

Summary
2-Methylthio-N6-isopentenyl modification of adenosine (ms2i6A) is an evolutionally conserved modification found in mitochondrial (mt)-tRNAs. Cdk5 regulatory subunit-associated protein 1 (CDK5RAP1) specifically converts N6-isopentenyladenosine (i6A) to ms2i6A at position A37 of four mt-DNA-encoded tRNAs, and the modification regulates efficient mitochondrial translation and energy metabolism in mammals. Here, we report that the ms2 conversion mediated by CDK5RAP1 in mt-tRNAs is required to sustain glioma-initiating cell (GIC)-related traits. CDK5RAP1 maintained the self-renewal capacity, undifferentiated state, and tumorigenic potential of GICs. This regulation was not related to the translational control of mt-proteins. CDK5RAP1 abrogated the antitumor effect of i6A by converting i6A to ms2i6A and protected GICs from excessive autophagy triggered by i6A. The elevated activity of CDK5RAP1 contributed to the amelioration of the tumor-suppressive effect of i6A and promoted GIC maintenance. This work demonstrates that CDK5RAP1 is crucial for the detoxification of endogenous i6A and that GICs readily utilize this mechanism for survival.
Subject Areas: Biological Sciences, Molecular Biology, Cell Biology, Stem Cells Research, Cancer
Graphical Abstract
Highlights
-
•
CDK5RAP1 is required to sustain the growth of GICs through ms2 modification of i6A
-
•
Deficit of CDK5RAP1 inhibits the growth of GIC through i6A accumulation
-
•
CDK5RAP1 detoxifies i6A by conversion into ms2i6A in the mitochondria of GICs
-
•
Mitochondria serve as antidotal machinery against i6A in GICs
Biological Sciences; Molecular Biology; Cell Biology; Stem Cells Res earch; Cancer
Introduction
In cancer biology, mitochondria are key organelles for understanding the behavior of cancer cells. Mitochondria are involved in fine-tuning cellular metabolism, oxygen consumption, and energy production, and they control cell death programs such as apoptosis (Cogliati et al., 2016, Pernas and Scorrano, 2016). Moreover, mitochondria play key roles in cancer stem cells (CSCs), which are believed to confer histopathological heterogeneity and drug and/or radiation resistance in cancer tissues. The structure and dynamics of mitochondria are related to CSC-related traits such as self-renewal capacity and tumorigenic potential (Guha et al., 2014, Xie et al., 2015), and the translation of mitochondrial (mt) DNA-encoded genes can be a potential target for anticancer drugs (Skrtić et al., 2011). However, our knowledge regarding the molecular mechanism underlying the mitochondrial control of CSC fate is limited.
Nucleoside modifications have two roles in cancer biology: as an input for cellular decisions and as an output from nucleotide metabolism. The former is known to control cell growth and differentiation through epigenetic regulation (Brien et al., 2016, Lacadie et al., 2016, Zaidi et al., 2014), whereas the latter is known as a by-product of nucleotide metabolism. However, previous studies have shown that modified nucleosides can regulate the fate of cancer cells. When artificially applied, N6-isopentenyladenosine (i6A), a modified nucleoside derived from tRNAs, induces cell-cycle arrest and cell death in many types of cancer cells, including glioblastoma cells (Castiglioni et al., 2013, Ciaglia et al., 2017, Laezza et al., 2009, Rajabi et al., 2010, Ranieri et al., 2018). Nevertheless, whether endogenous i6A exerts the same effect and inhibits tumor growth remains unknown. Endogenous i6A may also attenuate antitumor effects on cancer cells.
i6A modifications exist only on the adenosine at position 37, which is a nucleotide neighboring the anticodon region in tRNAs (Wei et al., 2015). TRIT1 is an isopentenyl transferase that converts A into i6A in mammals (Schweizer et al., 2017). Moreover, i6A is converted to 2-methylthio (ms2) i6A (ms2i6A) by cyclin-dependent kinase 5 regulatory subunit associated-protein 1 (CDK5RAP1) in the mitochondria of mammalian cells (Wei et al., 2015). CDK5RAP1, a mitochondria-localizing methylthio-modifying enzyme, is essential for the conversion of i6A to ms2i6A in mt-RNAs that read codons for Trp, Tyr, Phe, and Ser (Wei et al., 2015). This enzyme contributes to the maintenance of cellular respiration and metabolism in skeletal and cardiac muscles via the precise translation of mt-DNA-encoded genes. Therefore, CDK5RAP1 deficiency reduces OXPHOS-related protein levels and triggers myopathy in mice and humans (Wei et al., 2015). Because intramitochondrial translation is a key element for sustaining malignancy and CSC-related properties, we hypothesized that CDK5RAP1 contributes to intramitochondrial translation and is essential for malignancy.
Although the importance of mitochondrial translation in malignant tumors has been emphasized, the role of CDK5RAP1 in this process is not fully understood. A previous study suggested that CDK5RAP1 deficiency induces cell-cycle arrest and apoptosis in breast cancer via the ROS/JNK signaling pathway (Wang et al., 2015). In the present study, however, we demonstrated that CDK5RAP1 deficiency induced excessive autophagy but not apoptosis and was sufficient to repress glioma-initiating cell (GIC)-related capacities. Mechanistically, CDK5RAP1 deficiency caused an intracellular accumulation of i6A, which needs to be converted to ms2i6A to avoid the tumor-suppressive effects of i6A. The results of the present study revealed a novel role of mitochondria in cancer biology, that is, a modification of mitochondrial tRNAs functions as an antidotal machinery to sustain the GIC-related traits.
Results
CDK5RAP1 Is Required to Sustain GIC-Related Traits
Because GICs have been proposed to contribute to the malignant properties of glioma tissues (Lathia et al., 2015), we first examined whether CDK5RAP1 controlled the stemness of patient-derived GICs. In glioma biology, GIC-related traits are usually defined by self-renewal capacity measured using sphere formation assay, the expression of stem cell markers, and tumor-propagating potential assessed by xenograft tumor models (Suvà et al., 2014). We prepared three types of GIC cell lines categorized by their molecular subtype: JKGIC1 (mesenchymal), JKGIC2 (proneural), and JKGIC5 (proneural) (Figures S1A and S1B). Upon CDK5RAP1 knockdown by infection with lentiviruses containing shCDK5RAP1, the sphere-forming capacity of these GICs was significantly reduced (Figures 1A and S1C). Single cell-sphere formation assay showed that CDK5RAP1 was required for the in vitro self-renewal capacity of JKGIC2 and JKGIC5 (Figure 1B). CDK5RAP1 deficiency suppressed the protein and mRNA levels of the transcriptional factors essential for GIC (e.g., Sox2, Oig2, POU3F2, and SALL2, Suvà et al., 2014) and GIC-related markers (Figures 1C, S1D, S1F, and S1G). Immunofluorescence analyses revealed that CDK5RAP1 was required to sustain the undifferentiated state of GICs, as indicated by the loss of Sox2, Nestin, and CD133 expression in shCDK5RAP1-infected cells (Figures 1D and S1E).
