis: repeated-measures one-way ANOVA (F(1.030, 3.089) = 0.968, p = 0.3993). E, Application of the N-type (ω-Cgtx-GVIA,1 μm) or the P/Q-type Ca2+ channel (ω-AgaIVA, 1 μm) antagonists had no effect on the spike frequency of arcuate NPY neurons in Tg2576 brain slices. n = 4 cells from 3 mice per group. Statistical analysis: repeated-measures one-way ANOVA (F(1.947, 5.841) = 1.168, p = 0.3727). Data are mean ± SEM. **p < 0.01.
Arcuate NPY neurons from Tg2576 mice exhibit nimodipine-sensitive increases in cytoplasmic Ca2+ levels
Since the electrical activities in Tg2576 arcuate NPY neurons were sensitive to the L-type Ca2+ channel blocker nimodipine, we next used fura-2 AM to image and quantitate cytoplasmic-free Ca2+ levels in dissociated arcuate NPY neurons identified by GFP. Arcuate NPY neurons from Tg2576 mice had higher cytoplasmic-free Ca2+ levels at baseline compared with those from WT mice (Fig. 2A). Incubation with nimodipine (2 μm) decreased cytoplasmic-free Ca2+ levels in arcuate NPY neurons from Tg2576 mice but not in WT mice (Fig. 2A). Next, we incubated arcuate NPY neurons dissociated from WT mice with oligomeric Aβ1–42 (100 nm) and found a significant dose-related increase in cytoplasmic-free Ca2+ levels that was reversed by nimodipine (Fig. 2B).
Figure 2.
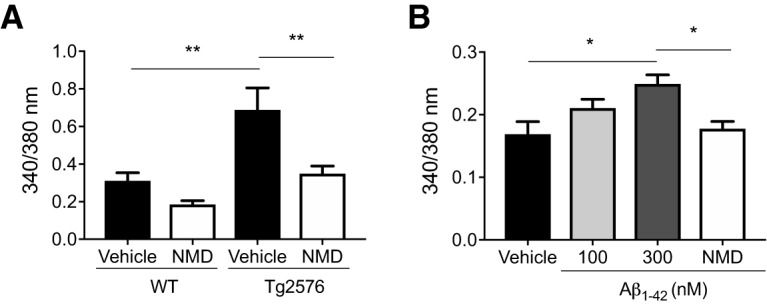
Arcuate NPY neurons from Tg2576 brain slices or exogenous Aβ1–42-treated WT brain slices have increased cytoplasmic-free Ca2+ levels that can be reversed by the L-type Ca2+ channel blocker nimodipine (NMD). A, Arcuate NPY neurons from Tg2576 mice have increased cytoplasmic-free Ca2+ levels compared with WT mice that are decreased by nimodipine. Cytoplasmic-free Ca2+ was measured using fura-2 AM in arcuate NPY neurons isolated by enzymatic digestion from WT or Tg2576 mice. After obtaining baseline levels, cells were perfused with nimodipine (2 μm). n = 18–21 cells from ≥ 3 mice per group. Statistical analysis: one-way ANOVA (F(3,74) = 9.738, p < 0.0001, followed by post hoc Tukey's multiple comparisons). **p < 0.01. B, Exogenous Aβ1–42-treated arcuate NPY neurons from WT mice have increased cytoplasmic-free Ca2+ levels that are decreased by nimodipine. Cytoplasmic-free Ca2+ was measured using fura-2 AM in arcuate NPY neurons isolated by enzymatic digestion from WT mice. After obtaining baseline levels, cells were first perfused with oligomeric Aβ1–42 (100 or 300 nm) followed by nimodipine (2 μm). n = 4 or 5 cells from 2 mice per group. Statistical analysis: one-way ANOVA (F(3,14) = 5.028, p = 0.0143, followed by post hoc Tukey's multiple comparisons). *p < 0.05. Data are mean ± SEM.
The peak L-type Ca2+ I–V curve of arcuate NPY neurons from Tg2576 mice is shifted to the left
Due to the increased cytoplasmic-free Ca2+ levels and altered activity in arcuate NPY neurons from Tg2576 and WT slices treated with Aβ1–42, we next sought to examine the role of Ca2+ currents in these neurons. Using whole-cell voltage-clamp recordings, voltage-gated calcium currents were elicited in the presence of the Na+ channel blocker TTX (1 μm) and the N-type Ca2+ channel blocker ω-Cgtx-GVIA (1 μm) (Fig. 3A). Interestingly, there was a left shift in the peak L-type Ca2+ current I–V curve of arcuate NPY neurons from Tg2576 brain slices compared with WT slices (Fig. 3B), an effect also observed in arcuate NPY neurons of WT slices incubated with oligomeric Aβ1–42 (100 nm) (Fig. 3C). The left shift in the I–V curves suggests a propensity for L-type Ca2+ channels to open at a lower voltage threshold, closer to the resting membrane potentials, resulting in increased Ca2+ influx into the neurons. Nimodipine (2 μm) blocked L-type Ca2+ currents at −10 mV in NPY neurons from brain slices of Tg2576 mice as well as WT brain slices incubated with oligomeric Aβ1–42. (Fig. 3D).
Figure 3.
