

Abstract
Regulators of G-protein signaling (RGS) proteins modulate receptor signaling by binding to activated G-protein α-subunits, accelerating GTP hydrolysis. Selective inhibition of RGS proteins increases G-protein activity and may provide unique tissue specificity. Thiadiazolidinones (TDZDs) are covalent inhibitors that act on cysteine residues to inhibit RGS4, RGS8, and RGS19. There is a correlation between protein flexibility and potency of inhibition by the TDZD 4-[(4- fluorophenyl)methyl]-2-(4-methylphenyl)-1,2,4-thiadiazolidine-3,5-dione (CCG-50014). In the context of a single conserved cysteine residue on the α4 helix, RGS19 is the most flexible and most potently inhibited by CCG-50014, followed by RGS4 and RGS8. In this work, we identify residues responsible for differences in both flexibility and potency of inhibition among RGS isoforms. RGS19 lacks a charged residue on the α4 helix that is present in RGS4 and RGS8. Introducing a negative charge at this position (L118D) increased the thermal stability of RGS19 and decreased the potency of inhibition of CCG-50014 by 8-fold. Mutations eliminating salt bridge formation in RGS8 and RGS4 decreased thermal stability in RGS8 and increased potency of inhibition of both RGS4 and RGS8 by 4- and 2-fold, respectively. Molecular dynamics simulations with an added salt bridge in RGS19 (L118D) showed reduced RGS19 flexibility. Hydrogen-deuterium exchange studies showed striking differences in flexibility in the α4 helix of RGS4, 8, and 19 with salt bridge–modifying mutations. These results show that the α4 salt bridge–forming residue controls flexibility in several RGS isoforms and supports a causal relationship between RGS flexibility and the potency of TDZD inhibitors.
SIGNIFICANCE STATEMENT
Inhibitor potency is often viewed in relation to the static structure of a target protein binding pocket. Using both experimental and computation studies we assess determinants of dynamics and inhibitor potency for three different RGS proteins. A single salt bridge–forming residue determines differences in flexibility between RGS isoforms; mutations either increase or decrease protein motion with correlated alterations in inhibitor potency. This strongly suggests a causal relationship between RGS protein flexibility and covalent inhibitor potency.
Introduction
Drug specificity is often considered to be like a key fitting into a complementary shaped lock. It has become clear recently that protein dynamics can play an important role in drug discovery (Feixas et al., 2014). Regulators of G-protein signaling (RGS) proteins bind to activated Gα subunits of G-proteins, thereby accelerating GTP hydrolysis and attenuating G-protein signaling. In regulating G-protein–coupled receptor (GPCR) signaling, RGS proteins play a role in the physiology of numerous systems. By inhibiting RGS proteins, signaling via a GPCR may be enhanced. There are twenty RGS isoforms, each with a different tissue distribution. The combination of GPCR agonists with inhibitors specific for a single RGS isoform should limit the effects on GPCR signaling to the subset of target tissues with intersecting distributions of the RGS isoform and GPCR. This has the potential of reducing agonist off-target effects, which makes RGS proteins an attractive target for modulation of GPCR signaling.
The potent RGS inhibitors discovered to date are all covalent modifiers of cysteine residues and are selective for RGS4 and RGS1 (Roman et al., 2010; Turner et al., 2012; Hayes et al., 2018). These proteins have four and three cysteines, respectively, in the RGS homology domain, which is more than most other RGS proteins. RGS4 has been linked to nervous system–related disease states in which RGS4 inhibition may be desirable, including seizures (Chen et al., 2012) and Parkinson’s disease (Lerner and Kreitzer, 2012; Blazer et al., 2015; Shen et al., 2015). Continued efforts to seek noncovalent inhibitors are worth pursuing, because the lower risk associated with noncovalent inhibitors is considered safer and may facilitate further development (Potashman and Duggan, 2009). In addition, it would be valuable to discover RGS inhibitors with other specificities since other RGS proteins that are not potently inhibited by covalent modifiers have been implicated as potential targets, including RGS17 in cancer (James et al., 2009; Bodle et al., 2013) and RGS19 in depression (Wang et al., 2014). To identify noncovalent inhibitors with novel specificities, it will be useful to understand what factors—apart from the number of cysteines in the RGS domain—drive the selectivity of RGS inhibitors.
The RGS homology domain contains nine α helices. A cysteine residue on α4, which faces the interior of the α4-α7 helical bundle, is conserved among 18 of the 20 RGS isoforms, with the exception of only RGS6 and RGS7 (Tesmer, 2009). Interestingly, when RGS proteins are mutated to contain only this single, shared cysteine, there are still dramatic differences in the potency by which different isoforms are inhibited (Shaw et al., 2018). RGS19, which contains only the shared α4 cysteine, is more potently inhibited than the single-cysteine versions of RGS4 and RGS8 (Mohammadiarani et al., 2018; Shaw et al., 2018).
