

Abstract
The Heck reaction is one of the most reliable and useful strategies for the construction of C–C bonds in organic synthesis. However, in contrast to the well-established aryl Heck reaction, the analogous reaction employing alkyl electrophiles is much less developed. Significant progress in this area was recently achieved by merging radical-mediated and transition-metal-catalyzed approaches. This review summarizes the advances in alkyl Heck-type reactions from its discovery early in the 1970s up until the end of 2018.
Keywords: Heck reaction, cross-coupling, alkyl halides, alkenes, transition-metal catalysis
Graphical Abstract
1. Introduction
The Mizoroki–Heck reaction1 is one of the most powerful approaches towards multisubstituted alkenes. This reaction represents the first example of Pd-catalyzed C–C bond-forming reactions,2 which follow a classical Pd(0)/Pd(II) catalytic cycle enabling coupling of aryl and vinyl electrophiles with olefins.
The Heck reaction has been exhaustively employed in organic synthesis, drug discovery, electronics, and in industry,3 which has led to its recognition with a Nobel Prize in 2010. Current research in this area is directed towards the use of decreased loadings of palladium,4 the employment of low-cost transition metals,5 and the development of asymmetric protocols.6 Historically, aryl halides/pseudohalides were the electrophiles of choice for all cross-coupling reactions, thus, not surprisingly, they were also extensively used in Heck reactions. Throughout years, this research has been summarized in many excellent reviews.7 In contrast, alkyl electrophiles were found to be more challenging coupling partners, mostly due to the competing β-H elimination process2 and the slower rates of the oxidative addition step.8 Nonetheless, under more recently developed conditions, alkyl halides9 were shown to be suitable partners for this transformation. However, the number of reports on alkyl Heck reactions remains scarce compared to those employing aryl substrates.
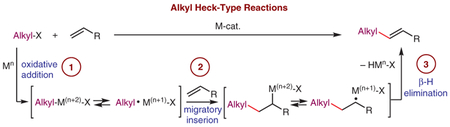
In general, alkyl Heck reactions feature the same mechanism as the classical Heck reaction between aryl halides and alkenes, involving: (1) oxidative addition, (2) migratory insertion, (3) β-hydrogen elimination, and (4) catalyst regeneration steps (Scheme 1). However, in contrast to the aryl Heck reaction, its alkyl version is often presumed to involve radical intermediates, thus operating via a hybrid organometallic–radical scenario. Mechanistic studies, such as radical trapping,10 radical clock experiments,11 as well as ESR studies, have proved the presence of radical species in some alkyl Heck reactions. The mechanisms of their formation, which may depend on the nature of the alkyl component, the leaving group, and the metal catalyst, are still not completely understood. It was shown that employment of visible light allows alkyl Heck reactions to be accomplished under milder conditions; over recent years this area of research has grown significantly. Various complexes of Co, Ir/Ru, Au, Pd, and other transition metals were found to catalyze this reaction under visible-light irradiation, with significantly expanded scope. This review highlights the advances in the field of alkyl Heck-type reactions of alkyl electrophiles with alkenes since its discovery in the early seventies.1d It is organized by the type of electrophile used, such as benzylic, activated (possessing a carbonyl or equivalents at the β-position), perfluorinated, and unactivated alkyl electrophiles. Related transformations, such as Heck-type reactions involving a β-X elimination step and cascade transformations commencing with radical addition to an alkene moiety are not discussed herein.12
Scheme 1.

Mechanism of transition-metal-catalyzed alkyl Heck-type reactions
2. Pd-Catalyzed Heck-Type Reactions
2.1. Benzylic Electrophiles
The first example of the coupling of an alkyl halide with an olefin was reported by Heck in his original seminal work in 1972 (Scheme 2).1d Benzyl chloride (1) reacted with methyl acrylate (2) in the presence of 1 mol% of Pd(OAc)2 and Bu3N as the base to deliver a regiomeric mixture of alkenes 3. In 1995, Zhuangyu and co-workers developed a base-free alkyl Heck reaction of benzyltris(n-butyl)ammonium bromide salts 4 toward exclusive formation of the conjugated products 6 (Scheme 2).13 Both electron-rich and electron-deficient alkenes 5 were efficiently benzylated under these conditions. Based on ESR studies, the authors suggested the involvement of benzyl radicals, which would form via the reductive cleavage of the benzyl quaternary ammonium salt in the presence of the palladium(0) species. Later, the coupling of benzyl chlorides with olefins was elegantly utilized by Kita to obtain the key intermediate 8 in the synthesis of beraprost, a vasodilator and antiplatelet agent (Scheme 2).14 In this work, the Heck reaction followed by hydrogenation was shown to be superior over other methods tested for installation of an alkyl chain at the benzylic position of compound 7.
Scheme 2.

