

Abstract
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease in which motor neurons degenerate, resulting in muscle atrophy, paralysis, and fatality. Studies using mouse models of ALS indicate a protracted period of disease development with progressive motor neuron pathology, evident as early as embryonic and postnatal stages. Key missing information includes concomitant alterations in the sensorimotor circuit essential for normal development and function of the neuromuscular system. Leveraging unique brainstem circuitry, we show in vitro evidence for reflex circuit-specific postnatal abnormalities in the jaw proprioceptive sensory neurons in the well-studied SOD1G93A mouse. These include impaired and arrhythmic action potential burst discharge associated with a deficit in Nav1.6 Na+ channels. However, the mechanoreceptive and nociceptive trigeminal ganglion neurons and the visual sensory retinal ganglion neurons were resistant to excitability changes in age-matched SOD1G93A mice. Computational modeling of the observed disruption in sensory patterns predicted asynchronous self-sustained motor neuron discharge suggestive of imminent reflexive defects, such as muscle fasciculations in ALS. These results demonstrate a novel reflex circuit-specific proprioceptive sensory abnormality in ALS.
SIGNIFICANCE STATEMENT Neurodegenerative diseases have prolonged periods of disease development and progression. Identifying early markers of vulnerability can therefore help devise better diagnostic and treatment strategies. In this study, we examined postnatal abnormalities in the electrical excitability of muscle spindle afferent proprioceptive neurons in the well-studied SOD1G93A mouse model for neurodegenerative motor neuron disease, amyotrophic lateral sclerosis. Our findings suggest that these proprioceptive sensory neurons are exclusively afflicted early in the disease process relative to sensory neurons of other modalities. Moreover, they presented Nav1.6 Na+ channel deficiency, which contributed to arrhythmic burst discharge. Such sensory arrhythmia could initiate reflexive defects, such as muscle fasciculations in amyotrophic lateral sclerosis, as suggested by our computational model.
Keywords: amyotrophic lateral sclerosis, dynamic clamp, motor neurons, persistent sodium, proprioceptive neurons, SOD1
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease in which motor neurons throughout the brain and spinal cord progressively degenerate. In this devastating disease, motor neuron (MN) degeneration leads to muscle paralysis and atrophy, and death ensues 3–5 years following clinical disease onset (Cleveland and Rothstein, 2001; Bruijn et al., 2004). Use of transgenic mouse models of ALS has provided key insights into presymptomatic mechanisms of disease development. These include central mechanisms encompassing glutamatergic excitotoxicity at MN synaptic terminals, diminished energy supply resulting from metabolite deficiency, and dysfunctional RNA metabolism, protein homeostasis, and aggregation (Bruijn et al., 2004; Taylor et al., 2016). Additional non–cell-autonomous contributors, such as dysfunctional astrocytes (Yamanaka et al., 2008), microglia (Boillée et al., 2006b), and oligodendrocytes (Lee et al., 2012), provide evidence that ALS disease mechanisms are not limited to MNs. Peripheral neural dysfunction, such as impairment in axonal transport (Williamson and Cleveland, 1999; Puls et al., 2003), and synaptic pruning/failure at the neuromuscular junctions (Pun et al., 2006; Casas et al., 2016), also plays a crucial role in initiating muscle paralysis. Importantly, these diverse aspects of early disease development collectively support the idea that additional neuronal pathways, such as early sensory input changes, are worth exploring as these may provide opportunities for identifying novel early biomarker and therapeutic targets.
Vulnerability to neurodegeneration is not the same for all MNs and within motor pools in ALS (Pun et al., 2006; Kanning et al., 2010). Both in normal aging and in ALS, motor unit physiology is a crucial determinant of preferential vulnerability, where fast fatigable motor units degenerate first, followed by fast fatigue resistant, whereas slow motor units are relatively spared (Pun et al., 2006; Hegedus et al., 2007). Second, MNs that control eye movements (oculomotor, trochlear, and abducens) and sphincter muscles (Onuf's nucleus), as well as gamma MNs that innervate intrafusal muscle fibers are selectively resistant to death (Iwata and Hirano, 1978; Ferrucci et al., 2010; Lalancette-Hebert et al., 2016). Comparative analyses have highlighted important differences in transcriptional profiles, electrical and synaptic properties, and neuromuscular biology and innervation patterns between the resistant and vulnerable MNs (Frey et al., 2000; Nimchinsky et al., 2000; Hedlund et al., 2010; Comley et al., 2015; Venugopal et al., 2015; Nijssen et al., 2017). These inherent differences indicate local and long-range defects in sensorimotor circuits of vulnerable MNs in ALS (Durand et al., 2006). For instance, a circuit-specific difference in the ALS-resistant oculomotor neurons involves a lack of Ia muscle spindle afferent proprioceptive inputs (Spencer and Porter, 1988), which are a principal source of glutamatergic excitation during muscle stretch reflexes. A lack of Ia spindle afferent inputs to spinal gamma MNs was suggested as a mechanism of disease resistance in ALS mouse models (Schneider et al., 2012). Consistent with that observation, reducing Ia proprioceptive muscle spindle afferents partly contributed to α-MN survival. Therefore, identification and characterization of circuit-specific vulnerability to disease progression in ALS (Brownstone and Lancelin, 2018) could help develop effective therapeutic strategies.
To test whether circuit-specific dysfunction involves proprioceptive sensory neurons, we leveraged the unique architecture of the brainstem trigeminal sensorimotor circuitry involved in jaw control. We examined the proprioceptive Ia afferents in the pontine mesencephalic nucleus (Mes V) in the SOD1G93A mouse model at P11 ± 3 when the jaw motor pools are reported to be dysregulated (Venugopal et al., 2015). Our results show that Mes V neurons present electrical abnormalities and arrhythmic burst discharge patterns, associated with a marked reduction in Nav1.6-type Na+ currents. Rescue of these Na+ currents restored normal rhythmic burst patterns (see also Venugopal et al., 2019). Concomitant examination of trigeminal ganglion neurons and retinal ganglion neurons (RGNs) confirmed exclusive changes only in the proprioceptive Mes V neurons. Using a computational modeling approach, we show a functional consequence of sensory abnormality on downstream motor integration predictive of imminent reflex dysfunction in ALS.
Materials and Methods
Transgenic mice expressing high levels of human SOD1G93A (mutant SOD1 or mSOD1) and their WT littermates were used for all the experiments (JAX strain: 002726 B6SJL-Tg (SOD1*G93A)1Gur/J). All animal protocols were approved by the Institutional Animal Care and Use Committee at UCLA. Experiments were performed at postnatal week 2 (8- to 14-d-old mice of either gender) when the rhythmic jaw movements and suckling behavior are fully developed (Turman, 2007). Genotype of mice was determined by standard PCR technique using tails samples (Laragen). Experimental preparations and techniques include the following: (1) live brainstem slices to conduct in vitro whole-cell current-clamp, voltage-clamp, and dynamic-clamp electrophysiology from Mes V sensory neurons; (2) acutely dissociated trigeminal ganglion neurons to conduct current-clamp experiments; (3) live whole retinal preparation to conduct current-clamp experiments; (4) fixed cryosectioned coronal pontine sections for Nav1.6 protein quantification; and (5) computational model of Mes V-TMN network to investigate a functional consequence of sensory abnormality on motor discharge.
In vitro patch-clamp electrophysiology
Brainstem slice preparation for Mes V electrophysiology
Brain slices were prepared and used for conducting whole-cell current-, voltage-, and dynamic-clamp electrophysiology in the brainstem primary sensory neurons of the trigeminal Mes V nucleus. Pups were anesthetized using isoflurane vapor inhalation and decapitated. The head was immediately immersed in carboxygenated (95% O2/5% CO2), ice-cold sucrose cutting solution composed of the following (in mm): 194 sucrose, 30 NaCl, 4.5 KCl, 1.2 NaH2PO4, 26 NaHCO3, 10 glucose, and 1 MgCl2. The pontine brainstem was rapidly extracted and adhered to the cutting chamber of a vibratome platform at the rostral end (DSK Microslicer; Ted Pella); the brainstem was vertically supported by an agar block. The cutting chamber was filled with ice-cold carboxygenated cutting solution. Beginning at the caudal level where the exit of the facial nerve was markedly visible, 3 or 4 coronal pontine slices (∼250 μm thick) were cut and placed in the carboxygenated incubation solution at room temperature, composed of the following (in mm): 124 NaCl, 4.5 KCl, 1.2 NaH2PO4, 26 NaHCO3, 10 glucose, 2 CaCl2, and 1 MgCl2 (Schurr et al., 1988). The pH of the incubation solution was maintained at 7.28 ± 0.2.
Trigeminal ganglia (TG) extraction and acute dissociation of trigeminal ganglion neurons (TGNs) for electrophysiology
To evaluate excitability changes in the nonproprioceptive neurons of the trigeminal system of the mSOD1 mice, we performed acute dissociation of the TG (Malin et al., 2007; Xu et al., 2010; Yamamoto et al., 2013). Pups were decapitated under isoflurane anesthesia similar to that in Brainstem slice preparation for Mes V electrophysiology. The TG were bilaterally removed with the aid of a dissection microscope and transferred into ice-cold (4°C) modified Tyrode's solution containing the following (in mm): 130 NaCl, 20 NaHCO3, 3 KCl, 4 CaCl2, 1 MgCl2, 10 HEPES, and 12 glucose, with antibiotic/antimycotic solution (0.5%; Thermo Fisher Scientific). The ganglia were then minced and incubated in collagenase (1 mg/ml, Type I; Thermo Fisher Scientific) for 40 min and then in collagenase with trypsin/EDTA (0.2%; Thermo Fisher Scientific) for another 40 min at 37°C. The TG cells were then washed twice with the modified Tyrode's solution and triturated gently using fire-polished Pasteur glass pipettes. Finally, the cell suspension was mixed with BSA (15%; Thermo Fisher Scientific) and centrifuged at 900 rpm for 10 min to remove myelin and debris. The pellet was resuspended with Neurobasal A (Thermo Fisher Scientific) containing B27 (2%; Thermo Fisher Scientific), l-glutamine (0.2%; Thermo Fisher Scientific), and antibiotic/antimycotic solution (0.1%), and cells were plated onto glass coverslips coated with poly-d-lysine/laminin (Thermo Fisher Scientific). The cells were then incubated at 37°C in a humidified 5% CO2 chamber, and whole-cell patch-clamp electrophysiology was conducted ∼24 h after plating (Chen et al., 2008; Marchenkova et al., 2016).
Retina extraction and preparation for electrophysiology
To ascertain whether visual sensory neurons are susceptible to early changes in excitability in ALS mice, we extracted whole retinas from mSOD1 and WT mice for patch-clamp electrophysiology. For retinal extraction, pups were deeply anesthetized using isoflurane inhalation, and the eyes were denucleated and placed in an ice-cold solution containing the following (in mm): in room temperature-oxygenated solution containing the following (in mm): 124 NaCl, 4.5 KCl, 1.2 NaH2PO4, 26 NaHCO3, 10 glucose, 2 CaCl2, and 1 MgCl2. The pH of the incubation solution was maintained at 7.28 ± 0.2, with osmolarity adjusted to 300 ± 5 mOsm. Each retina was exposed by a single cut along the ora serrata. An additional cut was made along the optic disk to dissect the eyecup into two halves (Wang et al., 1997; Qu and Myhr, 2011). The retina was isolated from pigment epithelium and was stored for 1 h in oxygenated incubation solution at room temperature before whole-cell patch-clamp recording.
Whole-cell current-clamp recording
Whole-cell current-clamp recording was performed in three sets of sensory neurons: the trigeminal proprioceptive Mes V neurons, trigeminal ganglion mechanoreceptive/nociceptive neurons, and the RGNs. Axopatch-1D patch-clamp amplifier (Molecular Devices) and pCLAMP acquisition software (version 9.2, Molecular Devices) were used. Silver chloride electrode (Ag/AgCl wire) and 3 mm KCl agar-bridge were used for suitable grounding and electrical isolation. Patch electrodes were fabricated from borosilicate glass capillary tubing (1.5 mm OD, 0.86 mm ID) using a model P-97 puller (Sutter Instrument). Tip resistance was 3–5 mΩ when filled with a pipette internal solution containing the following (in mm): 135 K-gluconate, 5 KCl, 0.5 CaCl2, 5 HEPES (base), 5 EGTA, 2 Mg-ATP, and 0.3 Na-ATP (Gupta et al., 2012), pH 7.25 ± 0.2, osmolarity adjusted to 290 ± 5 mOsm. The external recording solution consisted of the following (in mm): 140 NaCl, 4 KCl, 2 CaCl2, 10 HEPES (base), 2 MgCl2, and 10 glucose.
Mes V neuron identification.
The proprioceptive Mes V nucleus was identified bilaterally in the coronal slices as a dorsally located ellipsoid region, ventrolateral to the aqueduct in the caudal pontine slices at the level of VII nerve exit, and in the lateral periphery of the periaqueductal gray in further rostral pontine slices (Wu et al., 2001; Enomoto et al., 2006; Seki et al., 2017). The Mes V neurons were easily distinguished on the basis of their large, ellipsoid soma with ∼30–50 μm diameter (Henderson et al., 1982; Del Negro and Chandler, 1997; Enomoto et al., 2006). In a subset of experiments, sensory Mes V neurons were identified using anterograde labeling by injection of 1% AlexaFluor-568 (Thermo Fisher Scientific) in the jaw closer muscles (20 μl on each side) (see Fig. 1).
Figure 1.

