

Abstract
The genotype-to-phenotype relationship in health and disease is complex and influenced by both an individual’s environment and their unique genome. Personal genetic variants can modulate gene function to generate a phenotype either through a single gene effect or through genetic interactions involving two or more genes. The relevance of genetic interactions to disease phenotypes has been particularly clear in cancer research, where an extreme genetic interaction, synthetic lethality, has been exploited as a therapeutic strategy. The obvious benefits of unmasking genetic background-specific vulnerabilities, coupled with the power of systematic genome editing, have fueled efforts to translate genetic interaction mapping from model organisms to human cells. Here, we review recent developments in genetic interaction mapping, with a focus on CRISPR-based genome editing technologies and cancer.
INTRODUCTION
Our current knowledge of cancer cell function coupled with growing catalogues of genome sequence data for human tumours and cancer cells lines provides a rich foundation for precision oncology [e.g. The Cancer Genome Atlas (TCGA): https://cancergenome.nih.gov/; Catalogue of Somatic Mutations in Cancer (COSMIC) https://cancer.sanger.ac.uk/cosmic]. Genetically tailored therapeutics that take advantage of specific driver pathways can be designed to selectively kill cancer cells. For example, Trastuzumab, an antibody therapeutic that targets the HER2 receptor, is specific for HER2-positive breast cancers, while Imatinib, a tyrosine kinase inhibitor, targets the BCR-ABL fusion protein that drives most chronic myelogenous leukemias. Alternatively, therapies have been designed to exploit vulnerabilities generated by cancer cell-specific genetic variation. For example, BRCA1 and BRCA2-mutant breast and ovarian cancer cells are defective for DNA double strand break (DSB) repair, which renders the cancer cells hypersensitive to small molecules that inhibit the poly(ADP-ribose) polymerase 1 and 2 (PARP1/2) enzymes, which would otherwise initiate DSB repair through alternative mechanisms [1]. These examples illustrate the importance of understanding functional cancer genetics, yet current successes in the clinic that reflect decades of basic research have focused largely on a few key biological pathways. Clearly, there is much to be learned from unbiased systematic analyses of human gene function and genetic interactions (GIs), with a focus on unbiased identification of all biological pathways relevant to cancer cell division and discovering genetic variation that might be exploited to develop targeted therapeutics. In this article, we provide an overview of recent developments in the field of GI mapping with a particular focus on cancer.
CONTEXT-DEPENDENT GENE ESSENTIALITY AND THE CANCER PHENOTYPE
Recent improvements in genome editing technologies, most notably CRISPR (clustered regularly interspaced short palindromic repeats)-based methods, have accelerated the development of resources for genome-scale perturbation of genes in mammalian genomes, opening the door to systematic functional genomics analysis. The first and most widely used CRISPR technology pairs the Cas9 endonuclease with a gRNA (guide RNA) to target it to a given genomic site, where Cas9 induces a DNA double-strand break. Repair by non-homologous end-joining frequently results in insertions or deletions, leading to a functional knock-out of a gene of interest (Table 1). So far, efforts have largely focused on assessing the impact of individual gene perturbation on cell proliferation, a phenotypic read-out that reports on general cell physiology, is scalable and quantitative.
Table 1: Technologies for genetic interaction mapping in mammalian cells.
Overview of current and emerging technologies with potential to aid in genetic interaction mapping in mammalian cells. Corresponding references can be found in main text.
| Technology | Description | Application | Type | Variations | References (mentioned in main text) |
|---|---|---|---|---|---|
| CRISPR mutagenesis (CRISPRm) or cutting (CRISPRc) | genome editing through Cas enzyme-induced double-strand breaks and endogenous repair; targeting by gRNA | pooled and arrayed loss-of-function experiments; genome-scale screens; single-well mechanistic experiments | genomic (coding sequence) | compatible with multi-targeting approaches; homologous-recombination-mediated knock-in; various natural and engineered Cas enzymes with different properties | |
| CRISPR interference (CRISPRi) | transcriptional repression by Cas fusion proteins for targeting and repression | pooled and arrayed loss-of-function experiments; genome-scale screens; single-well mechanistic experiments | transcriptional (promoter region) | compatible with multi-targeting approaches | |
| CRISPR activation (CRISPRa) | transcriptional activation by Cas fusion proteins for targeting and repression | pooled and arrayed gain-of-function experiments; genome-scale screens; single-well mechanistic experiments | transcriptional (promoter region) |
compatible with multi-targeting approaches | |
| CRISPR base editing | mutagenesis of individual bases through modification (e.g. deamination) by Cas fusion proteins | arrayed and single-well experiments; forward-genetic screens | genomic (coding sequence) |
multiple Cas and deaminase versions for editing different bases; CRISPR-STOP for engineering stop codons | |
| epigenetic editing | alteration of chromatin states by Cas fusion proteins with chromatin modifying enzymes | arrayed (possibly also pooled) experiments; single-well mechanistic experiments | epigenomic, transcriptional | multiple modifiers and combinations possible | |
| combinatorial CRISPR | induction of multiple perturbations in the same cell by multiple gRNAs | pooled and arrayed genetic interaction experiments; genome-scale screens; single-well mechanistic experiments | genomic or transcriptional | combinations of CRISPR, CRISPRi and CRISPRa; orthologous promoters or Cas enzymes | |
| Perturb-Seq, CROP-Seq | CRISPR-mediated induction of perturbation coupled to single-cell RNA sequencing | pooled loss- or gain-of-function experiments | genomic (single cell) | different methodologies; compatible with different CRISPR systems | |
