

Abstract
Background
Scaffolding is an important step in genome assembly that orders and orients the contigs produced by assemblers. However, repetitive regions in contigs usually prevent scaffolding from producing accurate results. How to solve the problem of repetitive regions has received a great deal of attention. In the past few years, long reads sequenced by third-generation sequencing technologies (Pacific Biosciences and Oxford Nanopore) have been demonstrated to be useful for sequencing repetitive regions in genomes. Although some stand-alone scaffolding algorithms based on long reads have been presented, scaffolding still requires a new strategy to take full advantage of the characteristics of long reads.
Results
Here, we present a new scaffolding algorithm based on long reads and contig classification (SLR). Through the alignment information of long reads and contigs, SLR classifies the contigs into unique contigs and ambiguous contigs for addressing the problem of repetitive regions. Next, SLR uses only unique contigs to produce draft scaffolds. Then, SLR inserts the ambiguous contigs into the draft scaffolds and produces the final scaffolds. We compare SLR to three popular scaffolding tools by using long read datasets sequenced with Pacific Biosciences and Oxford Nanopore technologies. The experimental results show that SLR can produce better results in terms of accuracy and completeness. The open-source code of SLR is available at https://github.com/luojunwei/SLR.
Conclusion
In this paper, we describes SLR, which is designed to scaffold contigs using long reads. We conclude that SLR can improve the completeness of genome assembly.
Keywords: Scaffolding, Genome assembly, Sequence analysis, Pacific biosciences, Oxford Nanopore
Background
With the increasing availability of third-generation sequencing technologies, which include Single-Molecule Real-Time (SMRT) technology from Pacific Biosciences and Nanopore-based technology from Oxford Nanopore, many biological applications have been greatly improved. Compared with second-generation sequencing technologies, third-generation sequencing technologies produce longer reads with a higher sequencing error rate [1]. In the field of de novo genome assembly, a large number of assembly tools based on third-generation sequencing technologies have been presented to resolve the most prominent problem: repetitive regions. However, producing a complete and accurate assembly is still a challenging task. Scaffolding is an important step in the pipeline of genome assembly, and aims to orient and order contigs [2, 3]. Scaffolding generates scaffolds consisting of sequence fragments including oriented and ordered contigs. The gap between two adjacent contigs in a scaffold is filled with ’N’ characters. Scaffolding can significantly increase the continuity of assembly results and benefit downstream analyses such as those of gene order, comparative or functional genomics and patterns of recombination [4].
According to the kind of reads used for scaffolding, existing scaffolding tools generally fall into the following three categories:
(i) Using paired reads for scaffolding. The insert size of paired reads can reach a few thousands bases, so this technique can partially resolve the problem of repetitive regions. Such scaffolding tools, such as OPERA [5], SSPACE [6], BESST [7], ScaffMatch [8], SCARPA [9], ScaffoldScaffolder [10], and BOSS [11], usually use greedy heuristic algorithms to generate scaffolds based on a scaffold graph, in which a vertex denotes a contig and an edge represents the existence of paired reads that can be separately aligned to the two corresponding contigs. However, because the length of reads from second-generation technologies is commonly only a few hundred bases, the reads can usually be aligned with two or more positions in the contigs. Moreover, the region between the paired reads is unknown, and there are sequencing errors in the reads. Some spurious edges are usually introduced into a scaffold graph, which complicates the scaffolding task. Obtaining more accurate and contiguous scaffolding results based on paired reads is a difficult task.
(ii) Using long reads for scaffolding. This kind of scaffolding tool usually aligns the long reads against contigs first and then finds contigs that can be aligned with the same long read. Then, these tools use the local alignment result to infer the global order and orientation of contigs. For instance, SSPACE-LongRead [12] first aligns whole long reads with contigs using the alignment tool BLASR [13]. Next, contig pairs and multi-contig linkage information are obtained and used to order and orient the contigs and generate scaffolds. LINKS [14] does not align the whole long reads to the contigs; it first extracts the k-mer pairs in an interval from long reads. Afterwards, these k-mer pairs are aligned to the contigs, and the alignment results are used to link the contigs. Finally, LINKS selects a neighbour of a contig as its correct neighbour based on the number of links. SMSC [15] first aligns the long reads to the contigs using either Nucmer [16] or BLASR and then constructs a breakpoint graph in which a vertex is a contig and an edge is added to indicate a long read bridging two vertices. It transforms the scaffolding problem to a maximum alternating path coverage problem in the breakpoint graph and resolves this problem using a 2-approximation algorithm. RAILS [17] scaffolds contigs with long reads using the scaffolding engine originally developed for SSAKE [18] and LINKS. Based on the sequencing coverage of each contig, npScarf [19] classifies contigs into unique contigs and repetitive contigs. npScarf first bridges the unique contigs and generates scaffolds based on a greedy strategy and then fills the gaps by repetitive contigs. However, most contig sets used for scaffolding do not include information on sequencing coverage, which limits the application of npScarf.
(iii) Using optical mapping data or Hi-C data for scaffolding. Optical mapping data can serve as a unique "fingerprint" or "barcode" for genome sequences. By comparing optical mapping data with a restriction enzyme map of the contigs, the order and orientation of contigs can be inferred. Supernova [20], Architect [21], ARCS [22] and fragScaff [23] attempt to find pairs of contigs based on linked reads. The problem with using optical mapping data is that a barcode used to locate contigs may have many different alignment positions, which usually causes contradictions between contigs. Hi-C data are commonly sequenced by paired-end sequencing. The paired reads come from the interacting fragments between genomic loci that are nearby in three-dimensional space but may be separated by many nucleotides in the linear genome. Scaffolding using Hi-C data is the most challenging method, as the genomic distance between a given Hi-C-based read pair is highly variable and may span a few kilobases to megabases without any direct indication of the true distance [1].