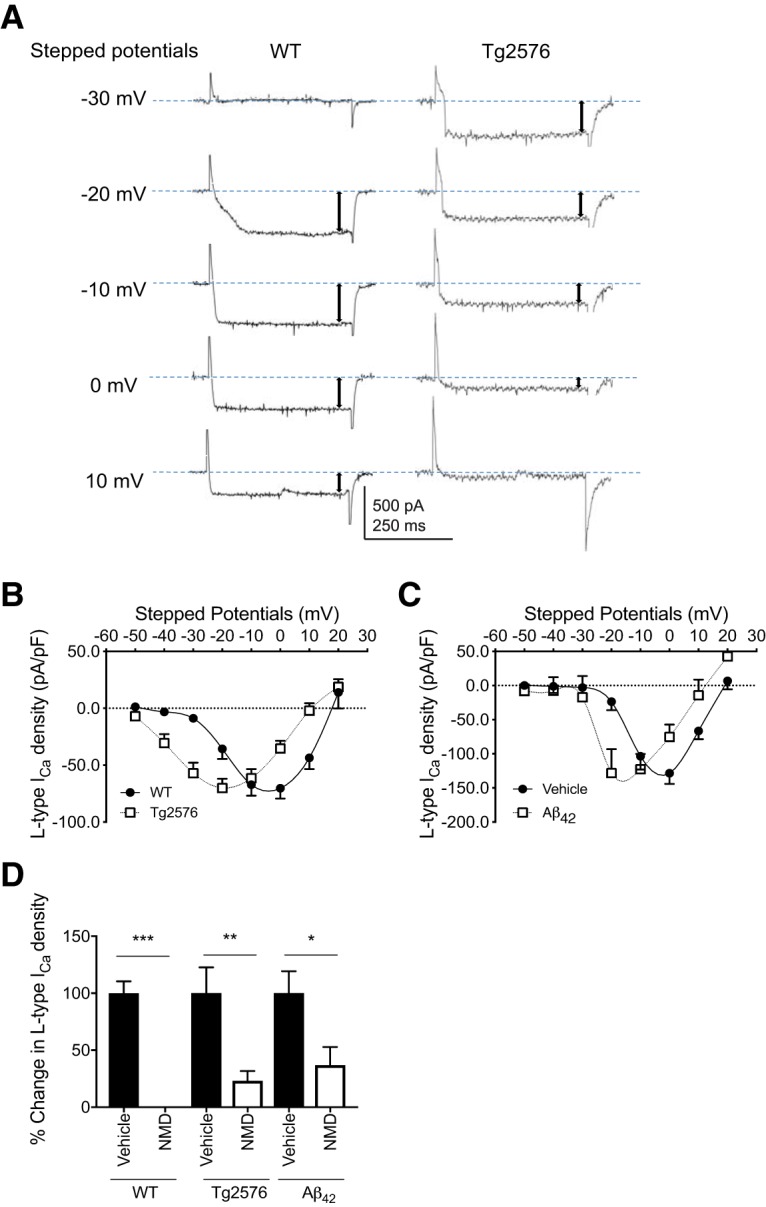
Arcuate NPY neurons from Tg2576 brain slices or exogenous Aβ1–42-treated WT brain slices have a left shift in the peak L-type Ca2+ current I–V curve that can be reversed by the L-type Ca2+ channel blocker nimodipine (NMD). A, L-type Ca2+ currents in arcuate NPY neurons from Tg2576 brain slices show a propensity for channel opening close to the resting potential compared with arcuate NPY neurons from WT brain slices. Representative traces are shown using the holding potential −60 mV to different stepped potentials from −50 to 20 mV (showing −30 to 10 mV). B, I–V relationships for L-type Ca2+ currents in arcuate NPY neurons from WT or Tg2576 brain slices. There was a left shift in the peak L-type Ca2+ current I–V curve in arcuate NPY neurons from Tg2576 brain slices compared with WT brain slices. The peak L-type Ca2+ current in the I–V curve occurred at −20 mV for arcuate NPY neurons from Tg2576 brain slices compared with 0 mV for arcuate NPY neurons from WT slices. At −20 mV, arcuate NPY neurons from Tg2576 brain slices have significantly higher L-type Ca2+ currents compared with arcuate NPY neurons from WT brain slices. Data are represented as L-type Ca2+ current density (pA/pF). n = 19–27 per group from ≥ 12 mice per group. Statistical analysis: two-tailed unpaired Student's t test (t = 2.832, df = 45, p = 0.0069). C, The peak L-type Ca2+ current I–V curve in arcuate NPY neurons from WT brain slices treated with oligomeric Aβ1–42 (100 nm) recapitulates the left shift seen in arcuate NPY neurons from Tg2576 brain slices. The peak L-type Ca2+ current in the I–V curve occurred between −10 mV and −20 mV for arcuate NPY neurons after application of oligomeric Aβ1–42 (100 nm) slices compared with 0 mV at baseline (vehicle treatment). At −20 mV, arcuate NPY neurons from oligomeric Aβ1–42 (100 nm)-treated WT brain slices have significantly higher L-type Ca2+ currents compared with arcuate NPY neurons from vehicle-treated WT brain slices. Data are represented as L-type Ca2+ current density (pA/pF). n = 6 per group from ≥ 4 mice per group. Statistical analysis: two-tailed paired Student's t test (t = 4.598, df = 5, p = 0.0059). D, L-type Ca2+ current induced at −10 mV were significantly decreased after application of nimodipine (2 μm) in arcuate NPY neurons from WT brain slices, Tg2576 brain slices, and WT brain slices treated with oligomeric Aβ1–42 (100 nm). n = 5–8 per group from ≥ 4 mice per group. Statistical analysis: two-tailed paired Student's t test (WT: t = 9.806, df = 7, p < 0.0001; Tg2576: t = 4.694, df = 5, p = 0.0054; Aβ1–42: t = 2.912, df = 4, p = 0.0436). Data are mean ± SEM. *p < 0.05. **p < 0.01. ***p < 0.001.
CaMKII and IP3 inhibitors can partially reverse the left shift in the L-type Ca2+ current I–V curve of arcuate NPY neurons from Tg2576 mice
To gain initial insight into the signaling pathways contributing to the Aβ-mediated changes in L-type Ca2+ currents, we investigated whether common modulators of intracellular Ca2+ signaling were involved (Gao et al., 2006; Zamponi et al., 2015; Zamponi, 2016). KN93 (10 μm), an inhibitor of calcium/calmodulin-dependent kinase II (CaMKII), and 2APB (50 μm), an inhibitor of IP3 and transient receptor potential channels, partially reversed the left shift in the peak L-type Ca2+ current I–V curve of Tg2576 slices, but the phospholipase C inhibitor U733122 (10 μm) had no effect (Fig. 4A–C). This observation suggests that the intracellular Ca2+ dyshomeostasis in arcuate NPY neurons caused by Aβ is mediated in part through CaMKII and IP3-dependent mechanisms.
Figure 4.

Pharmacologic inhibition of CaMKII or IP3 can partially reverse the left shift in the peak L-type Ca2+ current I–V curve in arcuate NPY neurons from Tg2576 brain slices. A, I–V relationships for L-type Ca2+ currents in arcuate NPY neurons from Tg2576 brain slices after application of the CaMKII antagonist KN93 (10 μm). The left-shifted I–V curve was partially reversed after application of KN93. The peak L-type Ca2+ current in the I–V curve occurred at −20 mV for arcuate NPY neurons from vehicle-treated Tg2576 brain slices compared with −10 mV after KN93 application. At −20 mV, arcuate NPY neurons from KN93 (10 μm)-treated Tg2576 brain slices have significantly lower L-type Ca2+ currents compared with vehicle-treated Tg2576 brain slices. Data are represented as L-type Ca2+ current density (pA/pF). n = 5 per group from 4 mice per group. Statistical analysis: two-tailed paired Student's t test (t = 2.854, df = 4, p = 0.0462). B, I–V relationships for L-type Ca2+ currents in arcuate NPY neurons from Tg2576 brain slices after application of the IP3 antagonist 2APB (50 μm). The left-shifted I–V curve was partially reversed after application of 2APB. The peak L-type Ca2+ current in the I–V curve occurred at −20 mV for arcuate NPY neurons from Tg2576 brain slices compared with −10 mV after 2APB application. At −20 mV, arcuate NPY neurons from 2APB (50 μm)-treated Tg2576 brain slices have significantly lower L-type Ca2+ currents compared with vehicle-treated Tg2576 brain slices. Data are represented as L-type Ca2+ current density (pA/pF). n = 4 per group from 3 mice per group. Statistical analysis: two-tailed paired Student's t test (t = 3.656, df = 3, p = 0.0353). C, I–V relationships for L-type Ca2+ currents in arcuate NPY neurons from Tg2576 brain slices after application of the phospholipase C antagonist U733122 (10 μm). The left-shifted I–V curve was unchanged after application of U733122. The peak L-type Ca2+ current in the I–V curve occurred at −20 mV for arcuate NPY neurons from Tg2576 brain slices before and after U733122 application. At −20 mV, arcuate NPY neurons from U733122 (10 μm)-treated Tg2576 brain slices have similar L-type Ca2+ currents compared with vehicle-treated Tg2576 brain slices. n = 5 cells per group from 3 mice per group. Statistical analysis: two-tailed paired Student's t test (t = 0.3566, df = 4, p = 0.7394). Data are mean ± SEM.