Impaired excitability in intrinsically bursting mSOD1 Mes V neurons. a, Schematic represents the Ia proprioceptive sensory and motor nuclei of the trigeminal jaw control circuit identified using anterograde/retrograde tracer injection in jaw closer muscles in the SOD1G93A mouse (or mSOD1). Left half of a coronal pontine section is shown. Mes V, Trigeminal mesencephalic sensory nucleus; Mot V, trigeminal motor nucleus. White dotted enclosures represent both nuclei. b, Left, Anterogradely labeled Mes V neurons (green). Middle, Dye-filled (red) Mes V neuron during patch-clamp recording. Right, A merged image highlighting the double-labeled neuron. c, Representative examples of discharge patterns in two classes of Mes V neurons. Left, A rhythmic bursting neuron (maroon). Right, A tonic spiking neuron (right). These discharge patterns are induced by a 200 pA depolarizing step current for 20 s duration. d, Bar charts represent the age distribution of the mice, including 24 WT mice (n = 32) and 21 mSOD1 mice (n = 26), where n is the number of cells for all the data presented. e, A single near-threshold spike in a WT (blue) and a mSOD1 (red) Mes V neuron generated in response to a brief (5 ms) step depolarization. The corresponding injected current threshold (Ithreshold) (bottom) and membrane voltage threshold (Vthreshold) for spike generation (top, horizontal lines and arrows) are highlighted (magenta). f, g, Dot plots represent Ithreshold (left) and Vthreshold (right) in bursting (f) and tonic (g) Mes V neurons. Blue and red horizontal lines indicate average group values. f, For bursting neurons, *p < 0.05 (above horizontal line), for Ithreshold and Vthreshold. g, For tonic spiking neurons, NS (above black horizontal line) indicates no statistically significant group differences between WT and mSOD1 cells. h, Spike frequency-injected current responses in bursting (left) and tonic spiking (right) Mes V neurons: blue represents WT; red represents mSOD1. *p < 0.05 (above horizontal line), between WT and mSOD1 cells for injected current ≥ 150 pA. i, Pie charts represent proportional distribution of bursting and tonic spiking neurons in WT and mSOD1 mice. *p < 0.05.
TGN identification.
The dissociated TGNs were plated on a coverslip, and cells were identified for whole-cell patch-clamp recording using differential interference contrast microscopy. Medium to large isolated cell bodies with diameter > 10 μm were selected. During initial experiments, the dissociated cells were labeled using NeuN stain (Neurotrace) for morphology identification and yield estimation. The pipette internal solution was filled with 1% Texas Red (Invitrogen) to label the recorded cells in a subset of experiments (see Fig. 7).
Figure 7.

Excitability of primary sensory neurons in the retina of mSOD1 mice. a, Schematic showing retina extraction for patch-clamp recording. b, Bar charts represent the age distribution of the mice, including 7 WT mice (n = 13) and 5 mSOD1 mice (n = 10), where n is the number of cells for all the data presented in c–h. c, Inset, Current (IThreshold) and voltage (VThreshold) thresholds for action potentials in WT (blue) and mSOD1 (red) RG neurons. Dot plots represent comparison of these properties between WT (blue) and mSOD1 (red) RG neurons tested. f, Representative membrane voltage responses to increasing levels of 1 s current injection (bottom) in repetitively firing WT and mSOD1 RG neurons. g, Spike frequency-injected current responses in all the RG neurons that showed repetitive firing. h, Autocorrelation function of membrane potential in WT (left) and mSOD1 (middle) RG neurons. Second peaks (arrows) are highlighted. Right, Dot plots represent the second peaks for all the cells with no significant difference between WT (blue) and mSOD1 (red) TG neurons. g, RG neurons were classified into three types: single spikers (SS), rate adapting (RA), and tonic spiking (for details, see Results). d, Percentage of the subtypes of RG neurons in WT and mSOD1 mice are not significantly different.
RGN identification.
A single retinal quarter was placed in the patch-clamp recording chamber with the ganglion cell layer on the top. Using a recording glass pipette tip, membrane of the surface layer was penetrated and removed by moving the pipette back and forth to expose the retinal ganglion cell layer. The internal limiting membrane encasing the ganglion cell layer was carefully dissected using an empty patch pipette, and access was gained to a selected cell for patch-clamp recording (Murphy and Rieke, 2006; Margolis and Detwiler, 2007). Larger cells in the ganglion cell layer (∼20 μm diameter) were targeted in the flat-mount retina, and recordings were performed in the dark. We did not test cellular response to light versus dark, and we performed all our recording in normal ACSF without any synaptic blockers.
Whole-cell voltage-clamp recording
Voltage-clamp experiments were performed in Mes V neurons to evaluate changes in voltage-gated Na+ currents in the mSOD1 mice, compared with their WT littermates as controls. Recordings were performed using a pipette internal solution composed of the following (in mm): 130 CsF, 9 NaCl, 10 HEPES, 10 EGTA, 1 MgCl2, 3 K2-ATP, and 1 Na-GTP. The external recording solution consisted of the following (in mm): 131 NaCl, 10 HEPES, 3 KCl, 10 glucose, 2 CaCl2, 2 MgCl2, 10 tetraethylammonium (TEA)-Cl, 10 CsCl2, 1 4-AP, and 0.3 CdCl2. Use of TEA, 4-AP, and CdCl2 allowed isolation of voltage-gated Na+ currents by pharmacologically blocking the voltage-gated K+ and Ca2+ currents (Del Negro and Chandler, 1997; Enomoto et al., 2006). For acceptable recordings, each cell's uncompensated series resistance (Rs) was monitored throughout, and only recordings with Rs < 10% of the input resistance (Rinp) were included. Voltage-clamp protocols were used to activate the Nav1.6-type persistent and resurgent voltage-gated Na+ currents, both of which are critical for Mes V neuron excitability (Wu et al., 2005; Enomoto et al., 2007). To activate the resurgent Na+ current, the voltage-clamp protocol consisted of a brief 3 ms step to 30 mV from an initial holding potential of −90 mV, followed by 100 ms steps from 0 to −90 mV in steps of 10 mV, and then a return to the holding potential of −90 mV (Raman and Bean, 2001; Enomoto et al., 2006). The slower low-voltage-activated persistent Na+ current protocol included a slow voltage ramp from −90 to 30 mV for 1 s, followed by a step to −40 mV (Do and Bean, 2003; Wu et al., 2005). Following these protocols, these Na+ currents were abolished using 0.5 μm TTX, and leak-subtracted Na+ current magnitude was quantified (see Fig. 3).
Figure 3.
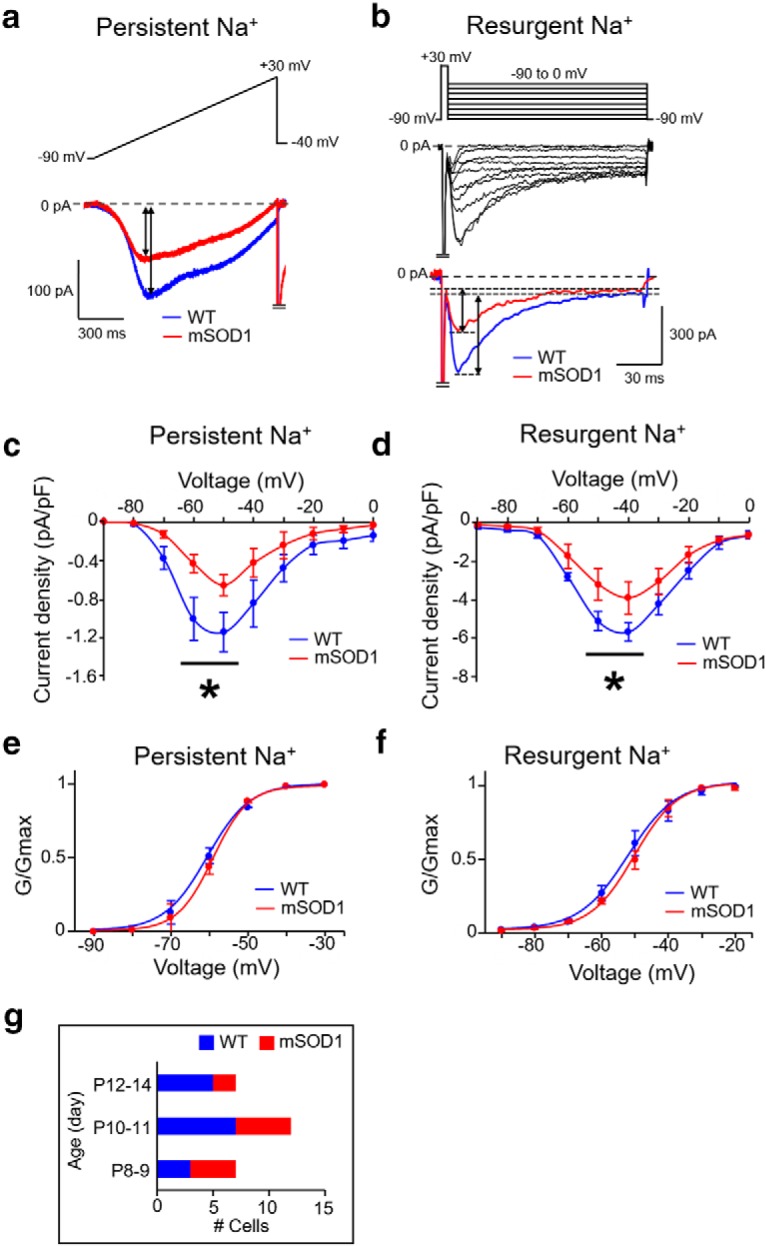
Reduced Nav 1.6-type Na+ currents in mSOD1 Mes V neurons. a, b, Representative current traces from whole-cell voltage-clamp experiments showing a reduction in TTX-sensitive persistent Na+ (a) and resurgent Na+ (b) currents in mSOD1 Mes V neuron (red), compared with WT (blue). Top, Voltage-clamp protocol. Bottom, Current responses. Voltage-clamp protocol in a consists of a ramp voltage from −90 to 30 mV at 100 mV/s, and in b, consists of a brief, 5 ms step from −90 to 30 mV, to evoke a transient Na+ current, followed by repolarization to voltages between −90 and 0 mV to elicit resurgent Na+ current due to open-channel unblock. c, d, Current-voltage relationships of the peak persistent (c) and resurgent (d) Na+ current densities in mSOD1 Mes V neurons (red). c, Asterisk (above the black horizontal line) indicates statistically significant reduction in peak current amplitudes at subthreshold activation voltages between −60 and −50 mV for persistent Na+ peaks in mSOD1 (red) compared with WT (blue). d, Asterisk (above the black horizontal line) indicates statistically significant reduction in peak current amplitudes at repolarization voltages of −50 and −40 mV for resurgent Na+ peaks in mSOD1 (red) compared with WT (blue). e, f, Boltzmann curves fit to the normalized peak conductances as a function of activation voltages for persistent (e) and resurgent (f) Na+ currents in WT (blue) and mSOD1 (red) Mes V neurons. g, Bar charts represent the age distribution of the mice, including 8 WT mice (n = 14) and 6 mSOD1 mice (n = 11), where n is the number of cells for all the data presented in c–f.
Real-time dynamic-clamp electrophysiology
Real-time closed-loop dynamic-clamp electrophysiology was adopted for in silico knock-in of Nav1.6-type Na+ currents in Mes V neurons. Briefly, the Linux-based Real-Time eXperimental Interface (RTXI version 1.3) was used to implement dynamic clamp, running on a modified Linux kernel extended with the Real-Time Applications Interface, which allows high-frequency, periodic, real-time calculations (Lin et al., 2010). The RTXI computer interfaced with the electrophysiological amplifier (Molecular Devices, Axopatch 200A in current-clamp mode) and the data acquisition PC via a PCIe-6251 board (National Instruments). Computation frequency was 20 kHz. Brainstem slices were perfused with oxygenated recording solution (∼2 ml/min) at room temperature (∼22°C–24°C) while secured in a glass-bottom recording chamber mounted on an inverted microscope with differential interface contrast optics (Carl Zeiss, Axiovert 10). Current-clamp (and dynamic-clamp) data were acquired and analyzed using custom-made software (G-patch, Analysis) with sampling frequency 10 kHz and cutoff filter frequency 2 kHz. The conductance-based Nav1.6-type Na+ current models were developed in our laboratory as detailed by Venugopal et al. (2019).
Experimental design and statistical analyses
Animals were divided into two main experimental groups: those carrying the human-SOD1G93A transgene (mSOD1) and their age-matched noncarrier WT littermate controls. Statistical analyses were performed using Excel (Microsoft) and Systat 13 (Systat Software). Groups were compared using two-tailed unpaired Student's t test, two-way repeated-measures ANOVA to analyze group (WT ve mSOD1), and treatment effects (e.g., different levels of current application), and χ2 proportionality test for comparison of distribution of cell types in the two animal groups (WT vs mSOD1). For two-way repeated-measures ANOVA, we assumed equal variance and normal distribution and first conducted a post hoc univariate F test for pairwise comparisons of treatment effect for each genotype. The statistical significance obtained using the F test, which is based on a regression model, was further verified against a two-tailed unpaired Student's t test, which indeed gave a more conservative estimate of the statistical significance we report here (for details, see results relating to Figs. 1h, 3c–f, 6g, and 7e). p < 0.05 was considered statistically significant.
Figure 6.

