

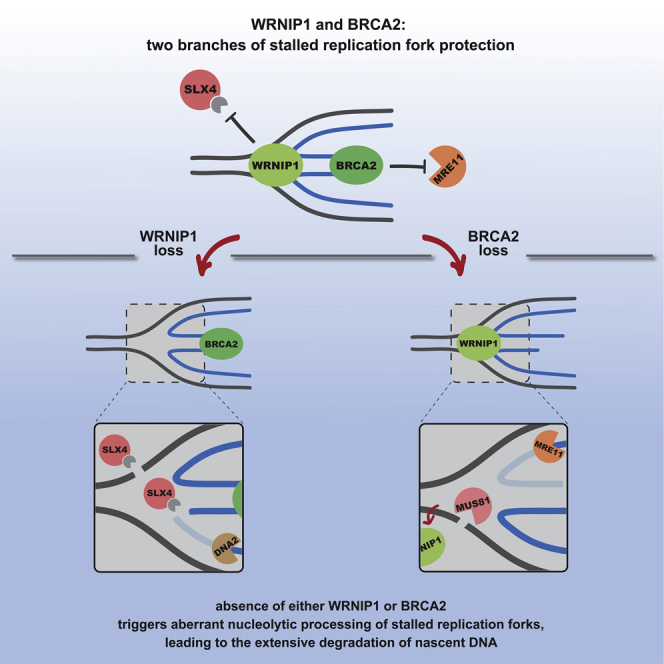
Summary
During DNA replication stress, stalled replication forks need to be stabilized to prevent fork collapse and genome instability. The AAA + ATPase WRNIP1 (Werner Helicase Interacting Protein 1) has been implicated in the protection of stalled replication forks from nucleolytic degradation, but the underlying molecular mechanism has remained unclear. Here we show that WRNIP1 exerts its protective function downstream of fork reversal. Unexpectedly though, WRNIP1 is not part of the well-studied BRCA2-dependent branch of fork protection but seems to protect the junction point of reversed replication forks from SLX4-mediated endonucleolytic degradation, possibly by directly binding to reversed replication forks. This function is specific to the shorter, less abundant, and less conserved variant of WRNIP1. Overall, our data suggest that in the absence of BRCA2 and WRNIP1 different DNA substrates are generated at reversed forks but that nascent strand degradation in both cases depends on the activity of exonucleases and structure-specific endonucleases.
Subject Areas: Biological Sciences, Molecular Biology, Cell Biology
Graphical Abstract
Highlights
-
•
WRNIP1, as BRCA2, protects stalled replication forks downstream of fork reversal
-
•
WRNIP1 and BRCA2 act in two different branches of the fork protection pathway
-
•
WRNIP1 protects from SLX4-mediated nucleolytic cleavage, possibly by direct binding
-
•
Fork protection function is specific to the shorter isoform of WRNIP1
Biological Sciences; Molecular Biology; Cell Biology
Introduction
During DNA replication, forks can encounter a plethora of situations that hinder their progression, such as a shortage of nucleotides, unrepaired DNA lesions, torsional stress, DNA secondary structures, transcription complexes, and RNA:DNA hybrids (Zeman and Cimprich, 2014). In response to virtually all such situations of DNA replication stress, stalled replication forks seem to get remodeled into four-way junctions (Zellweger et al., 2015), presumably to stabilize them until the fork-stalling obstacle is cleared (Neelsen and Lopes, 2015). During this process, termed replication fork reversal, DNA translocases, such as ZRANB3, SMARCAL1, and HLTF, drive the reannealing of the two parental DNA strands and the concomitant annealing of the newly synthesized strands to form a four-way junction (Achar et al., 2011, Achar et al., 2015, Bétous et al., 2012, Ciccia et al., 2012, Kile et al., 2015, Poole and Cortez, 2017, Vujanovic et al., 2017). In addition to these factors, the well-known recombination protein RAD51 is required for the formation of reversed forks (Zellweger et al., 2015), a function that is, in contrast to its role in homologous recombination, independent of its interaction partner BRCA2 (Mijic et al., 2017).
Although replication fork reversal is considered a protective measure, reversed replication forks, when not adequately stabilized, represent a source of genome instability, because they can become entry points for uncontrolled exonucleolytic degradation (Kolinjivadi et al., 2017, Lemaçon et al., 2017, Mijic et al., 2017, Taglialatela et al., 2017). BRCA2 plays an important role in the protection of reversed replication forks, presumably by assembling RAD51 filaments on the 3′-single-stranded (ss) DNA end of the reversed arm (Hashimoto et al., 2010, Mijic et al., 2017, Schlacher et al., 2011). A number of other proteins have been implicated in the protection of stalled replication forks from nucleolytic cleavage, such as BRCA1, FANCD2, REV1, BOD1L (Higgs et al., 2015, Schlacher et al., 2012, Yang et al., 2015), and more recently WRNIP1 (Leuzzi et al., 2016). Their mode of action in this context has however remained elusive, and it is unclear whether they exert their protective role at the level of the stalled or the reversed replication fork.
The AAA + ATPase WRNIP1 had initially been implicated in the maintenance of genome stability because of its physical interactions with DNA replication and repair proteins, such as the eponymous Werner helicase (WRN) and DNA polymerase delta (Kawabe et al., 2001, Tsurimoto et al., 2005). Moreover, it was found to colocalize with the replication fork remodeler ZRANB3 upon PCNA hyperubiquitination (Ciccia et al., 2012) and identified as a replication-associated repair protein enriched on nascent chromatin upon replication stress (Alabert et al., 2014, Dungrawala et al., 2015).
Here we provide evidence that, as BRCA2, WRNIP1 protects stalled replication forks that have undergone fork reversal. In contrast to BRCA2, however, WRNIP1 does not seem to protect the end of the protruded arm, but rather the junction point, of the reversed fork. Our data suggest that WRNIP1 may be able to do so by directly binding to reversed replication forks, thereby shielding them from SLX4-mediated cleavage.