Pd-catalyzed Heck reactions of benzyl halides; r.r. = regiomeric ratio
In 2000, Pan and co-workers observed an interesting Pd-catalyzed rearrangement in the vinylation reaction of α-chloromethylnaphthalene (9) (Scheme 2).15 In reactions with N-vinylimides, besides the expected Heck products 11, a product with an olefin attached at the peri position of the naphthalene ring was detected. This unusual rearrangement product 12 was proposed to form via the cyclopalladation intermediate 14, when the nitrogen-containing alkene served as a stabilizing ligand for benzylpalladium species 13. Later, Pan described the Pd-catalyzed cascade reaction of benzyl halides with N-allyl-N-(2-butenyl)-p-toluenesulfonamide (17) to furnish dihydropyrroles 18 with excellent regioselectivity (Scheme 2).16
In addition to benzyl halides, benzyl trifluoroacetates were also found to be compatible coupling partners for the Heck reaction. In 1999, Yamamoto reported that mixing benzyl trifluoroacetate with phosphine-coordinated Pd(0) led to the formation of an oxidative addition complex.17 In 2004, Shimizu showed that the reaction of this complex with ethyl acrylate under heating produced the corresponding Heck reaction product.18 Based on these initial discoveries, the catalytic benzylation of olefins with benzyl trifluoroacetates 19 was developed (Scheme 3).18 Moreover, the authors were able to achieve the benzylation of p-methylstyrene (22) with p-methoxybenzyl alcohol (21) using trifluoroacetic anhydride as an additive. Later, Zhou introduced an asymmetric Heck reaction of benzyl trifluoroacetates 19 with five-membered cyclic olefins 24.19 In the presence of Pd/phosphoramidite ligand 26, 2,3-, and 2,5-dihydrofurans, N-Boc-2,3-pyrroline, and cyclopentene (24a–c) were smoothly alkylated with electronically diverse benzyl trifluoroacetates leading to the corresponding products 25a–f in high yields, and with high degrees of regioselectivity and enantioselectivity (Scheme 3).
Scheme 3.

Pd-catalyzed Heck reactions of benzyl fluoroacetates; r.r. = regiomeric ratio
The first intramolecular Heck reaction of benzyl halides was developed by Negishi to access five- to seven-membered cyclic compounds (Scheme 4).20 The initial screening of the leaving group on the o-allylbenzyl electrophile 27 revealed that Cl, Br, and OMs were acceptable leaving groups, leading almost exclusively to the formation of 2-methyleneindane (28). In the cases of I and OCO2Me leaving groups, the cyclization occurred quite efficiently but with low regioselectivity, as the formation of regioisomer 29 was detected in significant amounts.
Scheme 4.

Intramolecular Pd-catalyzed Heck reactions of benzyl halides
Lastly, o-allylbenzyl acetate was practically unreactive. Interestingly, attempts to induce this cyclization under typical radical conditions led to the hydrostannation product 30 only, thus illustrating the superiority of the Heck reaction pathway for this type of cyclization. The developed intramolecular Heck reaction was also able to deliver six- and seven-membered cyclic compounds 31 and 32 from the corresponding benzyl chlorides in good yields and regioselectivity. However, attempts to obtain four- and eight-membered rings using this strategy (33 and 34, respectively) were unsuccessful (Scheme 4).
In 2008, Chen applied an intramolecular Heck-type approach for the synthesis of 3-alkyl-1H-quinolin-2-ones 35 via cyclization of benzyl halides with α,β-unsaturated amides.21 In 2011, Pan introduced another regioselective intra-molecular Heck-type coupling for the assembly of a biologically important core compounds, i.e., 4-aryldihydropyrroles 36.22 In this case, cyclization favored products with an endocyclic double bond, so that most of these dihydropyrroles were obtained as single regioisomers. In 2014, Gharpure disclosed a straightforward synthesis of isochromene derivatives 37 via the intramolecular Heck reaction of benzyl halides and vinylogous carbonates (Scheme 4).1b
2.2. α-Carbonyl Alkyl Halides
In the 1980s, Ban and Mori performed the initial study on the Pd(PPh3)4-catalyzed intramolecular alkylation of ole-fins using α-carbonyl alkyl halides 38.23 The reaction proceeded with low selectivity, delivering mixtures of atom transfer radical cyclization (ATRC) and Heck products (39 and 40, respectively) in moderate yields (Scheme 5). In 2003, Glorius reported an intermolecular Heck reaction of 2-chloroacetamides 41 with 2,3-dihydrofuran (24a), butyl vinyl ether (43) and styrenes 45 (Scheme 5, eq 1).24 Alkylation of 24a and 43 led to the exclusive formation of α-alkylated olefins 42 and 44, respectively, thus supporting the reaction mechanism involving palladium enolates rather than alkyl radical intermediates. Conversely, the reaction of bromoacetonitrile (47) with 24a produced β-alkylated 2,3-dihydrofuran 48 in 30% yield as a single product (Scheme 5, eq 2). This reaction was suggested to proceed via a radical pathway.
Scheme 5.

