

Abstract
Neurons in parasubiculum (PaS), presubiculum (PrS), and medial entorhinal cortex (MEC) code for place (grid cells) and head direction. Directional input has been shown to be important for stable grid cell properties in MEC, and PaS and PrS have been postulated to provide this information to MEC. In line with this, head direction cells in those brain areas are present at postnatal day 11 (P11), having directional tuning that stabilizes shortly after eye opening, which is before premature grid cells emerge in MEC at P16. Whether functional connectivity between these structures exists at those early postnatal stages is unclear. Using anatomical tracing, voltage-sensitive dye imaging and single-cell patch recordings in female and male rat brain slices between P2 and P61, we determined when the pathways from PaS and PrS to MEC emerge, become functional, and how they develop. Anatomical connections from PaS and PrS to superficial MEC emerge between P4 and P6. Monosynaptic connectivity from PaS and PrS to superficial MEC was measurable from P9 to P10 onward, whereas connectivity with deep MEC was measurable from P11 to P12. From P14/P15 on, reactivity of MEC neurons to parasubicular and presubicular inputs becomes adult-like and continues to develop until P28-P30. The maturation of the efficacy of both inputs between P9 and P21 is paralleled by maturation of morphological properties, changes in intrinsic properties of MEC principal neurons, and changes in the GABAergic network of MEC. In conclusion, synaptic projections from PaS and PrS to MEC become functional and adult-like before the emergence of grid cells in MEC.
SIGNIFICANCE STATEMENT Head direction information, crucial for grid cells in medial entorhinal cortex (MEC), is thought to enter MEC via parasubiculum (PaS) and presubiculum (PrS). Unraveling the development of functional connections between PaS, PrS, and MEC is key to understanding how spatial navigation, an important cognitive function, may evolve. To gain insight into the development, we used anatomical tracing techniques, voltage-sensitive dye imaging, and single-cell recordings. The combined data led us to conclude that synaptic projections from PaS and PrS to MEC become functional and adult-like before eye opening, allowing crucial head direction information to influence place encoding before the emergence of grid cells in rat MEC.
Keywords: learning, memory, ontogeny, parahippocampal region, spatial navigation
Introduction
Spatial navigation and memory strongly depend on computations in the hippocampal and parahippocampal domains (Nadel and O'Keefe, 1978; Eichenbaum and Fortin, 2005; Moser and Moser, 2008). The medial entorhinal cortex (MEC), parasubiculum (PaS), and presubiculum (PrS), all parts of the parahippocampal region, contain cells that code for position (grid cells) (Hafting et al., 2005; Boccara et al., 2010), direction (head direction cells) (Taube et al., 1990; Sargolini et al., 2006), borders (border cells) (Savelli et al., 2008; Solstad et al., 2008), or various forms of conjunctive representations (Sargolini et al., 2006; Solstad et al., 2008). Among the different theoretical conceptualizations on the emergence of grid cell properties, there is consensus that directional information is a necessary requirement (McNaughton et al., 2006; Burgess et al., 2007; Hasselmo et al., 2007; Burak and Fiete, 2009; Winter et al., 2015). Additionally, temporarily inhibiting hippocampal input exposes directional features in grid cells (Bonnevie et al., 2013). The entorhinal cortex itself lacks direct inputs from the vestibular system (Kerr et al., 2007) where directional input originates. The PaS and PrS are likely candidates to bridge the two systems. Both the PaS and PrS are established components of the head directional system, receiving vestibular inputs mediated through the anterior thalamic complex (Taube, 2007; Vann, 2010). Inactivation of the latter disrupts head direction and grid characteristics in MEC. Whether head direction information from PaS and PrS is also obligatory for the development of grid characteristics in MEC has not been assessed. Grid cells in MEC develop from postnatal day 16 (P16) onward, reaching adult-like stability after P19, whereas head direction cells in the anterior thalamic nucleus, MEC, PaS, and PrS are present at P11–P12, reaching stable head-directional tuning shortly after eye opening at P14–P15 (Langston et al., 2010; Wills et al., 2010, 2012; Ainge and Langston, 2012; Bjerknes et al., 2015; Tan et al., 2015). It has not yet been determined whether functional connectivity between the PaS, PrS, and MEC exists before grid cell formation at P14–P15. We provide evidence that premature-looking projections from PaS and PrS grow into a premature MEC network between P4 and P6. Using voltage-sensitive dye (VSD) imaging in rat brain slices with preserved connectivity between PaS, PrS, and MEC (Canto et al., 2012), we determined that the pathways from PaS and PrS to MEC become functional at ∼P9/P10 with responses in MEC looking premature. From P14/P15 on, reactivity of MEC neurons to PaS and PrS inputs becomes adult-like and continues to develop until P28–P30. The maturation of the efficacy of both inputs is paralleled by maturation of MEC morphological properties, changes in intrinsic properties of MEC principal neurons, and changes in the GABAergic network of MEC. These data show that immature monosynaptic inputs from PaS and PrS impact the MEC network before the emergence of premature grid cell properties in MEC at ∼P16 (Langston et al., 2010; Wills et al., 2010). Our data further indicate that the ongoing development of in vivo head direction and grid cell properties is paralleled by changes in the MEC network as well as in intrinsic physiological and morphological properties of individual neurons.
Materials and Methods
Animal experiments were performed in accordance with the rules and directives set by local governments and universities and the European Community on Animal Well-Being (European Directive 2010/63 on the Protection of Animals Used for Scientific Purposes).
Anatomical tracing
For anatomical tracing, 54 male and female Long–Evans rat pups P1–P23 of age (Taconic) were anesthetized with isoflurane (Isofane, Vericore) and injected with an anterograde tracer as previously reported (O'Reilly et al., 2015). Briefly, P0/P1 animals were placed in an isoflurane induction chamber, affixed to the top of the chamber with strips of adhesive putty, and aligned to a hole cut in the top of the chamber that allowed access to the head. Surgeries were performed with the animals maintained in the chamber that was placed on the stereotaxic frame. P2–P12 animals were placed in an isoflurane induction chamber until they could be moved to a stereotaxic frame and placed in a neonatal mask (Kopf, model 973-B), with the palate bar width reduced to better fit the mouth, and the head fixed with zygoma ear cups (Kopf, model 921). Older animals (P13–P23) were maintained under anesthesia with a small adult mask and head fixed with blunted ear bars. For all injections, a hole was drilled in the skull, the dura was punctured, and glass micropipettes with an outer diameter of 20–25 μm were lowered into the brain. The anterograde tracers biotinylated dextran amine (10,000 MW, Invitrogen) or Alexa-488-conjugated dextran amine (A488 DA; Invitrogen) were iontophoretically injected through the micropipettes (5–6 μA alternating currents, 6 s on/6 s off, for 5–12 min) into the PaS and PrS. Up to 50 μl/g body weight of saline was administered subcutaneously throughout the course of the surgery to avoid dehydration. Animals were also administered 5 μg/g body weight Rimadyl as an analgesic. After surgery, rat pups were allowed to recover to an awake state under a heating lamp. When fully awake, rat pups were returned to maternal care until the time of death, 24 h for P0–P13 rat pups and 24–48 h for rat pups from P14 onward.
Transcardial perfusion and tissue preparation for histology
Pups injected with an anatomical tracer were terminally anesthetized with isoflurane such that no reflexive responses (response to tail or foot pinch) were observed. Saline was transcardially perfused through the body from the left ventricle until the flow out of the right atrium was clear, followed by 4% PFA (Merck) until the body was sufficiently stiff. The brains were extracted and left in PFA for 24–48 h before being moved to a cryoprotective solution (20% glycerol, 2% DMSO in 125 mm PB, pH 7.4). Fifty-micrometer sections were then cut sagittally on a freezing microtome (Thermo Fisher Scientific). Sections were collected in six equally spaced series, and the first series was mounted on Superfrost Plus slides (Menzel-Gläser, Thermo Fisher Scientific) and dried overnight on a warming plate at 30°C for subsequent Nissl staining with cresyl violet. Sections were coverslipped using Entellan (Merck). The other series were placed in the cryoprotecting solution and stored at −20°C.
Immunohistochemistry and image acquisition
Tracing experiments.
To visualize biotinylated dextran amine, free-floating sections were washed in 125 mm PB three times for 10 min each. Afterward, the sections were washed in TBS-TX (50 mm Tris, 150 mm NaCl, 0.5% Triton X, pH 8.0) three times for 10 min each. Subsequently, the sections were incubated with Alexa-conjugated streptavidin (Alexa-488, S11223 or Alexa-546, S11225, Invitrogen) in a 1:200 solution with TBS-TX, overnight at 4°C. Then the sections were rinsed three times for 5 min in Tris-HCl and mounted on glass slides, dried overnight, cleared in xylene, and coverslipped with Entellan (Merck, catalog #107961). In some cases, Nissl staining was necessary to aid the delineation of brain regions. To do this, slides were soaked in xylenes overnight to remove the coverslips. Sections were rehydrated in decreasing ethanol solutions (100%, 100%, 90%, 80%, 70%), followed by 2 min in water. Subsequently, the sections were placed in cresyl violet for 3–6 min and rinsed three times in water, for 1 min each. The sections were then dehydrated in ethanol (70%, 80%, 90%, 96% EtOH containing acetic acid, 100%, 100%), cleared in xylenes, and coverslipped with Entellan (Merck). To assess the injection site, sections were assessed using fluorescence microcopy with the appropriate excitation wavelength (Carl Zeiss, Axio Imager M1/2). Sections of brains with successful injections were digitized using a slide scanner equipped for either brightfield or fluorescent imaging (Carl Zeiss, Mirax Midi; objective 20×; NA 0.8). For illustrative purposes, images were exported using Panoramic Viewer software (3DHistech) and processed in Photoshop and Illustrator (CS6, Adobe Systems).
Golgi staining.
We used a total of 11 pups of either sex to prepare our Golgi-stained library. All animals were perfused with Ringer as described above followed by a very brief flush with 1% PFA. The forebrain was dissected, left and right hemispheres separated, and all tissue was directly stained using a Rapid Golgi-Cox kit (FD NeuroTechnologies). The tissue staining varied between 1 and 3 weeks and was performed according the instructions provided by the vendor. Age at time of perfusion ranged from P0 to P22. In short, the tissue was immersed in Solutions A and B, placed in a foil-covered container, and stored for ∼1–3 weeks in the dark. The specimen was then transferred to Solution C and stored for an additional 3–7 d in the dark. Solutions were replaced on the second day of each immersion. Tissue was cut in the horizontal or sagittal plane in 100-μm-thick sections using a HM 450 sliding microtome (Thermo Fisher Scientific) and mounted directly to microscope slides (SuperFrost Plus adhesion slides). The tissue was dried in the dark at room temperature for several days. Slides with tissue were subsequently rinsed with distilled water for 8 min, then immersed for 10 min in a 1:1:2 ratio of Solution D, Solution E, and distilled water. Next, slides were washed in distilled water for 20 min, dehydrated and embedded in Entellan (Merck), and topped with a coverslip.
Slice preparation for VSD and single-cell patch recordings
For VSD and single-cell patch recordings with extracellular stimulation, 70 P4–P61 male and female Sprague Dawley pups (Taconic; Harlan) were anesthetized with isoflurane (Isofane, Vericore), subsequently decapitated, the brain quickly removed from the skull and placed in oxygenated (95% O2/5% CO2) ice-cold ACSF (in mm as follows): 126 NaCl, 3 KCl, 1.25 NaH2PO4, 3 MgSO4, 1 CaCl2, 10 glucose, and 26 NaHCO3. To maintain the connectivity between PaS, PrS, and MEC, 400-μm-thick semihorizontal slices were cut with an angle of 10–15 degrees while the brain was still perfused with saturated ice-cold ACSF (Vibratome 1000, Vibratome). This approach ensured that the connected mediodorsal and the lateroventral parts of the preparasubicular-entorhinal domains were in the same brain slice (Canto et al., 2012).
VSD imaging.
VSD imaging was performed at 32°C. Each slice was transferred onto a fine-mesh membrane filter (Omni pore membrane filter, JHWP01300, Millipore) held in place by a thin Plexiglas ring (11 mm inner diameter; 15 mm outer diameter; 1–2 mm thickness). Slices were maintained in a moist interface chamber, containing the previously used ACSF with 2 mm MgSO4 and 2 mm CaCl2, continuously supplied with a moistened mixture of the O2 and CO2 gas (Tominaga et al., 2000).
Single-neuron recordings.
Single-cell patch experiments were performed at 35°C. After cutting, slices were transferred to a submerged slice chamber with oxygenated ACSF containing the following (in mm): 126 NaCl, 3 KCl, 1.25 NaH2PO4, 2 MgSO4, 2 CaCl2, 10 glucose, and 26 NaHCO3. For all experiments, slices rested for at least 1 h until used one by one in the recording chamber superfused with ACSF.
Electrophysiological recordings
VSD imaging.
The slice was positioned under a fluorescence microscope (Axio Examiner, Carl Zeiss) and stained for 3 min with VSD RH-795 (0.5 mg/ml ACSF) (Koganezawa et al., 2008). Excitation light (filtered at 535 ± 25 nm bandpass) was reflected down onto the preparations by a dichroic mirror (half reflectance wave length of 580 nm). Epifluorescence through a long-wavelength pass filter (50% transmittance at 590 nm) was detected with a CMOS camera (MiCAM Ultima, BrainVision; 100 × 100 pixels array). When the optical recording was triggered, an electronically controlled shutter built into the light source (HL-151, BrainVision) was opened for 500 ms before the start of recording to avoid both mechanical disturbances caused by the shutter system and rapid bleaching of the dye. After starting, the optical baseline was allowed to stabilize for 50 ms before stimuli were delivered. For all experiments, 512 frames at a rate of 1.0 ms/frame were acquired. To represent the spread of neural activity, we superimposed color-coded optical signals on the bright-field image. In this procedure, we applied a color code to the fraction of the optical signal, which exceeded the baseline noise. To reduce baseline noise, we averaged eight identical recordings acquired with a 3 s interval directly in the frame memory. Optical signals were analyzed offline using BrainVision analyses software. Changes in membrane potential were evaluated for a ROI as fractional changes of fluorescence (ΔF/F). The ROI was chosen by a careful visual inspection of the optical signal. The region where the signal first entered MEC after PaS and PrS stimulation was chosen as our ROI. Four repetitive extracellular stimulations of 0.6 mA amplitude, a duration of 300 μs, and a frequency of 20 Hz were applied to PaS or PrS with a tungsten bipolar electrode with a tip separation of 150 μm. In vivo spike trains of head direction cells show 10–50 Hz firing (Boccara et al., 2010). We also applied 10, 40, 50, and 100 Hz stimulation, and the data look similar to data presented here.