Results
WRNIP1 Protects Reversed Replication Forks in a Mechanistically Different Way Than BRCA2
We first asked whether WRNIP1 was involved in the protection of reversed rather than stalled replication forks, similarly to what was recently reported for BRCA2 (Kolinjivadi et al., 2017, Lemaçon et al., 2017, Mijic et al., 2017, Taglialatela et al., 2017). To do so, we employed a widely used DNA fiber labeling approach in U2OS cells, in which we gave two consecutive pulses of CldU and IdU followed by a 5-h treatment with hydroxyurea to block DNA replication (Figure 1A; Table S1). In agreement with previous data (Leuzzi et al., 2016), in the absence of WRNIP1, nascent strands got degraded, resulting in a reduced IdU/CldU tract length ratio (Figure S1A). However, this effect could be completely reverted when either of the fork remodelers ZRANB3 or HLTF is codepleted with WRNIP1 (Figures 1B and S1B), suggesting that WRNIP1 acts downstream of fork reversal. Moreover, we also saw a complete restoration of fork stability when RAD51 was depleted on top of WRNIP1 (Figures 1B and S1B). Given RAD51's essential role in the promotion of fork reversal (Zellweger et al., 2015), this finding further suggests that WRNIP1, as BRCA2, protects stalled replication forks after they have undergone fork reversal.
Figure 1.

WRNIP1 Protects Reversed Replication Forks in a Different Way Than BRCA2
(A) Scheme of DNA fiber labeling assay.
(B) Replication fork degradation analysis upon knock-down of WRNIP1 and codepletion of factors involved in replication fork reversal.
(C) Replication fork degradation analysis upon knock-down of WRNIP1 (left) or BRCA2 (right) in the absence or presence of the MRE11 exonuclease inhibitor mirin, the MRE11 endonuclease inhibitor PFM01, or upon codepletion of MRE11.
Each dot represents an independent biological replicate. Values and gray bars indicate mean. Statistical analysis: one-way ANOVA with Sidak's correction for multiple comparisons (****, p < 0.0001; *, p < 0.1; ns, not significant). Scatterplots of one representative experiment and western blots showing depletion efficacies can be found in Figure S1. CldU, 5-chloro-2′-deoxyuridine; IdU, 5-iodo-2′-deoxyuridine; HU, hydroxyurea; RNAi, RNA interference; si, siRNA.
Since the inhibition of nucleases that are canonically involved in the processing of double-stranded DNA ends, such as MRE11, CtIP, and EXO1, can alleviate the fork degradation phenotype associated with knock-down of BRCA2 (Lemaçon et al., 2017, Schlacher et al., 2011), it is assumed that BRCA2 protects the protruded reversed arm, presumably by promoting the assembly of a RAD51 filament (Mijic et al., 2017). This notion is consistent with the observation that inhibition of MRE11's 3′–5′ exonuclease activity by mirin prevented the BRCA2 depletion-associated fork degradation phenotype to the same extent as a complete depletion of MRE11 by RNA interference, whereas the MRE11 endonuclease inhibitor PFM01 only led to a partial restoration of fork stability (Figure 1C, right panel; S1C, and S1D). In surprising contrast, fork degradation upon knock-down of WRNIP1 was prevented by depletion of MRE11 or inhibition of MRE11's endonuclease activity by PFM01, whereas inhibiting MRE11's exonuclease activity by mirin had only a very minor rescue effect (Figures 1C, left panel; S1C, and S1D). Our findings hence suggest that, in contrast to what has been proposed previously, WRNIP1's and BRCA2's fork protection functions seem to be mechanistically distinct.
WRNIP1 Protects Reversed Replication Forks from SLX4-Mediated Endonucleolytic Cleavage
In light of these observations, we then wondered whether WRNIP1, instead of protecting the protruded arm of a reversed fork, could rather shield its junction point, e.g., from the attack of structure-specific endonucleases. Over the last years, a number of endonucleases that are able to target four-way junctions have been discovered (Dehé and Gaillard, 2017). Several of them, namely SLX1, MUS81-EME1, and XPF-ERCC1, form a multiprotein complex with the scaffold protein SLX4 (Fekairi et al., 2009, Muñoz et al., 2009, Svendsen et al., 2009). In vitro, they have partially overlapping substrate preferences and can influence each other's biochemical activities (Wyatt et al., 2013, Wyatt et al., 2017). In vivo, however, they do not seem to form a constitutive complex throughout all cell cycle phases. Notably, cells synchronized in G1/S phase contain a pool of MUS81-EME1 that does not cofractionate with the rest of the complex on a sucrose gradient (Wyatt et al., 2017), suggesting the existence of functional subcomplexes. In addition to being able to cleave recombination-associated Holliday junctions, SLX4 has also been reported to promote endonucleolytic processing of replication forks (for review see Guervilly and Gaillard, 2018). Although this seems to primarily involve MUS81-EME1 and SLX1, SLX4-promoted fork processing by XPF-ERCC1 was recently reported (Bétous et al., 2018).
To assess whether WRNIP1 may be involved in preventing SLX4-driven unscheduled fork processing, we codepleted WRNIP1 and SLX4 and employed the same fiber labeling approach as described earlier. Depletion of SLX4 completely suppressed the fork degradation phenotype resulting from depletion of WRNIP1 (Figures 2A and S2A), suggesting that in the absence of WRNIP1, reversed forks can become the target of endonucleolytic cleavage. To address which of the SLX4-associated nucleases are involved in fork processing, we then individually codepleted SLX1, XPF, and MUS81 with WRNIP1. While codepletion of MUS81 had only a minor effect on fork stability, we could observe a complete alleviation of fork degradation upon codepletion of XPF and a partial alleviation upon codepletion of SLX1 (Figures 2A and S2A). These findings fit to previous observations that a major part of MUS81-EME1 is not associated with SLX4 during S phase (Wyatt et al., 2017) and suggest that in the absence of WRNIP1, reversed replication forks may be cleaved by SLX1 and XPF-ERCC1 in an SLX4-dependent manner.