Pd-catalyzed Heck reactions of α-carbonyl alkyl halides
More recently, Gevorgyan showed that activated tertiary alkyl bromides possessing an α-carbonyl moiety 49 can efficiently react with styrenes and electron-rich alkenes at room temperature (Scheme 5).25 Catalyzed by a Pd Xantphos G3 complex, the reaction furnished the Heck products 50a–d in moderate to high yields. A radical clock experiment suggested that the catalytic cycle might involve an alkyl Pd-radical hybrid species.
2.3. Fluoroalkyl Halides
In 1988, Chen reported the first Pd-catalyzed addition of perfluorinated alkyl iodides 51 to alkenes 52 leading to alkyl iodides 53, i.e., the atom transfer radical addition (ATRA) products (Scheme 6).26 The radical nature of this transformation was strongly supported by the mechanistic studies, which led the authors to propose the involvement of a Pd(0) complex in a radical initiation event. In 2012, Reutrakul developed the Pd(PPh3)4-catalyzed Heck reaction of (bromodifluoromethyl)sulfones 54 with alkenes (Scheme 6).27 The reaction proceeded smoothly in toluene at 100 °C delivering the coupling products 55a–f in moderate yields. Later, Zhang reported the first Heck reaction of perfluorinated alkyl halides with vinyl arenes/heteroarenes (57a–g), diene (57h), and electron-rich olefins (57i,j) (Scheme 6).28 The reaction features a quite general scope leading to valuable fluoroalkylated alkenes in good to excellent yields. Moreover, this method was shown to be effective for the synthesis of complex molecules possessing fluorinated fragments (57k,l). In follow-up work, the same group demonstrated that a similar catalytic system involving a Pd(II)-precatalyst and Xantphos as the ligand enabled a Heck-type coupling of secondary trifluoromethylated alkyl bromides (Scheme 6, products 59a–c).29 The performed mechanistic studies supported a hybrid Pd-radical mechanism and ruled out the possible involvement of the corresponding ATRA products (bromide-containing analogues of 53) for both reactions.28,29
Scheme 6.

Pd-catalyzed Heck reactions of fluoroalkyl halides
2.4. α-Functionalized Alkyl Halides
In 2014, Gevorgyan reported the Pd-catalyzed endo-selective Heck-type reaction of iodomethylsilyl ethers 61 employing ferrocene-derived bidentate phosphine ligand 64 (Scheme 7).30 The reaction was able to deliver seven-, eight-, and nine-membered siloxycycles 62a–i in good yields, which could further be converted into the corresponding allylic alcohols (see 63g,i as examples) via oxidation. Formally, this transformation provides a tool for the selective (Z)-hydroxymethylation of phenols and alkenols 60. The mechanistic studies suggested a hybrid Pd-radical mechanism for this Heck reaction. Also, the silicon atom was found to be crucial for the observed endo selectivity.
Scheme 7.

Pd-catalyzed Heck reactions of α-functionalized alkyl halides
In 2017, Gevorgyan’s group developed the first visible-light-induced Pd-catalyzed Heck reaction of alkyl halides at ambient temperature under exogenous photosensitizer-free conditions to furnish valuable allylic systems of diverse electronic nature (Scheme 7).31 Allylic silanes (66a,b,k–o), boronates (66c,j), germane (66d), stannane (66e), pivalate (66f), phosphonates (66g,p), phthalimide (66h), and tosylate (66i) derivatives were easily synthesized from primary and secondary α-functionalized alkyl halides and vinyl arenes/heteroarenes. The obtained allylic systems can be further modified, for example, via a Hosomi–Sakurai reaction (66j → 67). Later, the same group applied these photo-induced conditions for the Heck reactions of α-heteroatom-substituted tertiary alkyl iodides with styrene (Scheme 7, products 66q–s).25
Notably, in this case, presumably due to the insufficiently low reduction potentials of the activated tertiary substrates, activation by light was not necessary to obtain the Heck reaction products. The performed radical clock and radical trapping experiments described in these reports25,31 support a radical-type mechanism. It was also shown that Pd(0)Ln complexes were the single light-absorbing species in this reaction. The excited state is quenched by an alkyl halide, presumably via a SET event, which was calculated to be ‘barrierless’,12c to form the alkyl Pd hybrid species 68 that adds to an alkene producing the new radical species 69 (Scheme 8). Subsequent β-H elimination from the latter delivers the product 66, while the base regenerates the Pd(0) catalyst.
Scheme 8.

Gevorgyan’s mechanism for the visible-light-induced Pdcatalyzed Heck reaction
2.5. Unactivated Alkyl Electrophiles
The first Heck reaction of unactivated alkyl halides with alkenes was reported by Waegell and de Meijere in 1998 (Scheme 9).32a In this report, 1-bromoadamantane (70), a substrate which is not disposed to β-hydrogen elimination, was employed. Upon this Pd/C-catalyzed reaction at 120 °C, a number of substituted olefins 72 were obtained in low to moderate yields. In 2007, Fu introduced the first protocol for the Heck reaction of unactivated alkyl bromides and chlorides containing eliminable β-hydrogens (Scheme 10).32b The employment of the bulky NHC-ligand 75 on the Pd catalyst was the key for the success of this transformation, allowing an intramolecular insertion of the alkyl-Pd species into a double bond to proceed faster than the premature β-hydrogen elimination. Cyclopentane derivatives possessing an exo-alkene moiety 74a–g were obtained in high yields and regioselectivities from the corresponding unsaturated alkyl bromides and even chlorides 73 at elevated temperatures. The stereochemical outcome of this transformation supports the SN2 mechanism for the oxidative addition step, thus eliminating involvement of radical intermediates in this reaction.
Scheme 9.