| mutational scanning | high-density CRISPR-based mutagenesis | pooled loss- or gain-of-function experiments for single genes | genomic (coding sequence) | different mutagenesis methods possible | |
| unique molecular identifiers | single-cell barcoding (combined with CRISPR-mediated perturbations) | pooled and arrayed loss- or gain-of-function experiments; genome-scale screens; single-well mechanistic experiments; lineage tracking | genomic (single cell) | compatible with various CRISPR systems | |
| optical barcoding | imaging-based readouts coupled to barcoding and in situ sequencing | pooled and arrayed phenotypic experiments; genome-scale screens; single-well mechanistic experiments | genomic (single cell) | multiple phenotypes and various technical options conceivable | |
| protein barcoding (e.g. CITE-seq) | integration of protein-tags as barcodes, measured by antibody-mediated CyTOF technology | pooled and arrayed phenotypic experiments; genome-scale screens; single-well mechanistic experiments | genomic/proteomic (single cell) | multiple phenotypes possible |
Libraries of both RNAi (RNA interference) knock-down reagents and gRNAs for genome-scale CRISPR gene targeting have been applied to human cell lines to identify essential genes required for cell proliferation. These studies revealed a core set of essential genes required for viability in most cell lines, including highly conserved genes whose functions are maintained from yeast to humans, as well as genes that are required for viability only in a subset of cancer cell lines (reviewed in [2,3]). Mirroring the general findings from studies of gene deletion mutants in yeast, the core human cell essential gene set only includes a relatively small fraction (~10%) of the genes in the human genome, highlighting the extensive functional buffering inherent to eukaryotic genomes. Conversely, the variation inherent to specific cancer cell genomes results in context-specific essential genes whose mutation creates cell fitness defects only in a specific genetic background, presumably due to GIs. Importantly, genome-wide screens for GIs offer the potential to convert a given nonessential gene into a context-specific essential gene, and thereby define the genes and pathways that buffer the loss of function of any mutant query gene.
MAPPING GENETIC INTERACTIONS IN CANCER
Generally, a genetic interaction occurs when the fitness phenotype observed for a given double mutant deviates from the phenotype expected based on the two single mutant phenotypes [4]. If the double mutant grows better than expected, the gene pair is said to exhibit a positive GI. If the observed double mutant fitness is less than expected, the two genes display a negative interaction, ranging from synthetic sickness to the most extreme case, synthetic lethality (SL) (Fig. 1A).
Figure 1: Genetic interaction mapping.

(A) Schematic illustration of genetic interactions as measured by single mutant and double mutant fitness. Negative genetic interactions result in lower double mutant fitness than expected (synthetic sick, synthetic lethal), positive genetic interactions in greater fitness than expected (masking or suppressive). (B) Global pairwise genetic interaction network in yeast reveals functional clustering of genes with similar genetic interaction profiles and allows annotation of uncharacterized genes. (C) Expansion of the yeast functional genomics landscape by conditional and trigenic interactions. (D) CRISPR-mediated genetic interaction screens in mammalian cells. Top, gRNA representation in a cell line harbouring a cancer mutation is compared to a wild type cell line to identify synthetic lethal or suppressive interactions. Second, a limited number of defined mutants are generated in an isogenic cell line background and subjected to genome-scale CRISPR screening. Clustering by genetic interaction profile similarity reveals functional information. Third, instead of isogenic mutants, patient-derived cancer cell lines are used. In addition to CRISPR screening, genomic profiling is required to infer the “single mutant state” in those cell lines. Bottom, Direct assessment of pairwise genetic interactions between a limited set of genes by simultaneous delivery of two gRNAs into the same cell.
Efforts to broadly map GIs in human cells are particularly relevant to understanding cancer phenotypes for two reasons. First, GIs complicate our ability to predict phenotype from genotype, a major challenge that must be addressed to realize the promise of precision medicine for cancer. Second, GIs, and SL in particular, are important as a therapeutic concept in cancer. The idea of discovering and exploiting specific genetic vulnerabilities to kill cancer cells while sparing normal tissue was proposed more than 20 years ago [5]. Synthetic lethal interactions in cancer cells can be considered a form of context-dependent gene essentiality [2], although it should be noted that the number of modifiers associated with the GI could be complex rather than a simple digenic effect. SL as a therapeutic model has obvious benefits of potentially reducing side effects of cancer treatments, as well as the possibility of indirectly targeting “undruggable” mutations. These ideas have motivated massive academic and corporate cancer SL screening approaches in a vast range of cell systems, yielding thousands of SL interactions [6].
While the promise is huge, only one SL interaction has been translated into the clinical setting to date: as noted above, breast and ovarian cancer cells carrying mutations in BRCA1 or BRCA2 are highly sensitive to PARP inhibitors [1]. Even though many promising candidates have been identified in tumor cell models [1,7], most published SL interactions have not withstood pre-clinical evaluation. These failures may result from off-target effects, incomplete loss-of-function and poor reproducibility in RNAi screens [8,9], variable consistency of drug screens [10,11], incomplete penetrance [12], and context dependency [2]. These issues, coupled with the fact that GIs are rare, involving on the order of ~1% of tested gene pairs, suggest that efficient discovery of clinically actionable GIs will require a more global analysis of genetic networks that moves beyond individual genes and pathways.