Excitability of primary sensory neurons in the TG of mSOD1 mice. a, Schematic showing the trigeminal sensory nuclei along the midbrain-brainstem regions. Primary 1a afferent neurons in the TG (red dashed rectangle) were acutely dissociated, and whole-cell current-clamp recordings were performed. b, Top left, Example showing NeuN stain (green) used to identify the dissociated TG neurons. Top right, Example of a dissociated TG neuron filled with Texas red 568 dye during whole-cell recording. Bottom left, Merged image. Bottom right, Bright field (BF) image. Scale bars, 50 μm. c, TG neurons were classified into three types: Aβ, Aδ, and C (for criteria, see Results). Open arrows indicate membrane sag during a 1 s hyperpolarizing step pulse in Aβ and Aδ types. Black arrow indicates a hump in Aδ action potential. d, Percentage of the subtypes of TG neurons in WT and mSOD1 mice are not significantly different. e, Inset, The current (IThreshold) and voltage (VThreshold) thresholds for action potentials in WT (blue) and mSOD1 (red) TG neurons. Dot plots represent comparison of these properties between WT (blue) and mSOD1 (red) within the Aβ and Aδ groups with no statistical significance. f, Representative membrane voltage responses to increasing levels of 1 s current injection (bottom traces) in repetitively firing WT and mSOD1 TG neurons (both Aδ type). g, Spike frequency-injected current responses in all the Aδ TG neurons that showed repetitive firing. h, Autocorrelation function of membrane potential in WT (left) and mSOD1 (middle) TG neurons; second peaks (arrows) are highlighted. Right, Dot plots represent the second peaks for all the cells with no significant difference between WT (blue) and mSOD1 (red) TG neurons. i, Bar charts represent the age distribution of the mice, including 8 WT mice (n = 36) and 7 mSOD1 mice (n = 27), where n is the number of cells for all the data presented in e–h.
Data acceptance criteria and analytical approaches
Patch-clamp recording was performed in whole-cell configuration following rupturing of a giga-ohm seal. Cells with uncompensated series resistance > 10 mΩ and resting potential more positive than −50 mV were discarded. Further data acceptance criteria included input resistance ≥ 100 mΩ (measured as the slope, voltage/current at the end of 100 ms current pulses near rest Vrest ± 10 mV) and action potential height ≥ 80 mV (measured from spike threshold to peak). Spike characteristics, including height, half-width, after-hyperpolarization, and input threshold (Ithreshold), were determined using response to a 10 ms current pulse. Average spike frequencies used for frequency-current relationships were determined for increasing steps of 1 s current pulses, typically up to 3× Ithreshold. In our Mes V discharge patterns, we distinguished rhythmic bursting activity from tonic spiking by constructing the interevent interval (IEI) histogram with bin width of 1 to 5 ms. If the IEI histogram had a clear single peak at ∼20 ms, the cells were classified as tonic cells, whereas cells with additional peak(s) at larger IEI values ≥ 100 ms corresponding to the interburst intervals (IBIs) were grouped as bursting neurons. In the bursting Mes V neurons, we used 40 ms as the cutoff for IEI to further separate the interspike intervals (ISIs) from IBIs; all tonically spiking neurons had visibly regular IEIs < 50 ms with a sharp peak in the IEI histograms between 10 and 20 ms. These analyses were performed using Clampfit 10.5. Autocorrelation was used as a measure of rhythmicity of membrane voltage responses in Clampfit 10.5.
Nav1.6 fluorescent immunohistochemistry
Tissue preparation
Pups were deeply anesthetized using intraperitoneal injection of sodium pentobarbital (2.2 μl/g BW). Using a 25-gauge needle connected to a pressure-controlled perfusion system (∼200 mmHg), an adequate volume of freshly prepared, chilled 4% PFA (fixative) was transcardially perfused until clear drainage from the right atrium was noted. Subsequently, the mouse was decapitated, and the brain was extracted and incubated in 30% sucrose solution at 4°C for 48 h. The pontine brainstem block was cut and placed caudal side down within an RNase-free mold and embedded in optimal cutting temperature compound (Tissue TEK-OCT compound, Thermo Fisher Scientific), flash frozen using dry ice, and stored at −80°C.
Cryo-sectioning and fluorescent immunohistochemistry
Pontine brainstem was sectioned at 20 μm thickness beginning approximately at the level of the exiting VII nerve, and 6–8 sections per animal were mounted on glass slides (Thermo Fisher Scientific), ensuring inclusion of the rostro-caudal extent of the Mes V area. Fluorescent immunohistochemistry was immediately performed on slides by marking contours around each section using a MINI PAP PEN (Invitrogen). Every experiment was simultaneously run on WT and mSOD1 pair(s) to ensure similar tissue treatment for subsequent fluorescence quantification. Following three 10 min washes with 1× PBS, sections were incubated in a blocking buffer (10% normal donkey serum and 0.3% Tween-20 in 1× PBS) for 1 h at room temperature. Red fluorescence-conjugated Nav1.6 primary antibody derived from rabbit (Alomone Labs) was added (1:200 dilution) and incubated for 24 h at 4°C. Appropriate dilution was determined based on initial dilution series experiments (1:100, 1:200, and 1:500) with suitable negative controls. Following primary incubation, further wash steps (5 min, 3 times in 1× PBS) and green fluorescence-conjugated NeuN stain (Neurotrace) were added (1:200 dilution) for 30 min to enable identification of neuronal cell bodies. Sections were rinsed for 5–10 min with 1× PBS and coverslipped for imaging.
Imaging and quantification of Nav1.6 intensities
Imaging was performed using a fully automated epifluorescence microscope (Keyence, Bz 9000e). First, the Mes V nuclei were bilaterally identified with NeuN stain at low magnification (2×), ventrolateral to the periaqueductal gray. The identified cells within the nucleus were then imaged at 60× magnification (1/15 s exposure time for NeuN and 2 s exposure time for Nav1.6 with standard excitation settings), and z stacks were collected at 0.5 μm depth resolution with green (NeuN) and red (Nav1.6) filters. The z-projected images were then overlaid to generate a merged image to localize Nav1.6 label on Mes V neurons. Quantification of fluorescent intensities in Mes V neurons was performed using open-source ImageJ software (Schindelin et al., 2012). First, 12 ± 6 Mes V neurons with clearly visible NeuN-stained cell bodies and nuclei were manually traced as ROIs in each image. These ROIs were then overlaid on the z-projected Nav1.6 image (see Fig. 4). The mean ROI intensity and area were quantified for each cell/ROI using the ImageJ menu-driven functions.
Figure 4.

Immunofluorescent quantification of Nav1.6 protein expression in WT and mSOD1 Mes V cells. a, Representative images showing three rostro-caudal levels of coronal brainstem sections, stained with NeuN (left column of images). Scale bar, 500 μm. In these images, the Mes V nucleus is highlighted with dashed contours. At Level B, the subjacent trigeminal motor nucleus (mot V) is highlighted. For each level, comparative images consisting of Mes V neurons from WT and mSOD1 mice are shown at 60× magnification. Green represents NeuN. Red represents Nav1.6 protein. Merged images are shown enlarged with white arrows highlighting representative Mes V neurons. Left, Bottom, Boxed inset represents the pontine levels at which sections were collected. b, Representative image showing ROIs drawn at ∼12 Mes V neurons for immunofluorescence quantification. White arrows indicate three representative ROIs. c, Scatterplot showing WT (black circles) and mSOD1 (red circles) values of ROI area and mean intensity quantification. Trendlines show a positive linear regression. d, Box plots represent mean ROI intensity per cell for WT (black) and mSOD1 (red), respectively. n values indicate number of cells obtained from 4 WT and 4 mSOD1 mice across 9 and 8 sections, respectively. A two-tailed Student's t test was used for statistical comparison.
Computational model of simplified sensorimotor network
Our simplified sensorimotor network consisted of conductance-based models for sensory Mes V neuron and trigeminal MN (TMN). All model conductances followed the well-known Hodgkin-Huxley formalism (Hodgkin and Huxley, 1952). The model was implemented using XPPAUT (Ermentrout, 1997) script and will be made available upon request. The model consists of an excitatory network input from the Mes V to the TMN and the model assumptions are detailed below.
The Mes V neuron model consisted of our recently updated version consisting of both persistent and resurgent Na+ currents that were reduced in the mSOD1 mouse in this study (Venugopal et al., 2019). Briefly, the model incorporates a minimal set of ionic conductances essential for producing rhythmic bursting and for maintaining cellular excitability in these neurons (Wu et al., 2005). These include a potassium leak current Ileak, a 4-AP-sensitive delayed-rectifier type potassium current IK, and a Nav1.6-type sodium current INa with three separable components: fast, persistent, and resurgent currents. Each of these model currents is based on our previous experimental work (Chandler et al., 1984; Del Negro and Chandler, 1997; Wu et al., 2001, 2005; Enomoto et al., 2006; Venugopal et al., 2019).
The TMN model is based on well-established two-compartment MN models, which include dendritic Ca2+ currents (e.g., Booth et al., 1997; Venugopal et al., 2011b). This is based on known physiology of MNs, which richly express noninactivating dendritic L-type Ca2+ currents, also known as the persistent Ca2+ current (CaP) in both the brainstem and spinal cord (Lee and Heckman, 1998; Carlin et al., 2000; Theiss et al., 2007). We chose such a model to reproduce dendritic plateau potentials, membrane bistability (Booth et al., 1997), and also since the CaP plays a crucial role in synaptic amplification (Hultborn et al., 2003) and motor control (Lee et al., 2003; Heckman et al., 2005). The somatic compartment consisted of the action potential generating fast sodium, delayed-rectifier type potassium, high-voltage-activated N-type Ca2+ and N-type Ca2+-activated potassium conductances (Hsiao et al., 1998, 2005). The plateau-generating L-type persistent Ca2+ or CaP, CaP-activated potassium and a persistent sodium conductance were confined to the dendritic compartment. Both compartments also consisted of a potassium leak conductance.
A lumped synaptic variable modeled the excitatory synaptic input from the Mes V to TMN model. This assumption was based on known anatomy and physiology of Mes V-TMN connections. For example, nearly 80% of Ia muscle spindle afferents from the Mes V nucleus synapse on to proximal and distal dendrites of TMNs (Yoshida et al., 2017). To incorporate such dendritic excitation, we modeled the TMN with two electrically coupled morphological compartments (soma and lumped dendrite) (Booth et al., 1997; Venugopal et al., 2011b, 2012) and confined synaptic input to the dendritic compartment. Second, we tuned the strength of the model excitatory conductance to generate sensory-coupled spikes in the TMNs since Mes V neurons provide strong monosynaptic glutamatergic excitation to TMNs (Chandler, 1989; Del Negro and Chandler, 1998; Turman et al., 1999, 2000). For simplicity, we assumed no other form of synaptic excitation or inhibition. This allowed us to exclusively examine the consequence of irregular sensory patterns as observed in our data, on MN discharge. The lumped synaptic variable represented a classic excitatory synapse modeled as follows (Venugopal et al., 2011a):
where s is the synaptic variable, αs = 2 is the rate constant for fast rise of s, and βs = 0.05 is the decay rate constant for relatively slower decay of s (Heckman and Binder, 1991; Trueblood et al., 1996; Powers et al., 2012). The steady-state voltage-dependent function s∞(V) is given by the following:
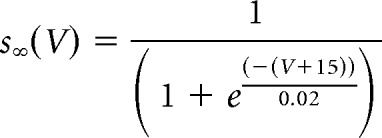 |
where, V is the voltage of the presynaptic neurons (here, Mes V neuron). The EPSC was included in the dendritic compartment of the TMN model and is given by the following conductance-based equation:
where Vd is the TMN dendritic voltage and Ve = − 20 mV is the reversal potential for an excitatory synaptic current. The value of maximal synaptic conductance, gsyn, was set such that presynaptic Mes V bursts generated postsynaptic TMN firing frequencies in the range observed in SOD1G93A TMNs (Venugopal et al., 2015).