Ghrelin and leptin fail to modulate cytoplasmic Ca2+ levels in arcuate NPY neurons of Tg2576 mice
Next, we tested whether a known activator (ghrelin) and inhibitor (leptin) of arcuate NPY neurons could modulate cytoplasmic-free Ca2+ levels in these neurons from WT and Tg2576 mice (McGuire and Ishii, 2016; Yanagi et al., 2018). Using Ca2+ imaging in arcuate NPY neurons dissociated from WT mice, we found that ghrelin (100 nm) increased the cytoplasmic-free Ca2+ levels (Fig. 5A). However, ghrelin had no effect on cytoplasmic-free Ca2+ levels of arcuate NPY neurons from Tg2576 mice (Fig. 5A). Similarly, leptin (100 nm) decreased the cytoplasmic-free Ca2+ levels in WT, but not in Tg2576 arcuate NPY neurons (Fig. 5B). We then sought to determine whether leptin regulates cytoplasmic-free Ca2+ levels in these neurons through L-type Ca2+ channels. Using whole-cell voltage-clamp recordings, we found that leptin decreased the peak L-type Ca2+ current amplitudes in WT, but not in Tg2576 arcuate NPY neurons (Fig. 5C,D), attesting to the leptin insensitivity of these neurons.
Figure 5.
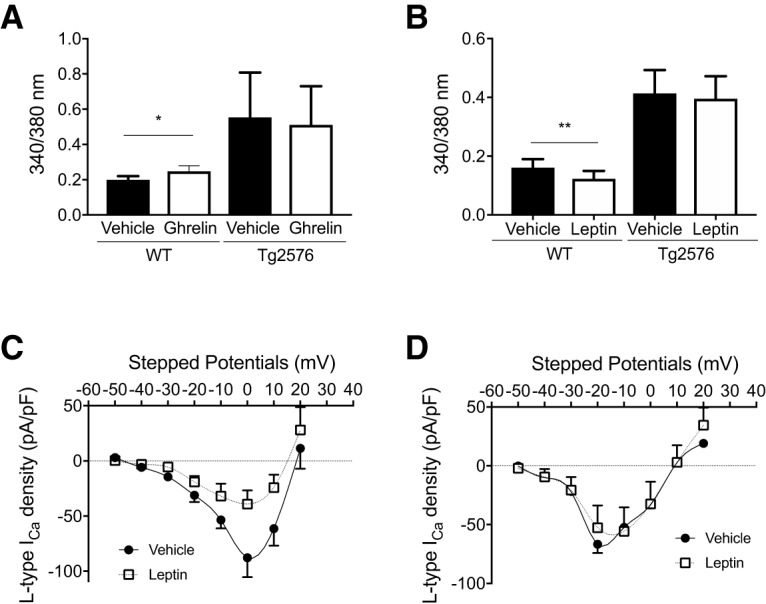
Ghrelin and leptin fail to modulate cytoplasmic-free Ca2+ levels in arcuate NPY neurons from Tg2576 mice. A, Ghrelin increases cytoplasmic-free Ca2+ levels in arcuate NPY neurons from WT but not Tg2576 mice. Cytoplasmic-free Ca2+ was measured using fura-2 AM in arcuate NPY neurons isolated by enzymatic digestion from WT or Tg2576 mice. After obtaining baseline levels, cells were perfused with ghrelin (100 nm). n = 34 WT and n = 7 Tg2576 cells from ≥ 3 mice per group. Statistical analysis: two-tailed paired Student's t test (WT: t = 2.664, df = 34, p = 0.0117; Tg2576: t = 1.020, df = 7, p = 0.3471). B, Leptin decreases cytoplasmic-free Ca2+ levels in arcuate NPY neurons from WT but not Tg2576 mice. Cytoplasmic-free Ca2+ was measured using fura-2 AM in arcuate NPY neurons isolated by enzymatic digestion from WT or Tg2576 mice. After obtaining baseline levels, cells were perfused with leptin (100 nm). n = 25 WT and n = 20 Tg2576 cells from ≥ 3 mice per group. Statistical analysis: two-tailed paired Student's t test (WT: t = 3.032, df = 24, p = 0.0057; Tg2576: t = 2.030, df = 19, p = 0.0566). C, I–V relationships for L-type Ca2+ currents in arcuate NPY neurons from WT brain slices after application of leptin (100 nm). Compared with vehicle treatment, leptin significantly decreased the peak L-type Ca2+ current at 0 mV. Data are represented as L-type Ca2+ current density (pA/pF). n = 7 cells per group. Statistical analysis at 0 mV: two-tailed paired Student's t test (t = 3.601, df = 6, p = 0.0114). D, I–V relationships for L-type Ca2+ currents in arcuate NPY neurons from Tg2576 brain slices after application of leptin (100 nm). Compared with vehicle treatment, there is no significant difference in L-type Ca2+ currents. Data are represented as L-type Ca2+ current density (pA/pF). n = 4 cells per group. Statistical analysis at −20 mV: two-tailed paired Student's t test (t = 1.379, df = 3, p = 0.2617). Data are mean ± SEM. *p < 0.05. **p < 0.01.
Aβ1–42 attenuates the neurophysiological responses to ghrelin in arcuate NPY neurons from WT mice, an effect mediated by L-type Ca2+ channels
Since the neurophysiological responses to ghrelin were attenuated in Tg2576 arcuate NPY neurons, we examined whether Aβ1–42 mediated this effect by L-type Ca2+ channels. Using whole-cell current-clamp recordings ghrelin could significantly depolarize the membrane potential and increase the spike frequency in WT arcuate NPY neurons, an effect that was attenuated when preincubated with oligomeric Aβ1–42 (100 nm) (Fig. 6A,B). Incubation with nimodipine (2 μm) restored the effects of ghrelin on the membrane potentials (Fig. 6A,B). Similarly, using Ca2+ imaging in arcuate NPY neurons dissociated from WT mice, the increased cytoplasmic-free Ca2+ levels mediated by ghrelin were attenuated when preincubated with oligomeric Aβ1–42 (100 nm) and subsequently restored with nimodipine (2 μm) (Fig. 6C).