Previously, using molecular dynamics (MD) simulations we found that RGS19 is more flexible than RGS4 and RGS8 (Shaw et al., 2018). In these modeling studies, we also found that salt bridge interactions were perturbed in response to inhibitor binding (Mohammadi et al., 2019). In this work, we sought to identify residue interactions responsible for flexibility differences among these isoforms and we predicted mutations that alter salt bridge interactions will both enhance RGS protein flexibility and increase the potency of RGS inhibitors such as 4-[(4- fluorophenyl)methyl]-2-(4-methylphenyl)-1,2,4-thiadiazolidine-3,5-dione (CCG-50014).
Materials and Methods
Materials.
The chemicals were purchased from Sigma-Aldrich (St. Louis, MO). The QuikChange II Mutagenesis Kit was purchased from Agilent (Santa Clara, CA). BL21(DE2) competent cells and the Protein Thermal Shift Dye Kit were purchased from Thermo Fisher Scientific (Watham, MA). Lumavidin Microspheres were purchased from Luminex (Austin, TX). CCG-50014 was synthesized as previously described (Blazer et al., 2011).
Protein Expression and Purification.
RGS proteins were produced as previously described (Shaw et al., 2018). Briefly, a his-tagged RGS domain of RGS8 in a pQE80 vector, a his-tagged RGS domain of RGS19 in a pET15b vector, and a his-tagged Δ51 N-terminally truncated RGS4 in a pET23d vector were transformed into BL21(DE3) competent Escherichia coli cells (Sigma-Aldrich). With an optical density at 600 nm of 2.0, protein production was induced by addition of 200 µM isopropyl-thio-D-galactopyranoside, and incubation was continued at 25°C for 16 hours. Cells were lysed and the protein was purified by nickel affinity chromatography. Mutations were induced with the QuikChange II Mutagenesis Kit (Agilent) and verified by Sanger sequencing. All RGS proteins, including those with mutations in salt bridge–forming residues, were produced on a single-cysteine background [wild-type (WT) RGS19, C160A RGS8, and C74A C132A C148A RGS4]. Gαo protein was expressed and purified as previously described (Lee et al., 1994).
Differential Scanning Fluorimetry.
Differential scanning fluorimetry was performed using the Protein Thermal Shift Dye Kit (ThermoFisher Scientific). Dye was added at 1X to 10 µM protein samples in 50 mM HEPES and 100 mM NaCl buffer (pH 7.4) in a volume of 20 µl. Fluorescence was read using a QuantStudio 7 Flex Real-Time PCR System while the temperature was ramped from 20 to 80°C at a rate of 0.05°C/s. Peak melting temperatures were defined as the point of fastest increase in fluorescence with respect to temperature. Data were analyzed using Protein Thermal Shift software version 1.3 (Thermo Fisher Scientific) and GraphPad Prism 7 (GraphPad Inc, LaJolla, CA).
Flow Cytometry Protein Interaction Assay.
Flow cytometry protein interaction assay was performed as previously described (Blazer et al., 2010) with minor modifications. RGS proteins were biotinylated by incubation at a 1:1 molar ratio with EZ-link NHS-LC-Biotin (ThermoFisher Scientific) for 2 hours on ice, and then excess biotin was removed using Amicon spin columns (Millipore, Burlington, MA). RGS proteins at 50 nM were incubated with xMAP LumAvidin beads (Luminex) while shaking at room temperature for 1 hour. Beads were washed and incubated with varying concentrations of CCG-50014, followed by addition of 50 nM Gαo labeled with AF-532 C5-maleimide (Life Technologies, Carlsbad, CA) (Blazer et al., 2010). Samples were read in a Luminex 200 flow cytometer as previously described (Blazer et al., 2010) and analysis was performed in GraphPad Prism 7.
Hydrogen-Deuterium Exchange.
Hydrogen-deuterium exchange (HDX) was performed as previously described (Chodavarapu et al., 2016; Shaw et al., 2018). Briefly, proteins were incubated on ice at 1.2 µM in 90% D2O solvent with 5 mM HEPES and 100 mM NaCl (pH 7.4) for the desired time (1, 3, 10, 30, or 100 minutes). Exchange was quenched by 1:1 addition of ice cold 1% formic acid. A Shimadzu pump was used to load 100 µl of each sample onto a pepsin column (Waters, Milford, MA) followed by incubation for 1 minute for digestion. Samples were then loaded to an Xbridge BEH C18 VanGuard trap column (Waters) and eluted and separated using an Ascentis Express Peptide ES-C18 column (Sigma-Aldrich) with a gradient of 0.1% formic acid to acetonitrile. All columns and solvents were maintained on ice. Peaks were detected with a Xevo G2-XS QToF mass spectrometer (Waters). Data were analyzed using MassLynx (Waters), HX-Express2 (Guttman et al., 2013), and GraphPad Prism 7.
Molecular Dynamics Simulation.