MAPPING GENETIC INTERACTIONS IN MODEL SYSTEMS: A TEMPLATE FOR GENETIC NETWORK ANALYSIS
Model organisms continue to be experimental test-beds for development and implementation of systematic GI studies due to their small genomes, genetic tractability and amenability to high-throughput analyses. Systematic genetics in the budding yeast Saccharomyces cerevisiae enabled assembly of the first comprehensive pairwise GI map surveying nearly all essential and non-essential yeast genes [13]. Most query genes display a number of different negative and positive interactions, and the set of GIs associated with a query gene forms a GI profile. Genes within the same biological pathway or protein complex have highly similar GI profiles, indicating that these profiles provide a quantitative measure of gene function. Indeed, the global map of yeast GI profiles models a powerful approach for annotating gene function, assembling a hierarchical map in which genes are grouped according to functional modules corresponding to pathways and complexes, biological processes, and cellular compartments [13](Fig. 1B). The global yeast GI network can be expanded to include additional layers of complexity, such as genetic suppression [14], trigenic interactions [15], and condition-specific GIs [16] (Fig. 1C). Similar mapping efforts, albeit less comprehensive, have been undertaken in multicellular eukaryotic model organisms, including cells derived from the fruit fly Drosophila melanogaster, whole-organism screens in the nematode Caenorhabditis elegans and the zebrafish Danio rerio.
Coherent sets of negative GIs often occur between genes in two pathways or complexes, which is referred to as a between pathway module (BPM) or within an essential pathway and complex, which is referred to as a within pathway module (WPM) [4], and these network structures of the yeast GI network motivated the development of a method to infer GIs from human GWAS studies in breast cancer [17]. Moreover, the existence of a synthetic lethal interaction in yeast increases the likelihood of finding an interaction between the human paralogues by 3- to 19-fold [18]. Recently, GIs identified in Drosophila cells have been used to guide hypotheses about Wnt signaling in human cancers [19], and a zebrafish GI system helped to establish a role for SPRED1 in mucosal melanoma [20]. These and other studies affirm the power of insights derived from experiments in model systems for guiding insightful human pre-clinical research.
THE CRISPR REVOLUTION IN MAMMALIAN CANCER GENETIC INTERACTION MAPPING
Since the first reports describing use of the bacterial CRISPR-Cas phage defence system for ectopic genome editing, the number of applications and expansions of the CRISPR ‘tool kit’ has exploded [21], revolutionizing the field of functional genomics and GI mapping (see Table 1). In addition to CRISPR-Cas9-mediated loss-of-function genome editing, systems for introducing point mutations and other targeted modifications, as well as for activating and repressing transcription, have been developed and used to interrogate GIs. Recent “classical” or loss-of-function CRISPR screens have identified new SL cancer targets (Fig. 1D) including: [1] sets of acute myeloid leukaemia-specific essential genes [22]; [2] ENL as a specific vulnerability in MLL-AF4-positive acute leukaemia [23]; and [3] interactions between RNF43 and FZD5 in pancreatic cancer [24]. In addition, a SL interaction of BAF-complex-mutant synovial sarcomas and malignant rhabdoid tumours with a non-canonical SWI/SNF complex has been described [25], and RB1-null small cell lung cancer cells are hyper-dependent on aurora kinase [26,27] (see Box 1 for pharmacogenetic interactions). These genetic insights add novel clinically testable hypotheses to the catalogue of cancer SL interactions.
Box 1: Pharmacogenetic interactions.
In addition to mapping gene-gene interactions, CRISPR genome editing has been used to dissect gene-drug or pharmacogenetic interactions. Pharmacogenetic interactions can mimic genetic interactions, but genetic interactions and networks can also be used as a template for interpreting drug-gene interactions, facilitating target identification (diagrammed below). Challenges include altered phenotypes relative to genetic perturbation due to drug off-target effects and inherent differences between pharmacologic inhibition and genetic ablation of a gene/protein of interest. Nonetheless, the identification or confirmation of a genetic interaction using a drug facilitates validation of a pharmacological target for pre-clinical investigations. If the drug displaying the desirable interaction is already in clinical development or approved, the subsequent development process will be accelerated considerably. Recently published pharmacogenetic screens include classic synthetic lethality screens [27] but also efforts using dual-gRNA systems to investigate potential drug synergies or resistance mechanisms [30,32]. On a larger scale, pharmacogenetic screens in hundreds of cancer cell lines are being performed within the framework of ongoing consortium projects listed below. These initiatives aim to produce a comprehensive view of pharmacogenetic interactions in cancer and may be expanded to organoid or in vivo systems.