Figure 6.
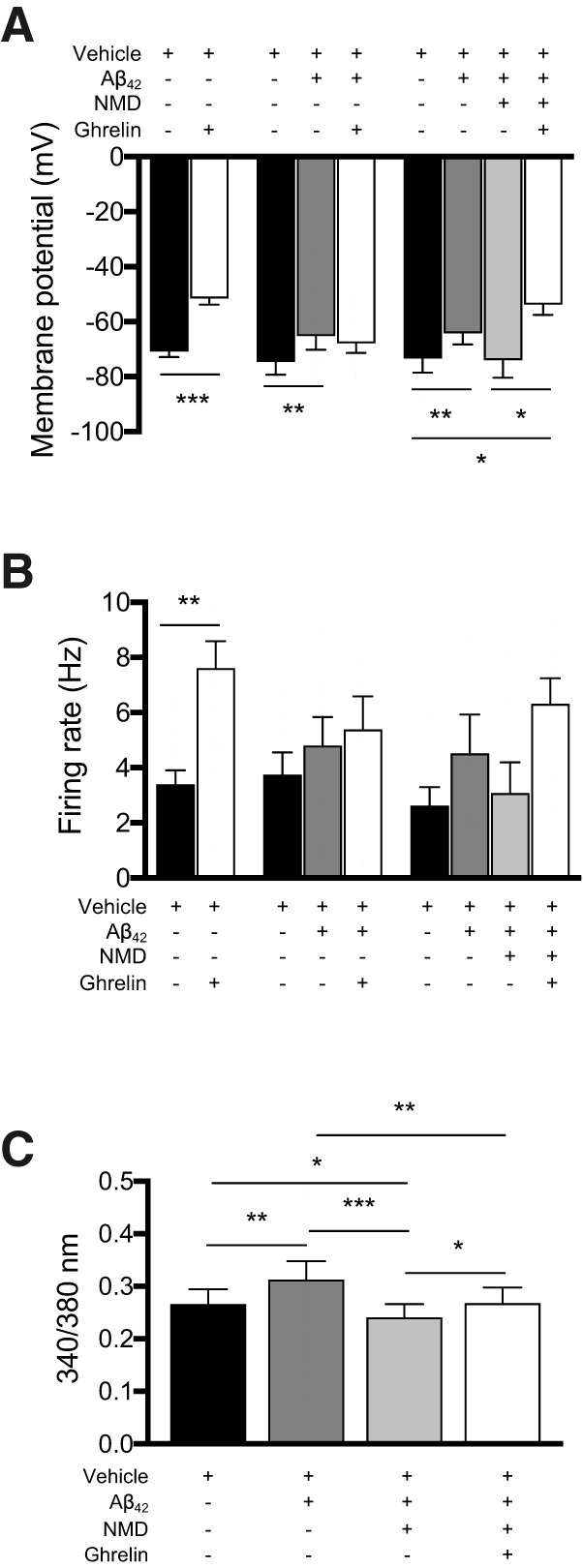
Nimodipine can restore the neurophysiological responses to ghrelin in arcuate NPY neurons treated with oligomeric Aβ1–42. A, B, Brain slices containing hypothalamic arcuate nucleus from young (3- to 4-month-old) NPY-GFP mice were used for whole-cell patch-clamp recordings. Application of ghrelin (100 nm) depolarized the membrane potential and increased spike frequency of arcuate NPY neurons. Exogenous oliogmeric Aβ1–42 (100 nm) depolarized the membrane potential of arcuate NPY neurons and inhibited its response to ghrelin (100 nm). Nimodipine (NMD, 2 μm) restored the ghrelin-mediated depolarization of arcuate NPY neurons treated with exogenous oligomeric Aβ1–42. Statistical analysis: two-tailed paired Student test for comparison of vehicle to ghrelin-treated arcuate NPY neurons (membrane potential: t = 6.444, df = 15, p < 0.001, n = 16 cells from 13 mice per group; spike frequency: t = 3.742, df = 13, p = 0.0025, n = 14 cells from 12 mice per group). Repeated-measures one-way ANOVA comparing vehicle, Aβ1–42, and ghrelin treatments (membrane potential: F(1.579, 12.63) = 7.577, p = 0.0096, n = 9 cells from 5 mice per group; spike frequency: F(1.365, 9.557) = 3.941, p = 0.0679, n = 8 cells from 5 mice per group), followed by post hoc Tukey's multiple comparisons. Repeated-measures one-way ANOVA comparing vehicle, Aβ1–42, nimodipine, and ghrelin treatments (membrane potential: F(1.883, 11.30) = 8.785, p = 0.0014, n = 7 cells from 4 mice per group; spike frequency: F(2.125, 12.75) = 3.220, p = 0.0716, n = 7 cells from 4 mice per group) followed by post hoc Tukey's multiple comparisons. *p < 0.05. **p < 0.01. ***p < 0.001. Data are mean ± SEM. C, Nimodipine restored the ghrelin-mediated increase in cytoplasmic-free Ca2+ levels in arcuate NPY neurons treated with exogenous oligomeric Aβ1–42. Cytoplasmic-free Ca2+ was measured using fura-2 AM in arcuate NPY neurons isolated by enzymatic digestion from young (3- to 4-month-old) WT NPY-GFP mice. After obtaining baseline levels, cells were perfused sequentially with oligomeric Aβ1–42 (100 nm), nimodipine (2 μm), and then ghrelin (100 nm). Statistical analysis: repeated-measures one-way ANOVA comparing vehicle, Aβ1–42, nimodipine, and ghrelin treatments (F(2.128, 44.69) = 13.90, p < 0.0001, n = 22 cells from 4 mice) followed by post hoc Tukey's multiple comparisons. *p < 0.05. **p < 0.01. ***p < 0.001. Data are mean ± SEM.
Ghrelin induces feeding behavior in WT, but not in Tg2576 mice, an effect mediated by L-type Ca2+ channels
We next tested whether the acute orexigenic effect of ghrelin, which is mediated by arcuate NPY neurons, was also altered in Tg2576 mice in vivo. Using a randomized controlled crossover design, young (3- to 5-month old) female Tg2576 and WT littermate mice were injected with a single bolus of ghrelin (0.5 mg per kg body weight, i.p.) or vehicle (PBS) in the first week and then switched to vehicle or ghrelin as appropriate in the second week. As expected, WT mice had an acute increase in food intake after ghrelin injection; however, Tg2576 mice had no significant increase in food intake after ghrelin injection (Fig. 7A). We then examined whether nimodipine, which readily crosses the blood–brain barrier (Ingwersen et al., 2018), could restore the acute orexigenic effects of ghrelin in the young Tg2576 mice. Pretreatment with nimodipine (10 mg per kg body weight, i.p.) led to a robust increase in food intake after ghrelin injection in Tg2576 mice that were similar to those seen in WT littermates, suggesting that blocking L-type Ca2+ channels could restore the orexigenic pathways in vivo in the Tg2576 mice (Fig. 7B). However, pretreatment of nimodipine did not restore the effects of ghrelin on feeding behavior in 10-month-old Tg2576 mice (Fig. 7C).