We performed two sets of classical all-atom and explicit-solvent MD simulations for single-cysteine RGS4 and RGS4 D90L, single-cysteine RGS8 and RGS8 E84L, and WT RGS19 and RGS19 L118D (Supplemental Table 1) using the NAMD software program (Phillips et al., 2005) on a high-performance computing cluster (Towns et al., 2014) using the CHARMM force field with CMAP correction (MacKerell et al., 1998, 2004). We used Visual Molecular Dynamics (VMD) for system creation and post simulation analysis (Humphrey et al., 1996). The initial coordinates were obtained from the Protein Data Bank (PDB) files with codes 1AGR (RGS4), 2DOE (RGS8), and 1CMZ (RGS19). Except for Cys95 in RGS4 and Cys89 in RGS8, all cysteines were changed to alanines. (It should be noted that amino acid numbering follows that for RGS8, isoform 2; National Center for Biotechnology Information reference sequence: NP_ 001095920.1. [https://www.ncbi.nlm.nih.gov/protein/NP_001095920.1/]) Each protein was then solvated in a simulation box of TIP3P water molecules (Jorgensen et al., 1983) and charge-neutralized with NaCl. The final solvated and ionized simulation domains contained 30,031 atoms (RGS4); 32,257 atoms (RGS8); and 25,077 atoms (RGS19). Each solvated and ionized system was energy minimized for ∼500–1000 cycles via conjugate-gradient optimization, and then equilibrated via 1-microsecond MD simulations conducted with a time step (Δt) of 2 fs. The constant number of atoms, pressure, and temperature (NPT) ensemble with a Langevin thermostat and a damping coefficient of 5 ps−1 was used for temperature control and the Nosé-Hoover barostat was used for pressure control. Periodic boundary conditions were used throughout; nonbonded interactions were accounted for with a cutoff of 10 Å where smooth switching was initiated at 8 Å. Long-range electrostatic interactions were handled using the particle mesh Ewald method.
Dynamic Cross-Correlation Analysis.
The dynamic cross-correlation maps of each system were calculated based on the Cα atoms of residues using the MD-TASK software package (Brown et al., 2017). Each cell value (Cij) in the matrix of the dynamic cross-correlation map was calculated using the following formula:
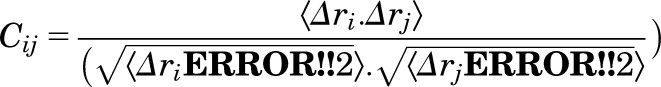 |
where Δri represents the displacement from the mean position of atom i, and the brackets denote the time average over the whole trajectory. Positive values of Cij show correlated motion between residues i and j, moving in the same direction, whereas negative values of Cij show anticorrelated motion between residues i and j, moving in the opposite direction.
Analysis of Salt Bridge Interactions.
Salt bridge interaction analysis was carried out using VMD based on a distance criterion uniformly applied to determine the existence of salt bridges for each frame in all trajectories (Schuster et al., 2019). Specifically, salt bridge interactions were considered to be formed if the distance between any of the oxygen atoms of acidic residues and the nitrogen atoms of basic residues were within a cutoff distance of 4 Å.
Statistical Analysis.
Statistical tests in this work are exploratory. Changes in thermal stability were analyzed by one-way ANOVA with Sidak’s multiple comparisons post test. Differences in deuterium incorporation were analyzed using two-way ANOVA with Sidak’s multiple comparisons post test. Error bars represent mean ± S.D.; except where otherwise indicated, all experimental biochemical data were done with n = 3 independent experiments, which was sufficient to demonstrate reproducibility. The resulting P values are descriptive rather than hypothesis testing. In saturation binding experiments, RGS-Gα inhibition was determined by fitting total and nonspecific binding. In functional inhibition experiments, the IC50 value was determined by fitting a four-parameter logistic curve. All curve fitting and statistical analyses were done using GraphPad Prism 7 (GraphPad Inc.).
Results
Comparison of the structures for RGS19 (PDB 1CMZ) (de Alba et al., 1999), RGS4 (PDB 1AGR) (Tesmer et al., 1997), and RGS8 (PDB 5DO9) (Taylor et al., 2016) shows that there are differing numbers of interhelical salt bridges on the exteriors of their α4-α7 helix bundles. Some of these may contribute to differences in stability and dynamics among the RGS isoforms.
RGS19 has only one interhelical salt bridge in this bundle, between E125 (α4) and K138 (α5) (Fig. 1, A and B). However, this salt bridge is well conserved among all three proteins (Fig. 1); therefore, it is unlikely to contribute to observed differences in flexibility (Shaw et al., 2018). A salt bridge network that connects α4, the α5-α6 interhelical loop, and α5 is present in RGS8 (E84-R119-E111) and RGS4 (D90-K125-E117) but absent in RGS19 (Fig. 1, A and B). The residues that form this network are present in seven of the 20 RGS protein family members, all in the R4 subfamily. Between the α5 and α6 helices, a salt bridge is present in RGS8 (D114-R132), but absent in both RGS4 and RGS19 (Fig. 1, A and C). Finally, a charged pair between the α6 and α7 helices is present in RGS8 (E91-K104) and RGS4 (D130-K155), but absent in RGS19 (Fig. 1, A and D).