Figure 1.

CDK5RAP1 Is Required to Sustain GIC-Related Traits
(A) Left: Quantification of primary and secondary spheres formed by JKGIC2-shControl, JKGIC2-shCDK5RAP1#1, JKGIC2-shCDK5RAP1#2 and JKGIC5-shControl, JKGIC5-shCDK5RAP1#1, JKGIC5-shCDK5RAP1#2 cells. CDK5RAP1 is required to sustain the anchorage-independent growth capacity of GICs. The data are presented as the number of spheres formed from 2,000 seeded cells. Each bar represents the SD value of four independent replicates. *p < 0.05. Sequence information of shRNAs is show in Table S1. Right: Representative phase contrast images of these cells. Scale bars, 100 μm.
(B) Quantification of primary spheres initiated from the single cells of JKGIC2-shControl, JKGIC2-shCDK5RAP1#1, JKGIC2-shCDK5RAP1#2, JKGIC2-shCDK5RAP1#5 and JKGIC5-shControl, JKGIC5-shCDK5RAP1#1, JKGIC5-shCDK5RAP1#2, JKGIC5-shCDK5RAP1#5. CDK5RAP1 is required for sustaining the sphere formation capacity of GICs. The data are presented as the percentage of spheres formed from 48 wells of single cells. Each bar represents the SD value of four independent replicates. *p < 0.05.
(C) Left: Representative immunoblotting images of GIC markers in shControl- and shCDK5RAP1-transfected JKGIC2 cells. Sox2, Olig2, POU3F2, and SALL2 levels were determined 4 days after infection with respective shRNA-encoding lentivirus. GAPDH served as a loading control. The experiment was repeated three times. The information of antibodies used in the present study is shown in Table S2. Right: Relative expression levels of CDK5RAP1 4 days after the lentiviral transduction of shRNAs. Each bar represents the SD value from three independent replicates. Sequence information of the primers for qPCR is shown in Table S1.
(D) Representative immunostaining images of JKGIC2-shControl and JKGIC2-shCDK5RAP1 cells. Transfection with shCDK5RAP1 reduced the number of Sox2-and Nestin-positive cells. Scale bars, 20 μm.
(E–G) CDK5RAP1 is required to sustain the tumorigenic potential of GICs. After shRNA induction, JKGIC1, JKGIC2, and JKGIC5 cells were subcutaneously injected (E, n = 3 per condition) and JKGIC2 were intracranially (F, n = 5 per condition) injected. Notably, CDK5RAP1 knockdown prolonged the overall survival of the mice with brain tumors (G). *p < 0.05. Scale bars: 1 cm in (E) and 2 mm in (F).
Also see Figure S1.
We then asked whether CDK5RAP1 was required to sustain tumor-propagating capacity in subcutaneous xenograft model in immunocompromised mice (BALB/c-nu). As shown in Figure 1E, CDK5RAP1 deficit significantly reduced tumor size in all types of xenograft models. For the orthotopic glioma model, we injected JKGIC2-shControl (JKGIC2 cells infected with lentiviruses containing pLKO.1-shControl) or JKGIC2-shCDK5RAP1 (pLKO.1-shCDK5RAP1#2) cells into the left cerebral hemisphere of ICR-nu mice. The loss of CDK5RAP1 attenuated tumor growth of JKGIC2 (Figure 1F) and prolonged the overall survival of mice with brain tumors (Figure 1G). These data clearly show that CDK5RAP1 is crucial for sustaining GIC-related characteristics.
CDK5RAP1 Controls GIC Properties Independently of Mitochondrial Translation, Dynamics, and Functions
Because mitochondrial function is essential for the maintenance of GIC properties (Seyfried et al., 2015) and because CDK5RAP1 is required for the efficient translation of mitochondrial proteins encoded by mitochondrial DNA (Wei et al., 2015), we hypothesized that the loss of GIC-related traits upon CDK5RAP1 knockdown was caused by mitochondrial dysfunction, probably by a deficiency in intramitochondrial translation (Figure 2A). To test this hypothesis, we analyzed the effect of CDK5RAP1 knockdown on mitochondrial function as measured by the oxygen consumption rate (OCR) in GICs. In GICs, differentiation cues (e.g., 10% FBS) induce a regulatory switch in mitochondrial functions (Figure S2A). For this assay, we prepared three types of JKGIC2 cells: JKGIC2-shControl, JKGIC2-shCDK5RAP1#2, and JKGIC2-shCDK5RAP1#5 (the knockdown efficiency of each shRNA is shown in Figure 2B). Surprisingly, CDK5RAP1 knockdown did not affect the OCR, indicating that CDK5RAP1 is not essential for mitochondrial respiration in GICs (Figure 2B). This was specific to GICs because we observed a significant change in the OCR in shCDK5RAP1-infected U87MG cells, a non-GIC line (Figure S2B). Moreover, CDK5RAP1 deficiency did not alter mitochondrial shape or dynamics, which are considered crucial for controlling GIC-related traits (Xie et al., 2015) (Figure 2C). Electron microscopy analysis showed that there was no significant difference in the mitochondrial shape or length between control and CDK5RAP1-deficient GICs (Figure 2D).
Figure 2.

CDK5RAP1 Deficiency Has No Effect on Mitochondrial Translation, Dynamics, or Function
(A) An initial hypothesis suggesting that the loss of GIC-related traits by CDK5RAP1 deficiency is caused by the failure of intramitochondrial translation.
(B) Left: Representative oxygen consumption rates in shControl- and shCDK5RAP1-transfected JKGIC2 cells. Respiratory coupling was not affected by CDK5RAP1 knockdown. n = 5 per condition. Right: Relative expression levels of CDK5RAP1 4 days after the lentiviral transduction of shRNAs. Each bar represents the SD value from three independent replicates.
(C) MitoTracker staining shows that mitochondrial shape was not altered upon CDK5RAP1 knockdown in JKGIC2.
(D) Left: Representative electron microscopy images of mitochondrial structures. Scale bars, 500 nm. Right: Mitochondrial length was not altered upon CDK5RAP1 knockdown. n = 38 for shControl and n = 87 for shCDK5RAP1#2.
(E) The level of MTCO1 encoded by mitochondrial DNA was determined 4 days after shRNA lentiviral infection. GAPDH served as a loading control. The same results were reproduced three times.
(F) Quantification of primary spheres formed from 2,000 JKGIC1 cells, which were transfected with shRNAs against CDK5RAP1, TUFM, or TSFM. Only shRNAs against CDK5RAP1 reduced the number of spheres formed. Each bar represents the SD value from four independent replicates. *p < 0.05.
Also see Figure S2.