Figure 2.

WRNIP1 Protects Reversed Replication Forks from SLX4-Dependent Nucleolytic Cleavage
(A) Replication fork degradation analysis upon knock-down of WRNIP1 and codepletion of SLX4 or associated nucleases. Each dot represents an independent biological replicate. Values and gray bars indicate mean. Statistical analysis: one-way ANOVA with Sidak's correction for multiple comparisons (****, p < 0.0001; **, p < 0.01; ns, not significant). Scatterplots of one representative experiment and western blots showing depletion efficacies can be found in Figure S2.
(B) EMSA to monitor binding of increasing amounts of WRNIP1 to ssDNA (left), dsDNA (second from left), a replication fork-like substrate (second from right), and a four-way junction substrate (right).
We then wondered whether WRNIP1 could protect reversed replication forks by directly binding to them. To address this possibility, we purified N-terminally Flag-tagged WRNIP1 from Sf9 insect cells (Figure S2B) and tested its ability to bind to a variety of DNA substrates in vitro. Using electrophoretic mobility shift assays (EMSAs), we could observe binding of purified WRNIP1 to a DNA structure mimicking a reversed replication fork, whereas it did not show any affinity for single-stranded or double-stranded DNA or for a substrate that resembles a canonical replication fork (Figure 2B). Importantly, WRNIP1's ability to bind to four-way junction substrates required neither an intact ubiquitin-binding zinc finger (UBZ) domain (Bish and Myers, 2007) nor a functional AAA + ATPase domain (Hishida et al., 2002), because the UBZ domain variant WRNIP1 D37A (Crosetto et al., 2008) and the Walker A and B variants K274A and E329Q were still able to bind to DNA (Figures S2B–S2D).
WRNIP1 Physically and Functionally Interacts with SLX1-SLX4
Given the observed restoration of fork stability upon codepletion of SLX4 with WRNIP1, and WRNIP1's ability to bind to four-way junction substrates, we next tested whether WRNIP1 was able to protect a four-way junction substrate from endonucleolytic cleavage in vitro. To do so, we titrated WRNIP1 into a nuclease assay with a four-way junction substrate and purified SLX1-SLX4. With increasing amounts of WRNIP1, the cleavage product decreased, suggesting that WRNIP1 can indeed protect four-way junctions from SLX1-SLX4 cleavage (Figure 3A). In line with their ability to bind to four-way junction substrates (Figure S2D), a similar effect was also observed when the UBZ or ATPase domain variants were added to SLX1-SLX4 cleavage reactions (Figure S3A).
Figure 3.

WRNIP1 Interacts Physically and Functionally with SLX1-SLX4
(A) Endonucleolytic cleavage assay with SLX1-SLX4 in the presence of increasing amounts of WRNIP1.
(B) Endonucleolytic cleavage assay with MUS81-EME1 in the presence of increasing amounts of WRNIP1.
(C) Coimmunoprecipitation experiment with purified His-tagged SLX1-SLX4 and purified Flag-tagged WRNIP1.
(D) Replication fork degradation analysis in WRNIP1-depleted cells, complemented with WRNIP1 variants. Endogenous WRNIP1 was depleted by siRNA targeting the 3′-UTR. Each dot represents an independent biological replicate. Values and gray bars indicate mean. Statistical analysis: one-way ANOVA with Sidak's correction for multiple comparisons (****, p < 0.0001; **, p < 0.01; ns, not significant). Scatterplots of one representative experiment and western blots showing depletion and expression efficacies can be found in Figure S3. 4-WJ, four-way junction; n-ds, nicked duplex DNA; WB, western blot; si, siRNA.
Strikingly, the protective effect of WRNIP1 seems to be specific for reactions with SLX1-SLX4, because titration of WRNIP1 into a nuclease assay with a nicked four-way junction substrate and purified MUS81-EME1 did not influence MUS81-EME1 cleavage (Figure 3B). Since WRNIP1 has a similar binding affinity for nicked four-way junctions than for intact four-way junctions (Figure S3B), we then wondered whether the observed specificity could stem from a direct interaction between WRNIP1 and SLX1-SLX4. To address this possibility, we immobilized purified SLX1-SLX4 on beads and added purified WRNIP1. In this coimmunoprecipitation experiment we could observe a robust direct interaction between WRNIP1 and the SLX1-SLX4 complex (Figure 3C).
Taken together, our data suggest that WRNIP1 protects the junction point of reversed replication forks from SLX4-dependent endonucleolytic cleavage, presumably through its ability to directly bind to these DNA structures and to interact with SLX1-SLX4. In contrast, WRNIP1 cannot protect from MUS81-EME1 cleavage in vitro, and depletion of MUS81 on top of WRNIP1 does not restore fork stability (Figures 2A and S2A). We further show that the UBZ and ATPase domains of WRNIP1 are dispensable for its protective function in vitro, which is in line with our observation that the UBZ and ATPase domain mutants are able to restore cellular fork stability to a similar extent as the wild-type construct (Figures 3D and S3C).