Waegell and de Meijere’s first Pd-catalyzed Heck reaction of unactivated alkyl halides
Scheme 10.

Pd-catalyzed intramolecular Heck reactions of unactivated alkyl halides; d.r. = diastereomeric ratio
In 2011, Alexanian developed a protocol for an intramolecular alkyl-Heck reaction, which relied on radical reactivity (Scheme 10).33 Following his previous work on the carbonylative Heck-type reaction,34 he showed that under increased CO pressure, Pd(PPh3)4 was capable of catalyzing the 5- or 6-exo-trig cyclization reaction of alkenyl iodides 76, leading to the exclusive formation of Heck reaction products 78a–e rather than the corresponding cycloalkenones 79. Interestingly, in the absence of CO, the reaction proceeded as well, albeit with lower efficiency and regioselectivity. In contrast to Fu’s work,32b mechanistic studies, including a radical trapping experiment, indicated the formation of radical species under these conditions. Accordingly, a hybrid Pd-radical mechanism was postulated. In 2017, the same group introduced a second-generation Pd-based catalytic system for an intramolecular Heck-type reaction, thus enabling efficient carbocyclization of unsaturated alkyl bromides 77 under CO-free conditions (Scheme 10).35 Moreover, in this work, the authors investigated the difference in the reactivity of alkyl iodides versus alkyl bromides in this cyclization. It was hypothesized that, in the case of alkyl bromides, the cyclization proceeds via auto-tandem catalysis (Scheme 11). Initiated by the Pd(0) complex, ATRC leads to the formation of alkyl bromide 86, which could be isolated from the reaction mixture. Subsequently, the Pd(0)-catalyzed dehydrohalogenation of 86 delivers the Heck reaction product. Alternatively, for alkyl iodides, the radical chain mechanism initiated by the Pd(0) catalyst was suggested as a more likely scenario.
Scheme 11.

Alexanian’s mechanism for the Pd-catalyzed carbocyclization of unactivated alkyl bromides via auto-tandem catalysis
In 2016, Liu reported a related Pd-catalyzed radical Heck-type cyclization utilizing alkyl iodides 80 possessing a 1-aryl-substituted alkene moiety (Scheme 10).36 The use of these specific substrates resulted in the exceptional endo-selective cyclization. This outcome is attributed to a much higher stability of the forming tertiary benzyl radical intermediate 81 versus a non-stabilized primary radical species 83, which would arise via an alternative 5-exo-trig cyclization. Thus, a number of 5-aryl-1,2,3,6-tetrahydropyridines 82, which are structural motifs found in a variety of natural products and pharmaceutical compounds, were synthesized using this approach.
In 2014, Alexanian developed an intermolecular version of alkyl Heck reaction of unactivated alkyl iodides (Scheme 12).37 By employing Pd(dppf)Cl2 as a catalyst, both primary and secondary alkyl iodides 88 reacted smoothly with sty-rene and acrylonitrile derivatives producing alkylated ole-fins 91a–k in moderate to excellent yields. Shortly after, Zhou applied a combination of a Pd(0) precatalyst and dppf as a ligand, which allowed intermolecular Heck reactions of styrenes to proceed with alkyl iodides 88, bromides 89 and even chlorides 90 (Scheme 12).38 The use of LiI as an additive in the reaction was crucial for achieving higher yields with halides 89 and 90, presumably due to in situ generation of more reactive iodide species. As a result, the corresponding Heck reaction products 91a,h–k were obtained in yields comparable to those reported by Alexanian employing alkyl iodides. Similar to the previously described Heck- type cyclization reactions, the mechanistic studies in both reports were consistent with a hybrid Pd-radical pathway, thus further illustrating the prominence of this pathway for overcoming premature β-hydrogen elimination in alkyl Heck reactions.
Scheme 12.