We then investigated whether CDK5RAP1 deficiency induced translation failure in mitochondrial proteins in GICs. As a positive control, we transduced shRNAs against the mitochondrial translation elongation factors TUFM or TSFM, which are crucial for the translation of mitochondrial proteins encoded by mitochondrial DNA (Belostotsky et al., 2012, Christian and Spremulli, 2012). In JKGIC1 cells, knockdown of TUFM or TSFM but not of CDK5RAP1 resulted in the profound loss of mitochondrially encoded cytochrome c oxidase I (MTCO1) (Figure 2E). Importantly, TUFM or TSFM knockdown had no effect on the sphere-forming capacity of GICs, whereas CDK5RAP1 knockdown significantly reduced the number of spheres formed (Figure 2F). We also confirmed these phenomena in JKGIC2 cells (Figures S2C and S2D). Interestingly, we noticed that CDK5RAP1 deficit reduced the level of NDUFB8 (a component of mitochondrial complex I encoded by nuclear gene) and thus induced the loss of complex I activity but did not attenuate the levels of mitochondrial proteins encoded by mitochondrial genome such as ND6, cytochrome b, and MTCO1 (Figures S2E and S2F). These data clearly show that intramitochondrial translation is not fundamental for sustaining GIC-related properties in glioblastoma cells and indicate that CDK5RAP1 deficiency affects the properties of GICs in an intramitochondrial translation-independent manner.
CDK5RAP1 Knockdown Induces Excessive Autophagy, which Critically Determines GIC Fate
A previous study showed that CDK5RAP1 knockdown induced cell-cycle arrest and apoptosis in breast cancer cell lines via the activation of the JNK signaling pathway (Wang et al., 2015). Therefore, we examined whether CDK5RAP1 knockdown induced apoptosis in GICs. However, CDK5RAP1 knockdown did not induce the activation of caspase 3 in JKGIC1, JKGIC2, and JKGIC5 cells (Figure 3A) and did not increase the number of TUNEL-positive cells in JKGIC2 cell lines (Figure S3A). Instead, we found that CDK5RAP1 knockdown induced autophagy as indicated by an increased presence of autophagosomes and autolysosomes in electron microscopy experiments (Figure 3B). We further validated the induction of the autophagic response in GICs upon CDK5RAP1 knockdown by immunofluorescence analysis of LC3 puncta formation and by immunoblotting analysis of LC3-II induction, AMPK activation, and mTOR inhibition (Figures 3C, 3D, S3B, and S3C).
Figure 3.

CDK5RAP1 Knockdown Induces Excessive Autophagy, Which Critically Determines GIC Fate
(A) CDK5RAP1 knockdown did not activate caspase-3 in JKGIC1, JKGIC2, and JKGIC5 cells. CDDP-treated cells served as a positive control of apoptotic status.
(B) Left: Representative electron microscopy images of the autophagic response. JKGIC2 cells were fixed after 4 days of lentiviral transduction of each shRNAs. Right: CDK5RAP1 knockdown increased the number of autophagosomes and autolysosomes. n = 5 per condition. *p = 0.0391 versus shControl. Scale bars, 1 μm.
(C) Representative immunostaining images for LC3 in JKGIC2 cells. CDK5RAP1 knockdown induces LC3 puncta formation. Rapamycin-treated cells served as a positive control of LC3 puncta formation. Scale bars, 10 μm.
(D) Immunoblotting analyses of mTOR, AMPK, and LC3 show that CDK5RAP1 knockdown activates the autophagic program in JKGIC2 cells.
(E) Left: mTOR inhibition with rapamycin triggers the autophagic response and decreases Nestin expression in JKGIC2 cells. n = 5 per condition. Right: Dose-dependent effect of rapamycin on the number of spheres formed from 2,000 JKGIC2 cells. Each bar represents the SD value from four independent replicates. *p < 0.05.
(F) Comparison of sphere formation by JKGIC2 and JKGIC5 cells transfected with shRNAs against CDK5RAP1 in the presence or absence of shRNAs against ATG5. ATG5 knockdown reduces the sphere-forming capacity but successfully rescues the shCDK5RAP1-mediated decrease in the anchorage-independent growth of GICs. Each bar represents the SD value from four independent replicates. *p < 0.05.
Also see Figure S3.
Previous studies have demonstrated that mTOR inhibition suppresses the stemness of GICs (Garros-Regulez et al., 2016, Sunayama et al., 2010). Indeed, rapamycin treatment attenuated the cellular growth and promoted the autophagic program in GICs (Figures 3E and S3D). We then investigated whether the autophagic response triggered by CDK5RAP1 knockdown could be rescued by knocking down Atg5, which is essential for the autophagy pathway. Although Atg5 knockdown slightly reduced the number of spheres formed by JKGIC2 and JKGIC5 cells, it successfully rescued the loss of anchorage-independent growth driven by CDK5RAP1 knockdown (Figures 3F and S3E). Excessive autophagy results in the growth inhibition of GICs (Ueda et al., 2012). In agreement with our previous study, CDK5RAP1 knockdown significantly inhibited the growth of JKGIC1 and JKGIC2 cells (Figure S3F). These data demonstrate that GICs require CDK5RAP1 to avoid excessive autophagy, which critically represses the GICs' cell growth.
Treatment with N6-Isopentenyladenosine (i6A) Induces Excessive Autophagy and Loss of GIC-Related Traits
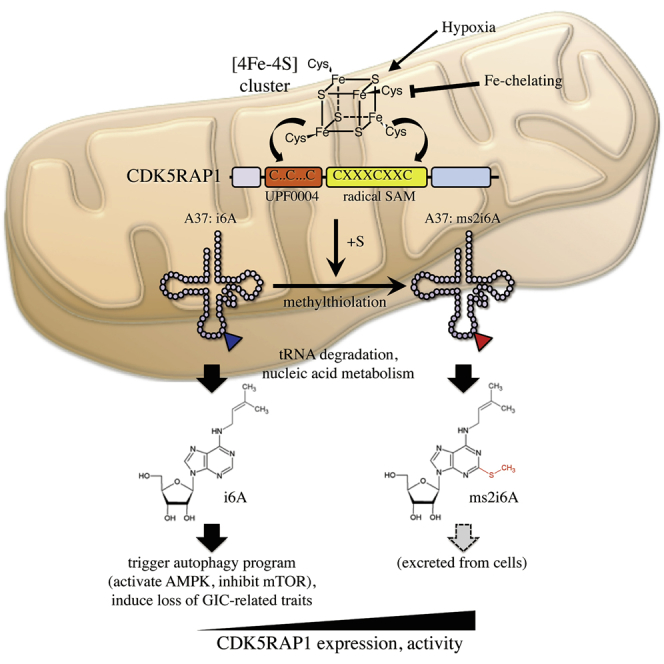
Thus far, the present study has revealed that CDK5RAP1 maintains GIC-related traits by inhibiting excessive autophagy but not by controlling intramitochondrial translation. How does CDK5RAP1 maintain the characteristics by inhibiting excessive autophagy? CDK5RAP1 is a mitochondrial enzyme that modifies nucleosides by adding methylthio groups and is crucial for the conversion of i6A to ms2i6A at A37 of mt-tRNAs (Wei et al., 2015). Consistent with this, the intracellular distribution of ms2i6A-containing tRNAs was limited to the mitochondria of GICs, as confirmed by immunofluorescence analysis with an anti-ms2i6A antibody, which specifically recognizes ms2i6A-containing tRNAs (Figure 4A). This result led us to hypothesize that CDK5RAP1 enhances the conversion of i6A to ms2i6A in mt-tRNAs of GICs, resulting in the inhibition of excessive autophagy.