Although some scaffolding tools based on long reads have made great progress, two primary issues still require more attention. (i) Scaffold graph construction: In a scaffold graph, each vertex refers to a contig, and an edge is created between two vertices if the two contigs can be aligned with the same long read. Due to the repetitive regions in contigs and the high sequencing error rate of long reads, the scaffold graph usually becomes very complicated, which has negative effects on the later scaffolding steps. Hence, simplifying the scaffold graph is a significant goal for scaffolding. (ii) Edge weighting: In the scaffold graph, most current methods prefer to weight each edge by the number of long reads that can be aligned with two vertices simultaneously. However, the length of the alignment between a long read and a contig can reflect the confidence level of the alignment, which is usually ignored by existing methods.
When a long read links the two flanking regions of a repetitive region, the problem of the repetitive region can be resolved because the order and orientation of the two flanking regions can be obtained directly. Moreover, a repetitive region can usually be aligned with more than one long read, and their 5’-end (or 3’-end) neighbour regions are not the same. After aligning the long reads against the contigs, we can identify whether contigs are repetitive based on their aligniment positions in the long reads. When constructing a scaffold graph, it is difficult to avoid spurious edges introduced by repetitive contigs and sequencing errors. We can identify spurious edges by detecting orientation and position contradictions in the scaffold graph [10, 11]. Using only non-repetitive contigs to construct a scaffold graph not only simplifies the complexity of the scaffold graph but also improves the accuracy of spurious edge detection.
In this paper, we present a scaffolding algorithm based on long reads and contig classification (SLR), which utilizes two new strategies to address the two issues above. For issue (i), SLR classifies the contigs into unique contigs and ambiguous contigs. SLR utilizes the unique contigs to construct a scaffold graph, which can decrease the complexity of the scaffold graph and simplify the following scaffolding steps. For issue (ii), SLR uses the alignment length to weight each edge in the scaffold graph. Moreover, SLR employs linear programming to detect and remove the contradictions in the scaffold graph, which guarantees that the scaffold graph includes only simple paths.
Based on these two new strategies, SLR determines the orientations and orders of the contigs. In experiments, SLR is compared with three popular scaffolding tools by scaffolding five long-read datasets with Pacific Biosciences and Oxford Nanopore technologies. The experimental results show that SLR produces better results in terms of accuracy and completion for most datasets.
Results
To evaluate the performance of SLR, we compared SLR with three popular scaffolding tools based on long reads, namely, SSPACE-LongRead (SSPACE-LR), LINKS and npScarf.
Datasets and metrics
Contig and long-read datasets for Escherichia coli (E. coli), Saccharomyces cerevisiae W303 (S. cerevisiae), and Human chromosome X (Chr X) were utilized as input for all tools. E. coli and S. cerevisiae include two different long-read datasets sequenced with Pacific Biosciences and Oxford Nanopore technologies and consist of two different contig sets assembled by different assemblers. The long reads for Chr X are from Pacific Biosciences. The details of the long-read datasets are shown in Table 1. The contig sets, which were evaluated by QUAST [24], are shown in Table 2. Then, these contig sets and long-read sets form nine datasets, shown in Table 3, were used for scaffolding, and each dataset included one contig set and one long-read set. We named the nine datasets as E. coli_1_SMRT, E. coli_2_SMRT, S. cerevisiae_1_SMRT, S. cerevisiae_2_SMRT, Chr X_1_SMRT, E. coli_1_ONT, E. coli_2_ONT, S. cerevisiae_1_ONT, and S. cerevisiae_2_ONT.
Table 1.
Details of long-read datasets
| E. coli | S. cerevisiae | Chr X | |||
|---|---|---|---|---|---|
| Genome size(Mbp) | 4.6 | 12.1 | 155.2 | ||
| Sequencing technology | SMRT | Nanopore | SMRT | Nanopore | SMRT |
| Read N50(bp) | 5,189 | 8,484 | 6,794 | 8,608 | 11,030 |
| Number of reads | 81,737 | 20,750 | 594,243 | 410,344 | 1,135,220 |
| Name | E. coli_SMRT | E. coli_ONT | S. cerevisiae_SMRT | S. cerevisiae_ONT | Chr X_SMRT |
Table 2.
Details of contig sets
| Contig set | Count | Errors | Genome | Mismatches | Indels | Largest | NA50 | NGA50 |
|---|---|---|---|---|---|---|---|---|
| Fraction(%) | alignment | |||||||
| E. coli_1 | 182 | 2 | 99.363 | 1.32 | 0.37 | 315,628 | 106,208 | 106,208 |
| E. coli_2 | 167 | 7 | 99.351 | 2.28 | 0.11 | 360,084 | 164,044 | 164,044 |
| S. cerevisiae_1 | 3179 | 35 | 96.688 | 79.03 | 8.42 | 233,103 | 47,994 | 52,239 |
| S. cerevisiae_2 | 6953 | 53 | 96.687 | 85.54 | 8.76 | 250,180 | 49,258 | 54,160 |
| Chr X_1 | 8623 | 41 | 97.037 | 2.40 | 1.28 | 793,618 | 76,506 | 71,372 |
Table 3.