Pd-catalyzed intermolecular Heck reactions of unactivated alkyl halides
Until recently, all the reported methods for the Heck reaction of unactivated alkyl halides required elevated temperatures. In 2017, Gevorgyan’s group demonstrated the possibility of achieving a room temperature Heck reaction of unactivated primary and secondary alkyl halides under visible-light-induced exogeneous photosensitizer-free conditions employing a Pd(0)/Xantphos catalytic system (Scheme 12, products 92a–c).31 Shortly after, the groups of Shang and Fu independently developed a similar method for the reactions of primary and secondary unactivated alkyl bromides with styrenes to provide the Heck reaction products 92d–h in excellent yields and E/Z ratios.39
Moreover, challenging tertiary alkyl bromides were also found to be capable coupling partners (products 92i–n). In addition to styrenes, unactivated tertiary alkyl bromides reacted with electron-deficient alkenes in a highly efficient manner (products 92o,p). The mechanistic studies of this work strongly supported a radical pathway, which was initiated by a SET event from the photoexcited Pd(0) complex to an alkyl halide (see Scheme 8). Furthermore, the performed X-ray photoelectron spectroscopy studies detected palladium in three oxidation states: Pd(0), Pd(I), and Pd(II), thus demonstrating that the Pd(0)–Pd(I)–Pd(II) catalytic cycle is a highly feasible scenario for these visible-light-induced conditions. Shortly after, Gevorgyan independently reported analogous efficient alkyl Heck reactions of unactivated tertiary alkyl iodides with styrenes and acrylonitrile (Scheme 12, products 92q–v).25
Very recently, Gevorgyan’s group reported a photoinduced, Pd-catalyzed radical relay Heck reaction at remote unactivated C(sp3)–H sites of aliphatic alcohols, which synergistically combines a C–H activation via a hydrogen atom abstraction (HAT) process and an alkyl Heck reaction (Scheme 13).40 The control of the β-, γ-, or δ-sites in this regioselective Heck reaction was achieved by the employment of easily installable/removable iodomethyl silyl tethers on alcohols 93. These tethers are known to engage in a SET process with the photoexcited Pd complex to form the Pd-radical hybrid species 95.30,31 Remarkably, 95 bypasses the potential side-reaction processes, such as hydrodehalogenation or premature Heck reaction (97), but rather undergoes a selective 1,5-, 1,6- or 1,7-HAT to produce the translocated Pd-radical hybrid species 96. Subsequently, the latter is able to couple with acrylonitrile, acrylate or styrene derivatives to afford the remote Heck reaction products 94a–m in good yields. Interestingly, the iodine-atom transfer intermediate 96′ was observed during this reaction, which presumably is formed via a reversible I-atom transfer from the Pd(I)I species.
Scheme 13.

Pd-catalyzed aliphatic radical relay Heck reaction at unactivated C(sp3)–H sites of alcohols
Lately, the scope of alkyl electrophiles in the Heck reaction has been expanded. Thus, in 2018, Zhou’s group reported a Heck-type reaction of cyclic (98) and acyclic (99) epoxides using a Pd(0)-Xantphos catalytic system (Scheme 14).41 Styrenes, conjugated dienes, and electron-deficient olefins were efficiently alkylated to give ring-opening products 100a–h and 101a,b with retention of the stereochemistry of the original epoxides. The authors proposed that the reaction proceeds via the in situ generation of β-iodohydrins 104 from the epoxide 98g and Et3N·HI, which undergoes a SET reduction by the Pd(0) complex to form the alkyl Pd-hybrid radical species 105 (Scheme 15). Subsequently, this species adds to the double bond of alkene to form a new radical species 106, which gives the reaction product via recombination with Pd(I) and subsequent β-H elimination. The measured redox potentials of the reaction components are consistent with this hypothesis, thus ruling out an alternative radical polar crossover pathway involving oxidation of benzyl radical species 106 by Pd(I) into the corresponding benzyl cation 108.
Scheme 14.

Pd-catalyzed intermolecular Heck reactions of epoxides and redox-active esters
Scheme 15.

Zhou’s mechanism for the Pd-catalyzed Heck reaction of epoxides
In addition to alkyl halides and epoxides, the redox-active esters have also been found to undergo an efficient Heck-type reaction. In 2018, the groups of Shang, Fu and Glorius independently discovered a photoinduced Pd-catalyzed Heck reaction of redox active esters, aliphatic N-(acyloxy)phthalimides 102, with styrenes (Scheme 14).42
The reactions of primary, secondary and tertiary substrates proceeded smoothly at room temperature (products 103a–s), thus providing an alternative approach towards alkylated alkenes starting from readily available carboxylic acids. Overall, the scope of these transformations was comparable with that of the visible-light-induced Heck reactions of alkyl halides. Likewise, mechanistic studies have proven a radical-type mechanism initiated by a SET event between a photoexcited Pd(0) complex and a redox-active ester, followed by its decarboxylative fragmentation towards an alkyl hybrid Pd-radical species. The latter would be engaged in the hybrid Pd-radical catalytic cycle, ultimately generating the corresponding Heck reaction products. Furthermore, photophysical studies revealed that the Pd complex is the only light-absorbing species in this transformation, while N-(acyloxy)phthalimides 102 showed no absorption in the visible light region.
3. Ni-Catalyzed Heck-Type Reactions
3.1. Benzylic Electrophiles
Following his work on the Ni-catalyzed allylic alkenylation,43 in 2011, Jamison introduced a protocol for Heck coupling of benzyl chlorides with terminal aliphatic alkenes. A number of branched Heck reaction products 110 were efficiently obtained at room temperature in the presence of Ni(COD)2, the monodentate phosphine ligand PCy2Ph and TESOTf as an additive (Scheme 16).44 The reaction was proposed to occur via a cationic Heck reaction pathway (Scheme 17). First, the oxidative addition complex 115 undergoes counteranion exchange with TESOTf to generate the cationic benzyl-Ni(II) species 116, followed by an olefin coordination and sterics-controlled migratory insertion. Subsequent β-H elimination of the formed alkyl-Ni(II) complex 117 affords the branched Heck reaction product 110 and HNi(II)OTf, which upon reduction to Ni(0) returns to the catalytic cycle. This protocol is unique for alkyl Heck reactions as it provides access to 1,1-dialkyl-substituted alkenes 110a–i in excellent yields and regioselectivities (>95:5 for most cases). Two years later, the same group discovered the precatalyst 111 that allows this reaction to proceed much faster and, most remarkably, to be carried out under open-flask conditions, where no exclusion of air, water, or degassing of solvents and reagents is required.45 Relying on these new conditions, a diverse range of allylbenzene derivatives 110h–j was efficiently synthesized in comparable yields to those reported under the previous protocol conditions (Scheme 16).
Scheme 16.