Pharmacogenomic Screening Consortia:
Genomics of Drug Sensitivity in Cancer (GDSC) –www.cancerrxgene.org
The Connectivity Map (CMAP) –www.broadinstitute.org/connectivity-map-cmap
The Genentech Cell Line Screening Initiative (gCSI) – [11]
The Cancer Therapeutics Response Portal (CTRP) – portals.broadinstitute.org/ctrp
The Cancer Cell Line Encyclopedia (CCLE) - portals.broadinstitute.org/ccle
The Cancer-Drug eXplorer (cDx) - cancerdrugexplorer.org
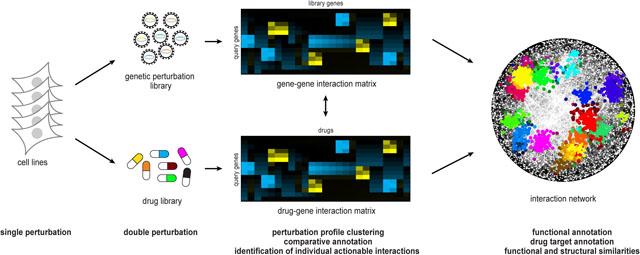
In order to interrogate reciprocal GIs between multiple genes (Fig. 1D), several combinatorial CRISPR-based screening platforms have been developed. For example, dual gRNA systems designed to enable simultaneous knock-out of two genes in the same cell have been used to: [1] interrogate all pairwise interactions of 73 cancer genes in multiple human cancer cell lines to reveal a druggable SL interaction network [28]; [2] survey barcoded dual gRNA combinations targeting 50 genes for their ability to inhibit proliferation of an ovarian cancer cell line [29] and; [3] measure pairwise interactions between ~21,000 druggable genes to generate a large matrix of ~500,000 measurements, identifying potential synergistic drug combinations [30]. Although dual gRNA screens are useful, concerns with respect to introduction of multiple DNA double-strand breaks in conventional Cas screening, as well as relatively poor screening efficiency, have prompted the development of combinatorial screening systems using CRISPR interference (CRISPRi), where endonuclease-deficient Cas9 is used to target transcriptional repressors to gene promoters to simultaneously repress multiple target genes [31]. In one study, CRISPRi was combined with CRISPR activation (CRISPRa), where transcriptional activators are used instead of repressors, by using two different Cas9 proteins from orthologous species to investigate directional GIs in a chronic myeloid leukaemia cell line [32]. A similar system of orthogonal CRISPR enzymes for activation and repression was used to perform GI screens between apoptosis genes, MAP kinases and AKT genes in multiple cancer cell lines, identifying a novel SL between BCL2L1 and MCL [33].
CRISPR tools have also been combined with single-cell technology for increased resolution and sensitivity of screens and an expanded repertoire of screenable phenotypes, such as cell lineage tracking using unique molecular identifiers (e.g. [34]). One recent study also used combinatorial CRISPR perturbations in single cells with transcriptomics as a readout [35], providing a useful avenue for larger scale interrogation of GIs in individual cancer cells. Similarly, CRISPR applications have greatly facilitated cancer mouse model generation, and have allowed direct in vivo screening for inhibitors of tumour growth, metastasis or interaction with the immune system and microenvironment, as reviewed elsewhere (e.g. [36]). These applications illustrate the power and versatility of CRISPR technology, but have not yet reached the capacity for unbiased, large-scale investigations of GIs.
TOWARDS COMPREHENSIVE GENETIC INTERACTION MAPPING IN HUMAN CELLS
The efficiency and ease of CRISPR technologies have catalyzed major efforts to systematically interrogate genetic dependencies in large panels of cancer cell lines like the Broad Institute’s “Cancer Dependency Map Project” (depmap.orgs) that currently contains screens in nearly 500 cancer cell lines. The lessons learned from RNAi applications with respect to quality of reagents, replication of screens, accuracy and robustness of measured phenotypes, as well as data analysis have been invaluable for the rapid development of CRISPR technologies and have led to increased awareness of potential pitfalls that are specific to CRISPR. For instance, multiple groups have reported that targeting amplified genomic regions results in gene-independent reduction of proliferation and viability, suggesting that an increased number of DNA double strand breaks generated by Cas9 causes toxicity [37–39]. However, bioinformatic algorithms can efficiently correct for these and other off-target effects [39,40], enabling integrated analysis of large collections of genome-scale CRISPR loss-of-function screening data. Not only are these datasets a valuable resource for hypothesis-driven data mining, especially with respect to uncovering novel SL interactions, but they also provide the basis for the first attempts towards generating comprehensive co-essentiality networks in human cells. Recent studies have integrated and re-analyzed multiple such datasets to derive cancer GI maps based on CRISPR screen and mutation data [41]. Similar to the analysis of GI profiles from the global yeast GI network, these efforts facilitated the annotation of protein complexes [42] and inspired the proposal that a hierarchical GI network for human cells can be extracted from cancer co-essentiality data [43,44] (Fig. 1D).
While CRISPR has virtually replaced RNAi in functional genomics applications, RNAi technology itself has seen recent technical improvements and computational off-target correction methods for large datasets [45–47]. This has catalyzed further projects to map cancer dependency maps using RNAi within the frameworks of “Project Achilles” and “Project DRIVE” [45,46], whose data are being integrated into the DepMap Project. These initiatives map genetic perturbations that cause a vulnerability in the form of a fitness defect in certain cancer cell lines, but do not directly measure GIs between defined pairs of genes. Nevertheless, systematic pairwise GI mapping in human cells with defined genetic backgrounds has been reported for a small set of cell lines [48,49] and pairwise GI mapping in hundreds to thousands of defined “single mutant” model cell lines is within reach (Fig. 1D). Recently, a first blueprint of a human GI landscape was produced based on experiments in two cancer cell lines [50]. The authors used CRISPRi to perturb over 200,000 gene pairs and used the resulting GI maps to annotate uncharacterized genes by functional clustering, an approach that mirrors GI mapping in yeast (see above). The project also identified novel cancer-relevant negative GIs, demonstrating that large-scale analysis in human cells is feasible and likely to provide rich functional information, facilitating annotation of the normal human and cancer cell genomes (Fig. 1D). Ultimately, the comparison of multiple CRISPR and RNAi datasets, coupled with experimental and clinical validation of the screening results, will provide the community with a clear view of the robustness of different technologies for a chosen biological question or clinical application.