Datasets used for scaffolding and evaluations for SLR and SLR1
| Dataset | Genome | Contig set | Long read set | Misassemblies | NGA50 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| SLR | SLR1 | SLR | SLR1 | |||||||
| E. coli_1_SMRT | E. coli | E. coli_1 | E. coli_SMRT | 4 | 12 | 723,879 | 295,999 | |||
| E. coli_2_SMRT | E. coli | E. coli_2 | E. coli_SMRT | 10 | 11 | 565,864 | 197,175 | |||
| S. cerevisiae_1_SMRT | S. cerevisiae | S. cerevisiae_1 | S. cerevisiae_SMRT | 52 | 57 | 374,744 | 232,712 | |||
| S. cerevisiae_2_SMRT | S. cerevisiae | S. cerevisiae_2 | S. cerevisiae_SMRT | 71 | 67 | 270,402 | 201,922 | |||
| Chr X_1_SMRT | Chr X | Chr X_1 | Chr X_SMRT | 83 | 82 | 2,390,483 | 2,165,615 | |||
| E. coli_1_ONT | E. coli | E. coli_1 | E. coli_ONT | 4 | 8 | 2,927,247 | 674,408 | |||
| E. coli_2_ONT | E. coli | E. coli_2 | E. coli_ONT | 9 | 14 | 733,062 | 361,345 | |||
| S. cerevisiae_1_ONT | S. cerevisiae | S. cerevisiae_1 | S. cerevisiae_ONT | 46 | 66 | 374,835 | 244,417 | |||
| S. cerevisiae_2_ONT | S. cerevisiae | S. cerevisiae_2 | S. cerevisiae_ONT | 68 | 85 | 270,362 | 201,066 | |||
Each dataset includes one contig set and one long-read set, and corresponds to one genome.
QUAST aligns the contigs (or scaffolds) to the reference genome and obtains some metrics. NG50 is the length of the longest contig (or scaffold) such that all the contigs (or scaffolds) of that length or longer cover at least half of the reference genome. N50 is the length of the longest contig (or scaffold) such that all the contigs (or scaffolds) of that length or longer cover at least half of the length of all contigs (or scaffolds). Misassemblies (Errors) is the number of positions (breakpoints) in the contigs or scaffolds in which errors (Translocation, Inversion, Relocation) occur. NGA50 is the NG50 of contigs or scaffolds after they have been broken at every breakpoint. Genome Fraction is the percentage of aligned bases in the reference genome. Usually, Misassemblies can represent the accuracy of the scaffolding result, and NGA50 and NA50 can reflect the completion and continuity of the scaffolding result. In the experiments below, we used QUAST to evaluate the scaffolding results for SSPACE-LR, LINKS, npScarf and SLR.
Evaluations on nine datasets
The long-read sets about first five datasets are obtained by SMRT technology. And, the long-read sets about last four datasets are obtained by Nanopore technology. All the scaffolding tools were run on these nine datasets, and detailed evaluation results from QUAST are shown in Additional file 1: Tables S1 and S2. Because NGA50 and Misassemblies are two important metrics for evaluating scaffolding tools, we show NGA50 vs Misassemblies in Fig. 1. The best scaffolding result can found in the top-left corner of each figure. Except in Fig. 1(b) and Fig. 1(i), SLR is in the top-left corner throughout Fig. 1, which indicates that SLR has lower Misassemblies and a higher NGA50. Although npScarf performs better in Fig. 1(b) and Fig. 1(i), the performance of SLR is close to it.
Fig. 1.
Nine figures plotting NGA50 vs Misassemblies. The results of SLR usually can be found in the top-left corner, which can illustrate the advantage of SLR
Running time and peak memory
Due to the high error rate in long reads, aligning long reads with contigs usually takes a long time. LINKS selects k-mer pairs from the long reads to link the contigs, which avoids long read alignment. However, LINKS requires more memory to store the k-mer pairs. As shown in Table 4, we find that LINKS consumes less time and more memory. SLR and npScarf have similar time consumption, because both use BWA-MEM [25] to align long reads against contigs. In all experiments, npScarf allocates a large memory despite the size of the dataset. When extracting alignment information from the BAM file, SLR keeps the alignment of one long read in memory and produces a local scaffold that is saved on the hard disk. After processing one long read, SLR processes the next long read, which can reduce the memory requirement. Compared with other tools, SSPACE-LR and SLR require less memory for scaffolding.
Table 4.
Running time and peak memory
| Dataset | Running time | Peak memory (G) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| SSPACE-LR | LINKS | npScarf | SLR | SSPACE-LR | LINKS | npScarf | SLR | |||
| E. coli_1_SMRT | 41m42s | 1m42s | 26m34s | 26m58s | 1.00 | 6.23 | 10.28 | 1.04 | ||
| E. coli_2_SMRT | 42m12s | 1m42s | 26m46s | 28m17s | 1.00 | 6.22 | 10.28 | 1.17 | ||
| S. cerevisiae_1_SMRT | 880m22s | 38m26s | 929m36s | 907m23s | 3.96 | 93.27 | 10.28 | 1.86 | ||
| S. cerevisiae_2_SMRT | 1162m28s | 40m35s | 1012m27s | 957m59s | 3.96 | 93.3 | 10.28 | 3.60 | ||
| Chr X_1_SMRT | 8413m53s | 41m2s | 6617m53s | 7782m13s | 12.56 | 114.2 | 12.82 | 3.98 | ||
| E. coli_1_ONT | 46m25s | 2m22s | 28m6s | 26m57s | 1.02 | 8.60 | 10.28 | 1.05 | ||
| E. coli_2_ONT | 47m57s | 2m25s | 28m17s | 28m42s | 1.01 | 8.60 | 10.28 | 1.11 | ||
| S. cerevisiae_1_ONT | 676m26s | 41m20s | 962m51s | 1001m25s | 3.43 | 117.97 | 10.28 | 1.84 | ||
| S. cerevisiae_2_ONT | 830m48s | 40m54s | 1046m16s | 1051m53s | 3.52 | 117.99 | 10.28 | 3.47 | ||
Effectiveness of contig classification
To verify the effectiveness of the contig classification method presented in this paper, we removed the step of contig classification from SLR and this new algorithm was named SLR1. Then, we benchmarked SLR with SLR1 on all datasets. The scaffolding results for SLR and SLR1 are shown in Table 3. We can see that SLR performs better than SLR1 in terms of Misassemblies and NGA50. Therefore, we can prove that our proposed contig classification method is effective.