Fig. 1.

(A) Alignment of RGS19, RGS4, and RGS8 sequences in the α4-α7 helix bundle. Charged residues that make interhelical contacts are indicated in red and blue. RGS19 has one, RGS4 has three, and RGS8 has four salt bridges. Structural alignments of α4-α5 (B), α5-α6 (C), and α6-α7 (D) helix pairs are shown, with highlighted residues in panel A rendered as sticks. RGS19 (PDB 1CMZ) is in green, RGS4 (PDB 1AGR) is in yellow, and RGS8 (PDB 5DO9) is in cyan. Black brackets in (A) indicate residues depicted in (B–D). Arrows show which panels depict each set of bracketed residues.
To estimate the relevance of each of these salt bridges in maintenance of helix bundle rigidity, the time each amino acid in a charged pair spent within a 4 Å of one another over the course of a long timescale (2 microseconds) MD simulation (Shaw et al., 2018) was measured. The α6-α7 salt bridge, which is present in RGS4 and RGS8 but absent in RGS19, occupied a salt bridge–forming distance for 31.5% of the simulation in RGS4 and 36.1% in RGS8. The salt bridge interaction between residues of the α4 and α5-α6 interhelical loop, also not present in RGS19, was maintained for 58.7% of time in RGS4 and 44.2% in RGS8 (Supplemental Table 2). The charged pair that is unique to RGS8 between the α5 and α6 helices remained in contact for 47.5% of the simulation.
We elected to make mutations that altered interhelical salt bridges to test their functional roles. There are two positions at which interhelical salt bridges are shared by RGS4 and RGS8 but are absent in RGS19: α4-α5 (Fig. 1B) and α6-α7 (Fig. 1D). In the α4 helix of RGS19, L118 was mutated to an aspartate to introduce the α4-α5 salt bridge found in RGS4 and RGS8 (Fig. 1B). In helix α7 of RGS19, Q183 was mutated to a lysine to introduce the α6-α7 salt bridge found in RGS4 and RGS8 (Fig. 1D). To eliminate confounding effects due to multiple cysteines in inhibitor potency experiments, all proteins (with and without salt bridge mutations) used a single-cysteine protein background. Each construct has only the conserved cysteine in helix α4 of the RGS domain.
To determine how disruption or addition of a salt bridge may alter protein structure or dynamics, thermal stability was measured by differential scanning fluorimetry. Addition of a salt bridge in RGS19 by the L118D mutation caused a 7°C increase in thermal stability compared with WT (Fig. 2A). In contrast, the Q183K mutation in RGS19 did not alter thermal stability or inhibitor potency (Supplemental Fig. 1). Removal of a salt bridge in RGS8 by the E84L mutation caused an 8°C decrease in thermal stability (Fig. 2B). Unexpectedly, RGS4 showed a more complex pattern in which the D90L mutation resulted in a biphasic melt curve and a 5°C increase in melting temperature rather than a decrease (Fig. 2C).
Fig. 2.

Thermal stability was determined by differential scanning fluorimetry. (A) The L118D mutation in RGS19 increased the melting temperature by 7°C compared with WT. (B) The E84L mutation in RGS8 decreased the melting temperature by 8°C. (C) The RGS4 D90L mutation introduced a biphasic melt curve and increased the melting temperature by 5°C. For each pair, the three replicate derivative melt curves are shown on the left and the average melt temperatures are shown on the right. Error bars represent S.D. (n = 3). Analyzed by one-way ANOVA with Sidak’s multiple comparisons test (****P < 0.0001).
To probe the molecular details of changes in structural flexibility in the mutant proteins, we conducted microsecond timescale classical MD simulations in explicit solvent for RGS19 L118D, RGS8 E84L, and RGS4 D90L. The root-mean-square deviations of these simulations are shown in Supplemental Fig. 2. To understand the effect of the mutations on the protein structures, particularly in helices in the vicinity of the mutated site, we computed the root-mean-square fluctuation per residue from two independent MD simulations of mutated and WT RGS19, RGS8, and RGS4. The calculated change in root-mean-square fluctuation per residue of the mutant RGS19 L118D from WT RGS19 revealed strong stabilization and a decrease in fluctuations of residues located in helices α4-α7 and in the interhelical loops between these helices. There was a particularly pronounced decrease in motion in the α5-α6 interhelical loop (Fig. 3A). We found a modest increase in fluctuation of residues in mutant RGS8 E84L versus the WT structure (Fig. 3B). These changes were in the loop region connecting helices α5 and α6, the α6 helix, and the loop connecting helices α6 and α7. Similar changes but of lesser extent were found in the mutant RGS4 D90L (Fig. 3C). Additionally, small decreases were observed in the root-mean-square fluctuation values of residues in helices α3 and α8 of the mutated RGS19 (Fig. 3A), but not in the mutated RGS8 and RGS4 (Fig. 3, B and C).
Fig. 3.