Figure 7.

Pretreatment with nimodipine can restore ghrelin-mediated feeding behavior in young (3–5 months), but not older (10 months), Tg2576 mice. A, Ghrelin (0.5 mg per kg body weight, i.p.) increased feeding in young (3–5 months) WT but not Tg2576 mice. n = 9 WT and n = 14 Tg2576 mice. Statistical analysis: two-tailed paired Student's t test (WT: t = 2.337, df = 8, p = 0.0476; Tg2576: t = 1.034, df = 13, p = 0.3201). B, Nimodipine (10 mg per kg body weight, i.p.) administered 1 h before ghrelin (0.5 mg per kg body weight, i.p.) restored the ghrelin-mediated feeding behavior in young (3–5 months) Tg2576 mice. n = 12 WT and n = 12 Tg2576 mice. Statistical analysis: two-tailed paired Student's t test (WT: t = 3.986, df = 11, p = 0.0021; Tg2576: t = 4.284, df = 11, p = 0.0013). C, Nimodipine (10 mg per kg body weight, i.p.) administered 1 h before ghrelin (0.5 mg per kg body weight, i.p.) failed to restore the ghrelin-mediated feeding behavior in older (10 months) Tg2576 mice. n = 12 WT and n = 14 Tg2576 mice. Statistical analysis: two-tailed paired Student's t test (WT: t = 4.128, df = 11, p = 0.0017; Tg2576: t = 1.754, df = 13, p = 0.103). Data are expressed as individual mice. *p < 0.05; **p < 0.01; paired t test. n.s. = not significant.
Hypothalamic neurons in the paraventricular nucleus from Tg2576 mice do not have increased cellular activity and have no change in L-type Ca2+ currents
Next, we sought to determine whether Aβ-mediated intracellular Ca2+ dysregulation is a general pathological process affecting all hypothalamic neurons. To this end, we selected random hypothalamic neurons in the paraventricular nucleus from WT and Tg2576 mice to determine whether these neurons were similarly affected as arcuate NPY neurons. Using whole-cell voltage clamp, we found that Tg2576 paraventricular hypothalamic neurons display baseline electrophysiological properties similar to those of WT mice and no changes in the amplitudes or the peak L-type Ca2+ current I–V curve (Fig. 8).
Figure 8.
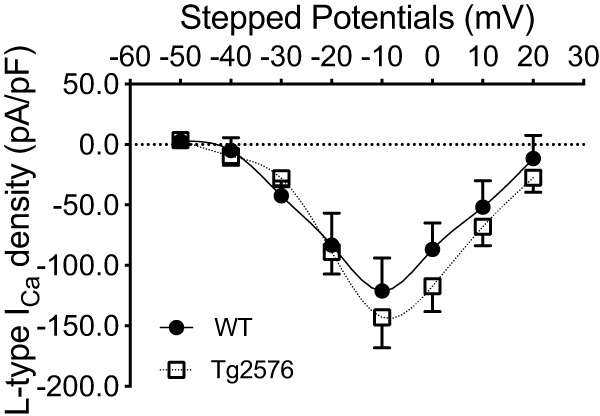
There are no significant differences in the L-type Ca2+ currents in hypothalamic paraventricular neurons from Tg2576 or WT mice. I–V curves for L-type Ca2+ currents in randomly selected hypothalamic paraventricular non-NPY neurons from WT or Tg2576 slices. There are no significant differences between hypothalamic paraventricular non-NPY neurons from WT and Tg2576 slices. The peak L-type Ca2+ current occurred at −10 mV for hypothalamic paraventricular neurons in both WT and Tg2576 brain slices with no significant differences in the amplitudes. Statistical analysis: two-tailed unpaired Student's t test (t = 0.2289, df = 6, p = 0.8266). Data are represented as L-type Ca2+ current density (pA/pF). n = 4 cells per group from ≥ 3 mice per group. Data are mean ± SEM.
Discussion
We investigated the cellular mechanisms underlying how Aβ mediates dysfunction of hypothalamic neurons that are essential for the regulation of body weight. Our prior studies had found that overexpression of APP or application of oligomeric Aβ1–42 in arcuate NPY neurons depolarized the membrane potential resulting in a lack of neurophysiological response to positive (ghrelin) and negative (leptin) modulators (Ishii et al., 2014). In this study, we found that the L-type Ca2+ channel antagonist nimodipine hyperpolarized the membrane potential, decreased the spike frequency, and reduced the intracellular Ca2+ levels due to overexpression of APP in arcuate NPY neurons. Furthermore, compared with arcuate NPY neurons from WT slices, there was a left shift in the peak L-type Ca2+ current I–V curve in arcuate NPY neurons from Tg2576 slices, an effect recapitulated in WT slices treated with oligomeric Aβ1–42. This left shift in the peak L-type Ca2+ current I–V curve suggests that Aβ can alter the L-type Ca2+ influx in arcuate NPY neurons to one that is low-voltage threshold activated with an increased propensity of channel opening around the resting membrane potential, leading to more frequent firing and disruption of intracellular Ca2+ homeostasis. These effects were due in part to CaMKII and IP3-dependent mechanisms. We further show evidence that leptin can decrease, whereas ghrelin can increase, cytoplasmic-free Ca2+ levels in arcuate NPY neurons from WT mice but not in Tg2576 mice. In vivo, pretreatment with nimodipine restored the acute hyperphagic effects of ghrelin in young (3–5 months), but not older (10 months), Tg2576 mice. Finally, not all hypothalamic neurons are affected, as paraventricular neurons from Tg2576 mice have L-type Ca2+ currents similar to those of WT mice. Together, these observations provide, for the first time, evidence that Aβ disrupts intracellular Ca2+ homeostasis and neurophysiological characteristics of select hypothalamic neurons by shifting from a high-threshold to a low-threshold activated L-type Ca2+ current.