Fork Resection in the Absence of Both WRNIP1 and BRCA2 Requires Endonucleolytic and Exonucleolytic Cleavage Steps
Our data so far suggest that WRNIP1 acts in a different branch of fork protection than BRCA2 by protecting the junction point, rather than the protruded arm, of a reversed fork. Unscheduled SLX4-mediated fork processing alone, however, cannot readily explain the extensive fork degradation phenotype associated with the depletion of WRNIP1. Since our data show that MRE11's exonuclease activity is not responsible for long-range fork resection in the absence of WRNIP1 (Figure 1C), we wondered whether the 5′–3′ exonuclease DNA2 that is known to resect unprotected forks upon BOD1L or CtIP depletion (Higgs et al., 2015, Przetocka et al., 2018) could play a role in fork resection upon WRNIP1 knock-down. Indeed, both the depletion of DNA2 and the inhibition of DNA2 by C5 restored fork stability in the absence of WRNIP1, whereas knock-down of the 5′–3′ exonuclease EXO1 had no effect on fork stability (Figures 4A and S4A). In the absence of WRNIP1, SLX4-associated endonucleases may hence target unprotected reversed forks and thereby generate a substrate for extensive DNA2-mediated resection.
Figure 4.

Fork Resection Requires Endonucleolytic and Exonucleolytic Cleavage Steps
(A) Replication fork degradation analysis upon knock-down of WRNIP1 in the absence or presence of the DNA2 inhibitor C5 or upon codepletion of DNA2 or EXO1.
(B) Replication fork degradation analysis upon knock-down of BRCA2 and codepletion of SLX4, MUS81, or EME1.
Each dot represents an independent biological replicate. Values and gray bars indicate mean. Statistical analysis: one-way ANOVA with Sidak's correction for multiple comparisons (****, p < 0.0001; ***, p < 0.001; ns, not significant). Scatterplots of one representative experiment and western blots showing depletion efficacies can be found in Figure S4. CldU, 5-chloro-2′-deoxyuridine; IdU, 5-iodo-2′-deoxyuridine; HU, hydroxyurea; RNAi, RNA interference; si, siRNA.
These data also underline the difference in fork protection between WRNIP1 and BRCA2, because neither depletion of DNA2 nor use of the DNA2 inhibitor C5 could alleviate the fork degradation phenotype of BRCA2-depleted cells (Lemaçon et al., 2017, Przetocka et al., 2018).
To further delineate these two apparently distinct branches of fork protection, we checked the effect of SLX4 and MUS81 depletion on fork stability in cells lacking BRCA2 (Figures 4B and S4B). If WRNIP1 and BRCA2 were to act independently of each other, also in the absence of BRCA2, WRNIP1 should presumably be able to protect the junction point of a reversed fork from SLX4-dependent cleavage. In agreement with this idea, we observed that fork degradation in BRCA2-depleted cells could not be alleviated by codepletion of SLX4 (Figures 4 and S4B).
On the other hand, codepletion of MUS81 or EME1 with BRCA2 could largely restore fork stability (Figures 4B and S4B). In this context, it should be noted that reversed forks are not a substrate for MUS81-EME1, which prefers nicked or resected four-way junctions. In the absence of BRCA2, however, MRE11 exonuclease activity may extensively resect reversed replication forks and, hence, convert them into substrates preferred by MUS81-EME1. Given our in vivo (Figure 2A) and in vitro (Figure 3B) data, we would not expect WRNIP1 to be able to protect resected reversed replication forks from MUS81-EME1 cleavage.
Collectively, our data indicate that BRCA2 and WRNIP1 are part of two distinct branches of the fork protection pathway. However, while their loss leads to the generation of different types of DNA structures that are targeted by distinct sets of nucleases, extensive fork resection seems to require, in both cases, an exonucleolytic and endonucleolytic cleavage step.
Only the Short Variant of WRNIP1 Is Able to Bind to DNA and Protect Stalled Replication Forks
In the cell, WRNIP1 transcripts are spliced into two major isoforms that give rise to a 640 amino acid-long protein (used throughout this study and referred to as WRNIP1) and a 665 amino acid-long protein (herein after referred to as WRNIP1L), respectively. These two variants differ only by a 25 amino acid-long insertion between the Walker B motif and the ATP-binding arginine finger (Figure 5A). Intriguingly, this insertion is highly conserved, with 84% of amino acid identity between human WRNIP1 and Escherichia coli MgsA, which is clearly above and beyond the, already remarkable, overall conservation (25% of identity) between the human and bacterial protein (Kawabe et al., 2001). Surprisingly, although the large majority of transcripts in the cell encode the long isoform (Figures S5A–S5C), WRNIP1L displayed poor binding to a four-way branched DNA structure (Figures 5B and S5D) and was unable to protect it from SLX1-SLX4-dependent cleavage when titrated into a nuclease assay (Figure 5C).
Figure 5.

WRNIP1, but Not WRNIP1L, Can Protect Reversed Replication Forks
(A) Alignment of WRNIP1's central ATPase domain from a variety of species. Highlighted in bold are the Walker B motif (left) and the arginine finger (right), shaded in gray is the 25 amino acid-long insertion found in WRNIP1L (665 amino acid protein), but not in WRNIP1 (640 amino acid protein). Symbols underneath reflect amino acid conservation, with asterisks marking identical amino acids and colons indicating conservation of chemical character. Hs, Homo sapiens; Mm, Mus musculus; Dd, Dictyostelium discoideum; Sc, Saccharomyces cerevisiae; Sp, Schizosaccharomyces pombe; Ec, Escherichia coli.
(B) EMSA to monitor binding of increasing amounts of WRNIP1L to a four-way junction substrate.
(C) Endonucleolytic cleavage assay of SLX1-SLX4 in the presence of increasing amounts of WRNIP1L. Dashed line indicates that lanes were left out for clarity.
(D) Replication fork degradation analysis upon knock-down of WRNIP1 and complementation with cDNAs expressing the short (WRNIP1) or long (WRNIP1L) isoform. On the right, knock-down of WRNIP1 (short*) or WRNIP1L (long*) using isoform-specific siRNAs. Each dot represents an independent biological replicate. Values and gray bars indicate mean. Statistical analysis: one-way ANOVA with Sidak's correction for multiple comparisons (****, p < 0.0001; ns, not significant). Scatterplots of one representative experiment and western blots showing depletion and expression efficacies can be found in Figure S5. 4-WJ, four-way junction; n-ds, nicked duplex DNA; si, siRNA.
