

Abstract
Light-driven [2+2] cycloaddition is the most direct strategy to build tetrasubstituted cyclobutanes, core components of many lead compounds for drug development. Significant advances in the chemoselectivity and enantioselectivity of [2+2] photocycloadditions have been made, but exceptional and tunable diastereoselectivity and regioselectivity (head-to-head vs. head-to-tail adducts), required for synthesis of bioactive molecules, have not yet been achieved. Here we show that colloidal quantum dots (QDs) serve as visible-light chromophores, photocatalysts, and reusable scaffolds for homo- and hetero-intermolecular [2+2] photocycloadditions of 4-vinylbenzoic acid derivatives, including aryl-conjugated alkenes, with up to 98% switchable regioselectivity and 98% diastereoselectivity for the previously minor syn-cyclobutane products, including the syn-head-to-tail cyclobutane, which has never before been accessed as the major product of a hetero-photocycloaddition. Transient absorption spectroscopy confirms that our system is the first example of catalysis triggered by triplet-triplet energy transfer from a QD. The precisely controlled triplet energy levels of QD photocatalysts facilitate efficient and selective heterocoupling, a major challenge in direct cyclobutane synthesis.
Light-driven [2+2] cycloadditions1–5, simple routes to complex, biologically relevant tetrasubstituted cyclobutanes6–8, follow several possible excited-state routes. Those that proceed through the triplet excited state of the reagent olefin are advantageous because (i) their scope is not limited by the electrochemical potentials of the substrate, (ii) triplets have long enough lifetimes to mediate intermolecular cycloadditions, and (iii) triplets are accessible with visible light through excitation of a triplet sensitizer, such as a transition metal complex or organic chromophore9, followed by triplet-triplet energy transfer (TT EnT). The TT EnT strategy minimizes deleterious side-reactions and has resulted in high-yield intra- and intermolecular [2+2] photocycloaddtions of quinolone1,10, cinnamate and derivatives11,12.
Pioneering work of Bach, Meggers, Yoon and others has demonstrated enantioselective intermolecular [2+2] photocycloadditions in the presence of hydrogen bonding templates1,2, chiral secondary amines3, chiral ligands of molecular catalysts4 and Lewis-acid co-catalysts5. Outstanding challenges include the competing fast cis/trans isomerization pathway of the substrates, and achieving selectivities for a particular regioisomer or diastereomer of the coupled product and for homo- vs. heterocoupling within a mixture of reactive olefins – the latter a priority because cyclobutane natural products and related compounds predominantly comprise two distinct olefins13. First, the anti diastereomer of the cyclobutyl product is sterically favored in untemplated reactions but the preference is often not significant enough to achieve efficient discrimination14. The syn diastereomer has been accessed only using templates that interact with substrates through covalent and non-covalent linkages, and through solid-state reactions. The syntheses of covalent scaffolds like paracyclophane15 – and, more importantly, the reactions used to cleave the addition product from the template – are however complex and often low-yielding, whereas non-covalent scaffolds (usually involving hydrogen bonds)16 have a very limited substrate scope and are often not rigid enough to template intermolecular reactions effectively. The solid-state reactions are a synthetically simpler approach to accessing syn diastereomers17, but do not offer a straightforward way to sensitize the reaction with visible light, or a way to achieve chemoselectivity and enantioselectivity.
Second, the distribution of head-to-head (HH) vs. head-to-tail (HT) regioisomers of [2+2] cycloadditions is highly dependent on the relative stability of the corresponding triplet biradical intermediates and therefore difficult to control extrinsically. Most HT homocoupled products are achieved through solid state reactions18, or through a select few liquid state methods that use templates or molecular cages19,20, but regioselective HT heterocoupled products have not been accessed with these methods.