Single-neuron recordings.
Whole-cell current-clamp recordings of MEC neurons were done under visual guidance using infrared differential interference contrast video microscopy. Recorded neurons were classified as principal neurons based on previously established physiological and morphological properties (Canto and Witter, 2012). Patch pipettes were pulled from standard-walled borosilicate capillaries (GC120F-10, Harvard Apparatus) with a resistance between 4 and 7 mΩ containing the following (in mm): 110 K-gluconate, 10 HEPES, 4 ATP-Mg, 0.3 GTP, 10 Na-phosphocreatine, 10 KCl, with 5 mg/ml biocytin, pH 7.3 adjusted with 1 m KOH, and an osmolarity of ∼290 m. The seal resistance was >1 GΩ. Recordings were made with a Multiclamp 700A Amplifier (Molecular Devices) in bridge mode. Capacitance compensation was maximal, and bridge balance was adjusted. The signal was low pass filtered at 3 kHz and acquired at a sampling rate of 5 kHz with an ITC-18 board (Instrutech). During recordings, neurons were filled with biocytin. The PaS or PrS was stimulated with a stimulation electrode that was covered with a glass pipette (tip diameter of ∼1–2 μm) filled with ACSF or a tungsten bipolar electrode with a tip separation of 150 μm. The place of stimulation was chosen to coincide with the area that showed high connectivity in VSD and previously performed experiments where we injected an anterograde tracer in PaS and PrS (Canto et al., 2012). Inputs from superficial PaS or PrS layers was stimulated locally with different lengths, strengths, and frequencies controlled by Igor, Master 8, and the stimulation isolator box itself. Each change in parameters was tested in response to 50 stimulus trains of 1 s duration. The parameters tested were as follows: 0 mV/10 μs, 0.9 mV/10 μs, 9 mV/10 μs, 22.5 mV/10 μs, 45 mV/10 μs, 67.5 mV/10 μs, and 90 mV/10 μs and as a control 0 mV/1 ms, 0 mV/0 s, and 90 mV/0 s positive pulses. Repetitive stimulation was performed with 90 mV/10 μs pulses after confirmation that there is no change in EPSP properties comparing strong and weak stimulations and no short-term-dependent plasticity, meaning equilibrium in EPSP size was reached. For the repetitive stimulation protocols, we show data in response to 20 Hz stimulation. We also used 100 Hz stimulation with single-cell recording experiments as an indicator for the monosynaptic nature of events and not orthodromically driven action potentials (APs). We also applied 10, 40, 50, and 100 Hz stimulation, and the data look similar to the data presented here. During patch experiments, intrinsic membrane properties were estimated from the voltage response to a series of current steps. Up to 10 alternating hyperpolarizing and depolarizing steps of 1 s duration incrementally increasing by 20 pA were applied to the neurons, starting with 0 pA and ending with ±200 pA, respectively. We tested how much positive current is needed for the neuron to reach threshold for spiking. We also checked whether the neurons showed membrane potential oscillations by bringing the membrane to a potential just below threshold for firing while recording the membrane fluctuations for at least 30 s. In addition, we induced oscillatory inputs at fixed currents, with the frequency increasing and afterward decreasing linearly in time (so-called ZAP protocol) (Hutcheon and Yarom, 2000). We injected for t = 28 s sinusoidal currents of Io = 40 pA and recorded the membrane voltage simultaneously. The injected current I(t) was ramped up from F0 = 0 Hz to the maximum frequency of fm = 20 Hz: I(t) = Io sin (2π f(t)t) with F(t) = F0 + (fm − f0)t/2T. The impedance amplitude profile (ZAP) functions allow characterizing the resonance properties.
Data analysis, experimental design and statistics used
VSD experiments.
Each pixel of the image sensor records the sum of the membrane potential changes of every membranous structure projected onto the pixel. Thus, fluctuations of the optical signal from baseline represent the sum of membrane potential changes. Assuming that in all instances the contribution of glia cells to the signal will be constant, we calculated the integrals of the curves as this measure represents the size of total membrane potential changes associated with neural activity (Grinvald et al., 1988). These measures, obtained using BrainVision analyses software (Koganezawa et al., 2008), were compared in the different stimulation protocols. Time of onset latencies was measured from the beginning of the stimulus artifact to the start of the fluorescent change response in MEC. Fitting a curve from 25 ms up to 400 ms after the last stimulus to the negative slope derived the decay times after the last applied stimulus. Signal sizes for the repeated-measures analysis (RMA) were calculated by measuring the distance between the baseline activity and the maximum deflection after each stimulus. Latencies and decay times were excluded for those cases where we could not detect any significant signal amplitude changes and significant decays.
Single-neuron recordings.
Synaptic events induced by extracellular stimulation were analyzed by taking an average trace of 50 sweeps of intracellular membrane potential changes in response to PaS and PrS stimulation, and rise and decay times were derived by fitting two decaying exponential functions to the evoked postsynaptic potentials (PSPs), starting at the beginning of the event. PSP latencies were measured from the beginning of the stimulus artifact to the evoked PSP start. The PSP amplitudes were calculated by measuring the distance between the PSP threshold and the maximum voltage deflection. Analysis of membrane properties of single neurons was done using custom-made procedures in Igor Pro Software (Wavemetrics) (Canto and Witter, 2012). A sum of two decaying exponential functions, one with positive, the other with negative amplitude, was fitted to the voltage response to a −200 pA step to reveal the input resistance. For the first step after inducing APs, the following parameters were calculated as described: First, we examined how much current was needed to induce APs in the neuron. Once APs were induced, we analyzed the AP threshold, which is the maximum in the second derivate of the voltage trace for the first AP in response to current injection; AP amplitude is the difference between the maximal amplitude of the AP and the AP threshold for the first AP in response to current injection. In addition, we calculated the AP half-width, which is the width of the AP at 50% amplitude measured from threshold to peak for the first AP and the AP rise time, which is the time to increase from 20% to 80% of the AP amplitude for the first AP in response to current injection. In case a neuron showed a depolarizing afterpotential (DAP) after an AP, the amplitude of DAP was calculated by measuring the difference between the maximal depolarizing voltage deflection after the AP induced and the amplitude of the threshold for firing. In those cases where an AP was induced due to threshold depolarization by DAP, we first analyzed all neurons and checked for the maximum DAP amplitude. For cases with an induced AP, we assigned this maximum DAP amplitude. We also calculated the interspike interval between pairs of subsequent APs. A point spread function was performed on current signals just below threshold activity to analyze membrane potential oscillations. Membrane oscillations were analyzed for power spectral density (Igor Pro Software procedure) using a fast Fourier transform (FFT) (Press et al., 1992). Regarding the ZAP protocol, an FFT was performed on voltage (V) and current (I) signals, and the impedance was calculated by Z(f) = FFT(V)/FFT(I). The resonance frequency is the peak in the impedance magnitude versus the frequency plot.
Statistical analysis.
Normality of the data was tested with Levene's test, and subsequently univariate ANOVA followed by post hoc Bonferroni tests were used to assess differences in characteristics between layers or age groups. If assumptions for a parametric test were not met (Levene's test, p < 0.05), Kruskal–Wallis followed by Mann–Whitney U tests were used. We applied a Bonferroni correction to adjust for multiple comparisons. Effects of application of bicuculline were tested with a paired t test or a sign test for paired comparisons. To analyze the changes in the VSD response and eEPSP amplitude within a train-of-four stimulation at different frequencies with different stimulation electrodes, an RMA was used followed by paired t tests. We used a Bonferroni correction to adjust for multiple comparisons. If Mauchly's test was significant, we used Greenhouse–Geisser correction values to test for significance.
Immunohistochemistry and image acquisition
VSD experiments.
To specify the area of stimulation and recording for VSD experiments, slices were postfixed in 4% PFA for up to 1 week and subsequently kept in PBS with 30% sucrose for >10 h and cut at 40–50 μm thickness using a freezing microtome. Mounted sections were Nissl-stained with cresyl violet and covered slipped using Entellan (Merck). Digital images of sections were combined with the optical imaging data using Photoshop (Adobe) to identify the region in which changes in neural firing occurred.
Single-neuron recordings.
During recordings, neurons were filled with biocytin. Subsequently, they were processed using standard procedures. After recordings, the slices were fixed in 4% PFA for at least 24 h and then placed in 2% DMSO/20% glycerin in PB overnight, pH 7.4. Slices were resectioned into 120-μm-thick sections with a freezing microtome, subsequently washed 3× for 10 min in 0.125 m PB, treated with 0.3% H2O2, followed by rinsing in TBS-TX 3× for 10 min. Afterward, sections were incubated in a solution of avidin-biotinylated-HRP complex (ABC, Vector Laboratories) in TBS-TX (24 h at room temperature or 48 h at 4°C, according to the specifications of the supplier). Peroxidase activity was visualized by incubation for 5–15 min in DAB-Ni in 0.125 PB and H2O2. After washing in Tris-HCl buffer 3 × 10 min, the sections were mounted, maintaining a fixed orientation on glass slides from a buffered gelatin solution (0.2% gelatin in Tris-HCl; 35°C) and carefully dried for at least 48 h. Sections were dehydrated and coverslipped with Entellan (Merck). Neurons were analyzed and, if needed, manually reconstructed with a Leica Microsystems or Carl Zeiss up-right microscope, using a 40 × objective and Neurolucida software (Neurolucida, MicroBrightField).
Results
Postnatal development of anatomical connectivity between PaS, PrS, and MEC
To assess when connections between PaS, PrS, and MEC develop, we labeled projections anterogradely during development (Table 1) (O'Reilly et al., 2015; Sugar and Witter, 2016). The age at which the first axons from PaS and PrS were present in superficial layers of MEC is ∼P4/P5 and P5/P6, respectively. In two representative experiments for PaS and PrS, the anterograde tracer biotinylated dextran amine in P5 animals labeled a few axons in MEC layers II and III, respectively. These axons were poorly branched, showed signs of the presence of growth cones, and very few varicosities were visible (Figs. 1A,A′, 2A,A′). Axons with varicosities are considered as having presynaptic terminals. In case of animals between P6 and P11 (N = 17; Table 1), we noticed a gradual increase in density of overall labeling, with an increasing level of branching and density of presumed synaptic contacts as estimated by the number of varicosities. The density of PaS axons increased specifically in LII, becoming more similar to the adult pattern. In contrast, the laminar terminal distribution of PrS fibers, typically seen in the adult, became only gradually apparent, such that even at P12 the branching of axons looked only weakly adult-like, showing axons primarily within LIII, with only some additional labeled fibers in LI (Figs. 1B,B′, 2B,B′). At the age of P12, the density of innervation of LII by PaS axons was higher than that seen in case of LIII innervation by PrS. This indicates that the development of PaS innervation precedes that of PrS innervation. This is in line with our electrophysiological data (see below). The adult-like dense innervation of LI and LIII in case of PrS (Köhler, 1984; Caballero-Bleda and Witter, 1993) became apparent only at ∼P20 (Figs. 1C,C′, 2C,C′). We conclude that, from P4/P5 onward, MEC neurons may potentially receive PaS and PrS inputs, but that the presynaptic morphology does not look adult-like until after P20 though the PaS laminar pattern is established before the PrS pattern. In our data on PrS inputs, we did not see indications of neuronal specific innervation patterns recently described in adult rats (Honda and Furuta, 2019), which is likely caused by the differences in single-cell resolution tracing used in the latter paper versus bulk injections of anterograde tracers used in the present paper.
Table 1.
Anterograde tracer injection per injection side and age of injectiona
| Age (P) | No. of injections |
Age (P) | No. of injections |
||||
|---|---|---|---|---|---|---|---|
| PaS | PrS | PaS/PrS | PaS | PrS | PaS/PrS | ||
| 2 | 2 | 0 | 0 | 13 | 1 | 1 | 0 |
| 3 | 1 | 1 | 0 | 14 | 1 | 0 | 0 |
| 4 | 3 | 1 | 0 | 15 | 0 | 1 | 0 |
| 5 | 1 | 1 | 0 | 16 | 0 | 1 | 0 |
| 6 | 2 | 0 | 0 | 17 | 0 | 1 | 0 |
| 7 | 2 | 1 | 0 | 18 | 1 | 3 | 0 |
| 8 | 1 | 3 | 0 | 19 | 0 | 3 | 0 |
| 9 | 0 | 0 | 1 | 21 | 2 | 3 | 0 |
| 10 | 0 | 0 | 1 | 22 | 1 | 1 | 0 |
| 11 | 2 | 3 | 1 | 23 | 1 | 0 | 0 |
| 12 | 0 | 5 | 0 | 24 | 1 | 0 | 0 |
aSummary of injections in either PaS or PrS, or combined involvement of both structures (PaS/PrS) per postnatal day.
Figure 1.

Postnatal development of projections from PaS to MEC. The presynaptic side. A–C, Representative grayscale inverted images of an area in MEC showing labeling of parasubicular axons in layers I-III in MEC at (A) P5, (B) P12, and (C) P20. A′, B′, C′, Higher-magnification images of areas indicated with white boxes in A–C. Indicated are examples of varicosities, likely reflecting presynaptic elements (white arrowheads) and axonal branching points (blue arrowheads). Insets, The anterograde tracer injection sites in PaS. Dashed lines indicate brain areas and layers. Scale bars: A–C, 500 μm; A′–C′, 100 μm. Sub, subiculum.
Figure 2.