Making use of this naturally occurring DNA binding-deficient variant we tested our hypothesis that WRNIP1 shields reversed replication forks by directly binding to them. In agreement with this prediction, degradation of replication forks caused by depletion of endogenous WRNIP1 could be prevented by expressing recombinant WRNIP1 but not WRNIP1L (Figure 5D and S5E). Moreover, fork degradation was observed only after the isoform-specific depletion of the short, but not the long, isoform of WRNIP1 (Figures 5D and S5E).
Taken together, these findings indicate that the short, but not the long, variant of WRNIP1 is involved in fork protection from SLX4-mediated cleavage and suggest that WRNIP1's ability to bind to branched DNA substrates is a prerequisite for this function.
Discussion
In this study, we investigate the molecular mechanism of how WRNIP1 can protect stalled replication forks and find that WRNIP1 exerts its protective function after stalled forks have undergone reversal. This conclusion is based on our observation that fork stability in WRNIP1-deficient cells is restored upon inactivation of the replication fork remodelers ZRANB3 and HLTF or the recombinase RAD51. It is of note, however, that the latter finding is in conflict with published data that show no alleviation of the WRNIP1 depletion–associated fork degradation phenotype upon knock-down of RAD51 (Leuzzi et al., 2016). In light of RAD51's dual role in the promotion of fork reversal and the protection of reversed forks (Mijic et al., 2017, Zellweger et al., 2015), we suspect that different knock-down efficiencies could be the explanation for the discrepancy between our data and the study by Leuzzi and colleagues. In agreement with this hypothesis, an incomplete knock-down of RAD51 was shown to cause a situation, in which fork reversal is still possible, but RAD51 levels are too low to efficiently protect reversed forks (Bhat et al., 2018). Conversely, a complete knock-down of RAD51 was reported to entirely block fork reversal (Mijic et al., 2017). In the aforementioned WRNIP1 study, a RAD51 inhibitor that selectively prevents RAD51 filament formation was also unable to prevent the WRNIP1 depletion-associated phenotype (Leuzzi et al., 2016). This finding is however not in contradiction with our data, as stable RAD51 filaments are not required for the formation of reversed replication forks (Mijic et al., 2017).
We further show that, even though WRNIP1, as BRCA2, acts downstream of replication fork reversal, the two proteins exert their protective function in different ways. Since mirin, an inhibitor of MRE11's exonuclease activity, was found to revert the fork degradation phenotype of BRCA2-deficient cells, it was proposed that BRCA2 and RAD51 protect the end of the regressed arm from MRE11 exonuclease-mediated resection (Kolinjivadi et al., 2017, Lemaçon et al., 2017, Mijic et al., 2017, Taglialatela et al., 2017). In contrast, we find that mirin has no effect on restoring stalled fork stability in WRNIP1-deficient cells. Instead, we show that depletion of SLX4 or its associated structure-specific endonucleases SLX1 and XPF-ERCC1 restores fork stability in the absence of WRNIP1, suggesting that WRNIP1 protects the junction point of a reversed fork, rather than the end of the protruded arm.
In line with this hypothesis, our in vitro data indicate that WRNIP1 can bind to DNA substrates that mimic reversed replication forks and protect them from endonucleolytic cleavage by SLX1-SLX4. In contrast to a previous study that reported a weak, ATP-dependent binding of WRNIP1 to primer/template junctions and replication fork-like structures (Yoshimura et al., 2009), we did not detect any binding to single- or double-stranded DNA nor replication fork-like structures. Moreover, in our experimental set-up, binding of WRNIP1 to four-way junction substrates was independent of the presence of ATP, and Walker A and B variants of WRNIP1 could bind to four-way junction substrates, and protect them from SLX1-SLX4 cleavage, to a similar extent as the wild-type protein. Accordingly, complementation of WRNIP1-depleted cells with Walker A and B domain mutants could restore fork stability to a similar extent as complementation with a wild-type construct. These findings are also in agreement with a previous study that reports restoration of fork stability with a WRNIP1 variant that has a single amino acid change in between the Walker A and B motifs (T294A) (Leuzzi et al., 2016).
Since protein ubiquitination plays an important role in the context of replication fork reversal (Vujanovic et al., 2017), we also addressed whether WRNIP1's UBZ domain (Bish and Myers, 2007) was required for its role in fork protection. The UBZ variant D37A WRNIP1 (Crosetto et al., 2008) proved, however, able to bind to DNA substrates in vitro, to protect four-way junctions from SLX1-SLX4 cleavage and to complement WRNIP1-depleted cells to a large extent, suggesting that WRNIP1's ability to bind to ubiquitin plays at most a minor role in the context of fork protection, possibly to help recruit WRNIP1 more efficiently to sites of DNA replication (Crosetto et al., 2008).
In contrast to what we found in cells lacking WRNIP1, replication fork stability could be restored in BRCA2-depleted cells by knocking-down MUS81 or EME1 but not SLX4. These different genetic interactions may be explained by the distinct substrate selectivity of MUS81-EME1 and SLX1-SLX4. While recombinant SLX1-SLX4 efficiently processes intact four-way junctions, recombinant MUS81-EME1 is much more active on four-way junctions that are nicked or contain one single-stranded arm (Gaillard et al., 2003, Wyatt et al., 2013). We hypothesize that the latter structure could be generated at reversed replication forks in the absence of BRCA2 as a result of MRE11-mediated resection of the protruded arm. Although such a scenario is consistent with a recently published study where MUS81-EME1 was found to drive unscheduled replication fork degradation in BRCA2-deficient cells (Rondinelli et al., 2017), it may not seem consistent at first glance with another recent study that contests a role of BRCA2 in protecting forks from MUS81-mediated cleavage (Lemaçon et al., 2017). It should be noted, however, that in the latter study a modified fiber labeling protocol was employed to exclude forks that are unable to restart after fork resection. Given MUS81's important role in replication fork restart, as shown in the same study, any restoration of fork stability in the absence of MUS81 could simply not be detected with this approach.