Third, selective heterocoupling is challenging to achieve even with a template or a solid-state reaction, because it requires selective co-localization of two different substrates. Reported examples are limited to coupling of two very similar, highly symmetric substrates, such as two monosubstituted stilbenes; this reaction has no regioselectivity21. Prior to this work, syn-HT heterocoupled products, the class of tetrasubstituted cyclobutanes that serve as scaffolds for many bioactive molecules, had never been formed as major products of a [2+2] photocycloaddition. In fact, there were no examples of tunable regioselectivity for any hetero-intermolecular [2+2] photocycloadditions. A strategy for extrinsic control of the configuration of the cyclobutane product is needed to counter the preferences driven by steric and electronic characteristics of the substrates.
Here, we use colloidal CdSe quantum dots (QDs) as visible light absorbers, triplet exciton donors, and self-assembly scaffolds to drive homo- and hetero-intermolecular [2+2] photocycloadditions of 4-vinylbenzoic acid derivatives. CdSe QDs donate energy to triplet states of organic molecules from triplet-like excitonic “dark” states, which lie <20 meV below their optically active “bright states”22–25. A QD’s triplet energy is tunable via size-dependent quantum confinement; here we adjust the size of the QD to selectively sensitize only one reagent olefin within a mixture, and thereby achieve efficient hetero-intermolecular couplings. The QD catalytic systems exhibit up to 98% switchable (between HH and HT) regioselectivity with up to 98% diastereoselectivity for the previously minor syn-HH or syn-HT configurations of the adducts, including those made from challenging stilbene derivatives (Figs. 1a,b). With one exception, highlighted below, the diastereomeric ratios (d.r.) we achieve are a factor of 5–10 higher than those reported with all other triplet sensitizers for similar systems11,12.
Fig. 1. Sensitization via the QD Photocatalyst and Mechanisms of Selectivity.

a, Sensitization of the triplet excited state of the substrate (T1) through TT-EnT from CdSe QDs, and schematic representation of syn-HH or syn-HT selectivity of QDs. A proposed mechanism of the route to each diastereomer is shown in Supplementary Scheme 2. The best regioselectivity is achieved with molecules where the black circle represents an aromatic group (Ar). b, Chemical structures of the molecular sensitizers, and the mechanism of anti-HH preference of reactions driven by these molecules. c, Ground state absorption spectra of a mixture of CdSe QDs (radius = 1.4 nm) and 1 in THF, before and after three 48-h illumination cycles. The differences between spectra at λ < 450 nm are due to consumption of 1. d, Decay dynamics of the exciton in CdSe QDs mixed with 250 eq of 1 in THF, upon photoexcitation at 470 nm, monitored at three wavelengths: 480 nm (pure electron dynamics), 900 nm (primarily electron dynamics), and 1300 nm (primarily hole dynamics). Electrons and holes decay with a time constant of 130±30 ps (where the error bar is the standard error) in the presence of 1; this time constant is absent if 1 is not present, see Supplementary Fig. 1, Supplementary Table 3. The cyclic voltammetry of 1 and the band-edge energies of the CdSe QDs are in the SI, Supplementary Fig. 4. “OD” ≡ optical density.
Results and Discussion
The Supplementary Information (SI) contains all details of synthesis and chemical analysis. The QDs do not etch, aggregate, or otherwise degrade for at least three 48 h reaction cycles (Fig. 1c). We readily separate the QD catalysts from reaction mixtures through addition of MeOH and centrifugation, and reuse them with no detectable decline in catalytic activity, Supplementary Table 1. No reactions occurred without QDs or without illumination, Supplementary Table 2. Through transient absorption spectroscopy, we observe coincident decays of the photoexcited electron and hole of the QD when the substrate is present, Fig. 1d. This result indicates that they are extracted from the QD as an electron-hole pair, and that the reaction is EnT-initiated rather than redox-initiated (and specifically TT EnT-initiated, since the singlet excited states of all substrates are too high-energy to access). The coupling yields of all reactions are ≥10x lower when we do not functionalize both substrates with carboxylate groups to reversibly bind to the QD (Supplementary Scheme 1), indicating that the reaction occurs at the surface of the particle. Carboxylates are “medium-strength” ligands for the surfaces of CdSe (kself-exchange ~500 s−1)26, so coupled products desorb from the QD surface to make room for new substrates.