Figure 4.

Treatment with N6-isopentenyladenosine (i6A) Induces Excessive Autophagy and Loss of GIC-Related Traits
(A) Representative images of immunostaining with an anti-ms2i6A antibody and MitoTracker in JKGIC2 cells. Note that the intracellular distribution of ms2i6A is limited to the mitochondria. Scale bars, 20 μm.
(B) Treatment with i6A, but not ms2i6A, for 24 h induces increases in LC3-II levels in JKGIC2 cells. GAPDH served as a loading control. The same results were reproduced three times.
(C) Treatment with i6A, but not ms2i6A, activates the autophagic program in JKGIC2 cells.
(D) Treatment with i6A for 24 h induces LC3 puncta formation in JKGIC2 cells.
(E) Quantification of primary spheres formed by JKGIC2 cells treated with i6A or ms2i6A. Treatment with i6A reduces the number of spheres, but this reduction is not rescued by the further addition of ms2i6A. Each bar represents the SD value from four independent replicates. *p < 0.05.
(F and G) Immunoblotting (F) and immunostaining (G) indicate that treatment with i6A, but not ms2i6A, decreases the protein levels of Nestin and Sox2 in JKGIC2 cells.
(H) Quantification of primary spheres formed by JKGIC2 cells (2,000/well) transfected with shRNAs against ATG5 and i6A. ATG5 knockdown successfully rescues the i6A-induced loss of stemness in JKGIC2 cells. Each bar represents the SD value from four independent replicates. *: p < 0.05.
(I and J) Exogenous transduction of murine Cdk5rap1 increases the amount of Nestin protein (I) and the number of spheres formed by JKGIC2 cells (2,000/well) (J). Each bar represents the SD value from four independent replicates. *p < 0.05.
(K) Ectopic expression of murine Cdk5rap1 prevents the loss of anchorage-independent growth ability in JKGIC2 due to exogenous treatment with 4 and 8 μM i6A. The data are presented as the number of spheres formed from 2,000 cells. Each bar represents the SD value from four independent replicates. *p < 0.05.
Also see Figure S4.
Because previous studies have shown that treatment with exogenous i6A induces tumor-suppressive effects such as apoptotic or autophagic cell death in cancer cells (Castiglioni et al., 2013, Ciaglia et al., 2017, Laezza et al., 2009, Rajabi et al., 2010, Ranieri et al., 2018), we examined whether i6A treatment induced excessive autophagy and consequent loss of GIC-related traits. In JKGIC2 cells, i6A treatment increased the intracellular concentration of i6A in a dose-dependent manner, and the intracellular concentration of i6A in the cells treated with 4 μM i6A reached the same level as that in JKGIC2 cells transfected with shCDK5RAP1 (Figure S4A). However, exogenous i6A did not induce a consequent increase in ms2i6A (Figure S4A), suggesting that CDK5RAP1 could not convert free i6A to ms2i6A. i6A treatment activated the autophagic program, as shown by LC3-II induction, AMPK activation, mTOR signaling pathway inhibition in JKGIC2 and JKGIC5 cells (Figures 4B, 4C, and S4B), and LC3 puncta formation in JKGIC2 cells (Figure 4D). i6A also attenuated the anchorage-independent cell growth and reduced the expression of GIC markers such as Sox2 and Nestin (Figures 4E–4G, S4B, and S4C). To validate that the loss of GIC-related traits driven by i6A treatment was in the context of autophagy, we prepared shControl- and shATG5-transfected JKGIC2 cells and treated them with variable concentrations of i6A. As expected, ATG5 knockdown successfully mitigated the phenotypic outcome of i6A (Figure 4H). In contrast, ms2i6A treatment induced neither autophagy nor loss of GIC-related traits and did not prevent the phenotypic outcomes triggered by i6A treatment (Figures 4B–4G). Moreover, N6-isopentenyladenine (i6Adenine) did not repress the anchorage-independent growth of JKGIC2 cells (Figure S4D). These data suggest that isopentenyl modification only on adenosine molecules elicits tumor-suppressive effects and that this increase in isopentenyl groups can be ameliorated via detoxification by CDK5RAP1-mediated methylthiolation (Figure S4E).
We next investigated whether the elevated expression levels of CDK5RAP1 conferred an antiautophagic phenotype to GICs. For this purpose, we transduced control red fluorescent protein (RFP) (pTomo-RFP) or murine Cdk5rap1 (pTomo-mCdk5rap1) into JKGIC2 cells (Figure 4I). Cdk5rap1 promoted the growth capacity of JKGIC2 and elevated the expression levels of the undifferentiated marker Nestin (Figures 4I and 4J). Exogenous Cdk5rap1 significantly attenuated the inhibitory effect of 3–4 μM i6A on the anchorage-independent growth ability and cell viability of JKGIC2 cells (Figures 4K and S4F). However, overexpression of Cdk5rap1 failed to attenuate the inhibitory effect of higher concentrations (>6 μM) of exogenous i6A (Figures 4K and S4F). Taken together, these results suggest that treatment with exogenous i6A induces excessive autophagy and loss of GIC-related traits. Cdk5rap1 overexpression induces the conversion of endogenous i6A to ms2i6A on tRNA species, resulting in a decrease in total i6A (endogenous + exogenous i6A) levels in GICs and consequent attenuation of the inhibitory effect of i6A on the anchorage-independent growth ability and cell viability of GICs when treated with 3–4 μM i6A. In contrast, when cells are treated with a high concentration (>6 μM) of i6A, the intracellular i6A level may reach the concentration corresponding to an antitumor effect. Therefore, the overexpression of Cdk5rap1 fails to attenuate the inhibitory effect of excessive exogenous i6A.
CDK5RAP1 Balances i6A and ms2i6A Concentrations in GICs in Response to the Microenvironment
To demonstrate whether CDK5RAP1 regulates endogenous concentrations of i6A and ms2i6A in GICs, we analyzed [i6A] and [ms2i6A] in mock-, shCDK5RAP1-, and mCdk5rap1-transduced JKGIC1, or JKGIC2 cells by mass spectrometry. The results showed that the ratio of total [i6A]/[i6A + ms2i6A] was increased in shCDK5RAP1-transfected cells but decreased in murine Cdk5rap1-overexpressing cells (Figures 5A and S5A).
Figure 5.

CDK5RAP1 Balances i6A and ms2i6A Concentrations in GICs in Response to the Microenvironment
(A) Mass spectrometry analysis of [i6A] and [ms2i6A] from the lysates of shControl- and shCDK5RAP1-, RFP-, mCdk5rap1-transfected JKGIC2 cells. Note that CDK5RAP1 knockdown induces an increase in the relative [i6A] in GICs, whereas CDK5RAP1 overexpression reduces the relative [i6A]. Each bar represents the SD value from three independent replicates. *p < 0.05.