Results
Impaired excitability of SOD1G93A proprioceptive Mes V neurons
First, we examined whether the electrical excitability of the jaw muscle proprioceptive neurons in the trigeminal Mes V nucleus shows abnormalities in the mSOD1 mice. Using suitable morphological and anatomical selection criteria, we conducted in vitro whole-cell patch-clamp recording and evaluated the basic membrane properties, action potential characteristics, and responses to increasing step depolarizations. The comparative membrane properties in mSOD1 and WT are summarized in Tables 1 and 2. In Figure 1a, we illustrate the location of the anterogradely labeled jaw closer muscle spindle afferents in the brainstem Mes V nucleus (green), obtained from injection of dye into the jaw closer muscles. Concomitantly, the jaw closer motor pools are also retrogradely labeled specifically in the dorsolateral trigeminal motor nucleus (Mot V) as shown in Figure 1a, ventrolateral to Mes V. In Figure 1b, we present an example of an anterogradely labeled Mes V sensory neuron, which is dye-filled during whole-cell patch-clamp recording. Figure 1d shows the age distribution of mice from which our dataset was obtained. In Figure 1c, we show representative examples of the two most commonly observed patterns of action potential discharges in our dataset from both WT and mSOD1 mice (Brocard et al., 2006; Enomoto et al., 2006). These included the following: (1) rhythmic burst discharge consisting of repetitive sequences of high-frequency spikes (∼80 Hz), followed by periods of quiescence on the order of 100–1000 ms (Wu et al., 2001; Brocard et al., 2006); and (2) a tonic spiking pattern with short ISIs, on the order of 10 ms. Our empirical criteria for classifying cells as bursters were based on the distributions of spike intervals (see Experimental design and statistical analyses). In the mSOD1 bursting Mes V neurons, we noted marked increase in the current and voltage thresholds for spike generation (Fig. 1e,f) compared with the WT. The mean ± SD for Ithreshold was 40.7 ± 5.4 pA and 57.1 ± 7.2 pA, for Vthreshold was −50.1 ± 2.1 mV and −44.6 ± 1.1 mV for WT and mSOD1, respectively. Corresponding p values from a two-sample Student's t test were 0.04 for Ithreshold and 0.008 for Vthreshold comparisons. The bursting neurons in the mSOD1 mice also showed diminished spike frequencies in response to increasing current injections (Fig. 1h). We quantified the average spike frequencies for the last 500 ms of 1 s step depolarization of increasing magnitude. A factorized two-way repeated-measures ANOVA was used to examine the effects of genotype (Factor 1), current injections (Factor 2), and any interaction effects on measured spike frequencies (dependent variable). We noted a significant between-subjects genotype effect (df = 1, F ratio = 28.996, p = 1.8E-05) and within subjects injected current effect (df = 5, F ratio = 38.484, p = 1.8E-016); there was no genotype × current interaction effect (df = 5, F ratio = 1.678, p = 0.145), suggesting that increasing current intensities similarly enhanced spike frequencies in both WT and mSOD1. A post hoc unpaired, two-tailed Student's t test confirmed significant reductions in spike frequencies for injected current magnitude ≥ 150 pA (Student's t test, 150 pA: p = 0.0091, 200 pA: p = 0.01, 250 pA: p = 0.0003, 300 pA: p = 0.01). Furthermore, a greater proportion of mSOD1 Mes V neurons presented burst-like activity (Fig. 1i). A χ2 proportionality test yielded p = 0.0074. Conversely, tonically spiking neurons were less perturbed and did not present any statistically significant Ithreshold or Vthreshold changes (Fig. 1e,g); however, at higher current injections ≥ 250 pA, these neurons also showed reduced firing frequencies similar to bursting neurons (Fig. 1h) (Student's t test, 250 pA: p = 0.008, 300 pA: p = 0.0174); the ANOVA statistics for between-subjects genotype differences was as follows: df = 1, F ratio = 11.8688, p = 0.0073, and for increasing current intensities was as follows: df = 5, F ratio = 27.202, p = 1.45E-012. We noted that these neurons also showed an increase in spike width (summary, Tables 1, 2). Moreover, the proportion of tonically firing cells was reduced in the mSOD1 Mes V dataset (Fig. 1i). Together, Mes V neurons in the SOD1G93A mouse are hypoexcitable with a large propensity toward burst formation in action potential trains.
Table 1.
Membrane properties of Mes V neurons
| Burst |
Tonic |
|||
|---|---|---|---|---|
| WT (n = 18) | mSOD1 (n = 19) | WT (n = 14) | mSOD1 (n = 7) | |
| RMP, mV | −56.9 ± 1.5 | −54.8 ± 1.0 | −53.6 ± 1.6 | −52.8 ± 1.4 |
| Rin, mΩ | 211.5 ± 25.9 | 253.1 ± 19.3 | 264.2 ± 16.9 | 300.1 ± 18.2 |
| Cm, pF | 49.2 ± 3.1 | 50.3 ± 2.7 | 42.0 ± 2.1 | 42.2 ± 2.2 |
Table 2.
Action potential properties of Mes V neuronsa
| Burst |
Tonic |
|||
|---|---|---|---|---|
| WT (n = 18) | mSOD1 (n = 19) | WT (n = 14) | mSOD1 (n = 7) | |
| AP height, mV | 89.3 ± 3.1 | 90.2 ± 4.3 | 92.5 ± 4.1 | 93.5 ± 3.8 |
| Half-width, ms | 1.3 ± 0.13 | 1.3 ± 0.10 | 1.2 ± 0.08 | 1.4 ± 0.09 (p = 0.025) |
| AP threshold, mV | −50.1 ± 2.1 | −44.6 ± 1.1 (p = 0.008) | −44.7 ± 2.1 | −41.4 ± 1.1 |
| AP current, pA | 40.7 ± 5.4 | 57.1 ± 7.2 (p = 0.04) | 41.4 ± 4.0 | 50.0 ± 7.7 |
| AHP peak, mV | −11.0 ± 0.9 | −8.1 ± 0.8 (p = 0.009) | −9.0 ± 0.7 | −8.5 ± 0.7 |
ap values are based on Student's t test.
Arrhythmic spike and burst patterns of SOD1G93A Mes V neurons
This overall reduction in action potential generation capability and increased burst discharge in the mSOD1 Mes V neurons was accompanied by further abnormalities in bursting properties. In Figure 2a, we illustrate a rhythmic burst pattern in a WT Mes V neuron (left) compared with irregular burst patterns in a mSOD1 Mes V neuron (right). The corresponding IEI time series highlights such irregularities in Figure 2b. In the rhythmic WT burst pattern, the IBIs are well demarcated from the ISIs. However, this distinction is less obvious in the mutant pattern; and as noted, the IBI regularity from one burst to another is also uncertain. The ISIs also presented significant irregularities observed for the shorter spike intervals. We used the autocorrelation function of the membrane voltage and compared its second peak between WT and mSOD1 cells using a time lag, τ = 100 ms to examine regularity of spikes (Rieke et al., 1999). This time window allowed examination of interspike irregularities within a typical burst. Figure 2c illustrates representative examples of the autocorrelation function for WT (left) and mSOD1 (middle) Mes V neurons. A two-sample unpaired Student's t test with assumed equal variance showed that the second peak of autocorrelation between the two groups was significantly different. A p = 0.0029 was noted with mean WT peak ± SD as 0.44 ± 0.04, and the mean mSOD1 peak ± SD as 0.30 ± 0.03. We also note that the subsequent peaks are almost completely suppressed, further highlighting irregularity in spike patterns in the mutant. Additionally, the burst duration and IBI lengths were reduced while the ISIs were increased (Fig. 2d): for WT and mSOD1, respectively, the mean burst duration ± SD were 0.41 ± 0.11 s and 0.06 ± 0.01 s, mean IBI ± SD were 0.89 ± 0.14 s and 0.29 ± 0.03 s, and mean ISI ± SD were 17.2 ± 0.34 ms and 26.4 ± 2.37 ms. Together, the proprioceptive Mes V neurons present early impairment in action potential generation and in the ability to sustain regular burst discharge in the SOD1G93A mouse.
Figure 2.

Arrhythmic burst discharge in mSOD1 Mes V neurons. a, Representative examples showing rhythmic bursting in a WT (blue) Mes V neuron (left), and arrhythmic bursting in a mSOD1 (red) Mes V neuron (right). b, Time series plots of IEIs corresponding to the examples shown in a highlight IEI irregularities (IEIs are shown on log scale). c, Autocorrelation function of the membrane voltage in a WT (left) and mSOD1 Mes V neuron (middle); second peak is highlighted. Right, Dot plots represent the second peaks in WT (blue) and mSOD1 (red) bursting neurons. d, Left, Inset, The timing properties of bursts. Dot plots represent significantly reduced burst duration (BD), IBIs, and increased ISIs in the mSOD1 (red) Mes V neurons compared with WT (blue). c, d, n = 14 (WT) and n = 16 (mSOD1), where n is the number of bursting cells. *p < 0.05 (above the black horizontal line).
Disrupted firing patterns of SOD1G93A proprioceptive Mes V neurons are linked to reduced Nav1.6 Na+ currents and channel protein
To identify the ionic basis for the observed impairments in action potential bursts, we tested whether the voltage-gated Na+ currents, which are essential for spike generation and burst control in these neurons, are compromised (Wu et al., 2005; Enomoto et al., 2006; Yang et al., 2009). Our previous report using the Nav1.6 subunit KO mouse demonstrated significant reductions in TTX-sensitive Na+ currents, which paralleled impairment in spike discharge (Enomoto et al., 2007). These Na+ currents, including the persistent and resurgent components, are important for generation and maintenance of burst discharge in Mes V neurons (Wu et al., 2005; Enomoto et al., 2006; Venugopal et al., 2019). Therefore, we examined whether the observed impairment in both excitability and burst discharge in a large majority of the mSOD1 Mes V neurons could be explained by reductions in the functional expression of these Nav1.6 Na+ channel currents. To test this, we used the in vitro voltage-clamp approach combined with suitable pharmacology and measured the Nav1.6 channel mediated persistent and resurgent Na+ currents in WT and mSOD1 Mes V neurons (see Cryo-sectioning and fluorescent immunohistochemistry). As shown in Figure 3 a–d, the mSOD1 Mes V neurons showed significantly reduced current density (pA/pF) of voltage-gated persistent and resurgent Na+ current components, compared with WT. As shown in Figure 3c, the peak persistent Na+ currents were downregulated at subthreshold activation voltages of −60 and −50 mV (mean ± SD current densities at −60 mV were −1.01 ± 0.22 pA/pF and −0.43 ± 0.10 pA/pF, and at −50 mV were 1.15 ± 0.21 pA/pF and −0.66 ± 0.11 pA/pF for WT and mSOD1 cells, respectively). Corresponding p values based on a Student's t test were 0.03 and 0.044 at −60 and −50 mV, respectively. Similarly, as shown in Figure 3d, the peak resurgent Na+ currents were also downregulated at −40 and −50 mV at which these currents are typically maximally activated (Enomoto et al., 2006, 2007) (mean ± SD current densities at −50 mV were −5.13 ± 0.282 pA/pF and −3.23 ± 0.51 pA/pF, and at −40 mV these values are −5.69 ± 0.84 pA/pF, and −3.93 ± 0.47 pA/pF for WT and mSOD1 cells, respectively). Corresponding p values for Student's t test were 0.0387 and 0.0424 at −50 and −40 mV, respectively. Such reductions in current density, however, were not accompanied by altered voltage-gating properties in the measured Na+ currents between WT and mSOD1 Mes V neurons, as noted by the normalized conductance Boltzmann curves (Fig. 3e,f).
Next, using fluorescent immunohistochemistry, we tested whether the observed decrease in the Nav1.6 Na+ currents in the mSOD1 mouse could result from a downregulation of Nav1.6 Na+ channels on Mes V neuronal membrane (Enomoto et al., 2007). We quantified the Nav1.6 protein expression in Mes V neuron cell bodies colocalized with NeuN stain. The NeuN-stained Mes V neurons were readily detected based on their oval cell bodies and rostro-caudal distribution in the dorsal pontine sections, ventrolateral to the periaqueductal gray (Fig. 4a, inset). As noted in the figure, the Nav1.6 protein expression was discernable in both WT and mSOD1 Mes V neurons. The three NeuN images (green) shown as level A–C illustrate the representative caudal-to-rostral sections used in our analysis. For immunofluorescent quantification, we manually outlined the NeuN-stained Mes V cell bodies (ROIs) in which the nuclei were clearly visible under high magnification (60×) (see Materials and Methods). These ROIs were then overlaid on the Nav1.6 protein-stained image (Fig. 4b) and the ROI areas, and mean intensities were quantified using the open-source ImageJ software (Schneider et al., 2012). Figure 4c shows a scatterplot of ROI areas and mean intensities for WT (black circles) and mSOD1 (red circles) Mes V neurons to illustrate that there was no sampling bias in our cell samples. There was an overall positive correlation between ROI/cell area and mean intensities in both WT and mSOD1 Mes V neurons (trend lines have positive slopes in Fig. 4c). However, such a correlation was weaker in the mutant. Statistical comparison of ROI areas and mean intensities between WT and mSOD1 revealed significantly diminished mean intensities in the mutant (mean ± SD: 36.02 ± 13.18 a.u. for WT and 27.01 ± 12.61 a.u. for mSOD1; p < 0.001 using two-sample Student's t test) (Fig. 4d; box plots show first, second, and the third quartiles, and error bars indicate 1.5× deviations from the median). However, the WT and mSOD1 ROI areas were not statistically different (data not shown), whereas the mean intensities per unit area (a.u./μm2) were also diminished in the mutant (p < 0.001). These results indicate that the reduced functional expression of Nav1.6 Na+ currents in the mSOD1 Mes V neurons are associated with a downregulation of Nav1.6 protein expression.
Rescue of Nav1.6 Na+ currents restored normal burst patterns and rhythmicity in SOD1G93A proprioceptive Mes V neurons
To test whether rescuing the Nav1.6 Na+ current impairment can restore the normal burst discharge in the mSOD1 Mes V neurons, we used an in vitro dynamic-clamp approach. This included real-time injection of conductance-based Nav1.6 Na+ currents into a mSOD1 Mes V neuron during whole-cell current-clamp recording of burst discharge (Fig. 5a). Figure 5b illustrates that addition of such realistic computer-generated Nav1.6 Na+ persistent and resurgent currents into an irregularly bursting mSOD1 Mes V neuron converted an irregular burst pattern to a more rhythmic pattern. The red traces in Figure 5b show the membrane voltage of the mSOD1 Mes V neuron during default burst discharge, and the black trace in between shows the modified burst discharge upon addition of the Nav1.6-type Na+ currents. To evaluate whether the rescue of Nav1.6-type Na+ currents in the mSOD1 neurons also restored the normal burst patterns similar to WT, we quantified the time intervals between spikes (IEIs) and demonstrate that the irregularities in mSOD1 IEIs were abolished (Fig. 5c) (Venugopal et al., 2019). Second, we measured the second peak of the autocorrelation function of the membrane potential and show that the rescue of Na+ currents restored the rhythmicity to WT values in a dose-dependent manner (Fig. 5d). Last, Na+ current addition also reinstated the time intervals and duration to WT values (Fig. 5e).
Figure 5.