Next, we combined the contig classification method with other scaffolding tools. SLR classified each contig set into a unique contig set and an ambiguous contig set. We first ran SSPACE-LR and LINKS on the unique contig set, generating some scaffolds. Then, we inserted the ambiguous contigs into the scaffolds. For this purpose, we should determine the order and orientation of the unique contigs in these scaffolds. BWA-MEM is used to align the unique contigs against these scaffolds. Only if a unique contig is completely aligned in a scaffold, the corresponding alignment is retained. Then, we can obtain the order and orientation of the unique contigs in these scaffolds. The final scaffolding results is shown in Fig. 2. SSPACE-LR-CC represents the method based on SSPACE-LR combined with contig classification. LINKS-CC represents the method based on LINKS combined with contig classification. According to Fig. 2, we find that SSPACE-LR-CC and LINKS-CC outperformed SSPACE-LR and LINKS in NGA50. This further confirms the effectiveness of the method of contig classification.
Fig. 2.
Contig classification combines with SSPACE-LR and LINKS
Compared with SLR, SSPACE-LR-CC outperformed SLR in NGA50 for E. coli_2_SMRT and Chr X_1_SMRT. For the remaining seven datasets, SLR performed better than SSPACE-LR-CC in NGA50. SLR performed better than LINKS-CC in NGA50 for all datasets. Meanwhile, SLR outperformed SSPACE-LR-CC and LINKS-CC in Misassemblies for all datasets.
The detailed evaluation results are provided in Additional file 1. Note that, because npScarf makes sequence consensus between contigs and long reads, it is difficult to identify the order of the unique contigs in the scaffolds. We did not use npScarf in the this experiment.
Evaluation using a repeat-aware evaluation framework
We also used a repeat-aware evaluation framework [26] to evaluate the performance of SSPACE-LR, LINKS, npScarf and SLR. For each original contig set, by aligning contigs with the reference genome, this framework splits contigs in misassembly events, and extracts repetitive sub-contig from original contigs. Then, it outputs a new contig set. The framework records the number of correct links, which is the number of correct contig joins. After a scaffolding tool runs on this new contig set and a long-read set, the framework computes the number of correctly predicted links. Therefore, we can compute precision, recall and F1-score for the scaffolding results. For the contig set about Chr X, the framework ran for more than one week and gave no new contig set. Hence, we processed only the remaining original contig sets. So, there are eight new datasets used for this experiment, which are named E. coli_1_SMRT_R, E. coli_2_SMRT_R, S. cerevisiae_1_SMRT_R, S. cerevisiae_2_SMRT_R, E. coli_1_ONT_R, E. coli_2_ONT_R, S. cerevisiae_1_ONT_R, and S. cerevisiae_2_ONT_R. The detailed evaluation results provided by the framework are shown in Additional file 1: Tables S9 and S10.
In addition, for these new datasets, we also evaluated the scaffolding results by QUAST, which are shown in Fig. 3. According to Fig. 3, SLR achieved the best NGA50 values for all the datasets. This experiment shows that SLR can identify repetitive contigs and overcome the problem of repeating regions.
Fig. 3.

NGA50 for datasets produced by repeat-aware evaluation framework
Discussion
npScarf utilizes sequencing coverage to classify contigs. However, most contigs used for scaffolding do not include information about sequencing coverage, which limits the application of npScarf. SLR can classify contigs without any additional information about the contig set. SSPACE-LR uses a greedy heuristic strategy to determine the neighbour of a contig based on the number of long reads that can be aligned. LINKS uses a strategy similar to that of SSPACE-LR to determine the neighbours by counting the number of k-mer pairs between two contigs. These two tools have difficulty identifying the correct neighbours when encountering complex repetitive regions.
Conclusion
With the development of third-generation high-throughput sequencing technologies, scaffolding methods based on long reads have undergone substantial improvement. A scaffold graph is the basis for inferring the orders and orientations of contigs. However, the problems introduced by repetitive regions and sequencing errors pose challenges in the process of constructing scaffold graphs. In this paper, we presented a novel scaffolder, SLR, for determining the orientations and orders of contigs based on long reads and contig classification. SLR employs a new contig classification procedure to overcome the problems associated with repetitive regions in scaffolding. SLR first produces local scaffolds based on the alignment between long reads and contigs. A local scaffold corresponds to a long read and the contigs that can be aligned with it. SLR classifies contigs into unique and ambiguous contigs based on local scaffolds. A scaffold graph including only unique contigs is constructed; this process can simplify the scaffold graph and improve the accuracy of detecting and removing contradictions. Experiments were conducted that included long reads obtained with SMRT-based and Nanopore-based technologies. The experimental results illustrated that SLR is superior in terms of continuity and accuracy. For larger genomes, such as the complete human genome, however, SLR is difficult to scale due to its long run time.
Method
A contig set C and a long read set LR are used as input data. The algorithm is composed of four steps: (i) producing local scaffolds; (ii) classifying contigs; (iii) constructing a scaffold graph; and (iv) generating scaffolds. In the first step, the alignment tool BWA-MEM is used to align LR against C. For each long read and set of contigs that can be aligned with it, SLR determines the orders and orientations of the contigs and forms a local scaffold. In the second step, SLR classifies the contigs into unique contigs and ambiguous contigs based on their positions in the local scaffolds. In the third step, SLR constructs a scaffold graph based on unique contigs and then detects and removes the contradictions in the scaffold graph. In the fourth step, SLR extracts the simple paths from the scaffold graph to yield a draft scaffold set. Next, SLR inserts the ambiguous contigs into the draft scaffolds. The details of each step are described below. Note that the long reads whose lengths are longer than Lr and the contigs whose lengthes are longer than Lc are used by SLR. Lr and Lc are two parameters that can be defined by users. In addition, if a contig is completely contained in other contigs, SLR will ignore it in the following scaffolding steps.