Overall, our data would be in agreement with a model in which WRNIP1 and BRCA2 act in two parallel branches of the fork protection pathway that are both required for the efficient protection of reversed replication forks (Figure 6). We envision that in the absence of WRNIP1, the junction point of a reversed fork would remain unprotected and become a target of SLX4-mediated endonucleolytic cleavage. After SLX4-mediated cleavage, the resulting broken fork could then serve as an entry point for DNA2-mediated nucleolytic degradation that would lead to the extensive nascent DNA degradation that we observe upon WRNIP1 depletion. In contrast, in the absence of BRCA2, the junction point of the reversed fork would initially be protected from SLX4-mediated cleavage by WRNIP1. The MRE11-mediated resection of the unprotected reversed arm, however, would generate a resected reversed fork and, hence, a substrate for MUS81-EME1. In agreement with our in vivo and in vitro data, WRNIP1 would not be able to prevent MUS81-EME1 cleavage. MUS81-EME1 would therefore be able to incise such a resected reversed fork and, thus, generate an entry point for further MRE11-mediated nucleolytic degradation.
Figure 6.

Model Illustrating the Two Branches of Fork Protection That Are Dependent on BRCA2 and WRNIP1, Respectively
See Discussion for details.
Taken together, we propose that in the absence of BRCA2 and WRNIP1 different DNA substrates are generated at reversed forks but that the extensive nascent strand degradation in both cases depends on the activity of exonucleases and structure-specific endonucleases. This model would imply that in the absence of WRNIP1 and BRCA2 the endpoint of fork degradation is similar, although WRNIP1 and BRCA2 are situated in two distinct branches of the fork protection pathway and protect replication forks in a mechanistically distinct manner. Accordingly, a previous study and our own data (not shown) demonstrate that codepletion of WRNIP1 and BRCA2 does not lead to an additive effect with respect to fork degradation (Leuzzi et al., 2016).
Interestingly, the long isoform of WRNIP1, WRNIP1L, which has a remarkably conserved 25 amino acid-long insertion in the ATPase domain, is unable to bind to DNA and protect it from SLX1-SLX4 cleavage. Moreover, fork stability in WRNIP1-depleted cells cannot be restored by WRNIP1L, suggesting that although WRNIP1L is the prevalent isoform in the cell, it is not the isoform that plays a role in fork protection. It remains to be seen whether, from an evolutionary point of view, the fork protection function of WRNIP1 is a new acquisition or whether in other species this highly conserved region does not interfere with WRNIP1's ability to protect replication forks. It is of note that WRNIP1 seems to be a moonlighting protein that, in addition to its function in the maintenance of genome stability, also plays a role in antiviral signaling of double-stranded RNA (Tan et al., 2017). Interestingly, in the cited study the long isoform was used to complement WRNIP1 knock-out cells, raising the possibility that the two isoforms could have entirely separate functions with WRNIP1 playing a role in fork protection and WRNIP1L having a function in innate antiviral immunity.
Limitations of the Study
Based on our DNA fibers analyses and in vitro experiments, which suggest that the junction point of a reversed fork becomes a target of SLX4-mediated endonucleolytic cleavage in the absence of WRNIP1, we propose a direct role for WRNIP1 in the protection of reversed replication forks.
One prediction of our model would be the formation of double-strand breaks at stalled replication forks in the absence of WRNIP1. Unexpectedly, however, we were not able to detect a significant increase in double-strand breaks upon WRNIP1 depletion, possibly because breaks are formed very transiently, and steady-state levels are therefore very low. More sensitive methods will have to be used in the future to address this issue.
It should also be stressed that DNA fiber assays have an intrinsically low resolution and can therefore only provide an endpoint result. In order to delineate the exact series of nucleolytic events taking place at the site of a stalled replication fork, additional approaches, such as the visualization of replication forks by electron microscopy, will be required to unravel further mechanistic details.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We are grateful to Lumir Krejci for the kind gift of purified MUS81-EME1, and we thank the whole Gari lab for helpful discussions. This project has received funding from the Swiss National Science Foundation (PP00P3_144784/1 and PP00P3_172959/1), the Human Frontier Science Program (CDA00043/2013-C), the Novartis Foundation for Medical-Biochemical Research (16A047), the “Stiftung für wissenschaftliche Forschung an der Universität Zürich”, and the University of Zurich. Work in the laboratory of P.H.L.G. was supported by an Institut National du Cancer, France PLBIO 2016-159 grant.
Author Contributions
B.P. and K.G. conceived the project and designed experiments. B.P., S.W., and S.K. performed experiments. S.S. and P.H.L.G. provided essential reagents and conceptual input. B.P. and K.G. analyzed and interpreted data, prepared figures, and wrote the manuscript with input from P.H.L.G. All authors read and edited the manuscript. K.G. supervised the study.
Declaration of Interests
The authors declare no competing interests.
Published: November 22, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.10.010.
Contributor Information
Bartlomiej Porebski, Email: bartlomiej.porebski@ki.se.
Kerstin Gari, Email: gari@imcr.uzh.ch.
Supplemental Information
4
See the corresponding excel file.