Rescue of Nav 1.6-type sodium currents in mSOD1 Mes V neurons using dynamic clamp. a, Schematic shows the dynamic-clamp setup used to introduce conductance-based models of Nav1.6-type Na+ currents into mSOD1 Mes V neurons in real time during whole-cell patch-clamp recording. Idyn is the computer-generated model Na+ current in combination with a step depolarization to drive the patched Mes V neuron. Vm is the measured membrane voltage. b, Representative traces showing the membrane voltage in a bursting mSOD1 Mes V neuron with control/default behavior (left, red), followed by addition of Nav1.6-type currents (middle, black), which restores WT-like rhythmic bursting; subsequent removal of added currents returns default mSOD1 behavior (right red) in this neuron. Bottom, Green trace represents Idyn. c, Time series plots of IEIs (log scale) for the three different conditions in b. Each dot represents an interval between two consecutive spikes. d, Autocorrelation function of the membrane voltage for default mSOD1 (left) and with addition of Nav1.6 currents (middle). The height of the second autocorrelation peak highlights rhythmicity (arrowhead). Right, Measured second peak values for 4 mSOD1 cells. Dashed horizontal line indicates average WT values. Gray shaded region represents ±SD. e, Treatment effects on burst characteristics, including burst duration (BD), IBIs, and ISIs within bursts (ISI) shown for various mSOD1 cells tested under the different conditions as in b. Dashed lines indicate average WT values. Gray regions represent ± SD.
Together, the muscle spindle afferent neurons in the mSOD1 Mes V nucleus present early impairment in excitability, including hypoexcitability and arrhythmicity, which are associated with reductions in the voltage-gated Na+ currents and Nav1.6 Na+ channels.
Mechanoreceptive and nociceptive trigeminal sensory neurons lack early excitability changes in the SOD1G93A mouse
Next, to assess whether the altered excitability is restricted to the proprioceptive muscle spindle afferents in the mSOD1 Mes V nucleus, we examined the nonproprioceptive primary sensory neurons in the TG. The sensory architecture of the trigeminal system segregates the Aα-type proprioceptive neurons in the Mes V and the Aβ mechanoreceptors, Aδ nociceptors, and C-fiber type neurons in the peripheral TG (Lazarov, 2002). Therefore, we examined whether the different types of sensory neurons in the TG were affected differently by SOD1 mutation. To conduct in vitro patch-clamp electrophysiology, we performed acute dissociation of the TG in P8-P14 WT and mSOD1 mice, age-matched with mice used to examine Mes V neurons (Fig. 6a,b; see also Trigeminal ganglia (TG) extraction and acute dissociation of trigeminal ganglion neurons (TGNs) for electrophysiology). In both WT and mSOD1 TG neurons, we were able to distinguish the Aβ from the Aδ nociceptive neurons based on action potential duration and presence of a hump, as shown in Figure 6c (Xu et al., 2010; Kim et al., 2011). A small proportion of C-fiber type neurons were also present in our dataset, which were distinguished from the A-type neurons based on a lack of membrane sag during the hyperpolarizing current injections (Fig. 6c: a white block arrow in the left two panels highlight membrane sag). However, these cells exhibited single spikes and were comparatively fewer in both WT and mSOD1 datasets to yield meaningful statistical assessment and, hence, were excluded from further analysis. Interestingly, there were no proportional shifts in the sample dataset among these different types of neurons between WT and mSOD1 mice as shown in Figure 6d. We compared the basic membrane properties and action potential characteristics of WT and mSOD1 TG neurons as shown in Tables 3 and 4, the spike threshold current and voltage as shown in Figure 6e. None of these properties was different between WT and mSOD1. The average spike frequency responses to increasing steps of depolarization also did not present any significant changes w.r.t. genotype (Fig. 6f,g; frequency calculation and statistical tests and assumptions were similar to Mes V neurons and p values were > 0.05 and F ratio ∼ 0). Last, the autocorrelation function of the membrane voltage (Fig. 6h) did not show arrhythmicity in the mSOD1 TG neurons compared with WT. The second peak of the autocorrelation function was similar, and subsequent peaks were also similarly discernable with the autocorrelation time lag of 100 ms. These results suggest that nonproprioceptive trigeminal sensory neurons are resistant to early excitability changes in the SOD1G93A mouse model for ALS.
Table 3.
Membrane properties of TG neurons
| Aβ |
Aδ |
C |
||||
|---|---|---|---|---|---|---|
| WT (n = 18) | mSOD1 (n = 13) | WT (n = 15) | mSOD1 (n = 11) | WT (n = 3) | mSOD1 (n = 3) | |
| RMP, mV | −53.2 ± 0.9 | −54.2 ± 0.9 | −50.8 ± 0.3 | −52.6 ± 1.1 | −62.5 ± 2.5 | −63.0 ± 3.0 |
| Rin, mΩ | 293.7 ± 86.4 | 164.6 ± 22.9 | 413.9 ± 50.1 | 373.2 ± 56.8 | 224.1 ± 50.6 | 308.8 ± 123.4 |
| Cm, pF | 32.8 ± 4.6 | 35.1 ± 4.2 | 20.3 ± 3.7 | 18.4 ± 4.8 | 21.8 ± 4.2 | 27.4 ± 8.8 |
Table 4.
Action potential properties of TG neurons
| Aβ |
Aδ |
C |
||||
|---|---|---|---|---|---|---|
| WT (n = 18) | mSOD1 (n = 13) | WT (n = 15) | mSOD1 (n = 11) | WT (n = 3) | mSOD1 (n = 3) | |
| AP height, mV | 107.8 ± 2.9 | 108.1 ± 3.8 | 113.8 ± 4.3 | 112.1 ± 2.6 | 104.5 ± 7.9 | 105.0 ± 8.0 |
| Half-width, ms | 1.3 ± 0.16 | 1.5 ± 0.25 | 2.6 ± 0.19 | 2.7 ± 0.33 | 2.3 ± 0.04 | 3.6 ± 0.9 |
| AP threshold, mV | −27.6 ± 1.3 | −26.6 ± 1.4 | −19.4 ± 1.4 | −20.3 ± 2.1 | −23.6 ± 7.8 | −20.4 ± 3.0 |
| AP current, pA | 546.7 ± 118.7 | 524.6 ± 103.2 | 212.7 ± 33.9 | 244.5 ± 75.5 | 210.0 ± 37.9 | 200.0 ± 61.1 |
| AHP peak, mV | −5.9 ± 0.8 | −5.1 ± 0.8 | −7.1 ± 0.7 | −6.3 ± 0.6 | −4.3 ± 1.2 | −3.1 ± 2.1 |
Early excitability changes are absent in the SOD1G93A RGNs
Last, we investigated whether the peripheral visual sensory RGNs show altered excitability in the mSOD1 mice. For these experiments, we used a whole retinal preparation and conducted whole-cell patch-clamp electrophysiology in situ in the RGNs in P8-P14 mice (Fig. 7a,b; see also Retina extraction and preparation for electrophysiology). The rationale for examining RGN excitability was threefold: (1) to test whether functional abnormalities were limited to muscle spindle afferent proprioceptive neurons while absent in the visual sensory neurons, similar to the mechanoreceptive and nociceptive TG neurons; (2) to ascertain whether excitability changes were not confounded by the type of preparation (in vitro dissociated TGNs vs in situ RGNs); and (3) to clarify whether postnatal excitability changes in other sensory neurons are equally immune in parallel with maturing peripheral sensory systems in mice (Cabanes et al., 2002; Chen et al., 2009). Similar to the Mes V and TG neurons, we compared the membrane properties (Tables 5, 6), action potential thresholds (Fig. 7c), spike frequency-injected current responses (Fig. 7d,e), rhythmicity/regularity of membrane voltage (Fig. 7f), and proportional distribution of apparent electrophysiological subtypes of RGNs in the WT and mSOD1 mice (Fig. 7g,h). We found that none of the above properties tested was different between the two groups (all the statistical tests were similar to Mes V neurons). These results confirm that the early excitability changes are limited to muscle spindle afferent proprioceptive neurons in the cranial sensory system of the SOD1G93A mouse model for ALS.
Table 5.
Membrane properties of retinal ganglion cells
| WT (n = 13) | SOD1 (n = 10) | |
|---|---|---|
| RMP, mV | −55.4 ± 2.0 | −54.1 ± 2.4 |
| Rin, mΩ | 462.3 ± 19.3 | 436.2 ± 24.4 |
| Cm, pF | 29.0 ± 3.3 | 29.3 ± 4.4 |
Table 6.
Action potential properties of retinal ganglion cells
| WT (n = 13) | mSOD1 (n = 10) | |
|---|---|---|
| AP height, mV | 67.6 ± 3.9 | 66.3 ± 5.3 |
| Half-width, ms | 3.8 ± 0.5 | 4.0 ± 0.7 |
| AP threshold, mV | −50.6 ± 1.5 | −51.6 ± 2.2 |
| AP current, pA | 22.7 ± 4.7 | 22.2 ± 4.6 |
| AHP peak, mV | −6.1 ± 0.4 | −6.4 ± 0.8 |
Computer modeling predicts motor dysfunction due to perturbed proprioceptive sensory gating
Difficulty in chewing and swallowing is a common clinical feature observed in ALS patients with both spinal and bulbar onset (Riera-Punet et al., 2018a,b). In the SOD1G93A mouse model, mastication is severely impaired early during disease development (Lever et al., 2009). During normal mastication, the Mes V neurons relay sensory feedback from the jaw muscle spindles to adjust the activity and force generated in the jaw-closer muscles. Nearly 80% of the glutamatergic projections from the Mes V neurons synapse on the jaw-closer trigeminal motor pools and provide muscle stretch reflex inputs to the TMNs (Yoshida et al., 2017). It is likely that the observed abnormalities in the mSOD1 Mes V excitability alter the jaw stretch reflex control. To examine how irregular discharge in the mSOD1 Mes V neurons might modify motor discharge, we used a computational model of a simplified sensorimotor network. Our model consisted of a realistic sensory Mes V neuron, which provides strong monosynaptic excitation to a postsynaptic TMN (Trueblood et al., 1996). We used an oversimplified, yet realistic, construct and tested how irregularities in ongoing burst patterns in the sensory Mes V neuron, such as during rhythmic jaw movements, could exclusively modulate the discharge patterns in a TMN. In Figure 8a, we first illustrate rhythmic burst discharge, such as in a WT Mes V neuron reproduced by the model. Such a regular pattern was converted into irregular/arrhythmic discharge observed in the mSOD1 Mes V neurons by a 25%–50% reduction in the Mes V persistent and resurgent Na+ conductances, and addition of subthreshold stochastic inputs to enhance burst irregularities (Fig. 8b) (see also Venugopal et al., 2019). We further assumed that the TMN model displays dendritic Ca2+ currents, which can mediate plateau potentials and membrane bistability, often observed in brainstem and spinal MNs (Hsiao et al., 1998, 2005; Lee and Heckman, 1998). The model Mes V directly excited the dendritic compartment of the TMN, which in turn depolarized the electrically coupled TMN soma. We further assumed that such depolarization drives dendritic Ca2+-mediated plateau potentials, as would occur during NMDA receptor activation (Hsiao et al., 2002; Manuel et al., 2012). Activation of Ca2+ plateau depolarized the TMN soma and enabled sensorimotor synchronization driving downstream motor discharge (Fig. 8c). However, when the sensory patterns were irregular, such synchronization was perturbed (Fig. 8d). In particular, the shorter burst intervals mimicking mSOD1 discharge patterns induced instances of asynchronous self-sustained discharge in the TMN (Fig. 8d, dashed boxes). In our model, this was due to inadequate deactivation of the plateau causing dendritic Ca2+ currents in the MN between sensory bursts. Although under in vivo conditions, other mechanisms, such as synaptic inhibition and Ca2+-activated K+ currents, might effectively regulate Ca2+ plateaus and jaw reflexes (e.g., Inoue et al., 1994; Hultborn et al., 2003; Li and Bennett, 2007; Venugopal et al., 2012), our results highlight that rhythmic sensory timing, or lack thereof, can modulate sensorimotor synchronization. Together, one putative consequence of proprioceptive sensory irregularities leads to an asynchronous sustained motor discharge and may partly contribute to looming dysfunctions, such as muscle fasciculations, which are common symptoms in ALS (Hirota et al., 2000; Vucic and Kiernan, 2006) (see summary in Fig. 8e).
Figure 8.

Computer-based model predicts irregular sensory-driven motor asynchrony. a, Rhythmic burst pattern in a WT Mes V sensory neuron (top) is reproduced in a conductance-based Mes V neuron model (bottom). Top and bottom right, Corresponding time series graphs of IEIs (green circles). b, Burst irregularities in a mSOD1 Mes V neuron (top trace) is reproduced in the model neuron (bottom) by a 25% reduction in persistent Na+ conductance and a 50% reduction in resurgent Na+ conductance together with a slight increase in membrane potential noise. Top and bottom right, Corresponding time series graphs of IEIs (green circles). c, d, Simulated postsynaptic membrane potentials in the motor dendrite (cyan) and soma (blue) for WT (c) and mSOD1 (d) sensory patterns (top, magenta). Dashed rectangles represent frequent occurrences of asynchronous self-sustained motor discharge patterns. e, Schematic showing putative motor asynchrony and dysfunctional discharge patterns resulting from burst irregularities in the primary Ia afferent neuron.
Discussion
In this study, we focused on SOD1G93A mutation-induced modifications to proprioceptive sensory neurons (Mes V cells), at an early time point when MN dysregulation was previously reported (Venugopal et al., 2015). There are four main findings: (1) an early circuit-specific reduced excitability exclusively in the trigeminal proprioceptive sensory neurons in comparison with mechanoreceptive, nociceptive, and visual sensory neurons in age-matched mSOD1 mice; (2) impaired bursting in the proprioceptive Mes V neurons associated with a downregulation of Nav1.6 Na+ currents and ion channels; (3) rescue of normal burst patterns in the mSOD1 Mes V neurons upon restoration of Na+ currents; and (4) computational model-based prediction of an effect of aberrant sensory gating on MN discharge patterns. Considering these results, we discuss the consequences on disease development and progression.