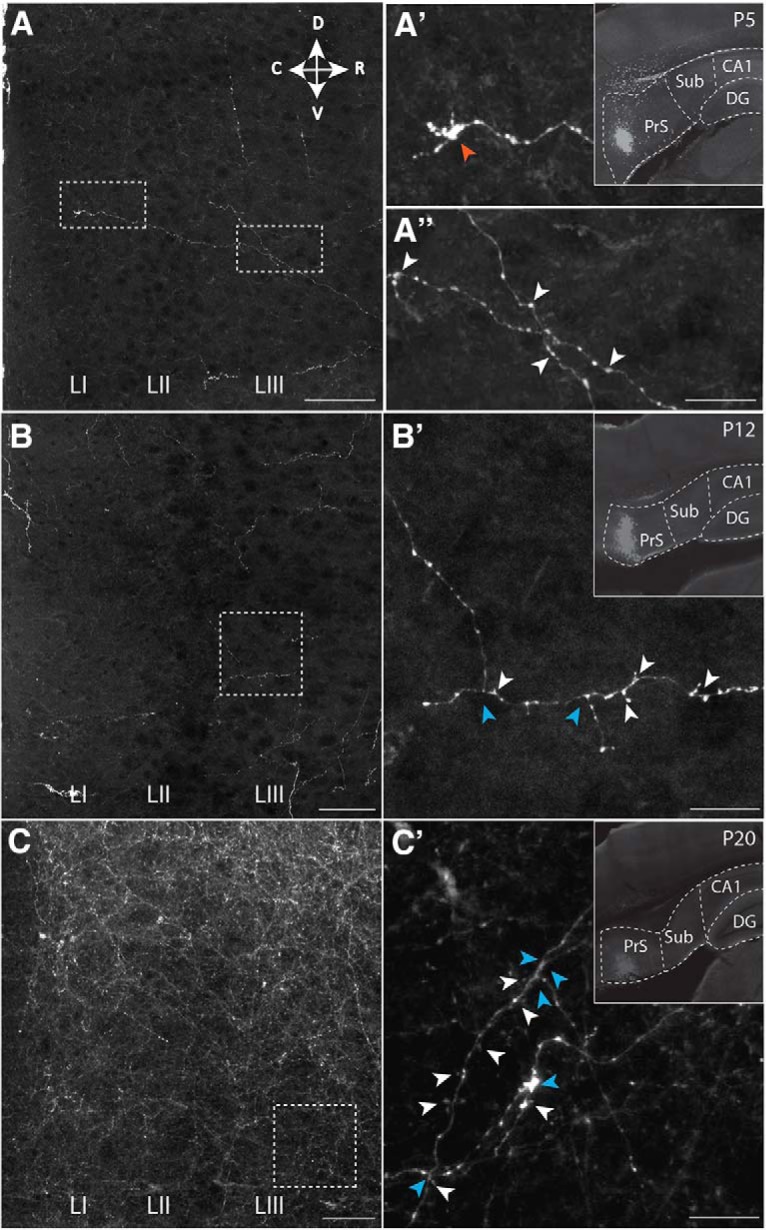
Postnatal development of projections from PrS to MEC. The presynaptic side. A–C, Representative area in MEC showing labeling of presubicular axons in layers I-III in MEC at (A) P5, (B) P12, and (C) P20. A′, B′, C′, Higher-magnification images of areas indicated with white boxes in A–C. Indicated are an example of a growth cone (red arrowhead), varicosities, likely reflecting presynaptic elements (white arrowheads), and axonal branching points (blue arrowheads). Insets, The anterograde tracer injection sites in PrS. Dashed lines indicate brain areas and layers. Scale bars: A–C, 500 μm; A′–C′, 100 μm. DG, dentate gyrus; CA1, field CA1 of the hippocampus; Sub, subiculum.
Postnatal development of MEC neurons
To evaluate whether neurites of MEC neurons are potentially able to receive inputs from PaS and PrS from P4/P5 onward, we studied Golgi silver-impregnated neurons (Fig. 3) and intracellularly filled MEC neurons from P2 onward (see all subsequent figures with electrophysiological data). The postnatal development of the membrane properties of the intrinsically filled neurons paralleled the development of the gross morphology.
Figure 3.
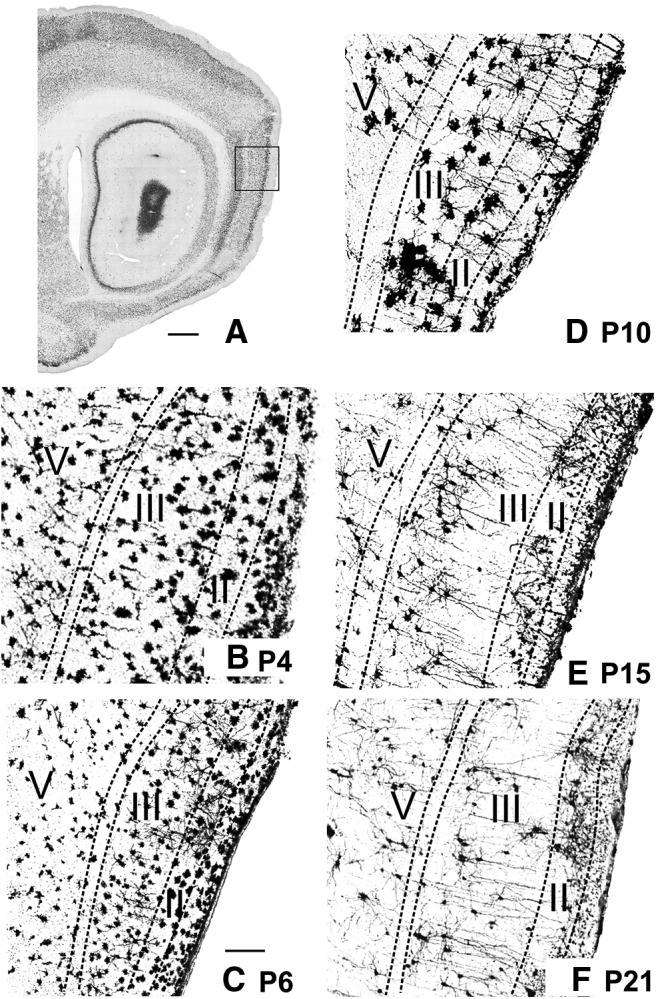
Postnatal development of morphological properties of neurons in MEC. The postsynaptic side. Golgi-stained material showing the neuronal morphology at different postnatal days. A, Sagittal section of a P20 animal stained with NeuN as a marker for neuronal somata. Square represents the approximate position of the Golgi images for B–F. Scale bar, 1.5 mm. High magnification of a piece of MEC stained with Golgi (for details, see Materials and Methods) of a P4 (B), P6 (C), P10 (D), P15 (E), and P21 (F) animal. The neurons appear immature from P4 to P10 and adult-like starting at P15. Apical dendrites can be seen radiating from the deep layers to the pial surface in the 2 older animals. Neurons in superficial layers of the youngest animals (B) appear substantially larger than in older animals; this is due to an incubation artifact resulting in too dense silver deposits. Both neuronal stains in younger tissue (Nissl) as well in the intracellular fill data corroborate that the neurons at these young postnatal ages are not larger. Scale bar: (in C), B–F, 200 μm.
Postnatal development of morphology of MEC neurons
Examination of Golgi silver-impregnated neurons revealed that the dendritic morphology of P2–P4 old MEC neurons was poorly developed (Fig. 3B), suggesting that the postsynaptic sides are still premature. Although stained somata could be observed in all MEC cell layers, the majority of somata with recognizable neurites were located in layers II and III. For all neurons, the number of neurites was low and the neurites were poorly developed, remaining mostly within the layer of origin. Dendrites showed sparse branching that did not extend over long distances. Spines were only occasionally visible (Fig. 3B). The morphology of the P2–P4 MEC neurons, together with the poor branching of parasubicular and presubicular projections makes functional connectivity unlikely at this early postnatal age. In young postnatal animals, the superficial part of layer I shows a high density of nicely aligned neurons, representing Cajal-Retzius cells. These cells are relevant to orchestrate correct axonal distribution and synaptic targeting. During development, the number of these neurons decrease strikingly at ∼P14 (Anstötz et al., 2015).
At ∼P6–P9/P10, neurons in layers II and III showed a more developed dendritic morphology (Fig. 3C,D). At P6/P7, LII and LIII principal neurons already had more widely spreading basal dendrites and apical dendrites occasionally reached the molecular layer, whereas the morphology of MEC layers V and VI neurons was still poorly differentiated (Fig. 3C). From P9/P10 onward, the morphology of neurons in MEC layers II and III looked more mature with neurites, but the neuritic tree morphology indicated that these neurons were still not adult-like (Fig. 3D). From P12–P15 onward, the gross neuronal morphology looked more adult-like (Fig. 3E). Intracellularly filled LV neurons had apical dendrites that occasionally reached the molecular layer. At ∼P15, dendrites of all neurons extended over substantial distances with spine density still maturing; and occasionally axons were visible, showing local collateral branching and varicosities (Burton et al., 2008). LV neurons had apical dendrites that crossed the lamina dissecans, displaying a feature of pyramidal morphology in which the apical dendrites clearly radiate toward the pial surface. Apical tufts in layers I/II from the LV dendrites were still rare at this age (Fig. 3E). By P21, most LV apical dendrites reached the molecular layer (Fig. 3F). At this age, all layers were populated with well-differentiated neurons showing adult-like dendritic branching, clearly developed spines, and in many neurons the axon could be followed to either distribute locally, to extend toward the deep white matter or both (Fig. 3F).
Postnatal development of physiology of MEC neurons
The development of the physiological properties of MEC neurons paralleled the morphological development and supported the morphological implication that functional connectivity becomes adult-like after P20. In vitro whole-cell recordings in pups ranging from P9 to P30 revealed that MEC neurons do not reach adult-like properties at ∼P21. Before P12, LII principal neurons showed no rhythmic oscillations. From P12–P14 onward, the first indications for rhythmic activity were present, but these rhythmic periods were still mixed with époques without oscillations (Table 2; Fig. 4A,B). From P15 on, the frequency of oscillations increased significantly, reaching theta range, and stayed constant until P28–P30 (Table 2; Fig. 4C,D). Regarding resonance frequencies, before P14, LII principal neurons had a resonance frequency lower than theta; whereas after P14, the frequency increased up to the theta range (Table 2; Fig. 4). We were not able to detect membrane potential oscillations, resonance properties, and sag potentials in LIII and LV principal neurons at any age, similar to other reports from studies using young adult animals (Gloveli et al., 1997; Canto and Witter, 2012).
Table 2.
Postnatal development of MEC LII neuronsa
| Layer II | P9–11 (N = 15) | P12–14 (N = 16) | P15–17 (N = 14) | P28–30 (N = 11) | Difference between age groups |
|---|---|---|---|---|---|
| Resonance frequency (Hz) | 1.57 ± 0.43 | 2.02 ± 0.53 | 3.62 ± 0.54 | 4.2 ± 0.48 | ANOVA df = 3, F = 7.19, p = 0.001 |
| *p = 0.029 | **p = 0.023 | ||||
| **p = 0.001 | |||||
| Membrane potential oscillations (Hz) | 0 | 0.58 ± 0.41 | 4.04 ± 0.44 | 4.54 ± 0.30 | ANOVA df = 3, F = 21.5, p = 0.000 |
| *p = 0.00 | *p = 0.000 | ||||
| **p = 0.00 | **p = 0.004 | ||||
| Input resistance (GΩ) | 416 ± 19 | 279 ± 46 | 216 ± 24 | 181 ± 28 | ANOVA df = 3, F = 3.56, p = 0.021 |
| **p = 0.022 | |||||
| Current to reach threshold firing (pA) | 35.7 ± 4.77 | 86.0 ± 11.4 | 90.0 ± 18.0 | 164 ± 16.1 | KW 18.1, df = 3, p = 0.000 |
| #p = 0.006 | **p = 0.000 | ||||
| *p = 0.008 | |||||
| **p = 0.000 | |||||
| Time constant (ms) | 25.7 ± 3.2 | 21.8 ± 3.95 | 19.9 ± 3.3 | 14.3 ± 3.46 | ANOVA df = 3, F = 10.3, p = 0.000 |
| *p = 0.016 | **p = 0.004 | ||||
| **p = 0.000 | |||||
| DAP (mV) | 1.64 ± 0.7 | 2.54 ± 0.5 | 3.24 ± 0.38 | 4.12 ± 0.4 | ANOVA df = 3, F = 0.58, p = 0.632 |
| AP rise time, (ms) | 0.36 ± 0.01 | 0.32 ± 0.01 | 0.27 ± 0.01 | 0.26 ± 0.01 | ANOVA df = 3, F = 10.0, p = 0.000 |
| *p = 0.000 | |||||
| **p = 0.000 | |||||
| AP half-width (ms) | 1.88 ± 0.13 | 2.18 ± 0.16 | 1.38 ± 0.16 | 1.34 ± 0.14 | ANOVA df = 3, F = 7.51, p = 0.000 |
| **p < 0.040 | *p = 0.005 | **p = 0.001 |
aN indicates the total number of neurons measured per age group. The rows represent average LII principal neuron characteristics of all neurons/age group ± SEM. The last column presents the results of the statistical test used to assess the differences between age groups. Differences between the data points are considered statistically significant at p < 0.05. Homogeneity was tested with Levene's test. Assumptions for parametric tests were not met for current to reach threshold firing, so these were tested with Kruskal–Wallis (KW) followed by Mann–Whitney U tests instead. The resonance frequency is the peak in the impedance magnitude versus the frequency plot calculated by dividing the voltage response by the current input. Membrane potential oscillation frequencies were analyzed for power spectral density. Input resistance is measured by dividing the amplitude of the voltage response with that of the current step injected by fitting a double exponential curve to a −200 pA voltage response. The current to reach threshold firing is the current that is needed for an AP to occur. The time constant (tau) is the time it takes for the voltage deflection in response to a negative current step to reach 63% of its maximal value. We measured it in response to a −200 pA step. The DAP was calculated by first measuring the maximal depolarizing voltage deflection after the first AP induced. Then the difference between the amplitude of the depolarizing afterpotential and the amplitude of the threshold for firing was calculated. The AP rise time is the time of the AP to increase from 20% to 80% for the first AP in response to a weak current injection. AP half-width is measured at 50% of the AP amplitude of the first AP in response to a weak current step.
**Significantly different from age group P28–P30.
*Significantly different from age group P15–P17.