Aβ-mediated intracellular Ca2+ dysregulation and the role of L-type Ca2+ channels
Accumulating evidence supports the calcium hypothesis of AD, which postulates that the disruption of mechanisms that normally regulate intracellular Ca2+ signaling plays a critical role in triggering the neuronal dysfunction and driving AD pathogenesis (Alzheimer's Association Calcium Hypothesis Workgroup, 2017). Multiple mouse models of Aβ pathology exhibit intracellular Ca2+ dyshomeostasis, including the Tg2576 mice (Lopez et al., 2008; Kastanenka et al., 2016). Furthermore, immunotherapy with aducanumab, an Aβ antibody developed for the treatment of AD, restored intracellular Ca2+ in Tg2576 mice, strengthening the hypothesis that Aβ directly disrupts intracellular Ca2+ homeostasis (Kastanenka et al., 2016). However, the mechanisms of these effects have remained elusive. Early studies have implicated L-type Ca2+ channels as having a significant role in the Aβ-mediated Ca2+ neurotoxicity particularly in cortical and hippocampal neurons (Weiss et al., 1994; Ueda et al., 1997; Fu et al., 2006). Additionally, whole-cell recordings from HEK293 cells recombinantly expressing L-type Ca2+ channels demonstrated that Aβ peptides can increase L-type Ca2+ currents (Scragg et al., 2005; Kim and Rhim, 2011). Here, we provided a mechanistic insight into this process by reporting that such Aβ-induced Ca2+ dysregulation could be attributed to a shift from high to low voltage-threshold activated L-type Ca2+currents.
Despite the significant evidence linking L-type Ca2+ channels to Aβ-mediated Ca2+ neurotoxicity in cortical and hippocampal neurons, to the best of our knowledge, nothing was known about its effects on hypothalamic neurons. We present the first evidence that Aβ can cause intracellular Ca2+ dyshomeostasis in select hypothalamic neurons, which could be an underlying mechanism for the early metabolic and noncognitive manifestations of AD mediated by hypothalamic dysfunction (Ishii and Iadecola, 2015). Additionally, we show that overexpression of APP or oligomeric Aβ1–42 in arcuate NPY neurons can trigger a shift from high to low voltage-threshold-activated L-type Ca2+ currents, leading to an increase in intracellular Ca2+ levels. This results in neuronal hyperactivity that likely destabilizes the firing synchrony of these neurons and disrupts the network similar to the effects of Aβ on hippocampal neurons (Frere and Slutsky, 2018; Zott et al., 2019). This effect appears to be selective since paraventricular neurons in the hypothalamus of Tg2576 mice were unaffected. However, it is likely that other neuronal populations are affected either independent of the arcuate NPY neuronal dysfunction or directly, such as the postsynaptic neurons in the parabrachial nucleus (Wu et al., 2009). Furthermore, the exact source of Aβ causing arcuate NPY neuronal dysfunction has not been elucidated. We have previously shown that APP overexpression can be detected in the hypothalamus of Tg2576 mice (Ishii et al., 2014); however, whether Aβ is expressed directly in arcuate NPY neurons is not known. Ex vivo experiments with application of exogenous Aβ1–42 to WT slices suggest that Aβ does not need to be expressed in arcuate NPY neurons to cause dysfunction of these neurons. Finally, ghrelin-mediated feeding was restored in young Tg2576 mice after pretreatment with nimodipine, but importantly, nimodipine could not restore ghrelin function in older Tg2576 mice. Therefore, neuronal dysfunction caused by Ca2+ overload through L-type Ca2+ channels may be reversible only early in Aβ pathology; however, it is unclear how early in the pathological process the dysfunction occurs.
How Aβ elicits a low voltage-threshold activated L-type Ca2+ current in arcuate NPY neurons remains elusive. Our studies did not specifically investigate the molecular mechanisms leading to this shift. L-type Ca2+ channels consist of five subunits with α1 subunit forming the main ion-conducting pore of the channel (Zamponi et al., 2015). There are two distinct L-type Ca2+ channel α1 subunits identified in the brain, resulting in the two major isoforms Cav1.2 and Cav1.3 (Zamponi et al., 2015). A major electrophysiological distinction between Cav1.2 and Cav1.3 is that Cav1.3 is activated at lower threshold potentials than Cav1.2, triggering subthreshold depolarization, high intracellular Ca2+ levels, spontaneous firings, and synaptic transmission (Koschak et al., 2001; Xu and Lipscombe, 2001; Zamponi et al., 2015; Pinggera and Striessnig, 2016). Therefore, one possibility is that Aβ upregulates Cav1.3 relative to Cav1.2. Alternatively, there could be a shift in cellular localization of Cav1.3, leading to a redistribution of the channel to the cell surface. This has been demonstrated in one study where the neurotoxic Aβ25–35 fragment increased L-type Ca2+ currents by upregulating Cav1.3 surface protein levels, whereas Cav1.3 mRNA and total protein levels remained unchanged (Kim and Rhim, 2011). It has also been demonstrated that the short Cav1.3 isoform, but not the long isoform, can associate to form cooperative clusters to help facilitate Ca2+ entry into neurons (Moreno et al., 2016). Therefore, Aβ could cause an increase in the short form of Cav1.3, leading to increased Ca2+ influx and subsequent neurotoxicity. Alternatively, Aβ pathology could modify the Cav1.2 protein, leading to changes in the electrophysiological properties of Cav1.2 that make it appear more Cav1.3-like. Future studies using genetic models will help to elucidate the contribution of Cav1.2 and Cav1.3 to Aβ-mediated Ca2+ neurotoxicity.
Finally, the upstream mechanisms contributing to the Aβ-mediated shift to low-threshold activated L-type Ca2+ currents in arcuate NPY neurons are not known, but our data suggest that they involve CaMKII and IP3-dependent pathways. Therefore, one hypothesis would be that Aβ first causes an increase in intracellular Ca2+ levels independent of L-type Ca2+ channels, such as through NMDA receptors, leading to activation of CaMKII and IP3 pathways. The activation of CaMKII and IP3 pathways could then in turn promote the switch to a low-threshold activated L-type Ca2+ current and further disruption of intracellular Ca2+ homeostasis by Ca2+-dependent facilitation in a positive-feedback mechanism.
L-type Ca2+ channels and the regulation of body weight
While the major focus of this study has been on understanding how Aβ pathology in the brain can lead to early systemic metabolic deficits, the results also highlight the role for L-type Ca2+ channels as modulators of arcuate NPY neuronal function and potential therapeutic targets in body weight disorders. Interestingly, L-type Ca2+ channels have been implicated in neuropsychiatric disorders, such as mood disorders that often have comorbid body weight disorders, suggesting a common molecular pathway linking these disorders (Treasure et al., 2015; Kabir et al., 2016). Therefore, modulation of L-type Ca2+ channels in hypothalamic neurons may serve as an important but underappreciated pathway for regulating body weight.