Table 1 shows a set of [2+2] photocycloadditions of 4-vinylbenzoic acid and derivatives that demonstrate the activity and diastereoselectivity of our QD photocatalyst when directly compared to tris(2,2’-bipyridine)ruthenium(II) (Ru(bpy)32+) or tris(2-phenylpyridinato)iridium (III) (Ir(ppy)3), which have been used to perform similar reactions11,12. The QD systems generate syn products with d.r. > 30:1, while the molecular complexes prefer anti products with much lower selectivity (d.r. < 3:1). The syn stereochemistry of the QD products is verified by X-ray crystallography of 8 (Supplementary Fig. 4) and Nuclear Overhauser Spectroscopy (NOESY NMR) of all products. For the heterocoupling of 3 and either 1 or 2, the QDs produce syn products with d.r. > 40:1, while the molecular complexes prefer anti products with d.r. < 2:1. For the stilbene derivatives 4 - 7, cycloadditions catalyzed by the molecular complexes do not occur with measureable yield, probably because of the known fast cis/trans isomerization of the excited-state substrates; we indeed observe a high concentration of cis isomers in the crude reaction mixtures after illumination. QDs, in contrast, photocatalyze reactions with excellent coupling yields, either, we propose, by accelerating the coupling step through co-localization of substrates or by slowing the isomerization by tethering the substrates on the QD surface.
Table 1 |.
Diastereoselectivity through Pre-organization on the QD Surface.
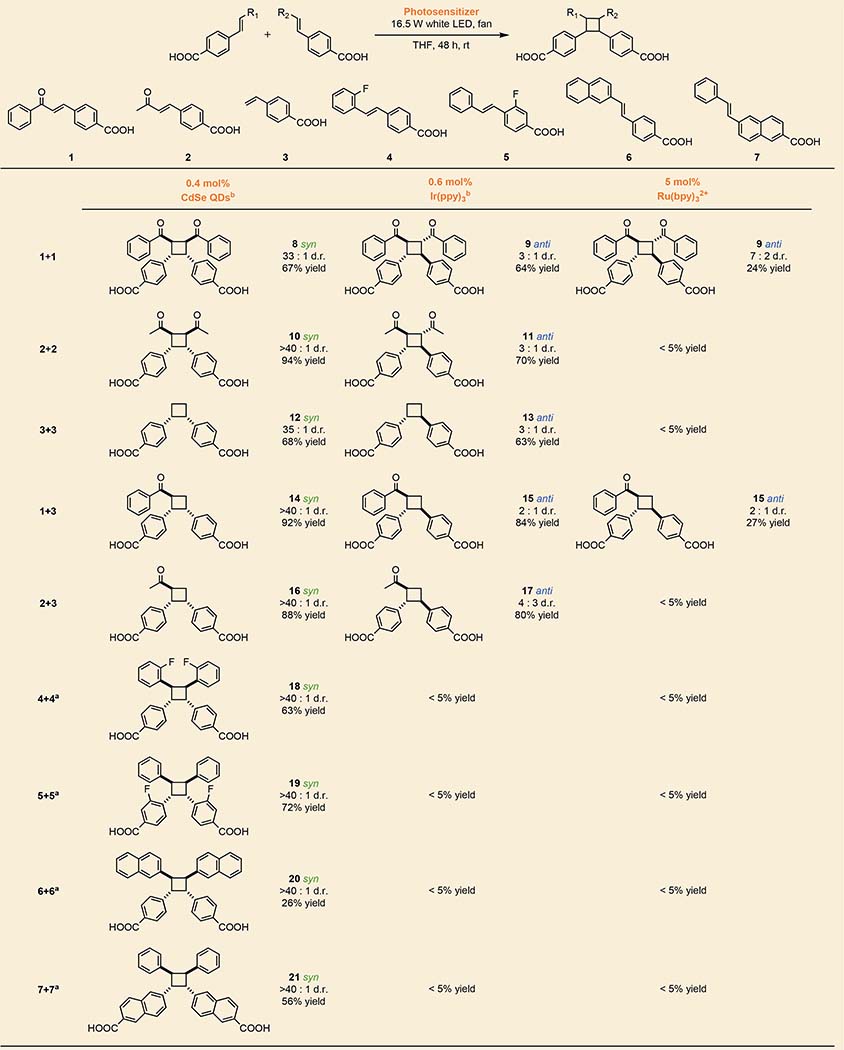 |
The triplet spectra of 1 and 2 shown in Supplementary Fig. 2. The yields listed are the isolated yields of the diastereomers drawn for QD systems (remaining mass≡starting material and its cis-isomer; zero HT product) and the NMR yields of the regioisomers drawn for Ir(ppy)3 and Ru(bpy)32+ systems (remaining mass≡starting material and its cis-isomer, minor unidentified side products), see the SI. Reactions of 1 were illuminated with >455 nm light so as not to excite the substrate directly.