(B) Schematic representation of the molecular characteristics of CDK5RAP1. Cysteine residues in the UPF0004 and radical SAM domains are crucial for the stabilization of the [4Fe-4S] clusters. The mitochondria localization signal (MLS) allows CDK5RAP1 to localize to the mitochondria. The TRAM domain is predicted to interact with tRNA species.
(C) The M.I. of ms2i6A corresponding to each mt-tRNA and tRNA expression level in JKGIC2 cells cultured in the presence of 21% and 1% O2. The M.I.s in all CDK5RAP1-targeted tRNA species are increased under hypoxic conditions, but tRNA expression is not. The data are presented as the M.I. relative to the cells cultured under normoxia. Each bar represents the SD value from two independent replicates. For the procedure regarding the measurement of M.I., also see Figure S5D.
(D) Hypoxic conditions decrease intracellular [i6A] and increase the secretion of ms2i6A by JKGIC2 cells. The data are presented as the percentage of the levels under normoxia. Each bar represents the SD value from three independent replicates. *p < 0.05.
(E and F) Hypoxic conditions significantly increased the expression levels of VEGF mRNA (E) but not CDK5RAP1 mRNA (F) in JKGIC2 cells. Each bar represents the SD value from four independent replicates. *p < 0.05.
Also see Figure S5.
Because the biochemical reaction of CDK5RAP1-mediated 2-methylthio conversion of i6A is strictly regulated within the mitochondria and is detected solely in mt-tRNAs (Fakruddin et al., 2017) and because ms2i6A is predominantly enriched in cell culture medium (∼9.6 times more than the concentration inside cells, Figure S5B), these data suggest that CDK5RAP1 decreases [i6A] by promoting the 2-methylthio conversion of i6A in the mitochondria and that the consequent ms2i6A derived from degraded mt-tRNAs is excreted (Figure S5C).
Two conserved domains in CDK5RAP1, namely, UPF0004 and the radical SAM domain, are essential for CDK5RAP1 activity (Fakruddin et al., 2017). The cysteine residues in these domains are required for the interaction with both [4Fe-4S] clusters, which provide the sulfur atoms for interactions with i6A molecules (Figure 5B). Given that [4Fe-4S] clusters are stable under anoxic conditions (Crack et al., 2007), we speculated that CDK5RAP1 was activated under hypoxic conditions. To test this possibility, we performed quantitative PCR measurements of tRNA 2-methylthio modifications. We extracted total RNA from GICs cultured in the presence of either 21% or 1% O2. The ms2i6A modifications in each mt-tRNA are represented as a modification index (M.I.) as reported previously (Figure S5D) (Xie et al., 2013). As expected, hypoxic culture conditions significantly increased the M.I. of each CDK5RAP1-targeted mt-tRNA, whereas hypoxic conditions had no effect on the expression of the tRNAs (Figures 5C and S5E). Consistent with this outcome, as observed in mCdk5rap1-overexpressing JKGIC1 and JKGIC2 cells, the ratio of total [i6A]/[i6A + ms2i6A] under hypoxic conditions was decreased, whereas the ratio of [ms2i6A]/[i6A + ms2i6A] was increased (Figures 5D and S5F). Hypoxia-inducible factor 1α (HIF-1α) is the inducible subunit of the HIF-1 transcription factor that regulates the expression of genes involved in the response to hypoxia. Vascular endothelial growth factor (VEGF) is one of the genes upregulated by HIF-1 (Lin et al., 2004). VEGF expression was significantly increased in GICs cells cultured under 1% O2 (Figures 5E, S5G, and S5I). In contrast, there was no difference in the expression level of CDK5RAP1 in cells cultured in the presence of 21% to 1% O2 (Figures 5F, S5H, and S5J). To further validate that [4Fe-4S] clusters are required for this reaction, we treated cells with deferoxamine (100 μM) to chelate intracellular Fe ions and measured the M.I. of the mt-tRNAs, and we found that Fe chelation reduced the M.I (Figure S5K). These data indicate that CDK5RAP1 activity is controlled by the microenvironment, such as hypoxic conditions and the concentration of intracellular Fe ions.
CDK5RAP1 Activity Is Upregulated in the Hypoxic Region of Human Glioblastoma and Is Required to Sustain the Sphere-forming Capacity of Malignant Cells but Not Normal Brain Cells
To gain insights into the contribution of CDK5RAP1 to human glioblastoma multiforme (GBM) pathophysiology, we harvested several specimens, including the “tumor core” and “peritumor normal area,” from three patients with GBM and analyzed the concentrations of i6A and ms2i6A by mass spectrometry. Intriguingly, [i6A] was significantly increased in the tumor core compared with that in the peritumor normal area in all cases (Figure 6A). Following the logic established by these observations and the above-mentioned data, we hypothesized that glioblastoma cells require CDK5RAP1 activity to detoxify i6A to protect cells from excessive autophagy. However, the expression level of CDK5RAP1 in the tumor core was the same as that in the peritumor normal area (Figure 6B). Because the GBM core is known to be hypoxic (Fujimura et al., 2013), we speculated that CDK5RAP1 activity was upregulated in the tumor core of GBM specimens. RT-PCR analysis of a HIF target gene, VEGF, indicated that the tumor core was more hypoxic than the peritumor normal area (Figure 6C). As expected, mass spectrometry analysis showed that [ms2i6A] in the tumor core was higher than that in the peritumor normal area (Figure 6D). Immunofluorescence analysis revealed that Nestin-positive tumor cells overlapped with ms2i6A-positive tumor cells in the tumor core area, whereas ms2i6A-negative tumor cells did not overlap with Nestin-positive cells (Figure 6E), suggesting that CDK5RAP1, upon activation under hypoxic conditions, promotes the maintenance of GIC-related traits.
Figure 6.

CDK5RAP1 Is Essential for Sustaining the GIC-Related Traits of GICs but Not That of Normal Brain Cells
(A) Left: Representative MR image and H&E staining of the brain tissue in a patient (case 1) with GBM. Tumor core and peritumor samples were harvested to extract RNA and nucleosides. Right: Mass spectrometry analysis of relative [i6A] shows that [i6A] is increased in the tumor core. Each bar represents the SD value from three independent replicates. *p < 0.05 versus the peritumor sample.
(B) Expression levels of CDK5RAP1 in human GBM specimens from the tumor core and peritumor areas are not significantly different. n = 5 per sample type.
(C) Expression level of VEGF in the peritumor and tumor core areas of patients with GBM. n = 5 per sample type.
(D) Mass spectrometry analysis of relative [ms2i6A] in the peritumor and tumor core areas of specimens from patients with GBM. Each bar represents the SD value from three independent replicates. *p < 0.05 versus the peritumor sample.
(E) Representative images of immunofluorescence analysis with anti-ms2i6A and anti-Nestin antibodies in the tumor core of patients with GBM. Note that the ms2i6A-positive cells and Nestin-positive cells overlap.