#Significantly different from age group P12–14.
Figure 4.

Postnatal development of physiological properties of principal neurons in MEC. The postsynaptic side. A–D, Response properties of MEC LII principal neurons. Voltage responses of one typical LII principal neuron per age group: (A) P9–P11, (B) P12–P14, (C) P15–P17, and (D) P28–P30. E–H, Response properties of MEC LIII principal neurons. Voltage responses of one typical LIII principal neuron per age group: (E) P9–P11, (F) P12–P14, (G) P15–P17, and (H) P28–P30. N indicates number of neurons measured per age group. A–H, First column, Voltage responses of typical principal neurons to a weak hyperpolarizing and depolarizing 1 s current step just reaching firing threshold. Insets, Zoom of 20 ms, displaying the AP afterpotentials. Second column, The voltage responses to a ±200 pA step of 1 s. Third column, A voltage response of the same neuron shown in the first two columns in response to a ZAP stimulus. Fourth column, Membrane fluctuations recorded just below firing threshold. In all subfigures, the average membrane potential is indicated right below the individual voltage traces.
With respect to firing properties, from P9 onward, LII principal neurons showed properties of intrinsic bursting neurons (Fig. 4A–D), with DAPs after the first spike (Fig. 4A–D, left, zoom-ins). The amplitude of DAPs did not increase significantly during development (Table 2). The rise time of APs increased significantly from P9–P11 to P12–P14 after which it stabilized (Table 2). The AP half-width was significantly smaller in age group P28–P30 compared with age groups P9–P11 and P12–P14 (Table 2). The input resistance showed a trend to decrease from P9 to P30 in both layer II and layer III neurons, with the current needed to reach AP threshold being lower in young LII principal neurons compared with old ones (Tables 2, 3). The time constant and AP half-width of layers II and V neurons decreased significantly from P9–P12 and P12–P14, respectively, to P28–P30 (Tables 2, 4). The AP half-width and intrinsic firing frequency of LIII principal neurons changed during development (Table 3; Fig. 4E–H). Weak depolarization led to significantly shorter interspike intervals in younger compared with P28–P30 old animals in response to weak and strong positive current (Table 3). LIII principal neurons changed from bursting to fast regular firing neurons at ∼P14.
Table 3.
Postnatal development of properties of MEC LIII neuronsa
| Layer III | P9–11 (N = 13) | P12–14 (N = 12) | P15–17 (N = 12) | P28–30 (N = 7) | Difference between age groups |
|---|---|---|---|---|---|
| Input resistance (GΩ) | 0.35 ± 0.47 | 0.34 ± 0.48 | 0.20 ± 0.01 | 0.18 ± 0.02 | KW 8.69, df = 3, p = 0.034 |
| *p = 0.029 | |||||
| **p = 0.029 | |||||
| Current to reach threshold firing (pA) | 30.7 ± 4.19 | 40 ± 5.39 | 30.8 ± 3.66 | 33.3 ± 6.6 | ANOVA df = 3, F = 0.82, p = 0.486 |
| Time constant (ms) | 24.0 ± 2.72 | 21.4 ± 2.11 | 23.9 ± 1.86 | 18.4 ± 2.13 | ANOVA df = 3, F = 0.31, p = 0.811 |
| AP rise time (ms) | 0.37 ± 0.03 | 0.34 ± 0.03 | 0.30 ± 0.03 | 0.34 ± 0.03 | ANOVA df = 3, F = 0.51, p = 0.674 |
| AP half-width (ms) | 1.92 ± 0.09 | 1.7 ± 0.08 | 1.47 ± 0.09 | 1.15 ± 0.1 | ANOVA df = 3, F = 12.2, p = 0.000 |
| *p = 0.049 | **p = 0.000 | ||||
| **p = 0.000 | |||||
| Interspike interval 1st and 2nd spike weak current step (s) | 0.08 ± 0.02 | 0.11 ± 0.31 | 0.16 ± 0.02 | 0.30 ± 0.03 | ANOVA df = 3, F = 6.56, p = 0.001 |
| **p = 0.001 | **p = 0.01 | ||||
| Interspike interval 1st and 2nd spike 200 pA current step (s) | 0.01 ± 0.00 | 0.01 ± 0.01 | 0.03 ± 0.00 | 0.04 ± 0.00 | KW 21.3, df = 3, p = 0.000 |
| *p = 0.003 | **p = 0.000 | ||||
| **p = 0.000 |
aN indicates the total number of neurons measured per age group. The rows represent average LIII principal neuron characteristics of all neurons/age group ± SEM. The last column presents the results of the statistical test used to assess the differences between age groups. Differences between the data points are considered statistically significant at p < 0.05. Homogeneity was tested with Levene's test. Assumptions for parametric tests were not met for input resistance and interspike interval 1st and 2nd spike 200 pA current steps, so differences were tested with Kruskal–Wallis (KW) followed by Mann–Whitney U tests. The interspike interval is calculated for the first and second spike in response to a weak and 200 pA step.
**Significantly different from age group P28–P30.
*Significantly different from age group P15–P17.
Table 4.
Postnatal development of properties of MEC LV neuronsa
| Layer V | P12–14 (N = 26) | P15–17 (N = 26) | P28–30 (N = 5) | Difference between age groups |
|---|---|---|---|---|
| Input resistance (GΩ) | 0.62 ± 0.04 | 0.65 ± 0.04 | 0.56 ± 0.04 | ANOVA df = 2, F = 1.23, p = 0.307 |
| Current to reach threshold firing (pA) | 40 ± 5.3 | 40.7 ± 3.4 | 43.3 ± 6.4 | ANOVA df = 2, F = 1.11, p = 0.344 |
| Time constant (ms) | 52.8 ± 12.7 | 40.4 ± 2.76 | 22.4 ± 2.43 | ANOVA df = 2, F = 2.09, p = 0.014 |
| **p = 0.050 | **p = 0.021 | |||
| AP rise time (ms) | 0.40 ± 0.04 | 0.34 ± 0.01 | 0.33 ± 0.02 | ANOVA df = 2, F = 1.34, p = 0.277 |
| AP half-width (ms) | 2.37 ± 0.21 | 1.75 ± 0.05 | 1.67 ± 0.20 | ANOVA df = 2, F = 3.80, p = 0.036 |
| **p = 0.04 | ||||
| Interspike interval 1st and 2nd spike weak current step (s) | 0.13 ± 0.02 | 0.13 ± 0.02 | 0.10 ± 0.05 | ANOVA df = 2, F = 0.03, p = 0.964 |
| Interspike interval 1st and 2nd spike 200 pA current step (s) | 0.02 ± 0.00 | 0.03 ± 0.01 | 0.03 ± 0.02 | ANOVA df = 2, F = 1.15, p = 0.332 |
aN indicates the total number of neurons measured per age group. The rows represent average LV principal neuron characteristics of all neurons/age group ± SEM. The last column presents the results of the statistical test used to assess the differences between age groups. Differences between the data points are considered statistically significant at p < 0.05. Homogeneity was tested with Levene's test. The interspike interval is calculated for the first and second spike in response to a weak and 200 pA step.
**Significantly different from age group P28–P30.
Postnatal development of functional connectivity between PaS, PrS, and MEC
We next studied and quantified the development of the functional connections from PaS and PrS with MEC to infer whether the ongoing development of in vivo head direction and grid cell properties is paralleled by changes in the PaS and PrS to MEC network ensemble. For this, we applied electrical stimulation with 1, 10, 20, 40, and 50 Hz (Boccara et al., 2010) to PaS and PrS and assessed MEC activity, making use of VSD imaging and single-neuron recordings. We have limited our descriptions and illustrations to the results of the 1 and 20 Hz stimulation frequencies because the results from these stimulation frequencies are representative of our observations at the range of frequencies noted above. For individual neuron recordings, we focused on principal neurons in layers II and III because this is where parasubicular and presubicular axonal collaterals terminate (Köhler, 1984; Caballero-Bleda and Witter, 1993). Based on current and published data (Langston et al., 2010; Couey et al., 2013), and on what is known about the functional development phases of the spatially modulated neuron types in vivo, we summarized data in six age groups (before P9, P9–P11, P12–P14, P15–P18, and P28–P30). Before P9, first anatomical but no monosynaptic physiological connections, measured with VSD imaging and single-neuron recordings, exist between PaS, PrS, and MEC. At P9–P11, the first physiologically functional monosynaptic connections between PaS and PrS with MEC develop. P12–P14 is defined by still closed eyes of the rats, and first head direction cells in vivo (Tan et al., 2015). P15–P18 is the age where inhibition within LII matures, where rats start exploring their own environment, have open eyes, where grid cells start to develop, and where we know that the head direction system matures notably with more reliable and directional head direction cells being present in the network (Ainge and Langston, 2012; Wills et al., 2013). Between P28 and P30, grid-cell and head-directional activity has stabilized. P59–P61 old animals were tested as a control to compare network activity in young adult rats with that in older rats.
Connectivity from PaS and PrS to MEC before P9
To establish the time point and laminar profile of head-directional input entering MEC, be it monosynaptic or polysynaptic, we first made use of VSD imaging of MEC from P7 onward while placing a bipolar stimulating electrode in either PaS or PrS. Such an electrode stimulates a large volume of tissue, and functional connectivity can thus be established. As described in the previous sections, anatomical connections are already present at this age and neuronal morphology of MEC neurons, particularly in superficial layers, such that MEC neurons could potentially respond to inputs. Even though we observed that, at P7, we are able to measure optical signals in MEC in response to bipolar stimulation of MEC itself (Fig. 5A), we do not see any optical signal within MEC before P9 in response to either weak or strong PaS and PrS stimulation (Fig. 5B,C; N = 3). The apparent lack of functional connectivity before P9 was corroborated by single-neuron recordings while stimulating either PaS or PrS (Fig. 5B′,C′); N = 4 for layer II and III, for PaS and PrS stimulation, and bipolar (strong) as well as glass electrode (weaker) stimulation. Regardless of stimulation strength, stimulation frequency, or duration, at P5/P6, no PSPs in MEC were observed. At P6/P7, the first indications for functional projections of PaS to dorsal MEC LII principal neurons were occasionally visible (N = 2), but only using strong extracellular pipette stimulation (45 mV). eEPSPs had a latency of 7.8 ± 0.4 ms and a failure rate of ∼95%, suggesting that the induced responses were not monosynaptic. At P7/8, the latencies for PaS to LII and PrS to LIII were short (latency 5.1 ± 0.2 ms; N = 4 and latency 5.34 ± 0.4 ms, respectively), yet the failure rate of synaptic events was high (80% and 60%, respectively) and no responses were visible at minimum stimulation, suggesting a not yet fully developed connectivity.
Figure 5.
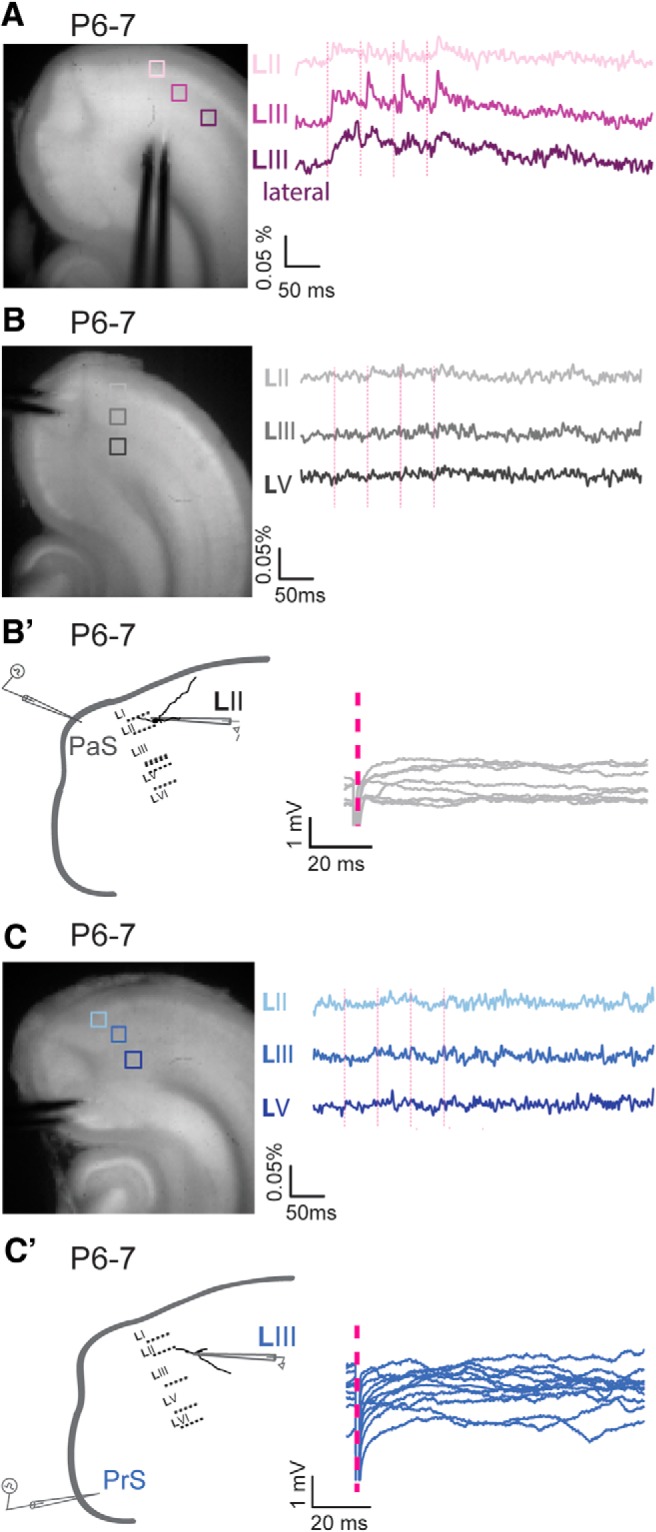
Before P9, monosynaptic membrane changes are not observed in MEC in response to PaS and PrS stimulation. A–C, Left, Semihorizontal brain section of a P6–P7 old animal with a stimulation electrode in (A) MEC, (B) PaS, and (C) PrS. Right, Optical signal traces after 20 Hz obtained from MEC LII, LIII, and LIII lateral or LV, respectively, of the boxed areas indicated to the left. B′, C′, Left, Drawings of a standard horizontal rat brain slice used to study contacts between (B′) PaS or (C′) PrS and MEC. The gray pipette stimulation electrode represents the stimulation position. A drawing of a representative principal neuron recorded from intracellularly (black pipette) is presented with dendrites and soma in black. No membrane changes are observed in MEC in response to (B′) PaS or (C′) PrS stimulation before P6/P7. Pink dotted lines indicate the time points of stimulation.