Reactions were illuminated for 4 days.
Listed loadings are for homocycloadditions; catalyst loadings for heterocycloadditions are 1.2 mol% QDs and 1.9 mol% molecular sensitizers.
The selective production of the kinetically disfavored syn configuration by the QDs, even in the case of two different coupling partners, is remarkable considering that the only templating chemistry is the reversible association of a carboxylate on the substrate with Cd2+ on the QD surface. The ability of intermolecular π-π interactions among rigid olefins to promote syn stereochemistry in solid-state reactions27–29 suggests that these same non-covalent interactions are responsible for the syn selectivity in the QD system, Fig. 1a.
Figure 2 illustrates the role of tunable QD size in achieving heterocoupling (over homocoupling) through selective TT EnT. The difficulty in controlling competition between homo- and heterocoupling has limited the scope of previous heterocouplings to substrates with large triplet energy differences. In this case, the triplet energies of 1 and 3 (2.25 eV and 2.51 eV respectively) are close enough such that Ir(ppy)3 indiscriminately sensitizes both substrates (Fig. 3B), resulting in 23% of the substrate 3 producing the undesired homocoupled product 9. In contrast, 1.4 nm CdSe QDs perform selective TT EnT to 1 without sensitizing 3, yielding highly efficient heterocoupling (92% yield of 14) with <5% of 3 participating in homocoupling. The comparatively unselective performance of the higher-energy 1.0 nm CdSe QDs illustrates that selective energetic overlap with a specific substrate is the key to efficient heterocoupling.
Fig. 2. Control of Homo- vs. Heterocycloaddition with QD Size.

a, Conditions for and possible products of [2+2] photocyloadditions of 1 and 3. b, Emission spectra of four photosensitizers, compared with the triplet energies of substrates 1 and 3, determined from phosphorescence spectra and calculations, see the SI. PL ≡ photoluminescence. c, Product distributions from reaction mixtures with the four photosensitizers. The yields listed are for the regioisomers indicated (drawn in a); the remaining mass is exclusively starting materials and the cis-isomer of 1 for the QD systems, and the same plus minor unidentified side products for the moleculecatalyzed reactions, see the SI. aThe yields in red are calculated in reference to substrate 3, which was added in excess.
Fig. 3. Regioselectivity through Substrate Affinity for the QD Surface.

a, Conditions and product distributions for the [2+2] photocycloadditions of 2 and 1, 4 to 7, 22 or 23. When 2 is coupled with 23, with no carboxylic acid linker, the distribution of regioisomer products matches that of Ir(ppy)3. The yields listed for QD reactions are isolated yields for the diastereomers drawn (the major products), except for the reaction with the control substrate 23. The yields listed for Ir(ppy)3 and Ru(bpy)32+ systems are the NMR yields of the regioisomers drawn. The remaining mass of the coupling of 2 with 22 is starting materials and their cis-isomers (>72%), the HH regioisomer, and the homocycloaddition products of both substrates for the QD system, and is the same plus minor unidentified side products for the Ir(ppy)3 system, see the SI. aReactions were catalyzed with 2.4 mol% QDs. bReactions were illuminated for 2 days. cThe reaction was illuminated with a blue LED. b, Transition densities for the first triplet excited states of 1 and 22 calculated using time-dependent density functional theory (TD-DFT).