Producing local scaffolds
SLR utilizes BWA-MEM to align LR against C, and the SAM file is converted to a BAM file by Bamtools [27]. Due to the high sequencing error rate in long reads, the alignment positions are usually different from the real positions. With the following method, SLR first revises the alignment positions and obtains reliable alignments.
For an alignment between the j-th long read lrj and the i-th contig ci, we assume that the region [ srij, erij] in lrj is aligned with the region [ scij, ecij] in ci. If srij<scij, srij′ = 0 and scij′ = scij - srij, else scij′ = 0 and srij′ = srij - scij. If LEN(lrj)−erij>LEN(ci)−ecij, erij′=erij+LEN(ci)−ecij and ecij′ = LEN(ci)−1, else ecij′=ecij+LEN(lrj)−erij and erij′=LEN(lrj)−1. LEN(lrj) and LEN(ci) are the lengths of lrj and ci respectively. [ srij′, erij′] and [ scij′, ecij′] are the real alignment regions. An example of the revision is shown in Fig. 4. After revision, the alignment will be reliable if the following hold: i) The mapping quality is higher than sm (a threshold with a default 20); ii) both the values erij−srij and ecij−scij are greater than lm (a threshold with a default 100); iii) for each of srij, erij, scij and ecij, the difference between its original position and its revised position is smaller than α (a threshold with a default 150). SLR retains only reliable alignments.
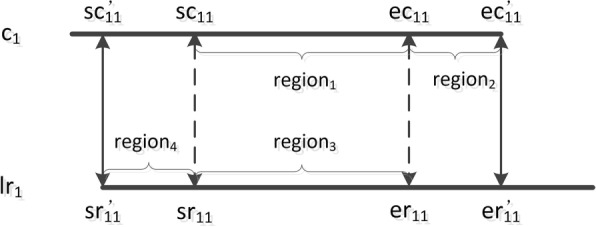
Fig. 4.

An example of alignment position revision. For an alignment given by the alignment tool, the region [ sr11, er11] (region3) in the long read lr1 is aligned with the region [ sc11, ec11] (region1) in the contig c1. Because sr11<sc11 and LEN(lr1)−er11>LEN(c1)−ec11, it means the region [0, sr11] (region4) in lr1 is not aligned with c1, and the region [ ec11,LEN(c1)−1] (region2) is not aligned with lr1. However, when lr1 is truely aligned with c1 and the alignment is reliable, region4 should be aligned with the region [ sc11−sr11, sc11] in c1, and region2 should be aligned with the region [ er11, er11+LEN(c1)−ec11]. Because of the high sequencing error rate in long reads, the alignment tool usually does not provide accurate alignment start and end positions. Then, SLR sets sc11′=sc11−sr11, sr11′=0, ec11′=LEN(c1)−1 and er11′=er11+LEN(c1)−ec11. When the alignment is reliable, the region [ sc11′, ec11′] in c1 is aligned with the region [ sr11′, er11′] in lr1
A local scaffold is composed of ordered and oriented contigs that can be aligned with the same long read. The i-th local scaffold lsi is represented by the vertex sequence si1,si2,...sim, where m is the number of contigs in the i-th local scaffold. sij is represented by a four-tuple (scij,scoij,scgij,sclij). scij refers to the j-th contig in lsi. scoij denotes the alignment orientation between the contig and the long read. scoij=1 represents forward alignment. scoij=0 represents reverse alignment. scgij denotes the gap distance between scij and sci(j+1). In particular, the gap distance of the last vertex is zero. sclij is the alignment length between scij and the long read. Note that if there are two or more contigs aligned with the same end of the long read, SLR keeps only the contig that has the greast alignment length. An example is shown in Fig. 5.
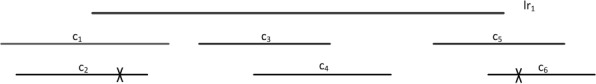
Fig. 5.

There are six contigs (c1,c2,c3,c4,c5,andc6) that can be aligned with the long read lr1. Because c1 and c2 are simultaneously aligned with the left end of lr1, SLR retains only contig c1 which has the greatest alignment length, and deletes the alignment information between c2 and lr1. Because c5 and c6 have been simultaneously aligned with the right end of lr1, we keep only c5, and delete the alignment information between c6 and lr1. Finally, SLR determines the orders and orientations of c1, c3, c4 and c5, which form a local scaffold
The contig scij is in the middle position of lsi if 1<j<m. scij and sci(j+1) are adjacent in lsi. If scoij=1, sci(j−1)(j>1) is the 5’-end neighbour contig of scij, and sci(j+1)(j<m) is the 3’-end neighbour contig of scij. If scoij=0, sci(j−1)(j>1) is the 3’-end neighbour contig of scij, and sci(j+1)(j<m) is the 5’-end neighbour contig of scij.
In this step, SLR finally obtains a local scaffold set LS. Due to the high sequencing error rate, a contig may not be aligned with the long read that connects its left and right neighbour contigs. To resolve this problem, SLR deletes some local scaffolds. For example, the local scaffold ls1 is (A,C), and the local scaffold ls2 is (B,C). If the sum of LEN(B) and the gap distance between B and C in ls2 is smaller than the gap distance between A and C in ls1 and there exists a local scaffold (A,B,C), SLR removes ls1.
Classifying contigs
Repetitive regions are the critical problem in the process of scaffolding. When constructing a scaffold graph, the 5’-end (or 3’-end) of a repetitive contig can usually be linked with two or more other contigs, which complicates the scaffold graph. Because repetitive contigs commonly emerge in many different local scaffolds, they have two or more distinct 5’-end (or 3’-end) neighbour contigs. When a contig is not in the middle position of any local scaffold, no long read can span the contig to link its two neighbour contigs, and this contig is usually a long unique contig. Although the contig has multiple 5’-end or 3’-end neighbour contigs, SLR uses contradiction removal step to identify its correct neighbour contigs. Hence, SLR can identify whether a contig is unique based on its positions in the local scaffolds.