Connectivity between P9 and P11
PaS stimulation
The first measurable optical signals in MEC in response to bipolar PaS stimulation were seen at P9 (Fig. 6A; N = 4). Optical signal changes were observed almost exclusively in layers II and III of MEC 5.6 ± 0.4 ms after stimulation. To validate potentially monosynaptic network events observed with VSD, we also performed recordings of individual neurons while stimulating PaS (Fig. 6B–E). Putative monosynaptic responses of individual MEC LII (N = 5) and LIII (N = 6) principal neurons in response to PaS pipette stimulation were also visible from P8/P9 and P9/P10 onward (PaS to MEC LII failure rate 2% PaS to LIII 5%; latencies LII 5.71 ± 0.16 ms and LIII 5.9 ± 0.2 ms; eEPSPs in response to minimum stimulation of 0.9 mV; able to follow 100 Hz stimulation; minimal jitter < 250 μs). From P9 to P11, the onset latencies for optical signals did not change, but the signal size increased significantly from 1916.2 ± 298.2 to 5136.2 ± 278.3 for LII and from 1131.3 ± 264.6 to 3774.5 ± 248.0 for LIII (Table 5 for age group P9–P11; and Fig. 6A). Repetitive PaS stimulation of 20 Hz led to significant facilitation of the optical signal and the eEPSP amplitudes in P9–P11 old LII principal neurons (for optical signals, Fig. 6A; Table 5, evoked response changes within repetitive stimulation: RMA LII, N = 27, df 3, F = 36.76, p = 0.000; for individual neuron data: RMA, df = 3, F = 3.812, p = 0.033; Fig. 6B,D,E; Table 6). Regarding deep layers, optical signals hardly outreached baseline activity at P9–P10. The first clear optical signals were visible at P11, with the onset latencies between layers II-V and LVI being significantly different (Table 5; Fig. 6E). The latter data are underlined by the fact that putative monosynaptic responses of individual MEC LV principal neurons can only be detected from P11/P12 onward (N = 4, failure rate 9%; latency 5.5 ± 0.3; eEPSPs in response to minimum stimulation of 0.9 mV; able to follow 100 Hz stimulation, data not shown; minimal jitter < 250 μs; Fig. 6E).
Figure 6.

Postnatal development of network and neuronal reactions in MEC in response to PaS stimulation. A, Left, Representative Nissl-stained slice illustrating the position of the bipolar stimulation electrode in PaS and the position of four pixels in LII-LVI (light to dark gray, respectively) from which the optical responses are collected. The pixel size is 0.03 mm2. Right, Averaged optical signal traces in MEC after 20 Hz PaS stimulation. Dashed vertical line indicates the onset of stimulation. B, Drawings of a standard horizontal rat brain slice used to study contacts between PaS and MEC. The gray pipette stimulation electrode indicates the stimulation position in superficial PaS. For each age group, a drawing of a representative principal neuron recorded from intracellularly (black pipette) is presented with dendrites and soma in black and axons in red. C, The average waveform of an eEPSP in MEC LII, in response to 1 Hz PaS stimulation at different age groups: left, P9–P12; second left, P12–P14; third left, P15–P17; right, P28–P30. Inset (right of traces), Zoom-in of the first 20 ms shown to the left. D, The average amplitudes ± SEM of eEPSPs in response to four repetitive stimulations. *Significance and the corresponding p value. E, Single eEPSPs in response to 1 Hz (top) or 20 Hz (bottom) PaS stimulation, as recorded in LII (light gray), LIII (gray), or LV (dark gray). The membrane potential of the neuron is written. Pink dotted lines indicate the time points of stimulation. N indicates number of neurons measured per age group.
Table 5.
Properties of VSD imaging signals to parasubicular stimulationa
| PaS | Signal size | Latency (ms) | Decay time (ms) | Difference between layers per age group | |||
|---|---|---|---|---|---|---|---|
| P9–P11 | LII | 4357 ± 382 | 5.0 ± 0.3 | 127 ± 56 | D: ANOVA df = 3, p = 0.395 | L: KW 5.20, df = 3, p = 0.004 | S: ANOVA df = 3, F = 41, p = 0 0.000 |
| (N = 29) | (N = 27) | (N = 10) | |||||
| LIII | 2936 ± 282 | 4.6 ± 0.4 | 96 ± 31 | ||||
| (N = 29) | (N = 27) | (N = 10) | |||||
| **p = 0.000 | |||||||
| LV | 1661 ± 420 | 4.6 ± 0.3 | 110 ± 62 | ||||
| (N = 29) | (N = 8) | (N = 5) | |||||
| **p = 0.000 | |||||||
| *p = 0.000 | |||||||
| LVI | 1141 ± 301 | 8.2 ± 0.9 | 101 ± 31 | ||||
| (N = 29) | (N = 8) | (N = 5) | |||||
| **p = 0.000 | **p = 0.000 | ||||||
| *p = 0.000 | *p = 0.000 | ||||||
| #p = 0.002 | |||||||
| P12–P14 | LII | 5036 ± 312 | 4.1 ± 0.2 | 82 ± 10, (N = 25) | D: KW 8.37, df = 3, p = 0.39 | L: ANOVA df = 3, F = 6.03, p = 0.000 | S: ANOVA df = 3, F = 8.79, p = 0.000 |
| (N = 32) | (N = 32) | ||||||
| LIII | 4224 ± 294 | 3.6 ± 0.3 | 138 ± 19 | ||||
| (N = 32) | (N = 32) | (N = 22) | |||||
| **p = 0.042 | **p = 0.020 | ||||||
| LV | 5500 ± 442 | 4.2 ± 0.2, | 111 ± 10 | ||||
| (N = 32) | (N = 32) | (N = 26) | |||||
| *p = 0.001 | **p = 0.015 | ||||||
| #p = 0.000 | |||||||
| LVI | 3639 ± 327 | 5.2 ± 0.2 | 96 ± 32 | ||||
| (N = 32) | (N = 32) | (N = 16) | |||||
| **p = 0.000 | **p = 0.007 | **p = 0.058 | |||||
| #p = 0.000 | *p = 0.000 | ||||||
| #p = 0.011 | |||||||
| P15–P17 | LII | 3361 ± 174 | 3.9 ± 0.3 | 40 ± 2 | D: KW 26.9, df = 3, p = 0.000 | L: ANOVA df = 3, F = 4.36, p = 0.006 | S: ANOVA df = 3, F = 21.2, p = 0.000 |
| (N = 29) | (N = 26) | (N = 26) | |||||
| LIII | 3021 ± 164 | 4.0 ± 0.2 | 80 ± 7 | ||||
| (N = 29) | (N = 26) | (N = 26) | |||||
| **p = 0.000 | |||||||
| LV | 4593 ± 269 | 3.6 ± 0.3 | 64 ± 4 | ||||
| (N = 29) | (N = 26) | (N = 26) | |||||
| **p = 0.000 | **p = 0.000 | ||||||
| *p = 0.000 | *p = 0.033 | ||||||
| LVI | 3061 ± 219 | 5.1 ± 0.3 | 63 ± 5 | ||||
| (N = 29) | (N = 26) | (N = 26) | |||||
| #p = 0.000 | **p = 0.008 | **p = 0.001 | |||||
| *p = 0.017 | |||||||
| #p = 0.001 | |||||||
| P28–P30 | LII | 1890 ± 199 | 5.3 ± 0.4 | 24 ± 4 | D: KW 12.8, df = 3, p = 0.005 | L: ANOVA df = 3, F = 1.5, p = 0.218 | S: ANOVA df = 3, F = 6.3, p = 0.001 |
| (N = 19) | (N = 14) | (N = 12) | |||||
| LIII | 1609 ± 155, | 4.2 ± 0.3, | 59 ± 8 | ||||
| (N = 19) | (N = 14) | (N = 11) | |||||
| **p = 0.003 | |||||||
| LV | 2063 ± 201 | 4.2 ± 0.4 | 65 ± 9 | ||||
| (N = 19) | (N = 14) | (N = 10) | |||||
| *p = 0.004 | **p = 0.000 | ||||||
| LVI | 1467 ± 172 | 5.0 ± 0.5 | 52 ± 8 | ||||
| (N = 19) | (N = 14) | (N = 10) | |||||
| **p = 0.007 | **p = 0.017 | ||||||
| #p = 0.000 | |||||||
| P59–P61 | LII | 2459 ± 507 | 4.1 ± 0.6 | 34 ± 5 | D: KW 10.6, df = 3, p = 0.014 | L: ANOVA df = 3, F = 0.6, p = 0.603 | S: ANOVA df = 3, F = 3.83, p = 0.024 |
| (N = 8) | (N = 8) | (N = 8) | |||||
| LIII | 2128 ± 362 | 4.5 ± 0.8 | 78 ± 12 | ||||
| (N = 8) | (N = 8) | (N = 8) | |||||
| **p = 0.006 | |||||||
| LV | 2355 ± 395 | 4.8 ± 1.2 | 42 ± 8 | ||||
| (N = 8) | (N = 8) | (N = 5) | |||||
| LVI | 1613 ± 273 | 5.3 ± 0.4 | 57 ± 6 | ||||
| (N = 8) | (N = 8) | (N = 5) | |||||
| **p = 0.005 | **p = 0.030 | ||||||
| #p = 0.012 | |||||||
aThe rows of the table represent five different age groups (P9–P11, P12–P14, P15–P17, P28–P30, and P59–P61). The last column presents the results of the statistical test used to assess the differences between layers. Homogeneity was tested with Levene's test. If assumptions for parametric tests were not met, Kruskal–Wallis (KW) followed by Mann–Whitney U tests were performed instead. N indicates the number of VSD responses analyzed. The rows represent average parameters of each layers of all age groups ± SEM. S, Signal size (the integrals of the optical signal curves, which indicate the sum of membrane potential changes); L, latency (measured from the beginning of the stimulus artifact to the start of the fluorescent change response); D, decay time (calculated by fitting a curve from 25 ms up to 400 ms after the last stimulus to the negative slope).
**Significantly different from LII.
*Significantly different from LIII.
#Significantly different from LV.
Table 6.
Properties of evoked PSPs in MEC LII in response to parasubicular stimulationa
| Layer II | P9–11 (N = 6) | P12–14 (N = 6) | P15–17 (N = 4) | P28–30 (N = 6) | Difference between age groups |
|---|---|---|---|---|---|
| eEPSP amplitude (mV) | 2.97 ± 0.63 | 3.44 ± 1.2 | 1.85 ± 0.34 | 1.84 ± 0.41 | ANOVA df = 2, F = 0.94, p = 0.536 |
| eEPSP latency (ms) | 5.69 ± 0.31 | 5.74 ± 0.31 | 5.98 ± 0.43 | 5.44 ± 0.43 | ANOVA df = 2, F = 0.91, p = 0.426 |
| eEPSP rise time (ms) | 4.85 ± 0.90 | 7.73 ± 0.95 | 6.64 ± 1.24 | 6.62 ± 0.41 | ANOVA df = 3, F = 2.02, p = 0.147 |
| eEPSP decay time (ms) | 79.0 ± 11.0 | 60.6 ± 20.5 | 24.1 ± 3.38 | 14.6 ± 2.41 | KW 14.6, df = 3, p = 0.002 |
| *p = 0.010 | **p = 0.004 | ||||
| **p = 0.002 | |||||
| eEPSP half-width (s) | 0.73 ± 0.09 | 0.60 ± 0.12 | 0.32 ± 0.02 | 0.22 ± 0.01 | KW 14.0, df = 3, p = 0.001 |
| *p = 0.010 | **p = 0.002 | ||||
| **p = 0.002 |
aThe columns of the table represent four different age groups (P9–P11, P12–P14, P15–P17, and P28–P30) starting at P9–P11, when the first indications for stable monosynaptic contacts between PaS and LII were found. N indicates the total number of neurons measured per age group. Data for each individual neuron entered in the analysis are the means of 50 replications for each condition. The last column presents the results of the statistical test used to assess the differences between age groups. The rows represent average evoked PSP parameters of all neurons/age group ± SEM. Assumptions for parametric tests were not met for all significant values in this table. Therefore, Kruskal–Wallis (KW) followed by Mann–Whitney U tests were performed and Bonferroni correction applied. eEPSP amplitude, measured from baseline to peak after single PaS stimulation; eEPSP latency, measured from the beginning of the artifact until start of the eEPSP; eEPSP rise time and decay time, calculated by fitting a double exponential curve to the eEPSP; eEPSP half-width, the width at 50% of the amplitude.
**Significantly different from age group P28–P30.
*Significantly different from age group P15–P17.
PrS stimulation
Projections from PrS to MEC became functional at approximately the same time (P9/P10) as projections from PaS to MEC. At P9/P10, optical signal changes were observed mainly in MEC LII and LIII (optical signal size per layer: P9/P10, N = 17: LII 578.6 ± 166.2, LIII 1049.2 ± 271.0, LV 347.7 ± 112.5, LVI 367.9 ± 109.4; Table 7; Fig. 7A), which was confirmed by the first putative monosynaptic signal changes in individual LII (N = 2) and LIII (N = 6) principal neurons from P8/P9 and P9/P10 onward (PrS to MEC LII failure rate 2%; PrS to LIII 9%; latencies LII 5.71 ± 0.16 ms; and LIII 5.31 ± 0.09; eEPSPs in response to minimum stimulation of 0.9 mV; able to follow 100 Hz stimulation, data not shown; minimal jitter < 250 μs). At P11, a significant increase in optical signal size occurred in deep layers (signal size per layer: P11, N = 11: LII 1137.7 ± 146.0, LIII 2027.7 ± 202.0, LV 1442.8 ± 255.5, LVI 1805.3 ± 340.7; Table 7; Fig. 7A) with onset latencies not being different between layers (Table 7). Also, individual MEC LV and LVI principal neurons showed a postsynaptic signal change in response to PrS stimulation from P11/P12 onward (N = 4, latency 4.5 ± 0.3, 9% failure rate; Fig. 7B–E; Table 8).