Ni-catalyzed Heck reactions of benzylic electrophiles
Scheme 17.

Jamison’s mechanism for the Ni-catalyzed Heck reaction of benzylic chlorides
In 2014, Jarvo developed a highly efficient, stereospecific, Ni-catalyzed intramolecular Heck reaction of chiral methyl benzyl ethers 112 to give methylenecyclopentanes 113 (Scheme 16).46 The stereochemical outcome of this reaction arose from inversion during the oxidative addition event, thus providing a single enantiomer of the key alkyl-Ni intermediate 114. The stereochemistry at this carbon center remained unchanged throughout the following steps.
Recently, Hu and Huang reported the Ni-catalyzed Heck reaction of benzylamines 118 proceeding via a C–N bond activation (Scheme 18).47
Scheme 18.

Ni-catalyzed Heck reaction of benzylamines
The reaction is believed to occur via a charge transfer (CT) complex 121, which undergoes an SET with the Ni(0) catalyst to form alkyl radical species 122. The latter can be captured with styrene derivatives to produce Heck reaction products 120a–i in moderate to good yields.
3.2. α-Carbonyl Alkyl Halides
In 2012, Lei reported the Ni-catalyzed Heck-type reaction of α-carbonyl-substituted alkyl halides 123 with styrenes to construct α-alkenyl carbonyl derivatives 124 (Scheme 19).48 Both amide- and ester-possessing secondary and tertiary alkyl bromides were reactive under Ni(PPh3)4/dppp-catalyzed conditions leading to the products 124a–k. Interestingly, the authors showed that a Ni(I)-complex such as Ni(PPh3)4Cl was able to catalyze this reaction, as well. Thus, the following radical-type mechanism has been proposed (Scheme 20). Presumably, the Ni(I) species is generated in situ from the Ni(0)-catalyst and an alkyl bromide via a SET event. The second SET event would produce the Ni(II)-complex and alkyl radical species 125, which undergoes radical addition to the alkene to form benzylic radical 126. A subsequent radical polar crossover process would regenerate the active Ni(I)-catalyst and produce cationic intermediate 127, which upon base-assisted deprotonation provides the final product. The involvement of a radical polar crossover pathway was supported by the presence of the lactam cyclization by-product in significant amounts in the reaction towards 124f (Scheme 19).
Scheme 19.

Ni-catalyzed Heck reaction of α-carbonyl alkyl halides
Scheme 20.

Lei’s mechanism for the Ni-catalyzed Heck reaction of α-carbonyl alkyl halides
3.3. Unactivated Alkyl Halides
The first example of Ni-catalyzed Heck reactions of un-activated alkyl halides was reported by Beletskaya in 1988 (Scheme 21). The Ni(PPh3)4Cl2/Zn/pyridine system was shown to be capable of catalyzing the reaction of unactivated primary and secondary alkyl bromides with styrene toward the Heck reaction products 129a–c in low to moderate yields.49
Scheme 21.