(F) Untreated NSCs, NSCs transformed with oncogenic lentiviruses, and mouse primary neurons transformed with oncogenic lentiviruses (all from Cdk5rap1fl/fl mouse brains) either with or without transduced adenoviruses harboring Cre recombinase were subjected to the sphere formation assay. Transformed cells but not NSCs require Cdk5rap1 to sustain their anchorage-independent growth capacity. The data are presented as the number of spheres formed from 2,000 cells. Each bar represents the SD value from four independent replicates. *p < 0.05.
(G) Representative data of the gliosphere initiation assay. Primary astrocytes from wild-type or Cdk5rap1fl/fl mouse brains were infected with Cre-inducible HRasG12V-expressing pTomo lentiviruses. The cells were further infected with adenoviruses harboring Cre recombinase to activate HRasG12V and recombine out the Cdk5rap1 alleles and were cultured in GIC medium for 7–10 days. The numbers of initiated spheres from 50,000 cells were counted. n = 4 per condition. For the procedure of the gliosphere initiation assay, see Figure S6. Cdk5rap1 is required to initiate the formation of gliospheres from primary astrocytes.
(H) Quantification of secondary, tertiary, and quaternary spheres formed by the transformed astrocytes harvested from the gliosphere initiation assay described in (G). Cdk5rap1 is required to sustain the self-renewal capacity of the transformed cells. The data are presented as the number of spheres formed from 2,000 cells. Each bar represents the SD value from four independent replicates. *p < 0.05.
Also see Figure S6.
To demonstrate whether CDK5RAP1 activity was required for the traits of only GICs but not normal neural stem cells (NSCs), we prepared NSCs and primary neurons from Cdk5rap1fl/fl mouse pups and infected these cells with control pTomo lentivirus or pTomo-shNF1-shp53 lentivirus. Dual knockdown of NF1 and p53 allowed the neural cells to acquire GIC-related properties within 1–2 weeks as previously reported (Friedmann-Morvinski et al., 2012). After several passages, we further infected the cells with adenoviruses harboring Cre recombinase to knockout the Cdk5rap1 alleles from these cells, after which the cells were subjected to the sphere formation assay. The Cdk5rap1 knockout resulted in a significant reduction of the anchorage-independent growth in the transformed NSCs and neurons, but not in normal NSCs (Figure 6F). We further investigated whether Cdk5rap1 was required for the acquisition of GIC-related traits (i.e., gliomagenesis). We transduced Cre-inducible HRasG12V/shp53 into astrocytes from wild-type and Cdk5rap1fl/fl mouse pups. The infected cells were subjected to sphere formation assays upon adeno-Cre infection, which further recombined out the loxP-RFP-loxP cassette to induce HRasG12V expression (Figure S6). In the astrocytes from Cdk5rap1fl/fl mice, adeno-Cre infection recombined the Cdk5rap1 alleles out. Using the gliosphere initiation assay, we found that wild-type astrocytes with HRas induction and p53 knockdown (HRas/shp53) formed numerous spheres, suggesting that the primary astrocytes acquired GIC-related properties. In contrast, the number of spheres in primary astrocytes from Cdk5rap1fl/fl mice was lower than that in HRas/shp53 astrocytes, suggesting that astrocytes required Cdk5rap1 to achieve gliomagenesis (Figure 6G). We then harvested and trypsinized the formed spheres and conducted the sphere formation assay. Similar to the transformed neurons, the anchorage-independent growth of the cells was reduced upon Cdk5rap1 deletion (Figure 6H). These data clearly demonstrate that CDK5RAP1 is essential for acquiring and sustaining the GIC-related traits of malignant and transformed cells but not that of normal NSCs.
Discussion
We previously reported that CDK5RAP1 was responsible for the ms2 modification of mammalian mt-tRNAs for the Ser (UCN), Phe, Tyr, and Trp codons (Wei et al., 2015). Under stress condition, deficiencies in ms2 modification impaired mitochondrial protein synthesis, OXPHOS activity, and ATP synthesis in normal tissues. Although the canonical role of CDK5RAP1 is to regulate the precise translation of mitochondrial DNA-encoded genes, even in the knockout mice the effect of loss-of-CDK5RAP1 can be observed mainly in complex I but not markedly in the other complexes in steady state (Wei et al., 2015). In the present study, we demonstrate that CDK5RAP1 deficiency induces excessive autophagy and the consequent loss of GIC-related traits. Importantly, we reveal that these phenomena are independent from the regulation of intramitochondrial translation by CDK5RAP1 in GICs, and moreover, we show that the intramitochondrial translation seems not to contribute to sustain the GIC-related traits (Figures 2 and S2). In GICs, CDK5RAP1 deficiency attenuates the antidotal effect of ms2i6A against i6A in the mitochondria, resulting in the promotion of tumor-suppressive effects of i6A. The present study suggests a novel function of CDK5RAP1-mediated ms2 modification corresponding to the detoxification of i6A in the mitochondria; GICs readily exploit this function to survive. We propose mitochondria as the sole antidotal machinery against i6A, which causes excessive autophagy and the consequent loss of GIC-related traits. The data presented here add a new layer of the function of chemical modification in mitochondrial tRNAs.
The present study showed that CDK5RAP1 was essential for acquiring and sustaining the GIC-related traits of malignant and transformed cells but not that of normal NSCs by the conversion of i6A to ms2i6A. A recent study showed lower proliferation and differentiation capabilities in NSCs in an adult rodent model of severe motor deprivation and a significant reduction in Cdk5rap1 expression in NSCs in the model (Adami et al., 2018). Although it is unclear that a reduction in Cdk5rap1 expression impairs proliferation and differentiation capabilities in NSCs, the canonical function and the regulation of precise translation of the mitochondrial protein may be involved in the proliferation and differentiation of NSCs. Thus, the functions of CDK5RAP1 may differ between NSCs and GICs.
In glioblastoma specimens, i6A was enriched in the tumor core area. Consistent with this, the expression levels of TRIT1, a mitochondrial tRNA isopentenyl transferase, were elevated in malignant tissues from the REMBRANDT cohort (REpository for Molecular BRAin Neoplasia DaTa, http://www.betastasis.com/glioma/rembrandt/). Such environmental conditions can promote excessive autophagy in GICs and consequently repress GIC-related properties, as demonstrated by the present study. To survive in this microenvironment, GICs require robust CDK5RAP1 activity. However, the expression of CDK5RAP1 in the tumor core was the same as that in the peritumoral normal area. Therefore, it may be important for GICs to survive under hypoxic conditions, such as those in the tumor core, to activate CDK5RAP1. CDK5RAP1 has two [4Fe-4S] clusters, which are essential for its enzymatic activity and are sensitive to oxygen (Wei et al., 2015). As shown in the present study, the activity of CDK5RAP1 was drastically increased under hypoxic conditions, although its expression was not induced under these conditions (Figures 5C–5F). The hypoxic area is known to harbor a subset of cells, known as CSCs, that have stem cell-like properties due to the induction of stemness genes such as Oct4, c-myc, and Nanog. Moreover, hypoxia induces drug resistance in GICs by activating several pathways mediated by COX-2, the PI3K pathway, AP1, c-Jun, Pim1, or Stat3 (Jalota et al., 2018). Our findings suggest a novel importance of the hypoxic environment for the survival and maintenance of GIC-related traits. Thus, GICs likely require continuous activation of CDK5RAP1.