Circuit-specific vulnerability in ALS
Selective vulnerability is a hallmark of neurodegenerative diseases (Double et al., 2010); however, the factors that determine such selectivity are elusive and remain poorly understood. In ALS, as also with normal aging (Kanning et al., 2010), MNs within a motor pool present selective and preferential vulnerability (Frey et al., 2000; Pun et al., 2006; Hegedus et al., 2007; Saxena and Caroni, 2011). Specifically, α-MNs forming the fast-fatigable motor units preferentially die followed by fast fatigue-resistant MNs, whereas the neighboring slow α-MNs and γ-MNs remain resistant to degeneration (Hegedus et al., 2007, 2008; Lalancette-Hebert et al., 2016). Although multiple intrinsic factors, such as cell size (Dukkipati et al., 2018), Ca2+ buffering capacities (von Lewinski and Keller, 2005), synaptic organization (Nimchinsky et al., 2000; Lorenzo et al., 2006), and gene and protein expression patterns (Brockington et al., 2013; Comley et al., 2015), could govern selective vulnerability, the underlying network architecture can be a crucial determinant. For instance, a lack of muscle spindle afferent terminals on γ- and ocular MNs has been suggested as a mechanism of disease resistance (Keller and Robinson, 1971; Lalancette-Hebert et al., 2016). These afferents relay the proprioceptive sensory feedback, which modulates MN activity (the stretch reflex) during muscle force generation via glutamatergic excitation (e.g., Chandler, 1989; Mentis et al., 2011). Ablation of such spindle afferents significantly delayed MN death and disease progression in the SOD1G93A mouse model (Lalancette-Hebert et al., 2016). Curiously, even in invertebrate SOD1-knock-in model systems, muscle spindle afferents act as an early trigger for MN degeneration (Held et al., 2019). This suggests the possibility that the proprioceptive feedback may indeed represent a phylogenetically conserved pathway of disease vulnerability. Furthermore, in a mouse model for spinal muscular atrophy, a related MN disease, proprioceptive inputs play a predominant role in triggering MN degeneration (Mentis et al., 2011). Advancing these findings, our comparative analysis of excitability of multiple sensory neurons revealed a SOD1 mutation-driven modification exclusively in the proprioceptive sensory neurons. Although non–motor and non–cell-autonomous triggers have been implicated for MN death in ALS (Boillée et al., 2006a), to date, there is no direct evidence for exclusive circuit elements showing intrinsic electrophysiological abnormalities. In combination with our previous report on early dysregulation of MN excitability (Venugopal et al., 2015), the present results implicate a defective sensorimotor network in ALS, which may represent a convergent neuroanatomical pathway across multiple neurodegenerative MN diseases.
Excitability change: an early disease compensation and a marker for vulnerable circuits
Neurodegenerative diseases involve a protracted phase of progressive decline in the functional homeostasis of vulnerable neurons (Roselli and Caroni, 2015). In mouse models of ALS, hyperexcitability represents the earliest form of homeostatic disruption in brainstem, spinal, and corticomotor neurons, which are vulnerable to degeneration (Pieri et al., 2003; Amendola et al., 2007; Bories et al., 2007; van Zundert et al., 2008; Vucic et al., 2009; Quinlan et al., 2011; Venugopal et al., 2015). These results prompted a hyperexcitability-driven excitotoxicity hypothesis for cell death in ALS (Durand et al., 2006; van Zundert et al., 2012). Although attractive, a causal relationship between hyperexcitability and subsequent neurodegeneration is not well supported (e.g., Leroy et al., 2014; Simon et al., 2016), and indeed recent evidence in MNs proposes a transition into hypoexcitability at adult stages (Delestrée et al., 2014; Martínez-Silva et al., 2018). Alternatively, it is likely that early hyperexcitability represents a compensatory mechanism of survival in normally low excitable cells since enhancing intrinsic excitability promotes neuroprotection in these MNs (Saxena et al., 2013). Early compensation is also possible in slow MNs, which normally show high excitability and are resistant to degeneration in ALS. Some of these slow MNs were hypoexcitable early in the SOD1G93A mouse (Venugopal et al., 2015). Such early hypoexcitability in surviving MNs likely represents a way to moderate energy demands for spike generation, which can be costly (e.g., Le Masson et al., 2014). This in turn could support cell survival in a disease background. A similar strategy could be used by the proprioceptive sensory neurons shown here, which are involved in ongoing reflex control. We further speculate that hypoexcitable shift in the set point operation of neurons within a vulnerable circuitry could be triggered by disease-induced changes in astroglial cells, which monitor extracellular ionic homeostasis (e.g., Ca2+ and K+) as well as bursting in Mes V neurons (Kadala et al., 2015; Morquette et al., 2015). Such crosstalk leading to compensatory intrinsic excitability changes needs to be further clarified. Notwithstanding a compensatory function, early excitability changes seem to be clear markers of vulnerable circuitry. For instance, disease-resistant MNs (e.g., oculomotor neurons) (Venugopal et al., 2015) or nonproprioceptive sensory neurons as shown here do not present early modifications in excitability. In contrast, a specific set of bursting proprioceptive Mes V neurons were hypoexcitable, which form a monosynaptic jaw stretch reflex circuitry with vulnerable TMNs. Such reduced presynaptic sensory excitability could also be a trigger for compensatory hyperexcitability in the postsynaptic TMNs (Venugopal et al., 2015). However, it is yet unclear whether and how such sensory alterations contribute to MN degeneration (see, e.g., Lalancette-Hebert et al., 2016). Together, early excitability changes are crucial markers of vulnerable circuitry and may represent an early compensation to meet energy demands and likely support cell survival early during disease development.
Altered activity patterns and ionic mechanisms as pathophysiological substrates
Altered phasic/burst patterns, reduced excitability, and loss of excitation are defining features of vulnerability to neurodegeneration (Roselli and Caroni, 2015). Here we present a unique result highlighting early irregular and impaired burst discharge in proprioceptive sensory neurons forming a vulnerable sensorimotor circuitry. One consequence of irregular bursting could affect sensory gating of postsynaptic motor discharge as shown by our computational model. Second, altered Ia afferents could directly alter static versus dynamic spindle inputs important for activation of slow versus fast persistent Ca2+ currents in MNs during a stretch reflex (Manuel et al., 2007). Moreover, impaired burst duration and frequencies together with reductions in persistent Na+ current could reduce extracellular Ca2+ and affect astrocyte-mediated Ca2+ buffering in the Mes V sensory nucleus (Morquette et al., 2015). Alternatively, reduced persistent Na+ and bursting could be a direct consequence of mSOD1 astrocyte dysfunction (Su et al., 2001). Curiously, retinal ganglion cells, which also rely on persistent Na+ current for repetitive spiking (Boiko et al., 2003; Van Wart and Matthews, 2006a,b), did not show altered rhythmicity as measured by the autocorrelation function. These observations suggest that the mutation could induce observed changes via altered extrinsic factors, such as astrocyte dysfunction or by altered intrinsic factors, such as abnormal protein synthesis contributing to the observed Na+ channelopathy in Mes V neurons. Furthermore, in the trigeminal system, the nonproprioceptive sensory neurons in the TG were resistant to abnormalities in the SOD1G93A mouse. Given that the TG and Mes V neurons share ontogeny (Lazarov, 2002), an exclusive change only in Mes V supports extrinsic triggers within the jaw stretch reflex circuitry causing abnormal excitability. However, we note that definitive studies measuring Mes V excitability under physiological conditions during natural movements, such as chewing, which dynamically modulate spindle activity would be essential to fully evaluate functional deficits resulting from proprioceptive afferent dysfunction.
Secondary effects of altered burst patterns could lead to recruitment of distinct types of voltage-dependent Ca2+ currents; resulting alterations in intracellular Ca2+ levels and kinetics can activate distinct CREB-dependent gene expression (Wheeler et al., 2012) and differentially activate nuclear translocation transcription factors, such as the NFAT family, important in immune response (Hernández-Ochoa et al., 2007). Importantly, reduced excitability can deplete intracellular ER Ca2+ stores and disrupt protein homeostasis (e.g., a ubiquitin-dependent degradation of transcription factors important for synaptic plasticity) (Lalonde et al., 2014). Together, we suggest that identifying presymptomatic excitability changes, their upstream physiological triggers (e.g., astrocyte dysfunction), and downstream effectors (e.g., CREB-mediated gene expression changes) can elucidate the cascade of events leading to neurodegeneration. Additionally, excitability changes also represent early markers of disease vulnerability in ALS.
Footnotes
This work was supported by National Institutes of Health/National Institute of Neurological Disorders and Stroke NS095157 to S.V., UCLA Faculty Research Grant to S.H.C., National Institutes of Health/National Heart, Lung, and Blood Institute 1R01HL134346 to R.O., the David Vickter Foundation and the Toeffler Family & Simon-Strauss Foundation to M.W.-P., National Institutes of Health R01CA196263 to I.S., and Knut and Alice Wallenberg Foundation to A.P. We thank Drs. Michael Levine, Sampath Alapakkam, Xia Yang, and their laboratory members for generous support on various aspects of this project; and Dr. Yatendra Mulpuri and Ravindu Udugampola for help with the histology experiments.
The authors declare no competing financial interests.
References
- Amendola J, Gueritaud JP, Lamotte d'Incamps B, Bories C, Liabeuf S, Allene C, Pambo-Pambo A, Durand J (2007) Postnatal electrical and morphological abnormalities in lumbar motoneurons from transgenic mouse models of amyotrophic lateral sclerosis. Arch Ital Biol 145:311–323. [PubMed] [Google Scholar]
- Boiko T, Van Wart A, Caldwell JH, Levinson SR, Trimmer JS, Matthews G (2003) Functional specialization of the axon initial segment by isoform-specific sodium channel targeting. J Neurosci 23:2306–2313. 10.1523/JNEUROSCI.23-06-02306.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boillée S, Vande Velde C, Cleveland DW (2006a) ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52:39–59. 10.1016/j.neuron.2006.09.018 [DOI] [PubMed] [Google Scholar]
- Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW (2006b) Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312:1389–1392. 10.1126/science.1123511 [DOI] [PubMed] [Google Scholar]
- Booth V, Rinzel J, Kiehn O (1997) Compartmental model of vertebrate motoneurons for Ca2+-dependent spiking and plateau potentials under pharmacological treatment. J Neurophysiol 78:3371–3385. 10.1152/jn.1997.78.6.3371 [DOI] [PubMed] [Google Scholar]
- Bories C, Amendola J, Lamotte d'Incamps B, Durand J (2007) Early electrophysiological abnormalities in lumbar motoneurons in a transgenic mouse model of amyotrophic lateral sclerosis. Eur J Neurosci 25:451–459. 10.1111/j.1460-9568.2007.05306.x [DOI] [PubMed] [Google Scholar]
- Brocard F, Verdier D, Arsenault I, Lund JP, Kolta A (2006) Emergence of intrinsic bursting in trigeminal sensory neurons parallels the acquisition of mastication in weanling rats. J Neurophysiol 96:2410–2424. 10.1152/jn.00352.2006 [DOI] [PubMed] [Google Scholar]
- Brockington A, Ning K, Heath P, Wood E, Kirby J, Fusi N, Lawrence N, Wharton S, Ince P, Shaw P (2013) Unravelling the enigma of selective vulnerability in neurodegeneration: motor neurons resistant to degeneration in ALS show distinct gene expression characteristics and decreased susceptibility to excitotoxicity Acta Neuropathol 125:95–109. 10.1007/s00401-012-1058-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownstone RM, Lancelin C (2018) Escape from homeostasis: spinal microcircuits and progression of amyotrophic lateral sclerosis. J Neurophysiol 119:1782–1794. 10.1152/jn.00331.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruijn LI, Miller TM, Cleveland DW (2004) Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci 27:723–749. 10.1146/annurev.neuro.27.070203.144244 [DOI] [PubMed] [Google Scholar]
- Cabanes C, López de Armentia M, Viana F, Belmonte C (2002) Postnatal changes in membrane properties of mice trigeminal ganglion neurons. J Neurophysiol 87:2398–2407. 10.1152/jn.2002.87.5.2398 [DOI] [PubMed] [Google Scholar]
- Carlin KP, Jones KE, Jiang Z, Jordan LM, Brownstone RM (2000) Dendritic L-type calcium currents in mouse spinal motoneurons: implications for bistability. Eur J Neurosci 12:1635–1646. 10.1046/j.1460-9568.2000.00055.x [DOI] [PubMed] [Google Scholar]