Ni-catalyzed Heck reactions of unactivated alkyl halides
In 2012, Álvarez de Cienfuegos and Cuerva introduced a Heck-type cyclization of alkyl iodides 130 under Ni/Ti-synergetic catalysis at room temperature (Scheme 21).50 Interestingly, depending on the conditions, the reaction could provide either normal or reductive Heck products (131a–d or 132, respectively). In the case of standard Heck reaction conditions, the Ni(I) complex is suggested to be the active catalyst that initiates the radical-type mechanism via SET with an alkyl iodide; whereas the role of Ti(III) is believed to execute a hydrogen atom transfer (HAT) from the intermediate 133. The latter hypothesis seems to be consistent with the results on the more efficient production of 132 in the reactions of less hindered alkenes. This outcome is attributed to the ability of Ti(III) to irreversibly trap less bulky radical intermediates 133, thus providing the corresponding reduced products upon acidic quenching. Recently, Alexanian developed the Ni-catalyzed intramolecular and intermolecular Heck reactions of alkyl bromides 134 (Scheme 21).51 Employing a Ni/Xantphos catalyst and Mn reductant, this reaction produced cyclized products 135a–d in good yields and enhanced alkene regioselectivity compared to the previously developed Pd-catalyzed carbocyclizations.35 Moreover, using these conditions, an intermolecular coupling of primary and secondary alkyl bromides with electron-deficient alkenes was accomplished (products 135e–g). Mechanistic studies suggested a radical-type scenario commenced by SET between Ni(0) and an alkyl bromide. In contrast to the Pd-catalyzed carbocyclization of alkyl bromides,35 noATRC product was observed in the course of this transformation.
4. Co-Catalyzed Heck-Type Reactions
The area of Co-catalyzed alkylation reactions has arisen after the seminal discovery by Tada on the generation of alkyl radicals from alkyl halides in the presence of cobaloxime(I) as the catalyst.52 A number of reports on intramolecular Co-catalyzed Heck reactions by Scheffold53 and others,54 as well as on the intermolecular version by Branchaud,55 appeared at the early stage of its development (Scheme 22). These initial protocols usually employed strong reductive conditions, and electrochemical or photoinduced activation of hydroxocobalamin (vitamin B12a) or cobaloxime complexes. In 2002, and his follow-up work in 2006, Oshima reported the Co-catalyzed Heck reaction of alkyl halides in the presence of Grignard reagents such as TMSCH2MgCl (Scheme 22).56 Alkyl iodides, bromides, and even chlorides reacted efficiently with styrene derivatives, producing the Heck reaction products 140a–i in good yields. Spectroscopic and crystallographic studies supported a SET process between the in situ generated Co(0) complex and an alkyl halide leading to the alkyl radical species engaged in the hybrid-radical mechanism. In 2004, the same group applied these conditions towards Heck reactions of epoxides 98 and 99 (Scheme 22).57 The reaction proceeded through in situ generation of 2-bromomagnesium methoxide 143 followed by the Co-catalyzed Heck reaction of alkyl bromides analogous to the prior work.56 The reaction was fairly effective for Heck reactions of symmetrical epoxides 141a–d, while asymmetrical substrates yielded a mixture of regioisomers 142a and 142a′.
Scheme 22.

Co-catalyzed Heck reactions using strong reducing reagents
In 2011, Carreira developed a room-temperature visible-light-induced intramolecular Heck reaction employing a cobaloxime catalyst 148 (Scheme 23).58 In contrast to the previously reported Co-catalyzed Heck reactions, the method relied on the Hunig’s base [N(i-Pr)2Et]-promoted Co(I)-catalyst turnover via deprotonation of the Co(III)–H intermediate. Therefore, strong reductants such as RMgX or Zn were no longer required, which significantly expanded the functional group compatibility of the Co-catalyzed Heck reaction. Excitingly, the authors were able to apply this mild protocol for the late-stage cyclization step in the total synthesis of (+)-daphmanidin.58 Later, Carreira reported an intermolecular version of this coupling using 2,2,2-trifluoroethyl iodide (146) and styrene derivatives (Scheme 23).59 The intramolecular and intermolecular Heck reactions proceeded uneventfully, generating the Heck products 145a–d and 147a–e in good to excellent yields. Sensitive functionalities such as amides, esters, ketones, and aldehydes were well tolerated. Recently, Wu and co-workers developed a room temperature Heck reaction of widely available alkyl carboxylic acids 149 using organo photoredox/cobaloxime dual catalysis (Scheme 24).60 The scope of this reaction was fairly broad as primary, secondary and tertiary alkyl radicals, generated from carboxylic acids, could be efficiently coupled with vinyl arenes/heteroarenes leading to the corresponding products 150a–g in good to excellent yields. Vinyl boronates and vinyl silanes (products 150h and 150i, respectively) were also shown to be competent alkene coupling partners in this reaction, which further extended the synthetic utility of this protocol.
Scheme 23.

Carreira’s Co-catalyzed Heck reaction using Hunig’s base
Scheme 24.

Wu’s Co-catalyzed Heck reaction of carboxylic acids
Moreover, under the developed conditions, an unprecedented three-component coupling of alkyl carboxylic acid, acrylates, and styrenes was demonstrated to afford highly functionalized vinyl arenes in good yields and as single regioisomers with exclusive E configuration (products 150j and 150k). Mechanistic studies, as well as DFT calculations, supported a radical-type mechanism (Scheme 25). According to which, a SET process from the carboxylate to the excited Mes-Acr+* species is followed by radical decarboxylation to produce the alkyl radical species 151. Subsequent trapping of the latter by an alkene, and then by the Co(II)-catalyst, leads to alkyl-Co(III) species 153, which undergoes β-H elimination towards the Heck reaction product 150 and a Co(III)-H species. Deprotonation of this species, followed by an SET with the reduced photocatalyst regenerates both active catalysts, Co(II)Ln, and Mes-Acr+. This represents the first method for the Heck reaction of unactivated alkyl carboxylic acids, with H2 and CO2 being the only by-products.
Scheme 25.

Wu’s mechanism for the Co-catalyzed Heck reaction of carboxylic acids
5. Cu-Catalyzed Heck-Type Reactions
In 2013, Chemler reported the first Cu-catalyzed alkyl Heck reaction (Scheme 26). In contrast to many alkyl Heck reactions, this reaction employed alkyl nucleophiles, such as alkyltrifluoroborates 154, as coupling partners for the alkenes.61
Scheme 26.