References
- Achar Y.J., Balogh D., Haracska L. Coordinated protein and DNA remodeling by human HLTF on stalled replication fork. Proc. Natl. Acad. Sci. U S A. 2011;108:14073–14078. doi: 10.1073/pnas.1101951108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achar Y.J., Balogh D., Neculai D., Juhasz S., Morocz M., Gali H., Dhe-Paganon S., Venclovas Č., Haracska L. Human HLTF mediates postreplication repair by its HIRAN domain-dependent replication fork remodelling. Nucleic Acids Res. 2015;43:10277–10291. doi: 10.1093/nar/gkv896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alabert C., Bukowski-Wills J.C., Lee S.B., Kustatscher G., Nakamura K., De Lima Alves F., Menard P., Mejlvang J., Rappsilber J., Groth A. Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nat. Cell Biol. 2014;16:281–291. doi: 10.1038/ncb2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bétous R., Mason A.C., Rambo R.P., Bansbach C.E., Badu-Nkansah A., Sirbu B.M., Eichman B.F., Cortez D. SMARCAL1 catalyzes fork regression and holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012;26:151–162. doi: 10.1101/gad.178459.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bétous R., Goullet de Rugy T., Pelegrini A.L., Queille S., de Villartay J.P., Hoffmann J.S. DNA replication stress triggers rapid DNA replication fork breakage by artemis and XPF. PLoS Genet. 2018;14:e1007541. doi: 10.1371/journal.pgen.1007541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat K.P., Krishnamoorthy A., Dungrawala H., Garcin E.B., Modesti M., Cortez D. RADX modulates RAD51 activity to control replication fork protection. Cell Rep. 2018;24:538–545. doi: 10.1016/j.celrep.2018.06.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bish R.A., Myers M.P. Werner helicase-interacting protein 1 binds polyubiquitin via its zinc finger domain. J. Biol. Chem. 2007;282:23184–23193. doi: 10.1074/jbc.M701042200. [DOI] [PubMed] [Google Scholar]
- Ciccia A., Nimonkar A.V., Hu Y., Hajdu I., Achar Y.J., Izhar L., Petit S.A., Adamson B., Yoon J.C., Kowalczykowski S.C. Polyubiquitinated PCNA recruits the ZRANB3 translocase to maintain genomic Integrity after replication stress. Mol. Cell. 2012;47:396–409. doi: 10.1016/j.molcel.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosetto N., Bienko M., Hibbert R.G., Perica T., Ambrogio C., Kensche T., Hofmann K., Sixma T.K., Dikic I. Human Wrnip1 is localized in replication factories in a ubiquitin-binding zinc finger-dependent manner. J. Biol. Chem. 2008;283:35173–35185. doi: 10.1074/jbc.M803219200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehé P.M., Gaillard P.H.L. Control of structure-specific endonucleases to maintain genome stability. Nat. Rev. Mol. Cell Biol. 2017;18:315–330. doi: 10.1038/nrm.2016.177. [DOI] [PubMed] [Google Scholar]
- Dungrawala H., Rose K.L., Bhat K.P., Mohni K.N., Glick G.G., Couch F.B., Cortez D. The replication checkpoint prevents two types of fork collapse without regulating replisome stability. Mol. Cell. 2015;59:998–1010. doi: 10.1016/j.molcel.2015.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fekairi S., Scaglione S., Chahwan C., Taylor E.R., Tissier A., Coulon S., Dong M.Q., Ruse C., Yates J.R., Russell P. Human SLX4 is a holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell. 2009;138:78–89. doi: 10.1016/j.cell.2009.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard P.-H.L., Noguchi E., Shanahan P., Russell P. The endogenous Mus81-Eme1 complex resolves Holliday junctions by a nick and counternick mechanism. Mol. Cell. 2003;12:747–759. doi: 10.1016/s1097-2765(03)00342-3. [DOI] [PubMed] [Google Scholar]
- Guervilly J.H., Gaillard P.H. SLX4: multitasking to maintain genome stability. Crit. Rev. Biochem. Mol. Biol. 2018;53:475–514. doi: 10.1080/10409238.2018.1488803. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y., Chaudhuri A.R., Lopes M., Costanzo V. Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 2010;17:1305–1311. doi: 10.1038/nsmb.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs M.R., Reynolds J.J., Winczura A., Blackford A.N., Borel V., Miller E.S., Zlatanou A., Nieminuszczy J., Ryan E.L., Davies N.J. BOD1L is required to suppress deleterious resection of stressed replication forks. Mol. Cell. 2015;59:462–477. doi: 10.1016/j.molcel.2015.06.007. [DOI] [PubMed] [Google Scholar]
- Hishida T., Iwasaki H., Ohno T., Morishita T., Shinagawa H. A yeast gene, MGS1, encoding a DNA-dependent AAA+ ATPase is required to maintain genome stability. Proc. Natl. Acad. Sci. U S A. 2002;98:8283–8289. doi: 10.1073/pnas.121009098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabe Y.I., Branzei D., Hayashi T., Suzuki H., Masuko T., Onoda F., Heo S.J., Ikeda H., Shimamoto A., Furuichi Y. A novel protein interacts with the Werner’s syndrome gene product physically and functionally. J. Biol. Chem. 2001;276:20364–20369. doi: 10.1074/jbc.C100035200. [DOI] [PubMed] [Google Scholar]