CONCLUSIONS AND OUTLOOK
CRISPR technologies have not only revolutionized the study of individual genes, but also large-scale investigations of gene essentiality and GIs, resulting in an ever-expanding toolbox of experimental and computational methods to find genetic vulnerabilities in cancer cells. Despite enormous technical progress, major challenges remain: the complexity and size of the human genome hampers the generation of global GI maps, benchmarks for robust and clinically relevant GIs remain to be developed, and the context-dependency of GIs in patients, tissues and cell lines represents an important hurdle for cogent mapping of genetic networks.
However, technology development will continue to drive innovations for human GI mapping, as has been the case for mapping of genetic networks in model systems. For example, the integration of other phenotypic readouts beyond cell proliferation holds great promise. Direct readouts include assays of protein-protein interactions or protein levels and modifications [51,52]. Quantitative data-based experimental pipelines that combine automated image analysis and systematic genetic perturbation have been productively used to explore bioprocess-specific GIs in yeast and fly [53,54] and comparable methods that involve optical barcoding and in situ sequencing have been recently developed for mammalian cells, enabling phenotypic profiling at single-cell resolution [55,56]. Single-cell technology will mature and allow more complex phenotyping, for instance by barcoding on the protein level and mass cytometry analyses [57]. A logical expansion of “deep-mutational-scanning”-like technologies, where individual genes are mutagenized to saturation, to the interrogation of allele-specific GIs should prove highly informative. Finally, the growing CRISPR toolbox will be exploited for engineering more precise genetic lesions for use in GI analyses by base editing or by using engineered or orthologous Cas enzymes with altered or expanded target site preferences [21]. These technologies will allow for conditional GI screens, similar to model systems [16], time resolved or gene dosage screens [54,58], and multiplexing for interrogation of higher-order GIs [59]. Importantly, most of the described applications are in principle also applicable to in vivo or 3D-culture systems such as organoids, albeit at smaller scale.
The application of diverse methods for genetic network analysis in cancer will require advanced strategies for data integration, which ultimately will result in improved coverage of gene (protein) interactions, as suggested by maps generated by combining genetic with protein-protein interaction and phosphoproteomics data [60,61]. The ever-growing repository of cancer genomics and GWAS data has sparked the development of approaches for data compilation and data mining or candidate prioritization [62], which will soon be enabled by powerful deep learning technologies to model cellular networks [63]. A global understanding of human genetic networks in healthy and diseased cells and tissues demands a systematic community effort, and will lead to new mechanistic insight into cancer, generate informed, clinically testable hypotheses and, ultimately better therapies.
ACKNOWLEDGEMENTS
We thank Michael Aregger and Michael Costanzo for valuable comments on the manuscript and for help with figures. Work on genetic networks in mammalian cell lines in the Andrews, Boone and Moffat labs is supported by grants from the Ontario Research Fund-Research Excellence (ORF-RE7 and -RE9) program, the Canadian Institutes of Health Research, the National Institutes of Health (R01HG005853) and the Canadian Foundation for Innovation. CB, BJA and JM are co-Director (CB) and Senior Fellows (BJA, JM) of the Genetic Networks Program in the Canadian Institute for Advanced Research. We apologize to colleagues whose work could not be cited due to space limitations for references.
REFERENCES
- 1.Ashworth A, Lord CJ: Synthetic lethal therapies for cancer: what’s next after PARP inhibitors? Nat Rev Clin Oncol 2018, 15:564–576. [DOI] [PubMed] [Google Scholar]
- 2.Rancati G, Moffat J, Typas A, Pavelka N: Emerging and evolving concepts in gene essentiality. Nat Rev Genet 2017, 19:34–49. [DOI] [PubMed] [Google Scholar]
- 3.Bartha I, di Iulio J, Venter JC, Telenti A: Human gene essentiality. Nat Rev Genet 2018, 19:51–62. [DOI] [PubMed] [Google Scholar]
- 4.VanderSluis B, Costanzo M, Billmann M, Ward HN, Myers CL, Andrews BJ, Boone C: Integrating genetic and protein–protein interaction networks maps a functional wiring diagram of a cell. Curr Opin Microbiol 2018, 45:170–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hartwell LH, Szankasi P, Roberts CJ, Murray AW, Friend SH: Integrating genetic approaches into the discovery of anticancer drugs. Science 1997, 278:1064–8. [DOI] [PubMed] [Google Scholar]
- 6.O’Neil NJ, Bailey ML, Hieter P: Synthetic lethality and cancer. Nat Rev Genet 2017, doi: 10.1038/nrg.2017.47. [DOI] [PubMed] [Google Scholar]
- 7.Shen JP, Ideker T: Synthetic Lethal Networks for Precision Oncology: Promises and Pitfalls. J Mol Biol 2018, doi: 10.1016/J.JMB.2018.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]; *Excellent recent review highlighting concepts, challenges and examples of synthetic lethal interactions in cancer research and therapy, also introduces the concept of synthetic lethal networks.