The results of the present study show that CDK5RAP1 deficiency induces excessive autophagy and the consequent loss of stemness in GICs but not in normal neurons or NSCs. The Warburg effect is a metabolic phenomenon characterized by increased glycolytic activity, decreased mitochondrial oxidative phosphorylation, and lactate production and is often observed in GICs (Koppenol et al., 2011). Functional differences in CDK5RAP1 between normal cells and GICs may be due to the difference in the metabolic phenotypes of the mitochondria in the cells.
A number of studies have shown that exogenous application of i6A has a strong antitumor effect in vitro (Castiglioni et al., 2013, Ciaglia et al., 2017, Laezza et al., 2009, Rajabi et al., 2010, Ranieri et al., 2018). In agreement with previous studies, the present study showed that i6A inhibited the GIC-related traits and induced autophagic cell death and that exogenous i6A was not converted to ms2i6A by CDK5RAP1. These results suggest that i6A is a promising therapeutic molecule to target GICs. However, a higher concentration (>4 μM) of exogenous i6A was required for the antitumor effect against GICs. The effective concentration of exogenous i6A seems to be much higher than endogenous i6A produced from degraded tRNAs. Importantly, the level of intracellular i6A when treated with 4 μM i6A was consistent with that in shCDK5RAP1-transfected JKGIC2 cells (Figure S4A). These results suggest that the membrane permeability of i6A is poor and/or that exogenous i6A is rapidly excreted after entering GICs. It may be important for clinical applications to develop delivery systems that can effectively deliver and maintain i6A in GICs. Moreover, i6A was significantly enriched in the tumor core where GICs are abundant, suggesting that i6A alone may be ineffective in combating GICs in the hypoxic tumor core that have constitutively active CDK5RAP1. Despite the antitumor effect of i6A in vitro, few studies have shown this effect in vivo. It may be important to combine i6A administration with CDK5RAP1 downregulation by either an iron chelator or shRNAs against CDK5RAP1.
In conclusion, CDK5RAP1-mediated modification of mitochondrial tRNAs is crucial for not only the precise translation of mitochondrial DNA-encoded proteins in normal tissues but also the detoxification of endogenous i6A in GICs. GICs readily utilize this mechanism to survive.
Limitations of the Study
In this study, we were unable to generate CDK5RAP1 knockout GICs because the cells could not grow. Therefore, we used the knockdown cells in all experiments.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Nobuko Maeda, Masayo Obata (Kumamoto University), and Masumi Furutani (Okayama University) for their technical assistance. This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Sciences, and Technology of Japan (17905074 and 18959602 to K.T., 16K18989 and 18K15241 to A.F.), the Japan Agency for Medical Research and Development (AMED) (17935694 to K.T., JP18cm0106143 to A.F.), and the Takeda Science Foundation (K.T.).
Author Contributions
T.Y., A.F., F.-Y.W., and K.T. designed the experiments and wrote the manuscript. T.Y. and A.F. carried out the experiments. N.S., J.K. and A.M. provided GBM samples.
Declaration of Interests
The authors declare no conflict of interest.
Published: November 22, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2019.10.012.
Supplemental Information
References
- Adami R., Pagano J., Colombo M., Platonova N., Recchia D., Chiaramonte R., Bottinelli R., Canepari M., Bottai D. Reduction of movement in neurological diseases: effects on neural stem cells characteristics. Front. Neurosci. 2018;12:336. doi: 10.3389/fnins.2018.00336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belostotsky R., Frishberg Y., Entelis N. Human mitochondrial tRNA quality control in health and disease: a channelling mechanism? RNA Biol. 2012;9:33–39. doi: 10.4161/rna.9.1.18009. [DOI] [PubMed] [Google Scholar]
- Brien G.L., Valerio D.G., Armstrong S.A. Exploiting the epigenome to control cancer-promoting gene-expression programs. Cancer Cell. 2016;29:464–476. doi: 10.1016/j.ccell.2016.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castiglioni S., Casati S., Ottria R., Ciuffreda P., Maier J.A. N6-isopentenyladenosine and its analogue N6-benzyladenosine induce cell cycle arrest and apoptosis in bladder carcinoma T24 cells. Anticancer Agents Med. Chem. 2013;13:672–678. doi: 10.2174/1871520611313040016. [DOI] [PubMed] [Google Scholar]
- Christian B.E., Spremulli L.L. Mechanism of protein biosynthesis in mammalian mitochondria. Biochim. Biophys. Acta. 2012;1819:1035–1054. doi: 10.1016/j.bbagrm.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciaglia E., Abate M., Laezza C., Pisanti S., Vitale M., Seneca V., Torelli G., Franceschelli S., Catapano G., Gazzerro P., Bifulco M. Antiglioma effects of N6-isopentenyladenosine, an endogenous isoprenoid end product, through the downregulation of epidermal growth factor receptor. Int. J. Cancer. 2017;140:959–972. doi: 10.1002/ijc.30505. [DOI] [PubMed] [Google Scholar]
- Cogliati S., Enriquez J.A., Scorrano L. Mitochondrial cristae: where beauty meets functionality. Trends Biochem. Sci. 2016;41:261–273. doi: 10.1016/j.tibs.2016.01.001. [DOI] [PubMed] [Google Scholar]
- Crack J.C., Green J., Cheesman M.R., Le Brun N.E., Thomson A.J. Superoxide-mediated amplification of the oxygen-induced switch from [4Fe-4S] to [2Fe-2S] clusters in the transcriptional regulator FNR. Proc. Natl. Acad. Sci. U S A. 2007;104:2092–2097. doi: 10.1073/pnas.0609514104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakruddin M., Wei F.Y., Emura S., Matsuda S., Yasukawa T., Kang D., Tomizawa K. Cdk5rap1-mediated 2-methylthio-N6-isopentenyladenosine modification is absent from nuclear-derived RNA species. Nucleic Acids Res. 2017;45:11954–11961. doi: 10.1093/nar/gkx819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann-Morvinski D., Bushong E.A., Ke E., Soda Y., Marumoto T., Singer O., Ellisman M.H., Verma I.M. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science. 2012;338:1080–1084. doi: 10.1126/science.1226929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimura A., Michiue H., Cheng Y., Uneda A., Tani Y., Nishiki T., Ichikawa T., Wei F.Y., Tomizawa K., Matsui H. Cyclin G2 promotes hypoxia-driven local invasion of glioblastoma by orchestrating cytoskeletal dynamics. Neoplasia. 