Figure 3 demonstrates the >90% tunable regioselectivity achievable for hetero-[2+2] photocycloadditions using our QD system. Most notably this system produces the disfavored heterocoupled HT regioisomers 25, 27 and 30. For untemplated reactions, i.e., those photosensitized by Ir(ppy)3 or by QDs when one substrate does not have a carboxylic acid substituent (gray shaded area), formation of the HH regioisomer is strongly favored through benzylic stabilization of the corresponding 1,4-diradical intermediate. In order to access the syn-HT heterocoupled products 25, 27 and 30, which have never been formed as a major product of [2+2] photocycloaddition, we tuned the regioselectivity of the QD-catalyzed reactions through the position of the carboxylate on each substrate (Fig. 3a). For coupling of 1, 4, 6 and 2, we achieve perfect HH regioselectivity with no HT products detected. When the carboxylate moiety is relocated to the opposite side of the substrate (5 vs. 4, 7 vs. 6, 22 vs. 1), the observed regioselectivity of the QD catalyst switches entirely, providing the HT product for the heterocoupling of 5, 7 and 2 with r.r. >40:1 and 22 and 2 with r.r. >10:1. The syn diastereoselectivity is preserved in these regioselective reactions: the heterocoupled products 24 - 28 have d.r. >37:1 and product 30 has d.r. >10:1.
The Ir(ppy)3 sensitizer produces the HH configuration as the major product regardless of whether 2 is coupled with 22 or 1 and the weak preference for the anti diastereomer remains in products 29 and 31 with d.r. up to 5:1. The loss of regioselectivity upon removing the carboxylate from one of the substrates (23) shows that the QD system achieves regioselective heterocoupling through pre-arrangement of substrates at the QD surface, a manner entirely independent of electronics of the reactive olefin. Moreover, the heterocoupling of stilbenes 4 - 7 with 2 by molecular substrates produces no measureable yield because of the same competing isomerization pathway described in reference to Table 1.
An examination of the unusually poor performance of the QD-catalyzed reaction between 22 and 2 shows that lower overall yield and selectivity is not due to side products (72% of the remaining yield can be accounted for by starting material), but, we believe, is limited by the location of triplet acceptor orbitals of the substrate (Fig. 3b). For 1, the transition to the triplet excited state (S0→T1) has good spatial overlap with the binding group of the substrate, and therefore good electronic coupling with the QD. For 22, there is a spatial gap between the QD surface and the triplet density; this gap makes TT EnT inefficient.
In summary, we have described a new and powerful approach to selective [2+2] photocycloadditions, and simultaneously presented the first example of catalysis triggered by triplet-triplet energy transfer from a quantum dot. By inducing self-assembly of substrate molecules through reversible association with the QD surface, we have accessed excellent diastereoselectivity and tunable regioselectivity – including previously inaccessible syn-HT photo-adducts – the first example of either selectivity type using the QD ligand shell, and the first examples of tunably regioselective hetero-intermolecular [2+2] photocycloadditions. The precisely controlled triplet energy levels of QD photocatalysts facilitate efficient and selective heterocoupling. Overall, QDs present a strategy for extrinsic control of the configuration of the cyclobutane product that counters the stereo-electronic preferences of the substrates, and have great potential to enhance any reaction that proceeds through a triplet excited state, including metal complex-catalyzed reactions. Improvement of the quantum efficiencies (moles product per incident photon) could be achieved by tuning the relative excited state energies of the QD and molecular substrate such that back-TT EnT is not competitive with the forward process30.
Data availability statement
Crystallographic data for the structures reported in this Article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 1900373. Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. All other data supporting the findings of this study are available within the Article (Figures 1–3 and Table 1) and its Supplementary Information (Supplementary Tables 1–8, Supplementary Scheme 1, Supplementary Figures 1–4, Supplementary Sections L, M), or from the corresponding author upon reasonable request.