- 8.Evers B, Jastrzebski K, Heijmans JPM, Grernrum W, Beijersbergen RL, Bernards R: CRISPR knockout screening outperforms shRNA and CRISPRi in identifying essential genes. Nat Biotechnol 2016, 34:631–3. [DOI] [PubMed] [Google Scholar]
- 9.Morgens DW, Deans RM, Li A, Bassik MC: Systematic comparison of CRISPR/Cas9 and RNAi screens for essential genes. Nat Biotechnol 2016, 34:634–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haibe-Kains B, El-Hachem N, Birkbak NJ, Jin AC, Beck AH, Aerts HJWL, Quackenbush J: Inconsistency in large pharmacogenomic studies. Nature 2013, doi: 10.1038/nature12831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haverty PM, Lin E, Tan J, Yu Y, Lam B, Lianoglou S, Neve RM, Martin S, Settleman J, Yauch RL, et al. : Reproducible pharmacogenomic profiling of cancer cell line panels. Nature 2016, 533:333–337. [DOI] [PubMed] [Google Scholar]
- 12.Ryan CJ, Bajrami I, Lord CJ: Synthetic Lethality and Cancer - Penetrance as the Major Barrier. Trends in cancer 2018, 4:671–683. [DOI] [PubMed] [Google Scholar]; *Interesting perspective on phenotypic penetrance as an understudied factor in human cancer synthetic lethality experiments.
- 13.Costanzo M, VanderSluis B, Koch EN, Baryshnikova A, Pons C, Tan G, Wang W, Usaj M, Hanchard J, Lee SD, et al. : A global genetic interaction network maps a wiring diagram of cellular function. Science 2016, 353. [DOI] [PMC free article] [PubMed] [Google Scholar]; **The first complete genetic interaction network of a eukaryotic cell.
- 14.van Leeuwen J, Pons C, Mellor JC, Yamaguchi TN, Friesen H, Koschwanez J, U aj MM, Pechlaner M, Takar M, U aj M, et al. : Exploring genetic suppression interactions on a global scale. Science (80-) 2016, 354:aag0839–aag0839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuzmin E, VanderSluis B, Wang W, Tan G, Deshpande R, Chen Y, Usaj M, Balint A, Mattiazzi Usaj M, van Leeuwen J, et al. : Systematic analysis of complex genetic interactions. Science 2018, 360:eaao1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Domingo J, Diss G, Lehner B: Pairwise and higher-order genetic interactions during the evolution of a tRNA. Nature 2018, 558:117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang W, Xu ZZ, Costanzo M, Boone C, Lange CA, Myers CL: Pathway-based discovery of genetic interactions in breast cancer. PLOS Genet 2017, 13:e1006973. [DOI] [PMC free article] [PubMed] [Google Scholar]; *Application of principles derived from yeast genetic interaction networks to discover genetic interactions in human breast cancer GWAS data.
- 18.Srivas R, Shen JP, Yang CC, Sun SM, Li J, Gross AM, Jensen J, Licon K, Bojorquez-Gomez A, Klepper K, et al. : A Network of Conserved Synthetic Lethal Interactions for Exploration of Precision Cancer Therapy. Mol Cell 2016, 63:514–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Billmann M, Chaudhary V, ElMaghraby MF, Fischer B, Boutros M: Widespread Rewiring of Genetic Networks upon Cancer Signaling Pathway Activation. Cell Syst 2017, 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ablain J, Xu M, Rothschild H, Jordan RC, Mito JK, Daniels BH, Bell CF, Joseph NM, Wu H, Bastian BC, et al. : Human tumor genomics and zebrafish modeling identify SPRED1 loss as a driver of mucosal melanoma. Science 2018, doi: 10.1126/science.aau6509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adli M: The CRISPR tool kit for genome editing and beyond. Nat Commun 2018, 9:1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao D, Lu X, Wang G, Lan Z, Liao W, Li J, Liang X, Chen JR, Shah S, Shang X, et al. : Synthetic essentiality of chromatin remodelling factor CHD1 in PTEN-deficient cancer. Nature 2017, doi: 10.1038/nature21357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Erb MA, Scott TG, Li BE, Xie H, Paulk J, Seo H-S, Souza A, Roberts JM, Dastjerdi S, Buckley DL, et al. : Transcription control by the ENL YEATS domain in acute leukaemia. Nature 2017, 543:270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Steinhart Z, Pavlovic Z, Chandrashekhar M, Hart T, Wang X, Zhang X, Robitaille M, Brown KR, Jaksani S, Overmeer R, et al. : Genome-wide CRISPR screens reveal a Wnt–FZD5 signaling circuit as a druggable vulnerability of RNF43-mutant pancreatic tumors. Nat Med 2016, 23:60–68. [DOI] [PubMed] [Google Scholar]
- 25.Michel BC, D’Avino AR, Cassel SH, Mashtalir N, McKenzie ZM, McBride MJ, Valencia AM, Zhou Q, Bocker M, Soares LMM, et al. : A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat Cell Biol 2018, doi: 10.1038/s41556-018-0221-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oser MG, Fonseca R, Chakraborty AA, Brough R, Spektor A, Jennings RB, Flaifel A, Novak JS, Gulati A, Buss E, et al. : Cells Lacking the RB1 Tumor Suppressor Gene are Hyperdependent on Aurora B Kinase for Survival. Cancer Discov 2018, doi: 10.1158/2159-8290.CD-18-0389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gong X, Du J, Parsons SH, Merzoug FF, Webster Y, Iversen PW, Chio L-C, Van Horn RD, Lin X, Blosser W, et al. : Aurora-A kinase inhibition is synthetic lethal with loss of the RB1 tumor suppressor gene. Cancer Discov 2018, doi: 10.1158/2159-8290.CD-18-0469. [DOI] [PubMed] [Google Scholar]
- 28.Shen JP, Zhao D, Sasik R, Luebeck J, Birmingham A, Bojorquez-Gomez A, Licon K, Klepper K, Pekin D, Beckett AN, et al. : Combinatorial CRISPR–Cas9 screens for de novo mapping of genetic interactions. Nat Methods 2017, doi: 10.1038/nmeth.4225. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This study and references 29–33 are the first examples of multiplexed or combinatorial genome editing to study genetic interactions in human cells.