Change in the root-mean-square fluctuation (RMSF) per residue (ΔRMSF) between wild-type RGS proteins and RGS proteins with mutation in the α4-α5 salt bridge–forming residue: L118D in RGS19 (A), E84L in RGS8 (B), and D90L in RGS4 (C). Data represent differences in the RMSF from two independent MD simulations of the mutated forms of RGS proteins.
To further investigate whether salt bridge–modifying mutations in RGS4, RGS8, and RGS19 affect residue-residue interactions, we calculated dynamic cross-correlation matrices for the Cα atoms in all MD trajectories. For WT RGS19, RGS8, and RGS4, there was a modest positive correlation between the motions of residues of the α4 helix and the residues of the α5 helix (Fig. 4). For the RGS19 L118D mutant, we found higher residue-residue correlations between helices α4 and α5 in comparison with unmutated RGS19 (see the arrows in Fig. 4A). For WT RGS8, we found that the motions of residues in the α4 helix [amino acids (aa) 79–93] and the α5 helix (aa 97–113) were marginally positively correlated (see the arrows in Fig. 4B). This positive correlation between the α4 and α5 helices remained in the RGS8 E84L mutant, but showed a modest shift in areas of correlation away from the loop connecting α4-α5 to the midregions of the α4 and α5 helices (see the arrows in Fig. 4B). There was no appreciable change between WT and mutant RGS4 (Fig. 4C).
Fig. 4.

Dynamic cross-correlation matrix calculated for the Cα atoms of RGS19/RGS19 L118D (A), RGS8/RGS8 E84L (B), and RGS4/RGS4 D90L (C). Horizontal dotted lines indicate the regions of the α4 helix, while vertical solid lines indicate the regions of the α5 helix for each protein. The color scheme ranges from anticorrelation (−1.0, blue), no correlation (0, green), and positive correlation (+1.0, red). Values are the average of two independent simulation runs.
To experimentally determine which regions in WT and mutant proteins were affected by the salt bridge mutations, HDX studies were performed. After exposure to solvent containing 90% D2O, proteins were digested with pepsin and deuterium incorporation (DI) was measured by mass spectrometry as previously reported (Shaw et al., 2018). In RGS19, mutation of L118 to a salt bridge–forming residue, aspartic acid, caused significant decreases in DI in both α4 helical fragments, aa 116–119 and aa 120–125. In the 116–119 fragment, WT RGS19 had reached 43.1% DI by 10 minutes, while the RGS19 L118D mutant showed less than one-half as much DI (18.7%). In fragment 120–125, WT RGS19 reached 18.5% DI at 10 minutes, while the RGS19 L118D mutant reached only 6.2%. Unlike RGS4 and RGS8, the RGS19 L118D mutant’s changes in DI were more restricted to fragments from helices neighboring the mutation site, and were most pronounced in the early (1–10 minutes) timescale (Fig. 5A). In RGS8, removal of the salt bridge–forming residue by the E84L mutation did not cause a significant change in DI in either of the fragments of the α4 helix but trended toward a global increase in DI throughout the protein (Fig. 5B). In RGS4, the fragment surrounding the salt bridge mutation site (aa 88–91) took up deuterium very slowly in both the WT and D90L mutant constructs, reaching 8.1% and 6.7% DI, respectively. However, the D90L mutation led to a substantial increase in deuterium exchange in the 92–97 fragment surrounding Cys95, from 17.5% to 37.0% DI. The RGS4 D90L mutant also trended toward increased DI across all protein fragments compared with WT RGS4, especially at higher timepoints (Fig. 5C).
Fig. 5.

Difference in percentage of deuterium incorporation (Δ% DI) between mutated and unmutated proteins in RGS19 L118D (A), RGS8 E84L (B), and RGS4 D90L (C) fragments, as measured by HDX. Red arrows indicate fragments containing mutated residue, and black arrows indicate fragments containing conserved α4 cysteine. Kinetics results of deuterium incorporation in these fragments for individual constructs are shown below (n = 3). Error bars represent S.D. Analyzed by two-way ANOVA with Sidak’s multiple comparisons test (*P < 0.05; **P < 0.01; ****P < 0.0001).
Finally, to assess the functional relevance of the α4 salt bridge–forming residues, we used a flow cytometry–based protein-protein interaction assay (Roman et al., 2007; Blazer et al., 2010) to measure the binding of RGS proteins to Gαo and the potency of inhibition by CCG-50014. The L118D mutation in RGS19 induced an increase in the negative logarithm of IC50 (pIC50) from −5.96 ± 0.23 log(M) (WT) to −5.08 ± 0.25 log(M) (L118D) (Fig. 6A). Conversely, removal of this charged α4 residue in RGS4 and RGS8 induced a decrease in IC50 (Fig. 6, B and C). CCG-50014 inhibited the RGS-Gα interaction with a pIC50 of −5.08 ± 0.16 log(M) for WT RGS4 and −5.63 ± 0.19 log(M) for the RGS4 D90L mutant. It showed a potency of −5.09 ± 0.69 log(M) for WT RGS8 and −5.29 ± 0.41 log(M) for the RGS8 E84L mutant. None of the mutations to salt bridge–forming residues on the α4 helix caused notable changes in affinity between Gαo and RGS proteins. The L118D mutation in RGS19 shifted the Kd value of the Gαo interaction from 20.5 ± 6.3 to 23.9 ± 5.3 nM, the E84L mutation in RGS8 shifted the Kd value from 3.9 ± 1.8 to 4.8 ± 0.3 nM, and the D90L mutation in RGS4 shifted the Kd value from 8.8 ± 3.1 to 6.7 ± 2.6 nM (Supplemental Table 3).