To reduce the negative effects of short repetitive contigs, SLR considers a contig whose length is shorter than Lca (a threshold that can be set by users) to be an ambiguous contig. These short contigs are temporally ignored in the local scaffolds. Next, the contigs longer than Lca are classified using the following method.
SLR identifies a contig as ambiguous if the following hold: i) The contig is in the middle position of one or more local scaffolds and ii) the number of 5’-end (or 3’-end) neighbour contigs of the contig is greater than one.
After all ambiguous contigs have been identified, the remaining contigs are considered unique contigs. In this way, the contigs are classified into unique contigs and ambiguous contigs by SLR. An example of such contig classification is shown in Fig. 6.
Fig. 6.

(a) There are six long reads: lr1, lr2, lr3, lr4, lr5, and lr6. The contigs c1 and c2 are aligned with lr1. c3, c4 and c5 are aligned with lr2. c6, c4 and c7 are aligned with lr3. c7, c8 and c9 are aligned with lr4. c10, c11 and c12 are aligned with lr5. c9, c11, c13 and c2 are aligned with lr6. We assume that all these alignments are forward, and all contigs are longer than Lca. (b) Based on the alignment result described in (a), SLR obtains six local scaffolds: ls1, ls2, ls3, ls4, ls5, and ls6. (c) The scaffold graph G1 is built using all contigs. We find that G1 is complicated. (d) Based on the contig classification method described in Section 2.2, the contigs can be divided into two categories. Because c4 is located in the middle position of ls2 and ls3 and has two distinct 3’-end neighbours and two distinct 5’-end neighbour contigs, it is identified as an ambiguous contig. c11 is also an ambiguous contig. The remaining contigs are identified as unique contigs. The scaffold graph G2 is built based on unique contigs and is thus less complicated than G1
Constructing a scaffold graph
A scaffold graph G is represented by a vertex set V and an edge set E. A vertex vi corresponds to a contig ci. An edge eij is denoted by a five-tuple (vi,vj,oij,gij,wij). Two vertices vi and vj are connected by eij. gij is the gap distance between vi and vj. oij is the relative orientation of vi and vj. There are four types of relative orientation between vi and vj: (i) the 3’-end of vi is connected to the 5’-end of vj; (ii) the 5’-end of vi is connected to the 3’-end of vj; (iii) the 5’-end of vi is connected to the 5’-end of vj, and (iv) the 3’-end of vi is connected to the 3’-end of vj. For types (i) and (ii), vi and vj are on the same strand. For the other two types, vi and vj are on the opposite strands. wij is the weight of the edge, which reflects its confidence.
Neglecting the ambiguous contigs and constructing scaffold graph G with only unique contigs will significantly simplify G and reduce the difficulties in inferring the orders and orientations of the unique contigs. Therefore, all unique contigs make up the vertex set V. Below, we describe how to create the edge set E. The superiority of constructing a scaffold graph using unique contigs is illustrated in Fig. 6.
Adding edges to the scaffold graph
First, SLR ignores the ambiguous contigs in all local scaffolds; therefore, some non-adjacent unique contigs may become adjacent in one local scaffold. Assume that the i-th local scaffold lsi(si1,si2,...sim) in LS includes two adjacent unique contigs scip and scis. If one or more ambiguous contigs exist between scip and scis, the gap distance between scip and scis is re-calculated by formula (1); otherwise, it is equal to scgip. Here, GD(scip,scis,lri) represents the gap distance between scip and scis in lsi. Moreover, SLR can obtain a weight value, which is the minimum value of sclip and sclis. The weight value can be used to evaluate the confidence level of the relation between scip and scis. As the weight value becomes larger, the order of the two unique contigs becomes more reliable.
| 1 |
We assume that scip is represented by ca and that scis is represented by cb. For ca and cb, SLR selects all local scaffolds in which ca and cb are adjacent. Next, SLR determines the relative orientation of the gap distance between and weight of ca and cb based on these local scaffolds. For two unique contigs, the relative order and orientation should be unique. If different values of oab are obtained from the local scaffolds, SLR keeps only the local scaffold set LSab for which the value of oab is the same, and the number of elements in LSab is the largest. The gap distance between ca and cb is calculated according to formula (2). In addition, we can obtain a weight value for each local scaffold in LSab. The final weight of ca and cb (denoted wab) can be obtained by seeking the maximum weight value obtained by the local scaffolds in LSab. Then, SLR adds an edge eab to G.
| 2 |
in which n is the number of elements in LSab, and lsi∈LSab.
After processing all pairs of unique contigs in LS, a draft scaffold graph G can be constructed by SLR for the subsequent steps.
Removing contradictions
Due to sequencing errors in long reads and complex repetitive regions, the scaffold graph G may still contain some spurious edges. Detecting and removing the spurious edges in G can be viewed as detecting and removing the orientation and position contradictions [10, 11]. BOSS utilizes an iterative strategy to detect and remove contradictions. BOSS first constructs a sub-graph that includes only edges with a high weight. Next, it iteratively adds the remaining edges to the sub-graph from high to low weight. Each iteration includes a sub-graph, and BOSS builds two linear programming models [28] to solve orientation and position contradictions in the sub-graph. SLR utilizes a revised method based on BOSS to remove contradictions. The difference in SLR compared to BOSS is that SLR adds all edges to the sub-graph in the first iteration. Hence, SLR completes contradiction removal within one iteration, while BOSS requires several iterations. The methods of building the linear programming model of BOSS and SLR are the same, as described below.
First, SLR detects and deletes orientation contradictions. For the edge eij∈G, if oi≠oj, SLR constructs constraint Eq. (3). If oi=oj, SLR constructs constraint Eq. (4).
| 3 |
| 4 |
in which ηij∈{0,1} is a variable that represents whether eij is spurious. 0i∈{0,1} is also a variable that denotes the orientation of vi. The objective function is .