- 29.Wong ASL, Choi GCG, Cui CH, Pregernig G, Milani P, Adam M, Perli SD, Kazer SW, Gaillard A, Hermann M, et al. : Multiplexed barcoded CRISPR-Cas9 screening enabled by CombiGEM. Proc Natl Acad Sci U S A 2016, 113:2544–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han K, Jeng EE, Hess GT, Morgens DW, Li A, Bassik MC: Synergistic drug combinations for cancer identified in a CRISPR screen for pairwise genetic interactions. Nat Biotechnol 2017, doi: 10.1038/nbt.3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Du D, Roguev A, Gordon DE, Chen M, Chen S-H, Shales M, Shen JP, Ideker T, Mali P, Qi LS, et al. : Genetic interaction mapping in mammalian cells using CRISPR interference. Nat Methods 2017, doi: 10.1038/nmeth.4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boettcher M, Tian R, Blau JA, Markegard E, Wagner RT, Wu D, Mo X, Biton A, Zaitlen N, Fu H, et al. : Dual gene activation and knockout screen reveals directional dependencies in genetic networks. Nat Biotechnol 2018, 36:170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Najm FJ, Strand C, Donovan KF, Hegde M, Sanson KR, Vaimberg EW, Sullender ME, Hartenian E, Kalani Z, Fusi N, et al. : Orthologous CRISPR-Cas9 enzymes for combinatorial genetic screens. Nat Biotechnol 2018, 36:179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Michlits G, Hubmann M, Wu S-H, Vainorius G, Budusan E, Zhuk S, Burkard TR, Novatchkova M, Aichinger M, Lu Y, et al. : CRISPR-UMI: single-cell lineage tracing of pooled CRISPR–Cas9 screens. Nat Methods 2017, 14:1191–1197. [DOI] [PubMed] [Google Scholar]
- 35.Adamson B, Norman TM, Jost M, Cho MY, Nuñez JK, Chen Y, Villalta JE, Gilbert LA, Horlbeck MA, Hein MY, et al. : A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell 2016, 167:1867–1882.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]; *The first combination of combinatorial CRISPR and single-cell technologies.
- 36.Winters IP, Murray CW, Winslow MM: Towards quantitative and multiplexed in vivo functional cancer genomics. Nat Rev Genet 2018, 19:741–755. [DOI] [PubMed] [Google Scholar]
- 37.Aguirre AJ, Meyers RM, Weir BA, Vazquez F, Zhang C-Z, Ben-David U, Cook A, Ha G, Harrington WF, Doshi MB, et al. : Genomic Copy Number Dictates a Gene-Independent Cell Response to CRISPR/Cas9 Targeting. Cancer Discov 2016, 6:914–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Munoz DM, Cassiani PJ, Li L, Billy E, Korn JM, Jones MD, Golji J, Ruddy DA, Yu K, McAllister G, et al. : CRISPR Screens Provide a Comprehensive Assessment of Cancer Vulnerabilities but Generate False-Positive Hits for Highly Amplified Genomic Regions. Cancer Discov 2016, 6:900–913. [DOI] [PubMed] [Google Scholar]
- 39.Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, et al. : Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet 2017, 49:1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]; **In addition to introducing an algorithm to correct copy number effects in CRISPR screens, this is the first “Cancer Dependency Map Project” study, containing data from 342 genome-scale cancer cell line screens, the largest CRISPR dataset to date.
- 40.Morgens DW, Wainberg M, Boyle EA, Ursu O, Araya CL, Kimberly Tsui C, Haney MS, Hess GT, Han K, Jeng EE, et al. : Genome-scale measurement of off-target activity using Cas9 toxicity in high-throughput screens. Nat Commun 2017, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rauscher B, Heigwer F, Henkel L, Hielscher T, Voloshanenko O, Boutros M: Toward an integrated map of genetic interactions in cancer cells. Mol Syst Biol 2018, 14:e7656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pan J, Meyers RM, Michel BC, Mashtalir N, Sizemore AE, Wells JN, Cassel SH, Vazquez F, Weir BA, Hahn WC, et al. : Interrogation of Mammalian Protein Complex Structure, Function, and Membership Using Genome-Scale Fitness Screens. Cell Syst 2018, 6:555–568.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]; *Interesting and extensive re-analysis of DepMap and human protein complex data.