Fig. 6.

Potency of inhibition of CCG-50014 against α4 is altered in salt bridge mutants of RGS proteins. (A) RGS19 negative logarithm of IC50 (pIC50): −5.96 ± 0.23 log(M); RGS19 L118D pIC50: −5.08 ± 0.25 log(M). (B) RGS8 pIC50: −5.09 ± 0.69 log(M); RGS8 E84L pIC50: −5.29 ± 0.41 log(M). (C) RGS4 pIC50: −5.08 ± 0.16 log(M); RGS4 D90L pIC50: −5.63 ± 0.19 log(M) (n = 3).
Discussion
A comparison of the crystal structures of the three RGS proteins studied here revealed several differences in charged residue contacts among the proteins. We first observed that RGS19 has fewer interhelical salt bridges in its α4-α7 helical bundle than RGS4 or RGS8. This may be responsible for the high flexibility previously observed in WT RGS19 (Shaw et al., 2018). RGS8 has four distinct interhelical salt bridges within the helical bundle, while RGS4 has three and RGS19 has one (Fig. 1A), correlating with previously observed flexibility differences. RGS19 is the most flexible, followed by RGS4 and RGS8 (Shaw et al., 2018). This further supports a role of salt bridges in RGS protein flexibility.
The changes in thermal stability in response to mutations in the α4 helix salt bridge–forming residues suggest that this location may be responsible for differences in stability and dynamics among the isoforms. This is supported by the increase in thermal stability in response to the L118D mutation in RGS19, and destabilization in RGS8 response to the E84L mutation. While the D90L mutation altered thermal stability in RGS4, it stabilized rather than destabilized the protein. The biphasic melt curves in D90L RGS4 make the thermal stability data difficult to interpret. HDX clarifies the effect of the D90L mutation in RGS4 by showing localized increases in the flexibility of the protein. The lack of effect on thermal stability with the Q183K mutation in RGS19 correlates with the observation that the α6-α7 salt bridges in RGS4 and RGS8 were less stably maintained in simulations than were the α4-α5 salt bridges. In light of these results, we found it unlikely that the difference between Q183 in α6 of RGS19 and the lysines found in RGS4 and RGS8 (K155 and K149, respectively) play a major role in the flexibility differences between these proteins. Rather, the salt bridge–forming residue on α4 is a stronger driver of differences in protein flexibility.
To determine the effects of mutations in salt bridge–forming residues on protein dynamics, both an in silico approach (all-atom MD simulations) and an experimental approach (hydrogen-deuterium exchange) were employed. In simulations, the increase in positive correlation between residues in the α4 and α5 helices in the RGS19 L118D mutant likely results from the introduced interhelical salt bridge. The decrease in DI in the α4 helix of RGS19 in the HDX studies is consistent with reduced solvent exposure. This is of particular interest given that the Cys123 target of the thiadiazolidinone compounds is located in that helix. Conversely, mutations that eliminated salt bridges in RGS4 and RGS8 increased DI in some fragments from their α4 helices (Fig. 5, A and B), suggesting that this results in increased solvent exposure and greater compound accessibility at the buried cysteine. Surprisingly, the RGS4 D90L mutant did not have increased DI in the fragment spanning the mutation site (Fig. 5C). In addition, the microsecond timescale MD simulations captured positive residue-residue (Cα-Cα) correlations between the α4 and α5 helices that were similar in WT and mutated RGS4 D90L. This fits with the thermal stability data and suggests that the effect of the D90L mutation in RGS4 is more complex than simple disruption of an ionic contact.
In MD simulations, the RGS4 D90L and RGS8 E84L mutations did not have as large an effect on the magnitude of residue fluctuations as did the L118D mutation in RGS19 (Fig. 3, A and B). This may be because differences become apparent on shorter timescales in RGS19 than in RGS4 and RGS8; therefore, simulations on microsecond timescales may not have captured all of the differences in dynamics caused by mutations in RGS4 D90L and RGS8 E84L. Indeed, in the HDX studies, stronger differences in DI were observed between RGS19 and RGS19 L118D at shorter timepoints (1 and 3 minutes) than in RGS4 D90L and RGS8 E84L (Fig. 5).