Table 7.
Properties of VSD imaging signals to presubicular stimulationa
| PrS | Signal size | Latency (ms) | Decay time (ms) | Difference between layers per age group | |||
|---|---|---|---|---|---|---|---|
| P9–P11 | LII | 880 ± 117 (N = 37) | 6.0 ± 1.1 (N = 15) | — | L: KW 0.77, df = 3, p = 0.508 | S: ANOVA df = 3, F = 8.12, p = 0.000 | |
| LIII | 1578 ± 182 (N = 37) | 4.1 ± 0.5 (N = 24) | 101 ± 19 (N = 9) | ||||
| **p = 0.000 | |||||||
| LV | 939 ± 171 (N = 37) | 4.3 ± 0.6 (N = 16) | — | ||||
| *p = 0.000 | |||||||
| LVI | 1144 ± 223 (N = 37) | 4.5 ± 0.5 (N = 17) | — | ||||
| *p = 0.000 | |||||||
| P12–P14 | LII | 1047 ± 96 (N = 42) | 5.6 ± 0.4 (N = 29) | — | D: KW 2.57, df = 2, p = 0.275 | L: KW 3.23, df = 3, p = 0.025 | S: KW 39.2, df = 3, p = 0.000 |
| LIII | 2456 ± 137 (N = 42) | 3.8 ± 0.3 (N = 42) | 163 ± 27 (N = 27) | ||||
| **p = 0.000 | **p = 0.018 | ||||||
| LV | 3857 ± 312 (N = 42) | 3.6 ± 0.2 (N = 39) | 95 ± 6 (N = 33) | ||||
| **p = 0.000 | **p = 0.006 | ||||||
| *p = 0.000 | |||||||
| LVI | 4361 ± 373 (N = 42) | 3.6 ± 0.2 (N = 38) | 126 ± 14 (N = 31) | ||||
| **p = 0.000 | **p = 0.009 | ||||||
| *p = 0.000 | |||||||
| P15–P17 | LII | 699 ± 64 (N = 37) | 4.7 ± 0.4 (N = 21) | — | D: KW 3.13, df = 2, p = 0.208 | L: KW 1.11, df = 3, p = 0.345 | S: KW 103, df = 3, p = 0.000 |
| LIII | 1606 ± 121 (N = 37) | 3.7 ± 0.4 (N = 32) | 120 ± 19 (N = 33) | ||||
| **p = 0.000 | |||||||
| LV | 3678 ± 297 (N = 37) | 3.4 ± 0.3 (N = 34) | 78 ± 6, (N = 29) | ||||
| **p = 0.000 | |||||||
| *p = 0.000 | |||||||
| LVI | 3943 ± 282 (N = 37) | 3.0 ± 0.3 (N = 34) | 89 ± 13 (N = 28) | ||||
| **p = 0.000 | |||||||
| *p = 0.000 | |||||||
| P28–P30 | LII | 466 ± 45 (N = 25) | 4.5 ± 0.9 (N = 11) | — | D: ANOVA df = 2, F = 0.072, p = 0.930 | L: KW 0.85, df = 3, p = 0.461 | S: KW 58.9, df = 3, p = 0.000 |
| LIII | 994 ± 82 (N = 25) | 4.1 ± 0.5 (N = 18) | 52 ± 12 (N = 10) | ||||
| **p = 0.000 | |||||||
| LV | 2018 ± 168 (N = 25) | 3.3 ± 0.4 (N = 19) | 59 ± 10 (N = 14) | ||||
| **p = 0.000 | |||||||
| *p = 0.000 | |||||||
| LVI | 2503 ± 223 (N = 25) | 4.1 ± 0.3 (N = 21) | 53 ± 10 (N = 18) | ||||
| **p = 0.000 | |||||||
| *p = 0.000 | |||||||
| P59–P61 | LII | 513 ± 155 (N = 9) | 5.0 ± 0.7 (N = 8) | — | D: ANOVA df = 2, F = 25.1, p = 0.782 | L: ANOVA df = 3, F = 1.7, p = 0.168 | S: ANOVA df = 3, F = 25.5, p = 0.000 |
| LIII | 926 ± 150 (N = 9) | 5.2 ± 0.9 (N = 8) | 55 ± 14, (N = 6) | ||||
| **p = 0.022 | |||||||
| LV | 1587 ± 213 (N = 9) | 3.8 ± 0.7 (N = 8) | 70 ± 15, (N = 6) | ||||
| **p = 0.000 | |||||||
| *p = 0.000 | |||||||
| LVI | 1835 ± 245 (N = 9) | 3.1 ± 0.5 (N = 8) | 55 ± 21, (N = 7) | ||||
| **p = 0.000 | |||||||
| *p = 0.000 | |||||||
aThe rows of the table represent five different age groups (P9–P11, P12–P14, P15–P17, P28–P30, and P59–P61). The last column presents the results of the statistical test used to assess the differences between layers. Homogeneity was tested with Levene's test. If assumptions for parametric tests were not met, Kruskal–Wallis (KW) followed by Mann–Whitney U tests were performed instead. N indicates the number of VSD responses analyzed. The rows represent average parameters of each layers of all age groups ± SEM.
**Significantly different from LII.
*Significantly different from LIII.
Figure 7.

Postnatal development of network and neuronal reactions in MEC in response to PrS stimulation. A, Left, Representative Nissl-stained slice illustrating the position of the bipolar stimulation electrode in PrS and the position of four pixels in LII-LVI (light to dark blue, respectively) from which the optical responses are collected. The pixel size is 0.03 mm2. Right, Averaged optical signal traces in MEC after 20 Hz PrS stimulation. Dashed vertical line indicates the onset of stimulation. B, Drawings of a standard horizontal rat brain slice used to study contacts between PrS and MEC. The gray pipette stimulation electrode indicates the stimulation position in superficial PrS. For each age group, a drawing of a representative principal neuron recorded from intracellularly (black pipette) is presented with dendrites and soma in black and axons in red. C, The average waveform of an eEPSP in MEC LIII, in response to 1 Hz PrS stimulation at different age groups: left, P9–P12; second left, P12–P14; third left, P15–P17; right, P28–P30. Inset (right of traces), Zoom-in of the first 20 ms shown to the left. D, The average amplitudes ± SEM of eEPSPs in response to four repetitive stimulations. *Significance and the corresponding p value. E, Single eEPSPs in response to 1 Hz (top) or 20 Hz (bottom) PrS stimulation, as recorded in LII (light blue), LIII (blue), or LV (dark blue). The membrane potential of the neuron is indicated. Pink dotted lines indicate the time points of stimulation. N indicates number of neurons measured per age group.
Table 8.
Properties of evoked PSPs in MEC LIII in response to presubicular stimulationa
| Layer III | P9–11 (N = 4) | P12–14 (N = 5) | P15–17 (N = 5) | P28–30 (N = 5) | Difference between age groups |
|---|---|---|---|---|---|
| eEPSP amplitude (mV) | 1.03 ± 0.47 | 1.08 ± 0.42 | 1.87 ± 0.23 | 2.96 ± 0.75 | ANOVA df = 3, F = 0.93, p = 0.380 |
| eEPSP latency (ms) | 5.90 ± 0.25 | 5.20 ± 0.27 | 5.40 ± 0.39 | 5.44 ± 0.60 | ANOVA df = 3, F = 1.51, p = 0.250 |
| eEPSP rise time (ms) | 3.70 ± 0.15 | 4.50 ± 1.01 | 4.68 ± 1.10 | 4.19 ± 0.76 | ANOVA df = 3, F = 0.25, p = 0.855 |
| eEPSP decay time (ms) | 357 ± 126 | 113 ± 25.7 | 390 ± 115 | 89.3 ± 3.76 | KW 8.46, df = 3, p = 0.037 |
| **p = 0.008 | |||||
| eEPSP half-width (s) | 1.63 ± 0.29 | 0.88 ± 0.17 | 2.48 ± 0.36 | 0.74 ± 0.38 | ANOVA df = 3, F = 10.1, p = 0.010 |
| *p = 0.002 | **p = 0.01 |
aThe columns of the table represent four different age groups (P9–P11, P12–P14, P15–P17, and P28–P30) starting at P9–P11, when the first indications for stable monosynaptic contacts between PrS and LIII were found. N indicates the total number of neurons measured per age group. Data for each individual neuron entered in the analysis are the means of 50 replications for each condition. The last column represents the results of the statistical test used to assess the differences between age groups. The rows represent average evoked PSP parameters of all neurons/age group ± SEM. Assumptions for parametric tests were not met for decay time values. Therefore, Kruskal–Wallis (KW) followed by Mann–Whitney U tests were performed and Bonferroni correction applied.
**Significantly different from age group P28–P30.
*Significantly different from age group P15–P17.
To summarize, our data indicate that superficial MEC layers receive functional putative monosynaptic inputs from PaS and PrS from P8/P9 onward. The input to deep MEC layers becomes significant and monosynaptic ∼2–3 d later at P11/P12.
Connectivity between P12 and P14
PaS stimulation
Compared with the previous age group, LII optical responses did not change (Fig. 6A; Table 5). We also analyzed responses of individual MEC LII principal neurons in response to PaS stimulation. Basic PSP parameters, such as amplitude, latency of onset, rise time, and half-width, did not differ between P9–P11 and P12–P14 animals (Table 6; Fig. 6B–E). LII shows facilitating optical responses and a trend for facilitating neuronal responses in response to 20 Hz stimulation (optical data, Fig. 6A;Table 5, evoked response changes within repetitive stimulation; optical data: RMA LII N = 32, df = 3, F = 0.002, p = 0.000; individual neuron data: RMA df = 3, F = 2.839, p = 0.083; Fig. 6D). The decay times of the optical and neuronal signals did not differ between layers (Tables 5, 6). LIII optical responses and responses in deep layers became stronger (Fig. 6A; Table 5). Within the layers, signal sizes in LVI were significantly smaller compared with signal sizes of LII and LV (Table 5). As in the previous age group, the latencies of responses were still significantly longer in LVI compared with the other layers (Table 5).
PrS stimulation
Between P12 and P14, optical responses were significantly weaker in LII compared with the strong and facilitating responses in LIII, LV, and LVI (evoked response changes within repetitive stimulation per layer: RMA LIII N = 42, df = 3, F = 65.92, p = 0.000, LV N = 39, df = 3, F = 51.87, p = 0.000; LVI N = 38, df = 3, F = 70.18, p = 0.000; Table 7; Fig. 7A). Induced optical signal sizes in LIII were smaller than the induced signal sizes observed in deep MEC (Table 7). In LIII, LV, and LVI, the optical signal sizes increased significantly compared with P9–P11 (Fig. 7A; Table 7). The latencies were significantly longer in LII compared with deep layers (Table 7) but did not differ from the previous age group. The decay times did not differ between layers (Table 7), with the average decay time of LII not being measured in response to PrS stimulation due to the small signals. Subsequently, we also compared the properties of the evoked PSPs in individual MEC LIII principal neurons in response to PrS stimulation and found that the PSP parameters did not differ (Table 8; Fig. 7B–E). LIII showed facilitating optical and neuronal responses to 20 Hz stimulation, but the neuronal responses did not reach significance (RMA df = 1.075, F = 0.930, p = 0.394; Fig. 7A,D; Table 7). The decay times of the optical and neuronal signal did not differ between layers (Tables 7, 8).
Connectivity between P15 and P17
PaS stimulation
In the P15–P17 age group, the latencies of the optical responses observed were similar to those of the P12–P14 group, and only LVI had a slightly longer latency (Table 5; Fig. 6A). All layers showed facilitation, including LIII, with decreasing decay times in the optical and individual neuron signal (evoked response changes within repetitive stimulation per layer, optical data: RMA, all layers have N = 26: LII df = 3, F = 9.163, p = 0.000, LIII df = 3, F = 58.35, p = 0.000, LV df = 3, F = 89.58, p = 0,000, LVI df = 3, F = 68.41, p = 0.000; individual neuron data: RMA df = 3, F = 1.268, p = 0.367; Tables 5, 6; Fig. 6A,C). The optical signal sizes of superficial layers were significantly smaller in this age group compared with the P12–P14 group, whereas no differences in the deep layers were observed between the two age groups. The largest decrease of optical signal sizes occurred in LII and LIII neurons from P12 to P13 (LII Kruskal–Wallis test χ2 12.49, df = 10 p = 0.000; Mann–Whitney U: P11 and P12–P13 and P14, p = 0.002, LIII Kruskal–Wallis test χ2 9.68, df = 10 p = 0.000; Mann–Whitney U: P12–P13 and P14, p = 0.034,). The decrease in signal amplitude observed in animals between P12 and P15 is most likely explained by an increase in GABAergic inhibition since optical signals represent only the net depolarizing signal. Interestingly, similar to the optical signals, the decay times of PSPs of individual LII principal neurons in response to PaS stimulation decreased significantly, also leading to a smaller eEPSP half-width and a tendency for a smaller EPSP amplitude (Table 6; Fig. 6C). These developments are thus in parallel with the developments in the optical signal and properties of individual neurons. All these data support the notion that, from P14 onward, the GABAergic inhibition matures vigorously. In line with this, application of the GABAa antagonist bicuculline (5 μm) increased the amplitudes of the optical signals in response to PaS stimulation in layers II and III only from P12 to P14 onward, whereas in deep layers GABAergic inhibition evolved later, starting to develop in ∼P15–P17 (normal ACSF vs bicuculline ACSF, paired t test: P7–P11, N = 6; sign test for paired comparisons: LII p = 0.062, LIII p = 0.062, LV p = 0.718, LVI p = 0.906; P12–P14, N = 6; LII p = 0.004, LIII p = 0.001, LV p = 0.670, LVI p = 0.950, P15–P17, N = 10; LII p = 0.000, LIII p = 0.000, LV p = 0.034, LVI p = 0.074).