- 43.Kim E, Dede M, Lenoir WF, Wang G, Srinivasan S, Colic M, Hart T: Hierarchical organization of the human cell from a cancer coessentiality network. bioRxiv 2018, doi: 10.1101/328880. [DOI] [Google Scholar]
- 44.Boyle EA, Pritchard JK, Greenleaf WJ: High-resolution mapping of cancer cell networks using co-functional interactions. Mol Syst Biol 2018, 14:e8594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. : Defining a Cancer Dependency Map. Cell 2017, 170:564–576.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This study and references 46–47 provide resources for large, improved RNAi datasets and novel cancer dependencies.
- 46.McDonald ER, de Weck A, Schlabach MR, Billy E, Mavrakis KJ, Hoffman GR, Belur D, Castelletti D, Frias E, Gampa K, et al. : Project DRIVE: A Compendium of Cancer Dependencies and Synthetic Lethal Relationships Uncovered by Large-Scale, Deep RNAi Screening. Cell 2017, 170:577–592.e10. [DOI] [PubMed] [Google Scholar]
- 47.McFarland JM, Ho ZV, Kugener G, Dempster JM, Montgomery PG, Bryan JG, Krill-Burger JM, Green TM, Vazquez F, Boehm JS, et al. : Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration. Nat Commun 2018, 9:4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vizeacoumar FJ, Arnold R, Vizeacoumar FS, Chandrashekhar M, Buzina A, Young JTF, Kwan JHM, Sayad A, Mero P, Lawo S, et al. : A negative genetic interaction map in isogenic cancer cell lines reveals cancer cell vulnerabilities. Mol Syst Biol 2013, 9:696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Blomen VA, Májek P, Jae LT, Bigenzahn JW, Nieuwenhuis J, Staring J, Sacco R, van Diemen FR, Olk N, Stukalov A, et al. : Gene essentiality and synthetic lethality in haploid human cells. Science 2015, 350:1092–6. [DOI] [PubMed] [Google Scholar]
- 50.Horlbeck MA, Xu A, Wang M, Bennett NK, Park CY, Bogdanoff D, Adamson B, Chow ED, Kampmann M, Peterson TR, et al. : Mapping the Genetic Landscape of Human Cells. Cell 2018, 0. [DOI] [PMC free article] [PubMed] [Google Scholar]; **The largest pairwise genetic interaction study in human cell lines to date, generating a first human draft genetic interaction network.
- 51.Diss G, Lehner B: The genetic landscape of a physical interaction. Elife 2018, 7:e32472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brockmann M, Blomen VA, Nieuwenhuis J, Stickel E, Raaben M, Bleijerveld OB, Altelaar AFM, Jae LT, Brummelkamp TR: Genetic wiring maps of single-cell protein states reveal an off-switch for GPCR signalling. Nature 2017, 546:307–311. [DOI] [PubMed] [Google Scholar]
- 53.Kraus OZ, Grys BT, Ba J, Chong Y, Frey BJ, Boone C, Andrews BJ: Automated analysis of high‐content microscopy data with deep learning. Mol Syst Biol 2017, 13:924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Heigwer F, Scheeder C, Miersch T, Schmitt B, Blass C, Pour Jamnani MV, Boutros M: Time-resolved mapping of genetic interactions to model rewiring of signaling pathways. Elife 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.de Groot R, Lüthi J, Lindsay H, Holtackers R, Pelkmans L: Large-scale image-based profiling of single-cell phenotypes in arrayed CRISPR-Cas9 gene perturbation screens. Mol Syst Biol 2018, 14:e8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Feldman D, Singh A, Schmid-Burgk JL, Mezger A, Garrity AJ, Carlson RJ, Zhang F, Blainey P: Pooled optical screens in human cells. bioRxiv 2018, doi: 10.1101/383943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wroblewska A, Dhainaut M, Ben-Zvi B, Rose SA, Park ES, Amir E-AD, Bektesevic A, Baccarini A, Merad M, Rahman AH, et al. : Protein Barcodes Enable High-Dimensional Single-Cell CRISPR Screens. Cell 2018, 175:1141–1155.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sack LM, Davoli T, Li MZ, Li Y, Xu Q, Naxerova K, Wooten EC, Bernardi RJ, Martin TD, Chen T, et al. : Profound Tissue Specificity in Proliferation Control Underlies Cancer Drivers and Aneuploidy Patterns. Cell 2018, 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zetsche B, Heidenreich M, Mohanraju P, Fedorova I, Kneppers J, Degennaro EM, Winblad N, Choudhury SR, Abudayyeh OO, Gootenberg JS, et al. : Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat Biotechnol 2017, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rosenbluh J, Mercer J, Shrestha Y, Oliver R, Tamayo P, Doench JG, Tirosh I, Piccioni F, Hartenian E, Horn H, et al. : Genetic and Proteomic Interrogation of Lower Confidence Candidate Genes Reveals Signaling Networks in β-Catenin-Active Cancers. Cell Syst 2016, 3:302–316.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Drake JM, Paull EO, Graham NA, Lee JK, Smith BA, Titz B, Stoyanova T, Faltermeier CM, Uzunangelov V, Carlin DE, et al. : Phosphoproteome Integration Reveals Patient-Specific Networks in Prostate Cancer. Cell 2016, 166:1041–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim J, Reardon B, et al. : Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173:371–385.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ma J, Yu MK, Fong S, Ono K, Sage E, Demchak B, Sharan R, Ideker T: Using deep learning to model the hierarchical structure and function of a cell. Nat Methods 2018, doi: 10.1038/nmeth.4627. [DOI] [PMC free article] [PubMed] [Google Scholar]