Supplementary Material
Acknowledgements
The authors thank Profs. Regan Thomson and Andrew Lee for helpful discussions. Research primarily supported by through the Air Force Office of Scientific Research (grant 9550-17-1-0271) (synthesis, photocatalysis, analytical chemistry), and by the Center for Bio-Inspired Energy Science, an Energy Frontier Research Center funded by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences (BES), under Award # DE-SC0000989 (calculations). C.R.R. thanks the International Institute for Nanotechnology at Northwestern University for a fellowship. This work made use of the IMSERC at Northwestern University, which has received support from the NIH (1S10OD012016-01 / 1S10RR01907101A1); Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205); the State of Illinois and International Institute for Nanotechnology (IIN).
Footnotes
Competing interests The authors declare no competing interests.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
References
- 1.Alonso R & Bach T A Chiral Thioxanthone as an Organocatalyst for Enantioselective [2+2] Photocycloaddition Reactions Induced by Visible Light. Angew. Chem 126, 4457–4460. [DOI] [PubMed] [Google Scholar]
- 2.Maturi MM & Bach T Enantioselective Catalysis of the Intermolecular [2+2] Photocycloaddition between 2-Pyridones and Acetylenedicarboxylates. Angew. Chem. Int. Ed 53, 7661–7664. [DOI] [PubMed] [Google Scholar]
- 3.Hörmann FM, Chung TS, Rodriguez E, Jakob M & Bach T Evidence for Triplet Sensitization in the Visible-Light-Induced [2+2] Photocycloaddition of Eniminium Ions. Angew. Chem. Int. Ed 57, 827–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hu N et al. Catalytic Asymmetric Dearomatization by Visible-Light-Activated [2+2] Photocycloaddition. Angew. Chem. Int. Ed 57, 6242–6246. [DOI] [PubMed] [Google Scholar]
- 5.Blum TR, Miller ZD, Bates DM, Guzei IA & Yoon TP Enantioselective Photochemistry through Lewis Acid–Catalyzed Triplet Energy Transfer. Science 354, 1391–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dembitsky VM Bioactive cyclobutane-containing alkaloids. J. Nat. Med 62, 1–33. [DOI] [PubMed] [Google Scholar]
- 7.Lee-Ruff E & Mladenova G Enantiomerically Pure Cyclobutane Derivatives and Their Use in Organic Synthesis. Chem. Rev 103, 1449–1484. [DOI] [PubMed] [Google Scholar]
- 8.Tsai I-L et al. New Cytotoxic Cyclobutanoid Amides, a New Furanoid Lignan and Anti-Platelet Aggregation Constituents from Piper Arborescens. Planta Med. 71, 535–542. [DOI] [PubMed] [Google Scholar]
- 9.Prier CK, Rankic DA & MacMillan DWC Visible Light Photoredox Catalysis with Transition Metal Complexes: applications in Organic Synthesis. Chem. Rev 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tröster A, Alonso R, Bauer A & Bach T Enantioselective Intermolecular [2 + 2] Photocycloaddition Reactions of 2(1H)-Quinolones Induced by Visible Light Irradiation. JACS 138, 7808–7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lei T et al. General and Efficient Intermolecular [2+ 2] Photodimerization of Chalcones and Cinnamic Acid Derivatives in Solution through Visible-Light Catalysis. Angew. Chem. Int. Ed 56, 15407–15410. [DOI] [PubMed] [Google Scholar]
- 12.Pagire SK, Hossain A, Traub L, Kerres S & Reiser O Photosensitised Regioselective [2+ 2]-Cycloaddition of Cinnamates and Related Alkenes. Chem. Commun 53, 12072–12075. [DOI] [PubMed] [Google Scholar]