PrS stimulation
Regarding presubicular to MEC connectivity, in the P15–P17 age group, LV and LVI showed the largest optical responses followed by responses in LIII and LII (Table 7; Fig. 7A). LIII responses were significantly weaker compared with P12–P14 animals (Fig. 7A). The latencies of responses in LII were no longer different from the other layers and were similar to the P12–P14 group (Table 7). LII did not show strong facilitation, whereas the optical signals of the other layers did (evoked response changes within repetitive stimulation per layer: RMA LII N = 22, df = 3, F = 5.025, p = 0.004; LIII N = 32, df = 2.388, F = 43.577, p = 0.000, LV N = 34, df = 3, F = 52.64, p = 0.000; LVI N = 34, df = 3, F = 55.92, p = 0.000; individual neuron data: RMA df = 1.009, F = 3.085, p = 0.153; Table 7; Fig. 7A). The decay time was similar in all cell layers, although we observed a trend that LIII had the longest decay time (Table 7). PSP properties of individual LIII neurons in response to PrS stimulation did not differ from the previous age groups, except for the PSP half-width, which was significantly longer compared with the previous age group, which is also in line with a trend in a longer decay time of the optical signals (Table 8; Fig. 7B–E). The decrease in signal amplitudes in LIII and deep layers after P14 in response to PrS stimulation may also be explained by the previously described increase in GABAergic inhibition. Application of bicuculline (5 μm) did not lead to an increase in signal amplitude in P9–P11 old rats (normal ACSF vs bicuculline ACSF, N = 6; paired t test: LII p = 0.054, LIII p = 0.116, LV p = 0.088, LVI p = 0.079), whereas bicuculline increased the amplitudes of the optical signals in deep layers from P12 to P14 onward. However, in superficial layers, the effect of bicuculline evolved at ∼P15–P17 (normal ACSF vs bicuculline ACSF, sign test for paired comparisons: P12–P14, N = 8; LII p = 0.164, LIII p = 0.070, LV p = 0.039, LVI p = 0.007; P15–P17, N = 9; LII p = 0.007, LIII p = 0.003, LV p = 0.003, LVI p = 0.007).
Connectivity between P28 and P30
PaS stimulation
From P28 to 30, which is the age group where head direction and grid cell activity stabilize (Langston et al., 2010; Wills et al., 2010), we observed a significantly smaller optical signal compared with age groups P12–P14 and P15–P17 (Table 5; Fig. 6A). The latencies of optical and neuronal signal onset were still similar between layers (Tables 5, 6). Responses in layers III and V showed only weak facilitation, whereas the optical and neuronal signals of LII and also the optical signals of LVI showed no facilitation (evoked response changes within repetitive stimulation per layer, optical data: RMA, all layers have N = 14, LII df = 1.484, F = 0.150, p = 0.798, LIII df = 3, F = 7.574, p = 0.000, LV df = 3, F = 18.09, p = 0.000, LVI df = 1.014, F = 1.430, p = 0.253; individual neuron data: RMA df = 3, F = 2.062, p = 0.176; Table 5; Fig. 6A–D). In this age group, as in the previous age group, optical and individual neuron signals in LII had the shortest decay time back to baseline after applied stimuli (Tables 5, 6), which led to a significantly shorter half-width of PSPs.
PrS stimulation
Age group P28–P30 showed smaller optical signals in all layers than P12–P14 and P15–P17, except for LII, where the signal was small at all ages (Table 7; Fig. 7A). The latencies of the optical and individual neuronal signal did not differ between layers and age groups (Tables 7, 8). As in the previous age group, LII showed the weakest facilitation in the optical signal, whereas the other layers still showed strong facilitation (evoked response changes within repetitive stimulation per layer; optical data: RMA LII N = 11, df = 3, F = 2.201, p = 0.108; LIII N = 18, df = 2.016, F = 6.798, p = 0.003, LV N = 19, df = 3, F = 12.124, p = 0.000; LVI N = 21, df = 2.26, F = 14, p = 0.000; individual neuron data: RMA df = 1.008, F = 3.311, p = 0.166; Table 7; Fig. 7A,D,E). The decay times were similar in all layers (Tables 7, 8).
Connectivity between P59 and P61
PaS stimulation
We further compared signal sizes of P28–P30 young adult rats with P59–P61 adult rats and found that there were no differences between the two age groups (Table 5; Fig. 8, left). Also, the latencies did not differ between layers and age groups (Table 5). Regarding facilitation, all layers showed only weak or no facilitation comparing the first with the last evoked response, similar to what we found for the previous age group (evoked response changes within repetitive stimulation per layer: RMA, all layers have N = 8, LII df = 1.579, F = 3.302, p = 0.083, LIII df = 3, F = 6.30, p = 0.003, LV df = 3, F = 6.293, p = 0.003, LVI df = 3, F = 8.912, p = 0.001; Table 5; Fig. 8, left). LII also showed the shortest decay time in these young adult rats, as in the previous age groups (Table 5).
Figure 8.

Network reactions in adult MEC. Averaged optical signal traces in MEC after 20 Hz PaS stimulation (left) and PrS stimulation (right) from age group P59–P61. Dashed vertical line indicates the onset of stimulation.
PrS stimulation
We further compared signal sizes of P28–P30 old rats with P59–P61 rats after PrS stimulation. We found that there were no differences between the two age groups (Table 7; Fig. 8, right). Furthermore, the latencies did not differ between layers and age groups (Table 7). As in the previous age groups, LII still showed the weakest facilitation, even though facilitation in deep layers was also weakened (evoked response changes within repetitive stimulation per layer: RMA, LII N = 6, df = 3, F = 0.557, p = 0.651; LIII N = 7, df = 3, F = 3.405, p = 0.040, LV N = 8, df = 3, F = 1.040, p = 0.395; LVI N = 8, df = 3, F = 2.657, p = 0.075; Table 7; Fig. 8, right). The decay times between layers were similar in this age group as well (Table 7).
Discussion
Our results show that axons from PaS and PrS evolve in MEC and have a potential to establish synaptic contacts with MEC neurons from ∼P4–P6 onward (Fig. 9). This potential greatly enhances during the subsequent week because the presynaptic axons show a marked morphological development after P4/P5. This development is accompanied by a parallel development of the postsynaptic targets; from P6/P7 onward, MEC neurons start to develop advanced dendritic branching patterns (Fig. 9). Morphologically, the network does not develop an adult-like connectivity matrix until P12–P15, and the complexity takes another 1–2 weeks to mature to adult levels. Our electrophysiological VSD imaging and single whole-cell recording experiments in rat brain slices from P4–P61 old animals are in line with this morphologically derived conclusion. Immature monosynaptic functional connections from both PaS and PrS to MEC (González-Burgos et al., 2000; Tolner et al., 2007) emerge at P8–P9. Both PaS and PrS to MEC connections develop over the course of a few days, and responses of MEC neurons to PaS and PrS stimulation become more reliable at ∼P10–P11. During this developmental period, PaS mainly interacts with neurons in LII, whereas PrS preferentially influences LIII. The functional innervation of deep MEC layers from both PaS and PrS begins at ∼P11–P12. From P12 onward, PaS and PrS activation leads to reliable monosynaptic activity in LII, LIII, and LV, coincident with the emergence of PrS head direction cells (Tan et al., 2015) (Fig. 9). From P14 onward, the level of network depolarization in MEC diminishes, likely due to an increase in inhibition, reaching levels shown in adult animals.
Figure 9.

Overview summarizing the development of functionality and functional projections from PaS and PrS to MEC in the rat. Top row, Head direction cells are present in PaS and PrS from P11 onward. P11 was the first day of recordings performed by Bjerknes et al. (2015). Polar plots showing distribution of firing rate for one representative head direction cell (peak firing rate indicated). Directional tuning stabilizes shortly after eye opening. Adapted with permission from Bjerknes et al. (2015). Second row, Grid cells are also present from the first day of recordings (P16), but grid regularity increases throughout the first 4–5 weeks of postnatal development. Rate maps of grids (top row) and spatial autocorrelations (bottom row) from P16 to P18 old rats show grid-like characteristics, but the regularity and specificity of the grid increase with age. Grid scores: blue represents r = −1; red represents r = 1. Adapted with permission from Langston et al. (2010) and Ainge and Langston (2012). Third row, Schematic overview of the development of the presynaptic (PaS and PrS) and postsynaptic (MEC) anatomy. Orange represents the somata of MEC neurons together with their neurites. The dendritic morphology of P4–P6 old MEC neurons is poorly developed. The majority of somata with recognizable neurites are located in layers II and III. For all neurons, the number of neurites is low and the neurites are poorly developed, remaining mostly within the layer of origin. Dendrites of mainly superficial layers show sparse branching that does not extend over long distances. Axons from PaS (gray tones) and PrS (blue tones) emerge from P4 to P6 but continue to develop over time. Indicated are varicosities (filled circles) and synapses (axonal branching points with synapse shaped terminals contacting the postsynaptic MEC neuron). From P9/P10 onward, the morphology of neurons in MEC layers II and III looks more mature, but the neuritic tree morphology together with the presynaptic morphology indicates that the connections are still not adult-like. From P12–P15 onward, the gross neuronal morphology looks more adult-like. Fourth row, The averaged optical signal traces in MEC after 20 Hz PaS stimulation (gray) and PrS stimulation (blue) from the different age groups. Bottom row, Averaged voltage trace of an eEPSP in MEC LII (gray; top) and LIII (blue; bottom), in response to 1 Hz PaS (gray; top) and PrS (blue; bottom) stimulation, respectively, at the different age groups: left, P9–P12; second left, P12–P14; third left, P15–P17; right, P28–P30.
Connectivity before P9
The majority of neurons in PaS, PrS, and MEC are formed before birth (Bayer, 1980a, b; Donato et al., 2017). However, 1 week postnatally, neurons are morphologically immature and axonal connections may not have formed yet. Projections from PaS and PrS to MEC can be anatomically traced from P4/P5 onward, yet the present data indicate that these projections are not functional before P9. Because stimulation of LV in MEC resulted in local activation of LII and LIII before P9, it appears that neurons in MEC can be electrically driven. Thus, although sporadic synaptic inputs from PaS and PrS do exist at P8, these inputs cannot yet reliably drive the existing local MEC network, similar to what was shown in mutant mice that lack the expression of Munc-18, a protein that is obligatory in vesicle exocytosis (Verhage et al., 2000; Bouwman et al., 2004). In those mutant mice, synaptic wiring of the brain may be structured normally, but without presence of synaptic release. Inputs from PaS and PrS may also differ from local connections in MEC in that the former inputs depend on the development of distal portions of the dendrite of their target cells. Our data indicate that the neuron morphology in such young animals is immature, lacking well-developed distal dendrites. How the lower spine density on P5 EC LII neurons compared with P14 neurons plays a role in development of these connections remains to be established.
Connectivity between P9 and P11
At this early age, PaS and PrS stimulation mainly evoked immature responses in LII and LIII, whereas responses in LV and LVI were weak or absent. As we showed, such weak responses cannot be explained by GABAergic inhibition at this developmental stage. In the case of MEC LV neurons, the weak to absent responses are more likely caused by the lack of fully developed basal and apical dendrites and the lack of spines.
Connectivity between P12 and P14
At ∼P12, the network and neuronal responses in MEC change in two main aspects. Upon PaS and PrS stimulation, responses in LV and LVI become more proficient; those in LII and LIII increase in amplitude and stabilize. The overall increased responsiveness of MEC deep layers is in line with what we found in P20–P30 old animals (Canto et al., 2012), which may reflect an overall increase in maturity of the physiology, the apical dendrite, and density of spines (Reboreda et al., 2007). The physiological maturation of LII and LIII principal neurons before P14 likely reflects the ongoing development of receptor expression (Burton et al., 2008). After P14, LII and LIII typical characteristics look adult-like (Canto and Witter, 2012), which suggests that integrative properties relying on intrinsic membrane properties (Losonczy et al., 2002; Garden et al., 2008) become stable around that age, even though it has been shown that profiles of individual LII neurons still show changes after P30 (Burton et al., 2008; Boehlen et al., 2010; Ray and Brecht, 2016).
We further observed changes in response properties of principal neurons after bicuculline application in all layers. This rather abrupt change in network sensibility to bicuculline likely relates to the so-called GABA shift that occurs in the cortex after birth (Cherubini et al., 1991; Ben-Ari et al., 2007).
Connectivity before and after P15
The current observations strongly support the notion that the development of projections from PaS and PrS to MEC is such that inputs from both structures exert functional effects on the MEC network before P15 but reach adult-like properties at ∼P15. That the signal amplitude shows a decrease until P28 is likely due to an increase of inhibition in the network still developing after P15 (Cherubini et al., 1991; Ben-Ari et al., 2007). Although cutting the axons may also cause this decrease of activation, we deem that unlikely since we checked connectivity in slices using anatomical tracing (Canto et al., 2012). The parallel development of GABAergic modulation and functional connectivity between PaS, PrS, and MEC support that both are key players in the development of functional cells, such as grid cells, in MEC (McNaughton et al., 2006; Burgess et al., 2007; Hasselmo et al., 2007; Burak and Fiete, 2009; Beed et al., 2013; Couey et al., 2013; Winter et al., 2015).
Development of PaS and PrS inputs to MEC match functional changes
Our results are the first to show that functional projections from PaS and PrS to MEC are present before immature grid cells have been observed in MEC at P16. These inputs to MEC show relatively mature properties at P15, around eye opening, which is the same time head direction cells stabilize in both MEC, PaS, and PrS, and long before grid cells have adult-like properties, which happens between P21 and P28–P30 (Langston et al., 2010; Wills et al., 2010; Tan et al., 2015). This developmental timeline is in agreement with the suggested hypothesis that head direction signals in MEC depend on inputs from at least PrS, but likely also on those from PaS. The latter hypothesis is supported by reports that manipulations of the head direction system at the level of the anterior thalamus in adults result in a disruption of grid cell firing (Winter et al., 2015). The early inputs from PaS and PrS, functional from P12, thus likely provide head directional codes to the developing MEC, several days before head direction or grid cells emerge in MEC. The stability of the signal in the head direction system dramatically increases within 24 h after eye opening (Tan et al., 2015), when the connectional matrix, as we reported, also reaches stable adult-like properties.