Finally, to determine how changes in protein flexibility affected the potency of inhibition by an RGS inhibitor, we used flow cytometry protein interaction assay to evaluate the inhibition of Gα binding by CCG-50014. Importantly, manipulation of RGS protein flexibility induced the expected changes in the potency of inhibition by thiadiazolidinone covalent modifiers. Thus, enhancing flexibility by removal of salt bridge–forming residues increased the potency of inhibition by CCG-50014 while reducing protein flexibility reduced potency of inhibition by CCG-50014. These results support a causal relationship between RGS protein flexibility and potency of inhibition.
In conclusion, differences in flexibility among RGS isoforms appear to drive differences in the potency of a covalent inhibitor, CCG-50014. The differences in isoform flexibility, in turn, are strongly influenced by the presence or absence of an α4-α5 salt bridge and manipulation of this salt bridge is sufficient to induce changes in inhibitor potency among single-cysteine RGS proteins. Developing a deeper understanding of these differences in flexibility may enable the development of a new generation of RGS inhibitors with novel specificities.
Acknowledgments
The authors acknowledge the Mass Spectrometry and Metabolomics Core Facility at the Research Technology Support Facility (Michigan State University). We are grateful for computational support in part from Trillian, a Cray XE6m-200 supercomputer supported by the National Science Foundation Major Research Instrumentation Program, and the NSF-supported Extreme Science and Engineering Discovery Environment Comet Resource at the San Diego Supercomputer Center.
Abbreviations
- aa
amino acids
- CCG-50014
4-[(4- fluorophenyl)methyl]-2-(4-methylphenyl)-1,2,4-thiadiazolidine-3,5-dione
- DI
deuterium incorporation
- GPCR
G-protein–coupled receptor
- HDX
hydrogen-deuterium exchange
- MD
molecular dynamics
- PDB
Protein Data Bank
- RGS
regulators of G-protein signaling
- WT
wild type
Authorship Contributions
Participated in research design: Shaw, Mohammadi, Vashisth, Neubig.
Conducted experiments: Shaw, Mohammadi, Quinn.
Performed data analysis: Shaw, Mohammadi, Quinn.
Wrote or contributed to the writing of the manuscript: Shaw, Mohammadi, Vashisth, Neubig.
Footnotes
This work was supported by the National Science Foundation [Grants 1507588 (to R.R.N.) and 1508595 (to H.V.)], the National Science Foundation Major Research Instrumentation Program [Grant PHY-1229408], the National Science Foundation–supported [Grant ACI-1053575] Extreme Science and Engineering Discovery Environment Comet Resource at the San Diego Supercomputer Center [Grant TG-MCB160183 (to H.V.)], and a National Institutes of Health Training Grant [Grant T32 GM092715 (to V.S.S. and R.R.N.)]. This work was also supported in part by the National Institutes of Health Centers of Biomedical Research Excellence Center of Integrated Biomedical and Bioengineering Research [Grant P20 GM113131] through an Institutional Development Award (to H.V.) from the National Institute of General Medical Sciences.
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Blazer LL, Roman DL, Muxlow MR, Neubig RR. (2010) Use of flow cytometric methods to quantify protein-protein interactions. Curr Protoc Cytom Chapter 13:Unit 13.11.1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazer LL, Storaska AJ, Jutkiewicz EM, Turner EM, Calcagno M, Wade SM, Wang Q, Huang X-P, Traynor JR, Husbands SM, et al. (2015) Selectivity and anti-Parkinson’s potential of thiadiazolidinone RGS4 inhibitors. ACS Chem Neurosci 6:911–919. [DOI] [PubMed] [Google Scholar]
- Blazer LL, Zhang H, Casey EM, Husbands SM, Neubig RR. (2011) A nanomolar-potency small molecule inhibitor of regulator of G-protein signaling proteins. Biochemistry 50:3181–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodle CR, Mackie DI, Roman DL. (2013) RGS17: an emerging therapeutic target for lung and prostate cancers. Future Med Chem 5:995–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DK, Penkler DL, Sheik Amamuddy O, Ross C, Atilgan AR, Atilgan C, Tastan Bishop Ö. (2017) MD-TASK: a software suite for analyzing molecular dynamics trajectories. Bioinformatics 33:2768–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Liu Y, Cottingham C, McMahon L, Jiao K, Greengard P, Wang Q. (2012) Neurabin scaffolding of adenosine receptor and RGS4 regulates anti-seizure effect of endogenous adenosine. J Neurosci 32:2683–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chodavarapu S, Jones AD, Feig M, Kaguni JM. (2016) DnaC traps DnaB as an open ring and remodels the domain that binds primase. Nucleic Acids Res 44:210–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Alba E, De Vries L, Farquhar MG, Tjandra N. (1999) Solution structure of human GAIP (Gα interacting protein): a regulator of G protein signaling. J Mol Biol 291:927–939. [DOI] [PubMed] [Google Scholar]