- Casas C, Manzano R, Vaz R, Osta R, Brites D (2016) Synaptic failure: focus in an integrative view of ALS. Brain Plast 1:159–175. 10.3233/BPL-140001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler SH. (1989) Evidence for excitatory amino acid transmission between mesencephalic nucleus of V afferents and jaw-closer motoneurons in the guinea pig. Brain Res 477:252–264. 10.1016/0006-8993(89)91413-3 [DOI] [PubMed] [Google Scholar]
- Chandler SH, Baker LL, Goldberg LJ (1984) Characterization of synaptic potentials in hindlimb extensor motoneurons during L-DOPA-induced fictive locomotion in acute and chronic spinal cats. Brain Res 303:91–100. 10.1016/0006-8993(84)90214-2 [DOI] [PubMed] [Google Scholar]
- Chen L, Liu C, Liu L (2008) The modulation of voltage-gated potassium channels by anisotonicity in trigeminal ganglion neurons. Neuroscience 154:482–495. 10.1016/j.neuroscience.2008.03.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Weng S, Deng Q, Xu Z, He S (2009) Physiological properties of direction-selective ganglion cells in early postnatal and adult mouse retina. J Physiol 587:819–828. 10.1113/jphysiol.2008.161240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland DW, Rothstein JD (2001) From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci 2:806–819. 10.1038/35097565 [DOI] [PubMed] [Google Scholar]
- Comley L, Allodi I, Nichterwitz S, Nizzardo M, Simone C, Corti S, Hedlund E (2015) Motor neurons with differential vulnerability to degeneration show distinct protein signatures in health and ALS. Neuroscience 291:216–229. 10.1016/j.neuroscience.2015.02.013 [DOI] [PubMed] [Google Scholar]
- Del Negro CA, Chandler SH (1997) Physiological and theoretical analysis of K+ currents controlling discharge in neonatal rat mesencephalic trigeminal neurons. J Neurophysiol 77:537–553. 10.1152/jn.1997.77.2.537 [DOI] [PubMed] [Google Scholar]
- Del Negro CA, Chandler SH (1998) Regulation of intrinsic and synaptic properties of neonatal rat trigeminal motoneurons by metabotropic glutamate receptors. J Neurosci 18:9216–9226. 10.1523/JNEUROSCI.18-22-09216.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delestrée N, Manuel M, Iglesias C, Elbasiouny SM, Heckman CJ, Zytnicki D (2014) Adult motoneurons are not hyperexcitable in a mouse model of inherited amyotrophic lateral sclerosis. J Physiol 592:1687–1703. 10.1113/jphysiol.2013.265843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do MT, Bean BP (2003) Subthreshold sodium currents and pacemaking of subthalamic neurons: modulation by slow inactivation. Neuron 39:109–120. 10.1016/S0896-6273(03)00360-X [DOI] [PubMed] [Google Scholar]
- Double KL, Reyes S, Werry EL, Halliday GM (2010) Selective cell death in neurodegeneration: why are some neurons spared in vulnerable regions? Prog Neurobiol 92:316–329. 10.1016/j.pneurobio.2010.06.001 [DOI] [PubMed] [Google Scholar]
- Dukkipati SS, Garrett TL, Elbasiouny SM (2018) The vulnerability of spinal motoneurons and soma size plasticity in a mouse model of amyotrophic lateral sclerosis. J Physiol 596:1723–1745. 10.1113/JP275498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand J, Amendola J, Bories C, Lamotte d'Incamps B (2006) Early abnormalities in transgenic mouse models of amyotrophic lateral sclerosis. J Physiol Paris 99:211–220. 10.1016/j.jphysparis.2005.12.014 [DOI] [PubMed] [Google Scholar]
- Enomoto A, Han JM, Hsiao CF, Wu N, Chandler SH (2006) Participation of sodium currents in burst generation and control of membrane excitability in mesencephalic trigeminal neurons. J Neurosci 26:3412–3422. 10.1523/JNEUROSCI.5274-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto A, Han JM, Hsiao CF, Chandler SH (2007) Sodium currents in mesencephalic trigeminal neurons from Nav1.6 null mice. J Neurophysiol 98:710–719. 10.1152/jn.00292.2007 [DOI] [PubMed] [Google Scholar]
- Ermentrout B. (1997) XPPAUT - The Differential Equations Tool, University of Pittsburgh, Pittsburgh. [Google Scholar]
- Ferrucci M, Spalloni A, Bartalucci A, Cantafora E, Fulceri F, Nutini M, Longone P, Paparelli A, Fornai F (2010) A systematic study of brainstem motor nuclei in a mouse model of ALS, the effects of lithium. Neurobiol Dis 37:370–383. 10.1016/j.nbd.2009.10.017 [DOI] [PubMed] [Google Scholar]
- Frey D, Schneider C, Xu L, Borg J, Spooren W, Caroni P (2000) Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci 20:2534–2542. 10.1523/JNEUROSCI.20-07-02534.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Elgammal FS, Proddutur A, Shah S, Santhakumar V (2012) Decrease in tonic inhibition contributes to increase in dentate semilunar granule cell excitability after brain injury. J Neurosci 32:2523–2537. 10.1523/JNEUROSCI.4141-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckman CJ, Binder MD (1991) Analysis of IA-inhibitory synaptic input to cat spinal motoneurons evoked by vibration of antagonist muscles. J Neurophysiol 66:1888–1893. 10.1152/jn.1991.66.6.1888 [DOI] [PubMed] [Google Scholar]
- Heckman C, Gorassini M, Bennett D (2005) Persistent inward currents in motoneuron dendrites: implications for motor output. Muscle Nerve 31:153–156. 10.1002/mus.20261 [DOI] [PubMed] [Google Scholar]
- Hedlund E, Karlsson M, Osborn T, Ludwig W, Isacson O (2010) Global gene expression profiling of somatic motor neuron populations with different vulnerability identify molecules and pathways of degeneration and protection. Brain 133:2313–2330. 10.1093/brain/awq167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegedus J, Putman CT, Gordon T (2007) Time course of preferential motor unit loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 28:154–164. 10.1016/j.nbd.2007.07.003 [DOI] [PubMed] [Google Scholar]
- Hegedus J, Putman CT, Tyreman N, Gordon T (2008) Preferential motor unit loss in the SOD1 G93A transgenic mouse model of amyotrophic lateral sclerosis. J Physiol 586:3337–3351. 10.1113/jphysiol.2007.149286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Held A, Major P, Sahin A, Reenan RA, Lipscombe D, Wharton KA (2019) Circuit dysfunction in SOD1-ALS model first detected in sensory feedback prior to motor neuron degeneration is alleviated by BMP signaling. J Neurosci 39:2347–2364. 10.1523/JNEUROSCI.1771-18.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson G, Pepper CM, Shefner SA (1982) Electrophysiological properties of neurons contained in the locus coeruleus and mesencephalic nucleus of the trigeminal nerve in vitro. Exp Brain Res 45:29–37. 10.1007/bf00235760 [DOI] [PubMed] [Google Scholar]
- Hernández-Ochoa EO, Contreras M, Cseresnyés Z, Schneider MF (2007) Ca2+ signal summation and NFATc1 nuclear translocation in sympathetic ganglion neurons during repetitive action potentials. Cell Calcium 41:559–571. 10.1016/j.ceca.2006.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota N, Eisen A, Weber M (2000) Complex fasciculations and their origin in amyotrophic lateral sclerosis and Kennedy's disease. Muscle Nerve 23:1872–1875. [DOI] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF (1952) A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol 117:500–544. 10.1113/jphysiol.1952.sp004764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao CF, Del Negro CA, Trueblood PR, Chandler SH (1998) Ionic basis for serotonin-induced bistable membrane properties in guinea pig trigeminal motoneurons. J Neurophysiol 79:2847–2856. 10.1152/jn.1998.79.6.2847 [DOI] [PubMed] [Google Scholar]
- Hsiao CF, Wu N, Levine MS, Chandler SH (2002) Development and serotonergic modulation of NMDA bursting in rat trigeminal motoneurons. J Neurophysiol 87:1318–1328. 10.1152/jn.00469.2001 [DOI] [PubMed] [Google Scholar]
- Hsiao CF, Wu N, Chandler SH (2005) Voltage-dependent calcium currents in trigeminal motoneurons of early postnatal rats: modulation by 5-HT receptors. J Neurophysiol 94:2063–2072. 10.1152/jn.00178.2005 [DOI] [PubMed] [Google Scholar]
- Hultborn H, Denton ME, Wienecke J, Nielson JB (2003) Variable amplification of synaptic input to cat spinal motoneurons by persistent inward current. J Physiol 552:945–952. 10.1113/jphysiol.2003.050971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Chandler SH, Goldberg LJ (1994) Neuropharmacological mechanisms underlying rhythmical discharge in trigeminal interneurons during fictive mastication. J Neurophysiol 71:2061–2073. 10.1152/jn.1994.71.6.2061 [DOI] [PubMed] [Google Scholar]
- Iwata M, Hirano A (1978) Sparing of the Onufrowicz nucleus in sacral anterior horn lesions. Ann Neurol 4:245–249. 10.1002/ana.410040309 [DOI] [PubMed] [Google Scholar]
- Kadala A, Verdier D, Morquette P, Kolta A (2015) Ion homeostasis in rhythmogenesis: the interplay between neurons and astroglia. Physiology (Bethesda) 30:371–388. 10.1152/physiol.00023.2014 [DOI] [PubMed] [Google Scholar]
- Kanning KC, Kaplan A, Henderson CE (2010) Motor neuron diversity in development and disease. Annu Rev Neurosci 33:409–440. 10.1146/annurev.neuro.051508.135722 [DOI] [PubMed] [Google Scholar]
- Keller EL, Robinson DA (1971) Absence of a stretch reflex in extraocular muscles of the monkey. J Neurophysiol 34:908–919. 10.1152/jn.1971.34.5.908 [DOI] [PubMed] [Google Scholar]
- Kim HY, Chung G, Jo HJ, Kim YS, Bae YC, Jung SJ, Kim JS, Oh SB (2011) Characterization of dental nociceptive neurons. J Dent Res 90:771–776. 10.1177/0022034511399906 [DOI] [PubMed] [Google Scholar]
- Lalancette-Hebert M, Sharma A, Lyashchenko AK, Shneider NA (2016) Gamma motor neurons survive and exacerbate alpha motor neuron degeneration in ALS. Proc Natl Acad Sci U S A 113:E8316–E8325. 10.1073/pnas.1605210113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde J, Saia G, Gill G (2014) Store-operated calcium entry promotes the degradation of the transcription factor Sp4 in resting neurons. Sci Signal 7:ra51. 10.1126/scisignal.2005242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarov NE. (2002) Comparative analysis of the chemical neuroanatomy of the mammalian trigeminal ganglion and mesencephalic trigeminal nucleus. Prog Neurobiol 66:19–59. 10.1016/S0301-0082(01)00021-1 [DOI] [PubMed] [Google Scholar]
- Lee RH, Heckman CJ (1998) Bistability in spinal motoneurons in vivo: systematic variations in persistent inward currents. J Neurophysiol 80:583–593. 10.1152/jn.1998.80.2.583 [DOI] [PubMed] [Google Scholar]
- Lee RH, Kuo JJ, Jiang MC, Heckman CJ (2003) Influence of active dendritic currents on input-output processing in spinal motoneurons in vivo. J Neurophysiol 89:27–39. 10.1152/jn.00137.2002 [DOI] [PubMed] [Google Scholar]
- Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, Liu Y, Tsingalia A, Jin L, Zhang PW, Pellerin L, Magistretti PJ, Rothstein JD (2012) Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 487:443–448. 10.1038/nature11314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Masson G, Przedborski S, Abbott LF (2014) A computational model of motor neuron degeneration. Neuron 83:975–988. 10.1016/j.neuron.2014.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy F, Lamotte d'Incamps B, Imhoff-Manuel RD, Zytnicki D (2014) Early intrinsic hyperexcitability does not contribute to motoneuron degeneration in amyotrophic lateral sclerosis. Elife 3:e04046. 10.7554/eLife.04046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lever TE, Gorsek A, Cox KT, O'Brien KF, Capra NF, Hough MS, Murashov AK (2009) An animal model of oral dysphagia in amyotrophic lateral sclerosis. Dysphagia 24:180–195. 10.1007/s00455-008-9190-z [DOI] [PubMed] [Google Scholar]
- Li X, Bennett DJ (2007) Apamin-sensitive calcium-activated potassium currents (SK) are activated by persistent calcium currents in rat motoneurons. J Neurophysiol 97:3314–3330. 10.1152/jn.01068.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin RJ, Bettencourt J, Wha Ite J, Christini DJ, Butera RJ (2010) Real-time experiment interface for biological control applications. Conf Proc IEEE Eng Med Biol Soc 2010:4160–4163. 10.1109/IEMBS.2010.5627397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo LE, Barbe A, Portalier P, Fritschy JM, Bras H (2006) Differential expression of GABAA and glycine receptors in ALS-resistant vs. ALS-vulnerable motoneurons: possible implications for selective vulnerability of motoneurons. Eur J Neurosci 23:3161–3170. 10.1111/j.1460-9568.2006.04863.x [DOI] [PubMed] [Google Scholar]
- Malin SA, Davis BM, Molliver DC (2007) Production of dissociated sensory neuron cultures and considerations for their use in studying neuronal function and plasticity. Nat Protoc 2:152–160. 10.1038/nprot.2006.461 [DOI] [PubMed] [Google Scholar]
- Manuel M, Meunier C, Donnet M, Zytnicki D (2007) Resonant or not, two amplification modes of proprioceptive inputs by persistent inward currents in spinal motoneurons. J Neurosci 27:12977–12988. 10.1523/JNEUROSCI.3299-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuel M, Li Y, Elbasiouny SM, Murray K, Griener A, Heckman CJ, Bennett DJ (2012) NMDA induces persistent inward and outward currents that cause rhythmic bursting in adult rodent motoneurons. J Neurophysiol 108:2991–2998. 10.1152/jn.00518.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchenkova A, van den Maagdenberg AM, Nistri A (2016) Loss of inhibition by brain natriuretic peptide over P2X3 receptors contributes to enhanced spike firing of trigeminal ganglion neurons in a mouse model of familial hemiplegic migraine type-1. Neuroscience 331:197–205. 10.1016/j.neuroscience.2016.06.034 [DOI] [PubMed] [Google Scholar]