- Kile A.C., Chavez D.A., Bacal J., Eldirany S., Korzhnev D.M., Bezsonova I., Eichman B.F., Cimprich K.A. HLTF’s ancient HIRAN domain binds 3’ DNA ends to drive replication fork reversal. Mol. Cell. 2015;58:1090–1100. doi: 10.1016/j.molcel.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolinjivadi A.M., Sannino V., De Antoni A., Zadorozhny K., Kilkenny M., Técher H., Baldi G., Shen R., Ciccia A., Pellegrini L. Smarcal1-Mediated fork reversal triggers mre11-dependent degradation of nascent DNA in the absence of Brca2 and stable Rad51 nucleofilaments. Mol. Cell. 2017;67:867–881.e7. doi: 10.1016/j.molcel.2017.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaçon D., Jackson J., Quinet A., Brickner J.R., Li S., Yazinski S., You Z., Ira G., Zou L., Mosammaparast N. MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun. 2017;8:860. doi: 10.1038/s41467-017-01180-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuzzi G., Marabitti V., Pichierri P., Franchitto A. WRNIP1 protects stalled forks from degradation and promotes fork restart after replication stress. EMBO J. 2016;35:1437–1451. doi: 10.15252/embj.201593265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mijic S., Zellweger R., Chappidi N., Berti M., Jacobs K., Mutreja K., Ursich S., Ray Chaudhuri A., Nussenzweig A., Janscak P. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun. 2017;8:859. doi: 10.1038/s41467-017-01164-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz I.M., Hain K., Déclais A.C., Gardiner M., Toh G.W., Sanchez-Pulido L., Heuckmann J.M., Toth R., Macartney T., Eppink B. Coordination of structure-specific nucleases by human SLX4/BTBD12 is required for DNA repair. Mol. Cell. 2009;35:116–127. doi: 10.1016/j.molcel.2009.06.020. [DOI] [PubMed] [Google Scholar]
- Neelsen K.J., Lopes M. Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 2015;16:207–220. doi: 10.1038/nrm3935. [DOI] [PubMed] [Google Scholar]
- Poole L.A., Cortez D. Functions of SMARCAL1, ZRANB3, and HLTF in maintaining genome stability. Crit. Rev. Biochem. Mol. Biol. 2017;52:696–714. doi: 10.1080/10409238.2017.1380597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przetocka S., Porro A., Bolck H.A., Walker C., Lezaja A., Trenner A., von Aesch C., Himmels S.F., D’Andrea A.D., Ceccaldi R. CtIP-mediated fork protection synergizes with BRCA1 to suppress genomic instability upon DNA replication stress. Mol. Cell. 2018;72:568–582.e6. doi: 10.1016/j.molcel.2018.09.014. [DOI] [PubMed] [Google Scholar]
- Rondinelli B., Gogola E., Yücel H., Duarte A.A., Van De Ven M., Van Der Sluijs R., Konstantinopoulos P.A., Jonkers J., Ceccaldi R., Rottenberg S. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat. Cell Biol. 2017;19:1371–1378. doi: 10.1038/ncb3626. [DOI] [PubMed] [Google Scholar]
- Schlacher K., Christ N., Siaud N., Egashira A., Wu H., Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145:529–542. doi: 10.1016/j.cell.2011.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K., Wu H., Jasin M. A distinct replication fork protection pathway connects fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell. 2012;22:106–116. doi: 10.1016/j.ccr.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svendsen J.M., Smogorzewska A., Sowa M.E., O’Connell B.C., Gygi S.P., Elledge S.J., Harper J.W. Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell. 2009;138:63–77. doi: 10.1016/j.cell.2009.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglialatela A., Alvarez S., Leuzzi G., Sannino V., Ranjha L., Huang J.W., Madubata C., Anand R., Levy B., Rabadan R. Restoration of replication fork stability in BRCA1- and BRCA2-deficient cells by inactivation of SNF2-family fork remodelers. Mol. Cell. 2017;68:414–430.e8. doi: 10.1016/j.molcel.2017.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan P., He L., Cui J., Qian C., Cao X., Lin M., Zhu Q., Li Y., Xing C., Yu X. Assembly of the WHIP-TRIM14-PPP6C mitochondrial complex promotes RIG-I-mediated antiviral signaling. Mol. Cell. 2017;68:293–307.e5. doi: 10.1016/j.molcel.2017.09.035. [DOI] [PubMed] [Google Scholar]
- Tsurimoto T., Shinozaki A., Yano M., Seki M., Enomoto T. Human Werner helicase interacting protein 1 (WRNIP1) functions as novel modulator for DNA polymerase δ. Genes Cells. 2005;10:13–22. doi: 10.1111/j.1365-2443.2004.00812.x. [DOI] [PubMed] [Google Scholar]
- Vujanovic M., Krietsch J., Raso M.C., Terraneo N., Zellweger R., Schmid J.A., Taglialatela A., Huang J.W., Holland C.L., Zwicky K. Replication fork slowing and reversal upon DNA damage require PCNA polyubiquitination and ZRANB3 DNA translocase activity. Mol. Cell. 2017;67:882–890.e5. doi: 10.1016/j.molcel.2017.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt H.D.M., Sarbajna S., Matos J., West S.C. Coordinated actions of SLX1-SLX4 and MUS81-EME1 for holliday junction resolution in human cells. Mol. Cell. 2013;52:234–247. doi: 10.1016/j.molcel.2013.08.035. [DOI] [PubMed] [Google Scholar]
- Wyatt H.D.M., Laister R.C., Martin S.R., Arrowsmith C.H., West S.C. The SMX DNA repair tri-nuclease. Mol. Cell. 2017;65:848–860.e11. doi: 10.1016/j.molcel.2017.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Liu Z., Wang F., Temviriyanukul P., Ma X., Tu Y., Lv L., Lin Y.F., Huang M., Zhang T. FANCD2 and REV1 cooperate in the protection of nascent DNA strands in response to replication stress. Nucleic Acids Res. 2015;43:8325–8339. doi: 10.1093/nar/gkv737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura A., Seki M., Kanamori M., Tateishi S., Tsurimoto T., Tada S., Enomoto T. Physical and functional interaction between WRNIP1 and RAD18. Genes Genet. Syst. 2009;84:171–178. doi: 10.1266/ggs.84.171. [DOI] [PubMed] [Google Scholar]
- Zellweger R., Dalcher D., Mutreja K., Berti M., Schmid J.A., Herrador R., Vindigni A., Lopes M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 2015;208:563–579. doi: 10.1083/jcb.201406099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman M.K., Cimprich K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014;16:2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
4
See the corresponding excel file.