Conclusion
In conclusion, we have demonstrated that Aβ disrupts intracellular Ca2+ homeostasis in arcuate NPY neurons by shifting from normally high to low voltage-threshold activated L-type Ca2+ currents. While additional studies are needed to elucidate the exact molecular mechanisms and whether Aβ can similarly affect nonhypothalamic neurons, our findings suggest that Aβ disruption of arcuate NPY neurons may contribute to the weight loss seen in the earliest stages of AD. Furthermore, our finding, that the L-type Ca2+ channel blocker nimodipine can restore ghrelin-mediated feeding in Tg2576 mice, suggests a therapeutic strategy against the early hypothalamic dysfunction caused by Aβ. Finally, since L-type Ca2+ channels may play a significant role in neurological and psychiatric disorders ranging from autism to Parkinson's disease (Zamponi, 2016), elucidating the mechanisms causing alterations in the function of L-type Ca2+ channels in the brain could lead to a better understanding in the role of these channels in a variety of pathophysiological conditions.
Footnotes
This work was supported by the National Institutes of Health Grant NS37853 to C.I. and Grant AG051179 to M.I. The support of Lauren and Andy Weisenfeld is gratefully acknowledged. We thank Dr. Anjali Rajadhyaksha for helpful discussions; Daniel Lee for technical support; and Alicia Savage-Nieves, Sophy Aguilar, Heather Blades, and Karen Carter for administrative support.
The authors declare no competing financial interests.
References
- Alzheimer's Association Calcium Hypothesis Workgroup (2017) Calcium hypothesis of Alzheimer's disease and brain aging: a framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement 13:178–182.e17. 10.1016/j.jalz.2016.12.006 [DOI] [PubMed] [Google Scholar]
- Baver SB, Hope K, Guyot S, Bjørbaek C, Kaczorowski C, O'Connell KM (2014) Leptin modulates the intrinsic excitability of AgRP/NPY neurons in the arcuate nucleus of the hypothalamus. J Neurosci 34:5486–5496. 10.1523/JNEUROSCI.4861-12.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman DE, Dall'Armi C, Voronov SV, McIntire LB, Zhang H, Moore AZ, Staniszewski A, Arancio O, Kim TW, Di Paolo G (2008) Oligomeric amyloid-β peptide disrupts phosphatidylinositol-4,5-bisphosphate metabolism. Nat Neurosci 11:547–554. 10.1038/nn.2100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley MA, Smith RG, Diano S, Tschöp M, Pronchuk N, Grove KL, Strasburger CJ, Bidlingmaier M, Esterman M, Heiman ML, Garcia-Segura LM, Nillni EA, Mendez P, Low MJ, Sotonyi P, Friedman JM, Liu H, Pinto S, Colmers WF, Cone RD, et al. (2003) The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 37:649–661. 10.1016/S0896-6273(03)00063-1 [DOI] [PubMed] [Google Scholar]
- Emmerzaal TL, Kiliaan AJ, Gustafson DR (2015) 2003–2013: a decade of body mass index, Alzheimer's disease, and dementia. J Alzheimers Dis 43:739–755. 10.3233/JAD-141086 [DOI] [PubMed] [Google Scholar]
- Frere S, Slutsky I (2018) Alzheimer's disease: from firing instability to homeostasis network collapse. Neuron 97:32–58. 10.1016/j.neuron.2017.11.028 [DOI] [PubMed] [Google Scholar]
- Fu H, Li W, Lao Y, Luo J, Lee NT, Kan KK, Tsang HW, Tsim KW, Pang Y, Li Z, Chang DC, Li M, Han Y (2006) Bis(7)-tacrine attenuates beta amyloid-induced neuronal apoptosis by regulating L-type calcium channels. J Neurochem 98:1400–1410. 10.1111/j.1471-4159.2006.03960.x [DOI] [PubMed] [Google Scholar]
- Gao L, Blair LA, Salinas GD, Needleman LA, Marshall J (2006) Insulin-like growth factor-1 modulation of CaV1.3 calcium channels depends on Ca2+ release from IP3-sensitive stores and calcium/calmodulin kinase II phosphorylation of the alpha1 subunit EF hand. J Neurosci 26:6259–6268. 10.1523/JNEUROSCI.0481-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, Nguyen JT, Hendrie HC, Unverzagt FW, Hake A, Smith-Gamble V, Hall K (2011) Accelerated weight loss and incident dementia in an elderly African-American cohort. J Am Geriatr Soc 59:18–25. 10.1111/j.1532-5415.2010.03169.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodison WV, Frisardi V, Kehoe PG (2012) Calcium channel blockers and Alzheimer's disease: potential relevance in treatment strategies of metabolic syndrome. J Alzheimers Dis 30 [Suppl 2]:S269–S282. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274:99–102. 10.1126/science.274.5284.99 [DOI] [PubMed] [Google Scholar]
- Ingwersen J, De Santi L, Wingerath B, Graf J, Koop B, Schneider R, Hecker C, Schröter F, Bayer M, Engelke AD, Dietrich M, Albrecht P, Hartung HP, Annunziata P, Aktas O, Prozorovski T (2018) Nimodipine confers clinical improvement in two models of experimental autoimmune encephalomyelitis. J Neurochem 146:86–98. 10.1111/jnc.14324 [DOI] [PubMed] [Google Scholar]
- Ishii M, Iadecola C (2015) Metabolic and non-cognitive manifestations of Alzheimer's disease: the hypothalamus as both culprit and target of pathology. Cell Metab 22:761–776. 10.1016/j.cmet.2015.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii M, Wang G, Racchumi G, Dyke JP, Iadecola C (2014) Transgenic mice overexpressing amyloid precursor protein exhibit early metabolic deficits and a pathologically low leptin state associated with hypothalamic dysfunction in arcuate neuropeptide Y neurons. J Neurosci 34:9096–9106. 10.1523/JNEUROSCI.0872-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DK, Wilkins CH, Morris JC (2006) Accelerated weight loss may precede diagnosis in Alzheimer disease. Arch Neurol 63:1312–1317. 10.1001/archneur.63.9.1312 [DOI] [PubMed] [Google Scholar]
- Kabir ZD, Lee AS, Rajadhyaksha AM (2016) L-type Ca(2+) channels in mood, cognition and addiction: integrating human and rodent studies with a focus on behavioural endophenotypes. J Physiol 594:5823–5837. 10.1113/JP270673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastanenka KV, Bussiere T, Shakerdge N, Qian F, Weinreb PH, Rhodes K, Bacskai BJ (2016) Immunotherapy with aducanumab restores calcium homeostasis in Tg2576 mice. J Neurosci 36:12549–12558. 10.1523/JNEUROSCI.2080-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Rhim H (2011) Effects of amyloid-β peptides on voltage-gated L-type Ca(V)1.2 and Ca(V)1.3 Ca(2+) channels. Mol Cells 32:289–294. 10.1007/s10059-011-0075-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno D, Gao HZ, Muroya S, Kikuyama S, Yada T (2003) Ghrelin directly interacts with neuropeptide-Y-containing neurons in the rat arcuate nucleus: Ca2+ signaling via protein kinase A and N-type channel-dependent mechanisms and cross-talk with leptin and orexin. Diabetes 52:948–956. 10.2337/diabetes.52.4.948 [DOI] [PubMed] [Google Scholar]