- Margolis DJ, Detwiler PB (2007) Different mechanisms generate maintained activity in ON and OFF retinal ganglion cells. J Neurosci 27:5994–6005. 10.1523/JNEUROSCI.0130-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Silva ML, Imhoff-Manuel RD, Sharma A, Heckman CJ, Shneider NA, Roselli F, Zytnicki D, Manuel M (2018) Hypoexcitability precedes denervation in the large fast-contracting motor units in two unrelated mouse models of ALS. Elife 7:e30955. 10.7554/eLife.30955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentis GZ, Blivis D, Liu W, Drobac E, Crowder ME, Kong L, Alvarez FJ, Sumner CJ, O'Donovan MJ (2011) Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron 69:453–467. 10.1016/j.neuron.2010.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morquette P, Verdier D, Kadala A, Féthière J, Philippe AG, Robitaille R, Kolta A (2015) An astrocyte-dependent mechanism for neuronal rhythmogenesis. Nat Neurosci 18:844–854. 10.1038/nn.4013 [DOI] [PubMed] [Google Scholar]
- Murphy GJ, Rieke F (2006) Network variability limits stimulus-evoked spike timing precision in retinal ganglion cells. Neuron 52:511–524. 10.1016/j.neuron.2006.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijssen J, Comley LH, Hedlund E (2017) Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathol 133:863–885. 10.1007/s00401-017-1708-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimchinsky EA, Young WG, Yeung G, Shah RA, Gordon JW, Bloom FE, Morrison JH, Hof PR (2000) Differential vulnerability of oculomotor, facial, and hypoglossal nuclei in G86R superoxide dismutase transgenic mice. J Comp Neurol 416:112–125. [DOI] [PubMed] [Google Scholar]
- Pieri M, Albo F, Gaetti C, Spalloni A, Bengtson CP, Longone P, Cavalcanti S, Zona C (2003) Altered excitability of motor neurons in a transgenic mouse model of familial amyotrophic lateral sclerosis. Neurosci Lett 351:153–156. 10.1016/j.neulet.2003.07.010 [DOI] [PubMed] [Google Scholar]
- Powers RK, Elbasiouny SM, Rymer WZ, Heckman CJ (2012) Contribution of intrinsic properties and synaptic inputs to motoneuron discharge patterns: a simulation study. J Neurophysiol 107:808–823. 10.1152/jn.00510.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puls I, Jonnakuty C, LaMonte BH, Holzbaur EL, Tokito M, Mann E, Floeter MK, Bidus K, Drayna D, Oh SJ, Brown RH Jr, Ludlow CL, Fischbeck KH (2003) Mutant dynactin in motor neuron disease. Nat Genet 33:455–456. 10.1038/ng1123 [DOI] [PubMed] [Google Scholar]
- Pun S, Santos AF, Saxena S, Xu L, Caroni P (2006) Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci 9:408–419. 10.1038/nn1653 [DOI] [PubMed] [Google Scholar]
- Qu J, Myhr KL (2011) The morphology and intrinsic excitability of developing mouse retinal ganglion cells. PLoS One 6:e21777. 10.1371/journal.pone.0021777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan KA, Schuster JE, Fu R, Siddique T, Heckman CJ (2011) Altered postnatal maturation of electrical properties in spinal motoneurons in a mouse model of amyotrophic lateral sclerosis. J Physiol 589:2245–2260. 10.1113/jphysiol.2010.200659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Bean BP (2001) Inactivation and recovery of sodium currents in cerebellar Purkinje neurons: evidence for two mechanisms. Biophys J 80:729–737. 10.1016/S0006-3495(01)76052-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieke F, Warland D, van Steveninck RR, Bialek W (1999) Spikes: exploring the neural code, Ed 1 Cambridge, MA: Massachusetts Institute of Technology. [Google Scholar]
- Riera-Punet N, Martinez-Gomis J, Paipa A, Povedano M, Peraire M (2018a) Alterations in the masticatory system in patients with amyotrophic lateral sclerosis. J Oral Facial Pain Headache 32:84–90. 10.11607/ofph.1882 [DOI] [PubMed] [Google Scholar]
- Riera-Punet N, Martinez-Gomis J, Willaert E, Povedano M, Peraire M (2018b) Functional limitation of the masticatory system in patients with bulbar involvement in amyotrophic lateral sclerosis. J Oral Rehabil 45:204–210. 10.1111/joor.12597 [DOI] [PubMed] [Google Scholar]
- Roselli F, Caroni P (2015) From intrinsic firing properties to selective neuronal vulnerability in neurodegenerative diseases. Neuron 85:901–910. 10.1016/j.neuron.2014.12.063 [DOI] [PubMed] [Google Scholar]
- Saxena S, Caroni P (2011) Selective neuronal vulnerability in neurodegenerative diseases: from stressor thresholds to degeneration. Neuron 71:35–48. 10.1016/j.neuron.2011.06.031 [DOI] [PubMed] [Google Scholar]
- Saxena S, Roselli F, Singh K, Leptien K, Julien JP, Gros-Louis F, Caroni P (2013) Neuroprotection through excitability and mTOR required in ALS motoneurons to delay disease and extend survival. Neuron 80:80–96. 10.1016/j.neuron.2013.07.027 [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open-source platform for biological image analysis. Nat Methods 9:676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW (2012) National Institutes of Health image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurr A, West CA, Rigor BM (1988) Lactate-supported synaptic function in the rat hippocampal slice preparation. Science 240:1326–1328. 10.1126/science.3375817 [DOI] [PubMed] [Google Scholar]
- Seki S, Chandler HS, Maheshwary R, Nisly H, Sampath PA, Olcese R, Wiedau-Pazos M, Venugopal S (2017) Pre-symptomatic abnormalities and associated channelopathies in spindle afferent trigeminal mesencephalic V neurons in a SOD1G93A mouse model for Amyotrophic Lateral Sclerosis. Society for Neuroscience 2017; Washington DC. [Google Scholar]
- Simon CM, Janas AM, Lotti F, Tapia JC, Pellizzoni L, Mentis GZ (2016) A stem cell model of the motor circuit uncouples motor neuron death from hyperexcitability induced by SMN deficiency. Cell Rep 16:1416–1430. 10.1016/j.celrep.2016.06.087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer RF, Porter JD (1988) Structural organization of the extraocular muscles. Rev Oculomot Res 2:33–79. [PubMed] [Google Scholar]
- Su H, Alroy G, Kirson ED, Yaari Y (2001) Extracellular calcium modulates persistent sodium current-dependent burst-firing in hippocampal pyramidal neurons. J Neurosci 21:4173–4182. 10.1523/JNEUROSCI.21-12-04173.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JP, Brown RH Jr, Cleveland DW (2016) Decoding ALS: from genes to mechanism. Nature 539:197–206. 10.1038/nature20413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theiss RD, Kuo JJ, Heckman CJ (2007) Persistent inward currents in rat ventral horn neurones. J Physiol 580:507–522. 10.1113/jphysiol.2006.124123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trueblood PR, Levine MS, Chandler SH (1996) Dual-component excitatory amino acid-mediated responses in trigeminal motoneurons and their modulation by serotonin in vitro. J Neurophysiol 76:2461–2473. 10.1152/jn.1996.76.4.2461 [DOI] [PubMed] [Google Scholar]
- Turman JE., Jr (2007) The development of mastication in rodents: from neurons to behaviors. Arch Oral Biol 52:313–316. 10.1016/j.archoralbio.2006.08.013 [DOI] [PubMed] [Google Scholar]
- Turman JE Jr, Ajdari J, Chandler SH (1999) NMDA receptor NR1 and NR2A/B subunit expression in trigeminal neurons during early postnatal development. J Comp Neurol 409:237–249. [DOI] [PubMed] [Google Scholar]
- Turman JE Jr, MacDonald AS, Pawl KE, Bringas P, Chandler SH (2000) AMPA receptor subunit expression in trigeminal neurons during postnatal development. J Comp Neurol 427:109–123. [DOI] [PubMed] [Google Scholar]
- Van Wart A, Matthews G (2006a) Expression of sodium channels Nav1.2 and Nav1.6 during postnatal development of the retina. Neurosci Lett 403:315–317. 10.1016/j.neulet.2006.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Wart A, Matthews G (2006b) Impaired firing and cell-specific compensation in neurons lacking nav1.6 sodium channels. J Neurosci 26:7172–7180. 10.1523/JNEUROSCI.1101-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zundert B, Peuscher MH, Hynynen M, Chen A, Neve RL, Brown RH Jr, Constantine-Paton M, Bellingham MC (2008) Neonatal neuronal circuitry shows hyperexcitable disturbance in a mouse model of the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J Neurosci 28:10864–10874. 10.1523/JNEUROSCI.1340-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zundert B, Izaurieta P, Fritz E, Alvarez FJ (2012) Early pathogenesis in the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J Cell Biochem 113:3301–3312. 10.1002/jcb.24234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venugopal S, Crook S, Srivatsan M, Jung R (2011a) Principles of Computational Neuroscience. In: Biohybrid systems: nerves, interfaces and machines (Ranu Jung. ed), pp 11–29. Weinheim, Germany: Wiley-VCH. [Google Scholar]
- Venugopal S, Hamm TM, Crook SM, Jung R (2011b) Modulation of inhibitory strength and kinetics facilitates regulation of persistent inward currents and motoneuron excitability following spinal cord injury. J Neurophysiol 106:2167–2179. 10.1152/jn.00359.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venugopal S, Hamm T, Jung R (2012) Differential contributions of somatic and dendritic calcium-dependent potassium currents to the control of motoneuron excitability following spinal cord injury Cogn Neurodyn 6:283–293. 10.1007/s11571-012-9191-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venugopal S, Hsiao CF, Sonoda T, Wiedau-Pazos M, Chandler SH (2015) Homeostatic dysregulation in membrane properties of masticatory motoneurons compared to oculomotor neurons in a mouse model for amyotrophic lateral sclerosis. J Neurosci 35:707–720. 10.1523/JNEUROSCI.1682-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venugopal S, Seki S, Terman DH, Pantazis A, Olcese R, Wiedau-Pazos M, Chandler SH (2019) Resurgent Na+ current offers noise modulation in bursting neurons. PLoS Comput Biol 15:e1007154. 10.1371/journal.pcbi.1007154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Lewinski F, Keller BU (2005) Ca2+, mitochondria and selective motoneuron vulnerability: implications for ALS. Trends Neurosci 28:494–500. 10.1016/j.tins.2005.07.001 [DOI] [PubMed] [Google Scholar]
- Vucic S, Kiernan MC (2006) Axonal excitability properties in amyotrophic lateral sclerosis. Clin Neurophysiol 117:1458–1466. 10.1016/j.clinph.2006.04.016 [DOI] [PubMed] [Google Scholar]
- Vucic S, Cheah BC, Kiernan MC (2009) Defining the mechanisms that underlie cortical hyperexcitability in amyotrophic lateral sclerosis. Exp Neurol 220:177–182. 10.1016/j.expneurol.2009.08.017 [DOI] [PubMed] [Google Scholar]
- Wang GY, Ratto G, Bisti S, Chalupa LM (1997) Functional development of intrinsic properties in ganglion cells of the mammalian retina. J Neurophysiol 78:2895–2903. 10.1152/jn.1997.78.6.2895 [DOI] [PubMed] [Google Scholar]
- Wheeler DG, Groth RD, Ma H, Barrett CF, Owen SF, Safa P, Tsien RW (2012) Ca(V)1 and Ca(V)2 channels engage distinct modes of Ca(2+) signaling to control CREB-dependent gene expression. Cell 149:1112–1124. 10.1016/j.cell.2012.03.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson TL, Cleveland DW (1999) Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci 2:50–56. 10.1038/4553 [DOI] [PubMed] [Google Scholar]
- Wu N, Hsiao CF, Chandler SH (2001) Membrane resonance and subthreshold membrane oscillations in mesencephalic V neurons: participants in burst generation. J Neurosci 21:3729–3739. 10.1523/JNEUROSCI.21-11-03729.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N, Enomoto A, Tanaka S, Hsiao CF, Nykamp DQ, Izhikevich E, Chandler SH (2005) Persistent sodium currents in mesencephalic v neurons participate in burst generation and control of membrane excitability. J Neurophysiol 93:2710–2722. 10.1152/jn.00636.2004 [DOI] [PubMed] [Google Scholar]
- Xu S, Ono K, Inenaga K (2010) Electrophysiological and chemical properties in subclassified acutely dissociated cells of rat trigeminal ganglion by current signatures. J Neurophysiol 104:3451–3461. 10.1152/jn.00336.2010 [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Ono K, Hitomi S, Harano N, Sago T, Yoshida M, Nunomaki M, Shiiba S, Watanabe S, Nakanishi O, Inenaga K (2013) Endothelin receptor-mediated responses in trigeminal ganglion neurons. J Dent Res 92:335–339. 10.1177/0022034513478428 [DOI] [PubMed] [Google Scholar]
- Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW (2008) Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci 11:251–253. 10.1038/nn2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Xing JL, Wu NP, Liu YH, Zhang CZ, Kuang F, Han VZ, Hu SJ (2009) Membrane current-based mechanisms for excitability transitions in neurons of the rat mesencephalic trigeminal nuclei. Neuroscience 163:799–810. 10.1016/j.neuroscience.2009.07.007 [DOI] [PubMed] [Google Scholar]
- Yoshida A, Moritani M, Nagase Y, Bae YC (2017) Projection and synaptic connectivity of trigeminal mesencephalic nucleus neurons controlling jaw reflexes. J Oral Sci 59:177–182. 10.2334/josnusd.16-0845 [DOI] [PubMed] [Google Scholar]