Footnotes
This work was supported by the Dutch research council grant ALW-VENI 863.14.005 to C.B.C, the Kavli Foundation, European Union 7th Framework Spacebrain Grant 200873, Centre of Excellence Grants 223262 and 145993, and Norwegian Research Council Equipment Grant 181676 and Research Grants 191929 and 227769. We thank Jonathan J. Couey for advice in the initial phase of the experiments.
The authors declare no competing financial interests.
References
- Ainge JA, Langston RF (2012) Ontogeny of neural circuits underlying spatial memory in the rat. Front Neural Circuits 6:8. 10.3389/fncir.2012.00008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anstötz M, Huang H, Marchionni I, Haumann I, Maccaferri G, Lübke JHR (2015) Developmental profile, morphology, and synaptic connectivity of Cajal-Retzius cells in the postnatal mouse hippocampus. Cereb Cortex 26:855–872. 10.1093/cercor/bhv271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer SA. (1980a) Development of the hippocampal region in the rat: I. Neurogenesis examined with 3H-thymidine autoradiography. J Comp Neurol 190:87–114. 10.1002/cne.901900107 [DOI] [PubMed] [Google Scholar]
- Bayer SA. (1980b) Development of the hippocampal region in the rat: II. Morphogenesis during embryonic and early postnatal life. J Comp Neurol 190:115–134. 10.1002/cne.901900108 [DOI] [PubMed] [Google Scholar]
- Beed P, Gundlfinger A, Schneiderbauer S, Song J, Böhm C, Burgalossi A, Brecht M, Vida I, Schmitz D (2013) Inhibitory gradient along the dorsoventral axis in the medial entorhinal cortex. Neuron 79:1197–1207. 10.1016/j.neuron.2013.06.038 [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Gaiarsa JL, Tyzio R, Khazipov R (2007) GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev 87:1215–1284. 10.1152/physrev.00017.2006 [DOI] [PubMed] [Google Scholar]
- Bjerknes TL, Langston RF, Kruge IU, Moser EI, Moser MB (2015) Coherence among head direction cells before eye opening in rat pups. Curr Biol 25:103–108. 10.1016/j.cub.2014.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccara CN, Sargolini F, Thoresen VH, Solstad T, Witter MP, Moser EI, Moser MB (2010) Grid cells in pre- and parasubiculum. Nat Neurosci 13:987–994. 10.1038/nn.2602 [DOI] [PubMed] [Google Scholar]
- Boehlen A, Heinemann U, Erchova I (2010) The range of intrinsic frequencies represented by medial entorhinal cortex stellate cells extends with age. J Neurosci 30:4585–4589. 10.1523/JNEUROSCI.4939-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnevie T, Dunn B, Fyhn M, Hafting T, Derdikman D, Kubie JL, Roudi Y, Moser EI, Moser MB (2013) Grid cells require excitatory drive from the hippocampus. Nat Neurosci 16:309–317. 10.1038/nn.3311 [DOI] [PubMed] [Google Scholar]
- Bouwman J, Maia AS, Camoletto PG, Posthuma G, Roubos EW, Oorschot VMJ, Klumperman J, Verhage M (2004) Quantification of synapse formation and maintenance in vivo in the absence of synaptic release. Neuroscience 126:115–126. 10.1016/j.neuroscience.2004.03.027 [DOI] [PubMed] [Google Scholar]
- Burak Y, Fiete IR (2009) Accurate path integration in continuous attractor network models of grid cells. PLoS Comput Biol 5:e1000291. 10.1371/journal.pcbi.1000291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess N, Barry C, O'Keefe J (2007) An oscillatory interference model of grid cell firing. Hippocampus 17:801–812. 10.1002/hipo.20327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton BG, Economo MN, Lee GJ, White JA (2008) Development of theta rhythmicity in entorhinal stellate cells of the juvenile rat. J Neurophysiol 100:3144–3157. 10.1152/jn.90424.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero-Bleda M, Witter MP (1993) Regional and laminar organization of projections from the presubiculum and parasubiculum to the entorhinal cortex: an anterograde tracing study in the rat. J Comp Neurol 328:115–129. 10.1002/cne.903280109 [DOI] [PubMed] [Google Scholar]
- Canto CB, Witter MP (2012) Cellular properties of principal neurons in the rat entorhinal cortex: II. The medial entorhinal cortex. Hippocampus 22:1277–1299. 10.1002/hipo.20993 [DOI] [PubMed] [Google Scholar]
- Canto CB, Koganezawa N, Beed P, Moser EI, Witter MP (2012) All layers of medial entorhinal cortex receive presubicular and parasubicular inputs. J Neurosci 32:17620–17631. 10.1523/JNEUROSCI.3526-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherubini E, Gaiarsa JL, Ben-Ari Y (1991) GABA: an excitatory transmitter in early postnatal life. Trends Neurosci 14:515–519. 10.1016/0166-2236(91)90003-D [DOI] [PubMed] [Google Scholar]
- Couey JJ, Witoelar A, Zhang SJ, Zheng K, Ye J, Dunn B, Czajkowski R, Moser MB, Moser EI, Roudi Y, Witter MP (2013) Recurrent inhibitory circuitry as a mechanism for grid formation. Nat Neurosci 16:318–324. 10.1038/nn.3310 [DOI] [PubMed] [Google Scholar]
- Donato F, Jacobsen RI, Moser MB, Moser EI (2017) Stellate cells drive maturation of the entorhinal-hippocampal circuit. Science 355:eaai8178. 10.1126/science.aai8178 [DOI] [PubMed] [Google Scholar]
- Eichenbaum H, Fortin NJ (2005) Bridging the gap between brain and behavior: cognitive and neural mechanisms of episodic memory. J Exp Anal Behav 84:619–629. 10.1901/jeab.2005.80-04 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garden DL, Dodson PD, O'Donnell C, White MD, Nolan MF (2008) Tuning of synaptic integration in the medial entorhinal cortex to the organization of grid cell firing fields. Neuron 60:875–889. 10.1016/j.neuron.2008.10.044 [DOI] [PubMed] [Google Scholar]
- Gloveli T, Schmitz D, Empson RM, Dugladze T, Heinemann U (1997) Morphological and electrophysiological characterization of layer III cells of the medial entorhinal cortex of the rat. Neuroscience 77:629–648. 10.1016/S0306-4522(96)00494-0 [DOI] [PubMed] [Google Scholar]
- González-Burgos G, Barrionuevo G, Lewis DA (2000) Horizontal synaptic connections in monkey prefrontal cortex: an in vitro electrophysiological study. Cereb Cortex 10:82–92. 10.1093/cercor/10.1.82 [DOI] [PubMed] [Google Scholar]
- Grinvald A, Frostig RD, Lieke E, Hildesheim R (1988) Optical imaging of neuronal activity. Physiol Rev 68:1285–1366. 10.1152/physrev.1988.68.4.1285 [DOI] [PubMed] [Google Scholar]
- Hafting T, Fyhn M, Molden S, Moser MB, Moser EI (2005) Microstructure of a spatial map in the entorhinal cortex. Nature 436:801–806. 10.1038/nature03721 [DOI] [PubMed] [Google Scholar]
- Hasselmo ME, Giocomo LM, Zilli EA (2007) Grid cell firing may arise from interference of theta frequency membrane potential oscillations in single neurons. Hippocampus 17:1252–1271. 10.1002/hipo.20374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda Y, Furuta T (2019) Multiple patterns of axonal collateralization of single layer III neurons of the rat presubiculum. Front Neural Circuits 13:45. 10.3389/fncir.2019.00045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutcheon B, Yarom Y (2000) Resonance, oscillation and the intrinsic frequency preferences of neurons. Trends Neurosci 23:216–222. 10.1016/S0166-2236(00)01547-2 [DOI] [PubMed] [Google Scholar]
- Kerr KM, Agster KL, Furtak SC, Burwell RD (2007) Functional neuroanatomy of the parahippocampal region: the lateral and medial entorhinal areas. Hippocampus 17:697–708. 10.1002/hipo.20315 [DOI] [PubMed] [Google Scholar]
- Koganezawa N, Taguchi A, Tominaga T, Ohara S, Tsutsui KI, Witter MP, Iijima T (2008) Significance of the deep layers of entorhinal cortex for transfer of both perirhinal and amygdala inputs to the hippocampus. Neurosci Res 61:172–181. 10.1016/j.neures.2008.02.007 [DOI] [PubMed] [Google Scholar]
- Köhler C. (1984) Morphological details of the projection from the presubiculum to the entorhinal area as shown with the novel PHA-L immunohistochemical tracing method in the rat. Neurosci Lett 45:285–290. 10.1016/0304-3940(84)90240-4 [DOI] [PubMed] [Google Scholar]
- Langston R, Ainge J, Couey J, Canto C, Bjerknes T, Witter M, Moser E, Moser M (2010) Development of the spatial representation system in the rat. Science 328:1576–1580. 10.1126/science.1188210 [DOI] [PubMed] [Google Scholar]
- Losonczy A, Zhang L, Shigemoto R, Somogyi P, Nusser Z (2002) Cell type dependence and variability in the short-term plasticity of EPSCs in identified mouse hippocampal interneurones. J Physiol 542:193–210. 10.1113/jphysiol.2002.020024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNaughton BL, Battaglia FP, Jensen O, Moser EI, Moser MB (2006) Path integration and the neural basis of the “cognitive map.” Nat Rev Neurosci 7:663–678. 10.1038/nrn1932 [DOI] [PubMed] [Google Scholar]
- Moser EI, Moser MB (2008) A metric for space. Hippocampus 18:1142–1156. 10.1002/hipo.20483 [DOI] [PubMed] [Google Scholar]
- Nadel L, O'Keefe J (1978) The hippocampus as a cognitive map. Oxford: Oxford UP. [Google Scholar]
- O'Reilly KC, Flatberg A, Islam S, Olsen LC, Kruge IU, Witter MP (2015) Identification of dorsal-ventral hippocampal differentiation in neonatal rats. Brain Struct Funct 220:2873–2893. 10.1007/s00429-014-0831-8 [DOI] [PubMed] [Google Scholar]
- Press WH, Teukolsky SA, Vetterling WT (1992) Power spectrum estimation using the FFT. In: Numerical recipes in C: the art of scientific computing, Ed 2 Cambridge: Cambridge UP. [Google Scholar]
- Ray S, Brecht M (2016) Structural development and dorsoventral maturation of the medial entorhinal cortex. Elife 5:e13343. 10.7554/eLife.13343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reboreda A, Raouf R, Alonso A, Séguéla P (2007) Development of cholinergic modulation and graded persistent activity in layer V of medial entorhinal cortex. J Neurophysiol 97:3937–3947. 10.1152/jn.01233.2006 [DOI] [PubMed] [Google Scholar]
- Sargolini F, Fyhn M, Hafting T, McNaughton BL, Witter MP, Moser MB, Moser EI (2006) Conjunctive representation of position, direction, and velocity in entorhinal cortex. Science 312:758–762. 10.1126/science.1125572 [DOI] [PubMed] [Google Scholar]
- Savelli F, Yoganarasimha D, Knierim JJ (2008) Influence of boundary removal on the spatial representations of the medial entorhinal cortex. Hippocampus 18:1270–1282. 10.1002/hipo.20511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solstad T, Boccara CN, Kropff E, Moser MB, Moser EI (2008) Representation of geometric borders in the entorhinal cortex. Science 322:1865–1868. 10.1126/science.1166466 [DOI] [PubMed] [Google Scholar]
- Sugar J, Witter MP (2016) Postnatal development of retrosplenial projections to the parahippocampal region of the rat. Elife 5:e13925. 10.7554/eLife.13925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan HM, Bassett JP, O'Keefe J, Cacucci F, Wills TJ (2015) The development of the head direction system before eye opening in the rat. Curr Biol 25:479–483. 10.1016/j.cub.2014.12.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taube JS. (2007) The head direction signal: origins and sensory-motor integration. Annu Rev Neurosci 30:181–207. 10.1146/annurev.neuro.29.051605.112854 [DOI] [PubMed] [Google Scholar]
- Taube JS, Muller RU, Ranck JB (1990) Head-direction cells recorded from the postsubiculum in freely moving rats: I. Description and quantitative analysis. J Neurosci 10:420–435. 10.1523/JNEUROSCI.10-02-00420.1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolner EA, Frahm C, Metzger R, Gorter JA, Witte OW, Lopes da Silva FH, Heinemann U (2007) Synaptic responses in superficial layers of medial entorhinal cortex from rats with kainate-induced epilepsy. Neurobiol Dis 26:419–438. 10.1016/j.nbd.2007.01.009 [DOI] [PubMed] [Google Scholar]
- Tominaga T, Tominaga Y, Yamada H, Matsumoto G, Ichikawa M (2000) Quantification of optical signals with electrophysiological signals in neural activities of di-4-ANEPPS stained rat hippocampal slices. J Neurosci Methods 102:11–23. 10.1016/S0165-0270(00)00270-3 [DOI] [PubMed] [Google Scholar]
- Vann SD. (2010) Re-evaluating the role of the mammillary bodies in memory. Neuropsychologia 48:2316–2327. 10.1016/j.neuropsychologia.2009.10.019 [DOI] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Südhof TC (2000) Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science 287:864–869. 10.1126/science.287.5454.864 [DOI] [PubMed] [Google Scholar]
- Wills TJ, Cacucci F, Burgess N, O'Keefe J (2010) Development of the hippocampal cognitive map in preweanling rats. Science 328:1573–1576. 10.1126/science.1188224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills TJ, Barry C, Cacucci F (2012) The abrupt development of adult-like grid cell firing in the medial entorhinal cortex. Front Neural Circuits 6:21. 10.3389/fncir.2012.00021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills TJ, Muessig L, Cacucci F (2013) The development of spatial behaviour and the hippocampal neural representation of space. Philos Trans R Soc Lond B Biol Sci 369:20130409. 10.1098/rstb.2013.0409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter SS, Clark BJ, Taube JS (2015) Spatial navigation: disruption of the head direction cell network impairs the parahippocampal grid cell signal. Science 347:870–874. 10.1126/science.1259591 [DOI] [PMC free article] [PubMed] [Google Scholar]