- Koizumi K, Hattori Y, Ahn SJ, Buendia I, Ciacciarelli A, Uekawa K, Wang G, Hiller A, Zhao L, Voss HU, Paul SM, Schaffer C, Park L, Iadecola C (2018) Apoε4 disrupts neurovascular regulation and undermines white matter integrity and cognitive function. Nat Commun 9:3816. 10.1038/s41467-018-06301-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koschak A, Reimer D, Huber I, Grabner M, Glossmann H, Engel J, Striessnig J (2001) Alpha 1D (Cav1.3) subunits can form l-type Ca2+ channels activating at negative voltages. J Biol Chem 276:22100–22106. 10.1074/jbc.M101469200 [DOI] [PubMed] [Google Scholar]
- Loh K, Herzog H, Shi YC (2015) Regulation of energy homeostasis by the NPY system. Trends Endocrinol Metab 26:125–135. 10.1016/j.tem.2015.01.003 [DOI] [PubMed] [Google Scholar]
- Lopez JR, Lyckman A, Oddo S, Laferla FM, Querfurth HW, Shtifman A (2008) Increased intraneuronal resting [Ca2+] in adult Alzheimer's disease mice. J Neurochem 105:262–271. 10.1111/j.1471-4159.2007.05135.x [DOI] [PubMed] [Google Scholar]
- Mattson MP. (2007) Calcium and neurodegeneration. Aging Cell 6:337–350. 10.1111/j.1474-9726.2007.00275.x [DOI] [PubMed] [Google Scholar]
- McGuire MJ, Ishii M (2016) Leptin dysfunction and Alzheimer's disease: evidence from cellular, animal, and human studies. Cell Mol Neurobiol 36:203–217. 10.1007/s10571-015-0282-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM (1984) Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA work Group under the auspices of department of health and human services task force on Alzheimer's disease. Neurology 34:939–944. 10.1212/WNL.34.7.939 [DOI] [PubMed] [Google Scholar]
- Moreno CM, Dixon RE, Tajada S, Yuan C, Opitz-Araya X, Binder MD, Santana LF (2016) Ca(2+) entry into neurons is facilitated by cooperative gating of clustered CaV1.3 channels. Elife 5:e15744. 10.7554/eLife.15744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinggera A, Striessnig J (2016) Cav 1.3 (CACNA1D) L-type Ca2+ channel dysfunction in CNS disorders. J Physiol 594:5839–5849. 10.1113/JP270672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scragg JL, Fearon IM, Boyle JP, Ball SG, Varadi G, Peers C (2005) Alzheimer's amyloid peptides mediate hypoxic up-regulation of L-type Ca2+ channels. FASEB J 19:150–152. 10.1096/fj.04-2659fje [DOI] [PubMed] [Google Scholar]
- Treasure J, Zipfel S, Micali N, Wade T, Stice E, Claudino A, Schmidt U, Frank GK, Bulik CM, Wentz E (2015) Anorexia nervosa. Nat Rev Dis Primers 1:15074. 10.1038/nrdp.2015.74 [DOI] [PubMed] [Google Scholar]
- Ueda K, Shinohara S, Yagami T, Asakura K, Kawasaki K (1997) Amyloid β protein potentiates Ca2+ influx through L-type voltage-sensitive Ca2+ channels: a possible involvement of free radicals. J Neurochem 68:265–271. 10.1046/j.1471-4159.1997.68010265.x [DOI] [PubMed] [Google Scholar]
- Ueno N, Dube MG, Inui A, Kalra PS, Kalra SP (2004) Leptin modulates orexigenic effects of ghrelin and attenuates adiponectin and insulin levels and selectively the dark-phase feeding as revealed by central leptin gene therapy. Endocrinology 145:4176–4184. 10.1210/en.2004-0262 [DOI] [PubMed] [Google Scholar]
- van den Pol AN, Yao Y, Fu LY, Foo K, Huang H, Coppari R, Lowell BB, Broberger C (2009) Neuromedin B and gastrin-releasing peptide excite arcuate nucleus neuropeptide Y neurons in a novel transgenic mouse expressing strong Renilla green fluorescent protein in NPY neurons. J Neurosci 29:4622–4639. 10.1523/JNEUROSCI.3249-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JH, Wang F, Yang MJ, Yu DF, Wu WN, Liu J, Ma LQ, Cai F, Chen JG (2008) Leptin regulated calcium channels of neuropeptide Y and proopiomelanocortin neurons by activation of different signal pathways. Neuroscience 156:89–98. 10.1016/j.neuroscience.2008.04.079 [DOI] [PubMed] [Google Scholar]
- Wang L, Saint-Pierre DH, Taché Y (2002) Peripheral ghrelin selectively increases Fos expression in neuropeptide Y-synthesizing neurons in mouse hypothalamic arcuate nucleus. Neurosci Lett 325:47–51. 10.1016/S0304-3940(02)00241-0 [DOI] [PubMed] [Google Scholar]
- Weiss JH, Pike CJ, Cotman CW (1994) Ca2+ channel blockers attenuate β-amyloid peptide toxicity to cortical neurons in culture. J Neurochem 62:372–375. 10.1046/j.1471-4159.1994.62010372.x [DOI] [PubMed] [Google Scholar]
- White H, Pieper C, Schmader K (1998) The association of weight change in Alzheimer's disease with severity of disease and mortality: a longitudinal analysis. J Am Geriatr Soc 46:1223–1227. 10.1111/j.1532-5415.1998.tb04537.x [DOI] [PubMed] [Google Scholar]
- Wu Q, Boyle MP, Palmiter RD (2009) Loss of GABAergic signaling by AgRP neurons to the parabrachial nucleus leads to starvation. Cell 137:1225–1234. 10.1016/j.cell.2009.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Lipscombe D (2001) Neuronal Ca(V)1.3alpha(1) L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J Neurosci 21:5944–5951. 10.1523/JNEUROSCI.21-16-05944.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagi S, Sato T, Kangawa K, Nakazato M (2018) The homeostatic force of ghrelin. Cell Metab 27:786–804. 10.1016/j.cmet.2018.02.008 [DOI] [PubMed] [Google Scholar]
- Zamponi GW. (2016) Targeting voltage-gated calcium channels in neurological and psychiatric diseases. Nat Rev Drug Discov 15:19–34. 10.1038/nrd.2015.5 [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Striessnig J, Koschak A, Dolphin AC (2015) The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol Rev 67:821–870. 10.1124/pr.114.009654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zott B, Simon MM, Hong W, Unger F, Chen-Engerer HJ, Frosch MP, Sakmann B, Walsh DM, Konnerth A (2019) A vicious cycle of β amyloid-dependent neuronal hyperactivation. Science 365:559–565. 10.1126/science.aay0198 [DOI] [PMC free article] [PubMed] [Google Scholar]