Second, SLR detects and deletes position contradictions. For the edge eij∈G, SLR constructs constraint Eq. (5).
| 5 |
in which pi is a variable that represents the assigned position of vi. ϕij is a slack variable in the range [0,1] that reflects the consistency between gij and |pj−pi|. The objective function is . For an edge, if the gap distance computed by the assigned position is far from the original one, the edge is deemed spurious one, and then SLR deletes it from G.
After eliminating the orientation and position contradictions, if there are two or more edges linking the same end of a vertex, SLR keeps only the edge with the highest weight and removes the others. Consequently, the scaffold graph G contains only simple paths.
Generating scaffolds
Each simple path in G refers to a scaffold, and SLR selects all simple paths and constructs a draft scaffold set. For any two adjacent vertices in the draft scaffold, SLR scans the local scaffold set LS again and finds local scaffolds that contain them. If ambiguous contigs exist between these vertices in a local scaffold, these ordered and oriented ambiguous contigs correspond to a path. If there are two or more different paths, SLR selects the one with the greatest number of local scaffolds that support it and then inserts it between the two vertices. Note that an ambiguous contig may occur two or more times in the scaffolds.
Next, SLR selects local scaffolds that contain the first contig of a scaffold. SLR constructs a scaffold graph based on these local scaffolds. If a simple path starts from the first contig in the scaffold graph, it is merged with the head of the scaffold. In the same way, SLR extends the tail of the scaffold. Once the first t contigs of a scaffold are the same as the last t contigs of another scaffold (t is a threshold set by users), SLR will merge them together to form a new scaffold. In the same way, SLR will reverse a scaffold and detect whether it can be merged with other scaffolds. Finally, SLR outputs the scaffolds as the final result.
Supplementary information
Additional file 1 It includes seven sections: (i) Datasets; (ii) Command lines; (iii) Scaffolding results about nine datasets; (iv) Different values of Lca for scaffolding; (v) Scaffolding results about SLR1 and SLR2; (vi) SSPACE-LR and LINKS combined with contig classification method; (vii) Scaffolding results based on repeat-aware evaluation framework.
Acknowledgements
The authors would like to thank professor Zhiheng Wang, anonymous reviewers, and editors for their helpful comments and suggestions.
Abbreviations
- Chr X
Human chromosome X
- E. coli
Escherichia coli
- S. cerevisiae
Saccharomyces cerevisiae W303
- SLR
Scaffolding algorithm based on Long Reads and contig classification
- SMRT
Single-Molecule Real-Time
- SSPACE-LR
SSPACE-LongRead
Authors’ contributions
JW participated in the design of the study and the analysis of the experimental results. MN, XH, and RR performed the implementation. HM and CK wrote the manuscript. All authors have read and approved the final manuscript for publication.
Funding
This work was supported in part by the National Natural Science Foundation of China under Grant No.61602156, No.61972134, No.61802113, No.61772557, and No.61433012, Henan Provincial Department of Science and Technology Research Project under Grant No.192102210118, Doctor Foundation of Henan Polytechnic University under Grant No.B2018-36, Henan Science and Technology Innovation Outstanding Youth Program under Grant No.184100510009, Henan University Scientific and Technological Innovation Team Support Program under Grant No.19IRTSTHN012. The funding bodies did not play any role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Availability of data and materials
All datasets used in this paper and command lines for all scaffolding tools are provided in Additional file 1.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Junwei Luo, Email: luojunwei@hpu.edu.cn.
Mengna Lyu, Email: 211809020031@home.hpu.edu.cn.
Ranran Chen, Email: 211709020027@home.hpu.edu.cn.
Xiaohong Zhang, Email: xh.zhang@hpu.edu.cn.
Huimin Luo, Email: luohuimin@henu.edu.cn.
Chaokun Yan, Email: ckyan@henu.edu.cn.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12859-019-3114-9.
References
- 1.Sedlazeck FJ, Lee H, Darby CA, Schatz MC. Piercing the dark matter: bioinformatics of long-range sequencing and mapping. Nature Rev Genet. 2018;19(6):329. doi: 10.1038/s41576-018-0003-4. [DOI] [PubMed] [Google Scholar]
- 2.Junwei L, Jianxin W, Zhen Z, Fang-Xiang W, Min L, Yi P. Epga: de novo assembly using the distributions of reads and insert size. Bioinformatics. 2015;31(6):825–33. doi: 10.1093/bioinformatics/btu762. [DOI] [PubMed] [Google Scholar]
- 3.Luo J, Wang J, Li W, Zhang Z, Wu FX, Li M, Pan Y. Epga2: memory-efficient de novo assembler. Bioinformatics. 2015;31(24):3988–90. doi: 10.1093/bioinformatics/btv487. [DOI] [PubMed] [Google Scholar]
- 4.Hunt M, Newbold C, Berriman M, Otto TD. A comprehensive evaluation of assembly scaffolding tools. Genome Biol,15,3(2014-03-03) 2014;15(3):42. doi: 10.1186/gb-2014-15-3-r42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao S, Sung WK, Nagarajan N. Opera: Reconstructing optimal genomic scaffolds with high-throughput paired-end sequences. J Comput Biol. 2011;18(11):1681–91. doi: 10.1089/cmb.2011.0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marten B, Christiaan V H, Hans J J, Derek B, Walter P. Scaffolding pre-assembled contigs using sspace. Bioinformatics. 2011;27(4):578–9. doi: 10.1093/bioinformatics/btq683. [DOI] [PubMed] [Google Scholar]