2013;15:1272–1281. doi: 10.1593/neo.131440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garros-Regulez L., Aldaz P., Arrizabalaga O., Moncho-Amor V., Carrasco-Garcia E., Manterola L., Moreno-Cugnon L., Barrena C., Villanua J., Ruiz I. mTOR inhibition decreases SOX2-SOX9 mediated glioma stem cell activity and temozolomide resistance. Expert Opin. Ther. Targets. 2016;20:393–405. doi: 10.1517/14728222.2016.1151002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guha M., Srinivasan S., Ruthel G., Kashina A.K., Carstens R.P., Mendoza A., Khanna C., van Winkle T., Avadhani N.G. Mitochondrial retrograde signaling induces epithelial-mesenchymal transition and generates breast cancer stem cells. Oncogene. 2014;33:5238–5250. doi: 10.1038/onc.2013.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalota A., Kumar M., Das B.C., Yadav A.K., Chosdol K., Sinha S. A drug combination targeting hypoxia induced chemoresistance and stemness in glioma cells. Oncotarget. 2018;9:18351–18366. doi: 10.18632/oncotarget.24839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppenol W.H., Bounds P.L., Dang C.V. Otto Warburg's contributions to current concepts of cancer metabolism. Nat. Rev. Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- Lacadie S.A., Ibrahim M.M., Gokhale S.A. Divergent transcription and epigenetic directionality of human promoters. FEBS J. 2016;283:4214–4222. doi: 10.1111/febs.13747. [DOI] [PubMed] [Google Scholar]
- Laezza C., Caruso M.G., Gentile T., Notarnicola M., Malfitano A.M., Di Matola T., Messa C., Gazzerro P., Bifulco M. N6-isopentenyladenosine inhibits cell proliferation and induces apoptosis in a human colon cancer cell line DLD1. Int. J. Cancer. 2009;124:1322–1329. doi: 10.1002/ijc.24056. [DOI] [PubMed] [Google Scholar]
- Lathia J.D., Mack S.C., Mulkearns-Hubert E.E., Valentim C.L.L., Rich J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015;29:1203–1217. doi: 10.1101/gad.261982.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C., McGough R., Aswad B., Block J.A., Terek R. Hypoxia induces HIF-1alpha and VEGF expression in chondrosarcoma cells and chondrocytes. J. Orthop. Res. 2004;22:1175–1181. doi: 10.1016/j.orthres.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Pernas L., Scorrano L. Mito-morphosis: mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu. Rev. Physiol. 2016;78:505–531. doi: 10.1146/annurev-physiol-021115-105011. [DOI] [PubMed] [Google Scholar]
- Rajabi M., Signorelli P., Gorincioi E., Ghidoni R., Santaniello E. Antiproliferative activity of N6-isopentenyladenosine on MCF-7 breast cancer cells: cell cycle analysis and DNA-binding study. DNA Cell Biol. 2010;29:687–691. doi: 10.1089/dna.2010.1073. [DOI] [PubMed] [Google Scholar]
- Ranieri R., Ciaglia E., Amodio G., Picardi P., Proto M.C., Gazzerro P., Laezza C., Remondelli P., Bifulco M., Pisanti S. N6-isopentenyladenosine dual targeting of AMPK and Rab7 prenylation inhibits melanoma growth through the impairment of autophagic flux. Cell Death Differ. 2018;25:353–367. doi: 10.1038/cdd.2017.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer U., Bohleber S., Fradejas-Villar N. The modified base isopentenyladenosine and its derivatives in tRNA. RNA Biol. 2017;14:1197–1208. doi: 10.1080/15476286.2017.1294309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyfried T.N., Flores R., Poff A.M., D'Agostino D.P., Mukherjee P. Metabolic therapy: a new paradigm for managing malignant brain cancer. Cancer Lett. 2015;356:289–300. doi: 10.1016/j.canlet.2014.07.015. [DOI] [PubMed] [Google Scholar]
- Skrtić M., Sriskanthadevan S., Jhas B., Gebbia M., Wang X., Wang Z., Hurren R., Jitkova Y., Gronda M., Maclean N. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell. 2011;20:674–688. doi: 10.1016/j.ccr.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunayama J., Matsuda K., Sato A., Tachibana K., Suzuki K., Narita Y., Shibui S., Sakurada K., Kayama T., Tomiyama A., Kitanaka C. Crosstalk between the PI3K/mTOR and MEK/ERK pathways involved in the maintenance of self-renewal and tumorigenicity of glioblastoma stem-like cells. Stem Cells. 2010;28:1930–1939. doi: 10.1002/stem.521. [DOI] [PubMed] [Google Scholar]
- Suvà M.L., Rheinbay E., Gillespie S.M., Patel A.P., Wakimoto H., Rabkin S.D., Riggi N., Chi A.S., Cahill D.P., Nahed B.V. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell. 2014;157:580–594. doi: 10.1016/j.cell.2014.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda Y., Wei F.Y., Hide T., Michiue H., Takayama K., Kaitsuka T., Nakamura H., Makino K., Kuratsu J., Futaki S., Tomizawa K. Induction of autophagic cell death of glioma-initiating cells by cell-penetrating D-isomer peptides consisting of Pas and the p53 C-terminus. Biomaterials. 2012;33:9061–9069. doi: 10.1016/j.biomaterials.2012.09.003. [DOI] [PubMed] [Google Scholar]
- Wang H., Wei L., Li C., Zhou J., Li Z. CDK5RAP1 deficiency induces cell cycle arrest and apoptosis in human breast cancer cell line by the ROS/JNK signaling pathway. Oncol. Rep. 2015;33:1089–1096. doi: 10.3892/or.2015.3736. [DOI] [PubMed] [Google Scholar]
- Wei F.Y., Zhou B., Suzuki T., Miyata K., Ujihara Y., Horiguchi H., Takahashi N., Xie P., Michiue H., Fujimura A. Cdk5rap1-mediated 2-methylthio modification of mitochondrial tRNAs governs protein translation and contributes to myopathy in mice and humans. Cell Metab. 2015;21:428–442. doi: 10.1016/j.cmet.2015.01.019. [DOI] [PubMed] [Google Scholar]
- Xie P., Wei F.Y., Hirata S., Kaitsuka T., Suzuki T., Suzuki T., Tomizawa K. Quantitative PCR measurement of tRNA 2-methylthio modification for assessing type 2 diabetes risk. Clin. Chem. 2013;59:1604–1612. doi: 10.1373/clinchem.2013.210401. [DOI] [PubMed] [Google Scholar]
- Xie Q., Wu Q., Horbinski C.M., Flavahan W.A., Yang K., Zhou W., Dombrowski S.M., Huang Z., Fang X., Shi Y. Mitochondrial control by Drp1 in brain tumor initiating cells. Nat. Neurosci. 2015;18:501–510. doi: 10.1038/nn.3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi S.K., Grandy R.A., Lopez-Camacho C., Montecino M., van Wijnen A.J., Lian J.B., Stein J.L., Stein G.S. Bookmarking target genes in mitosis: a shared epigenetic trait of phenotypic transcription factors and oncogenes? Cancer Res. 2014;74:420–425. doi: 10.1158/0008-5472.CAN-13-2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.