- Feixas F, Lindert S, Sinko W, McCammon JA. (2014) Exploring the role of receptor flexibility in structure-based drug discovery. Biophys Chem 186:31–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Weis DD, Engen JR, Lee KK. (2013) Analysis of overlapped and noisy hydrogen/deuterium exchange mass spectra. J Am Soc Mass Spectrom 24:1906–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes MP, Bodle CR, Roman DL. (2018) Evaluation of the selectivity and cysteine dependence of inhibitors across the regulator of G protein-signaling family. Mol Pharmacol 93:25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. (1996) VMD: visual molecular dynamics. J Mol Graph 14:33–38. [DOI] [PubMed] [Google Scholar]
- James MA, Lu Y, Liu Y, Vikis HG, You M. (2009) RGS17, an overexpressed gene in human lung and prostate cancer, induces tumor cell proliferation through the cyclic AMP-PKA-CREB pathway. Cancer Res 69:2108–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935. [Google Scholar]
- Lee E, Linder ME, Gilman AG. (1994) Expression of G-protein α subunits in Escherichia coli. Methods Enzymol 237:146–164. [DOI] [PubMed] [Google Scholar]
- Lerner TN, Kreitzer AC. (2012) RGS4 is required for dopaminergic control of striatal LTD and susceptibility to parkinsonian motor deficits. Neuron 73:347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell AD, Jr, Bashford D, Bellott M, Dunbrack RL, Jr, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, et al. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- Mackerell AD, Jr, Feig M, Brooks CL., III (2004) Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem 25:1400–1415. [DOI] [PubMed] [Google Scholar]
- Mohammadi M, Mohammadiarani H, Shaw VS, Neubig RR, Vashisth H. (2019) Interplay of cysteine exposure and global protein dynamics in small-molecule recognition by a regulator of G-protein signaling protein Proteins 87:146–156 DOI: 10.1002/prot.25642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadiarani H, Shaw VS, Neubig RR, Vashisth H. (2018) Interpreting hydrogen-deuterium exchange events in proteins using atomistic simulations: case studies on regulators of G-protein signaling proteins. J Phys Chem B 122:9314–9323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. (2005) Scalable molecular dynamics with NAMD. J Comput Chem 26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potashman MH, Duggan ME. (2009) Covalent modifiers: an orthogonal approach to drug design. J Med Chem 52:1231–1246. [DOI] [PubMed] [Google Scholar]
- Roman DL, Blazer LL, Monroy CA, Neubig RR. (2010) Allosteric inhibition of the regulator of G protein signaling–Gα protein–protein interaction by CCG-4986. Mol Pharmacol 78:360–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman DL, Talbot JN, Roof RA, Sunahara RK, Traynor JR, Neubig RR. (2007) Identification of small-molecule inhibitors of RGS4 using a high-throughput flow cytometry protein interaction assay. Mol Pharmacol 71:169–175. [DOI] [PubMed] [Google Scholar]
- Schuster KD, Mohammadi M, Cahill KB, Matte SL, Maillet AD, Vashisth H, Cote RH. (2019) Pharmacological and molecular dynamics analyses of differences in inhibitor binding to human and nematode PDE4: implications for management of parasitic nematodes. PLoS One 14:e0214554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw VS, Mohammadiarani H, Vashisth H, Neubig RR. (2018) Differential protein dynamics of regulators of G-protein signaling: role in specificity of small-molecule inhibitors. J Am Chem Soc 140:3454–3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Plotkin JL, Francardo V, Ko WKD, Xie Z, Li Q, Fieblinger T, Wess J, Neubig RR, Lindsley CW, et al. (2015) M4 muscarinic receptor signaling ameliorates striatal plasticity deficits in models of L-DOPA-induced dyskinesia. Neuron 88:762–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor VG, Bommarito PA, Tesmer JJG. (2016) Structure of the regulator of G protein signaling 8 (RGS8)-Gαq complex: molecular basis for Gα selectivity. J Biol Chem 291:5138–5145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesmer JJG. (2009) Structure and function of regulator of G protein signaling homology domains. Prog Mol Biol Transl Sci 86:75–113. [DOI] [PubMed] [Google Scholar]
- Tesmer JJG, Berman DM, Gilman AG, Sprang SR. (1997) Structure of RGS4 bound to AlF4−-activated Giα1: stabilization of the transition state for GTP hydrolysis. Cell 89:251–261. [DOI] [PubMed] [Google Scholar]
- Towns J, Cockerill T, Dahan M, Foster I, Gaither K, Grimshaw A, Hazlewood V, Lathrop S, Lifka D, Peterson GD, et al. (2014) XSEDE: accelerating scientific discovery. Comput Sci Eng 16:62–74. [Google Scholar]
- Turner EM, Blazer LL, Neubig RR, Husbands SM. (2012) Small molecule inhibitors of regulator of G protein signalling (RGS) proteins. ACS Med Chem Lett 3:146–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Terauchi A, Yee CH, Umemori H, Traynor JR. (2014) 5-HT1A receptor-mediated phosphorylation of extracellular signal-regulated kinases (ERK1/2) is modulated by regulator of G protein signaling protein 19. Cell Signal 26:1846–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]