- 13.Gutekunst WR & Baran PS Applications of C–H Functionalization Logic to Cyclobutane Synthesis. J. Org. Chem 79, 2430–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poplata S, Tröster A, Zou Y-Q & Bach T Recent Advances in the Synthesis of Cyclobutanes by Olefin [2 + 2] Photocycloaddition Reactions. Chem. Rev 116, 9748–9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zitt H, Dix I, Hopf H & Jones P 4,15-Diamino[2.2]paracyclophane, a Reusable Template for Topochemical Reaction Control in Solution. Eur. J. Org. Chem 2002, 2298–2307. [Google Scholar]
- 16.Bassani DM, Darcos V, Mahony S & Desvergne J-P Supramolecular Catalysis of Olefin [2 + 2] Photodimerization. JACS 122, 8795–8796. [Google Scholar]
- 17.Elacqua E et al. A Supramolecular Protecting Group Strategy Introduced to the Organic Solid State: Enhanced Reactivity through Molecular Pedal Motion. Angew. Chem. Int. Ed 51, 1037–1041. [DOI] [PubMed] [Google Scholar]
- 18.Zhang P et al. Enantioselective Biomimetic Total Syntheses of Katsumadain and Katsumadain C. Org. Lett 14, 162–165. [DOI] [PubMed] [Google Scholar]
- 19.Skiredj A et al. Spontaneous Biomimetic Formation of (±)-Dictazole B under Irradiation with Artificial Sunlight. Angew. Chem. Int. Ed 53, 6419–6424. [DOI] [PubMed] [Google Scholar]
- 20.Pattabiraman M, Natarajan A, Kaliappan R, Mague JT & Ramamurthy V Template Directed Photodimerization of Trans-1, 2-bis (n-pyridyl) Ethylenes and Stilbazoles in Water. Chem. Commun, 4542–4544. [DOI] [PubMed] [Google Scholar]
- 21.Bučar D-K, Sen A, Mariappan SVS & MacGillivray LRA [2+2] Cross-Photodimerisation of Photostable Olefins via a Three-Component Cocrystal Solid solution. Chem. Commun 48, 1790–1792. [DOI] [PubMed] [Google Scholar]
- 22.Mongin C, Garakyaraghi S, Razgoniaeva N, Zamkov M & Castellano FN Direct Observation of Triplet Energy Transfer from Semiconductor Nanocrystals. Science 351, 369–372. [DOI] [PubMed] [Google Scholar]
- 23.Huang Z, Simpson DE, Mahboub M, Li X & Tang ML Ligand Enhanced Upconversion of Near-Infrared Photons with Nanocrystal Light Absorbers. Chem. Sci 7, 4101–4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Efros AL et al. Band-edge Exciton in Quantum Dots of Semiconductors with a Degenerate Valence Band: Dark and Bright Exciton States. Phys. Rev. B 54, 4843. [DOI] [PubMed] [Google Scholar]
- 25.Nirmal M et al. Observation of the “Dark Exciton” in CdSe Quantum Dots. Phys. Rev. Lett 75, 3728. [DOI] [PubMed] [Google Scholar]
- 26.Fritzinger B, Capek RK, Lambert K, Martins JC & Hens Z Utilizing Self-Exchange to Address the Binding of Carboxylic Acid Ligands to CdSe Quantum Dots. J. Am. Chem. Soc 132, 10195–10201. [DOI] [PubMed] [Google Scholar]
- 27.MacGillivray LR et al. Supramolecular Control of Reactivity in the Solid State: From Templates to Ladderanes to Metal−Organic Frameworks. Acc. Chem. Res 41, 280–291. [DOI] [PubMed] [Google Scholar]
- 28.Ito Y Solid-state Organic Photochemistry of Mixed Molecular Crystals. Vol. 3 (Dekker: New York, 1999). [Google Scholar]
- 29.Natarajan AB, B. R in Supramolecular Photochemistry. Controlling Photochemical Processes (ed. Ramamurthy V, Inoue Y) 175–228 (Wiley, 2011). [Google Scholar]
- 30.Mongin C, Moroz P, Zamkov M & Castellano FN Thermally Activated Delayed Photoluminescence from Pyrenyl-Functionalized CdSe Quantum Dots. Nature Chem. 10, 225. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Crystallographic data for the structures reported in this Article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 1900373. Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. All other data supporting the findings of this study are available within the Article (Figures 1–3 and Table 1) and its Supplementary Information (Supplementary Tables 1–8, Supplementary Scheme 1, Supplementary Figures 1–4, Supplementary Sections L, M), or from the corresponding author upon reasonable request.