- 7.Sahlin K, Vezzi F, Nystedt B, Lundeberg J, Arvestad L. Besst - efficient scaffolding of large fragmented assemblies. Bmc Bioinformatics. 2014;15(1):281. doi: 10.1186/1471-2105-15-281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mandric I, Zelikovsky A. Scaffmatch: Scaffolding algorithm based on maximum weight matching. Bioinformatics. 2015;31(16):2632–8. doi: 10.1093/bioinformatics/btv211. [DOI] [PubMed] [Google Scholar]
- 9.Nilgun D, Michael B. Scarpa: scaffolding reads with practical algorithms. Bioinformatics. 2013;29(4):428–34. doi: 10.1093/bioinformatics/bts716. [DOI] [PubMed] [Google Scholar]
- 10.Bodily PM, Fujimoto MS, Snell Q, Dan V, Clement MJ. Scaffoldscaffolder: solving contig orientation via bidirected to directed graph reduction. Bioinformatics. 2016;32(1):17. doi: 10.1093/bioinformatics/btv548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luo J, Wang J, Zhang Z, Li M, Wu FX. Boss: a novel scaffolding algorithm based on an optimized scaffold graph. Bioinformatics. 2016;33(2):169. doi: 10.1093/bioinformatics/btw597. [DOI] [PubMed] [Google Scholar]
- 12.Boetzer M, Pirovano W. Sspace-longread: scaffolding bacterial draft genomes using long read sequence information. Bmc Bioinformatics. 2014;15(1):211–1. doi: 10.1186/1471-2105-15-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chaisson MJ, Tesler G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (blasr): application and theory. Bmc Bioinformatics. 2012;13(1):238. doi: 10.1186/1471-2105-13-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Warren RL, Yang C, Vandervalk BP, Behsaz B, Lagman A, Jones SJM, Birol I. Links: Scalable, alignment-free scaffolding of draft genomes with long reads. GigaScience,4,1(2015-08-04) 2015;4(1):1–11. doi: 10.1186/s13742-015-0076-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu S, Chen DZ, Emrich SJ. Single molecule sequencing-guided scaffolding and correction of draft assemblies. BMC genomics. 2017;18(10):879. doi: 10.1186/s12864-017-4271-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. Versatile and open software for comparing large genomes. Genome Biol. 2004;5(2):12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Warren RL. Rails and cobbler: Scaffolding and automated finishing of draft genomes using long dna sequences. J Open Source Softw. 2016;1(7):116. [Google Scholar]
- 18.René L W, Granger G S, Steven J M J, Robert A H. Assembling millions of short dna sequences using ssake. Bioinformatics. 2007;23(4):500–1. doi: 10.1093/bioinformatics/btl629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao MD, Nguyen SH, Ganesamoorthy D, Elliott AG, Cooper MA, Coin LJM. Scaffolding and completing genome assemblies in real-time with nanopore sequencing. Nature Commun. 2017;8:14515. doi: 10.1038/ncomms14515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weisenfeld NI, Kumar V, Shah P, Church DM, Jaffe DB. Direct determination of diploid genome sequences. Genome Res. 2017;27(5):757–67. doi: 10.1101/gr.214874.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuleshov V, Snyder MP, Batzoglou S. Genome assembly from synthetic long read clouds. Bioinformatics. 2016;32(12):216–24. doi: 10.1093/bioinformatics/btw267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yeo S, Coombe L, Chu J, Warren RL, Birol I. Arcs: Scaffolding genome drafts with linked reads. Bioinformatics. 2018;34(5):725–31. doi: 10.1093/bioinformatics/btx675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andrew A, Kitzman JO, Burton JN, Riza D, Akash K, Lena C, Mostafa R, Sasan A, Kevin LG, Steemers FJ. In vitro, long-range sequence information for de novo genome assembly via transposase contiguity. Genome Res. 2014;24(12):2041–9. doi: 10.1101/gr.178319.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gurevich A, Saveliev V, Vyahhi N, Tesler G. Quast: quality assessment tool for genome assemblies. Bioinformatics. 2013;29(8):1072–5. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 2013. arXiv preprint arXiv:1303.3997.
- 26.Mandric I, Knyazev S, Zelikovsky A. Repeat aware evaluation of scaffolding tools. Bioinformatics. 2017;34(15):2530–7. doi: 10.1093/bioinformatics/bty131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barnett DW, Garrison EK, Quinlan AR, Stromberg MP, Marth GT. Bamtools: a c++ api and toolkit for analyzing and managing bam files. Bioinformatics. 2011;27(12):1691–2. doi: 10.1093/bioinformatics/btr174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berkelaar M, Eikland K, Notebaert P. lp_solve 5.5, open source (mixed-integer) linear programming system. Software. May 1 2004.
- 29.Lee H, Gurtowski J, Yoo S, Marcus S, Mccombie WR, Schatz M. Error correction and assembly complexity of single molecule sequencing reads. Biorxiv. 2014:006395.
- 30.Goodwin S, Gurtowski J, Ethe-Sayers S, Deshpande P, Schatz MC, Mccombie WR. Oxford nanopore sequencing, hybrid error correction, and de novo assembly of a eukaryotic genome. Genome Res. 2015;25(11):1750. doi: 10.1101/gr.191395.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu G-C, Xu T-J, Zhu R, Zhang Y, Li S-Q, Wang H-W, Li J-T. Lr_gapcloser: a tiling path-based gap closer that uses long reads to complete genome assembly. GigaScience. 2019;8(1):giy157. doi: 10.1093/gigascience/giy157. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1 It includes seven sections: (i) Datasets; (ii) Command lines; (iii) Scaffolding results about nine datasets; (iv) Different values of Lca for scaffolding; (v) Scaffolding results about SLR1 and SLR2; (vi) SSPACE-LR and LINKS combined with contig classification method; (vii) Scaffolding results based on repeat-aware evaluation framework.
Data Availability Statement
All datasets used in this paper and command lines for all scaffolding tools are provided in Additional file 1.