Chemler’s Cu-catalyzed Heck reaction of alkyl trifluoroborates
The outcome of TEMPO-trapping and radical clock experiments indicated the radical nature of this transformation. The authors proposed that, under oxidative conditions, generation of alkyl radical species 157 and a Cu(I) species occurred via homolysis of the Cu–C bond in the complex 156, which formed via transmetalation of the Cu(II) catalyst with alkyl trifluoroborate 154 (Scheme 27). Addition of 157 to an alkene, followed by subsequent oxidation and deprotonation produced the Heck reaction product 155. Yet, to regenerate the catalyst, the Cu(I) species had to be oxidized by an external oxidant such as MnO2. This oxidative transformation features a quite general scope with regards to the trifluoroborate salt, as benzylic, unactivated primary, and secondary alkyl substrates could efficiently react with styrene derivatives to afford the Heck products 155a–i in good yields.
Scheme 27.

Chemler’s mechanism for the Cu-catalyzed Heck reaction of alkyl trifluoroborates
In the same year, Nishikata developed the Cu(I)/triamine (161)-catalyzed Heck reaction of activated tertiary alkyl bromides 159 with styrenes (Scheme 28).62 Ester-, nitro-, or keto-group-possessing tertiary alkyl bromides were demonstrated to couple with electron-rich styrenes (products 160a–e) under mild conditions, presumably via the formation of ATRA intermediate 162. A TEMPO-trapping experiment confirmed the intermediacy of a tertiary alkyl radical species in this reaction. Later, the same group introduced a modified Cu(I)/triamine-based catalytic system to accomplish the Heck reaction of amido-possessing alkyl bromides, which were unreactive under the previous conditions,62 with both electron-rich and electron-deficient styrene derivatives (Scheme 28).63 The reaction required higher temperatures and tri-(2-picolyl)amine (TMPA) as a ligand to furnish products 160f–k in reasonable yields. In 2014, Lei developed the Cu(I)/1,10-phen-catalyzed Heck reaction of primary benzylic and activated secondary and tertiary alkyl bromides with electron-rich styrenes (Scheme 28, products 163a–k).64 An EPR experiment supported the SET event between the Cu(I) catalyst and an alkyl bromide.
Scheme 28.

Cu-catalyzed Heck reactions of activated alkyl halides
In 2014, Chu and Tong synthesized CuB23 alloy short nanotubes, which were found to be capable of catalyzing the Heck reaction of unactivated alkyl iodides 88 under ligand-free conditions (Scheme 29).65 Under these conditions, unactivated primary and secondary substrates reacted smoothly, presumably via a radical pathway (products 164a–e). This method provided slightly superior yields compared to the Pd-catalyzed Heck reaction.37 Moreover, the catalyst showed a great recycling performance.
Scheme 29.

Cu-catalyzed Heck reaction of unactivated alkyl halides
Regioselectivity has been an issue in the Heck reactions of unactivated aliphatic olefins due to the undifferentiated C–H sites for β-H elimination. Very recently, the first regio-selective Heck reaction of alkyl bromides with unactivated aliphatic olefins was reported by Bi and co-workers (Scheme 30).66
Scheme 30.

Fu, Guan and Bi’s directed Cu-catalyzed Heck reaction of unactivated olefins
The high reactivity and regioselectivity of this transformation were governed by the aminoquinolone directing group (AQ) on the alkene 165, coordination of which to the Cu(I) catalyst activated the double bond (167) and also provided control of the regioselectivity for the β-H elimination step (168). Detailed mechanistic studies and DFT calculations indicated a radical pathway involving a dimethyl sulf-oxide assisted concerted H–Br elimination event from a conformationally strained Cu(III) cyclic transition state. Activated primary, secondary and tertiary alkyl bromides were suitable substrates in this reaction leading to products 166a–h.
6. Other Metals in Heck-Type Reactions
In 2003, Kambe reported the titanocene-catalyzed Heck-type reaction of alkyl halides with vinyl arenes (Scheme 31).67 The reaction proceeded efficiently at 0 °C for unactivated primary and secondary alkyl bromides and even chlorides (products 169a–e), however, considerable amounts of by-products were observed in some cases. The Ti(III)-complex formed in situ from the Ti(IV)-catalyst and the Grignard reagent is believed to generate alkyl radicals, as well as to trap the benzylic radical intermediate towards an alkyl Ti(IV) species 170, which upon β-H elimination delivers the Heck product 169.
Scheme 31.

Ti-catalyzed Heck reaction
In 2013, Lei’s group applied Rh- and Ir-photoredox catalysis for the Heck reaction of activated alkyl bromides with vinyl arenes (Scheme 32).68 The reaction operates via a radical-polar crossover pathway, where a photoredox catalyst is involved in SET with an alkyl bromide, and in oxidation of the benzylic radical intermediate towards the carbocation 172. Activated tertiary and secondary alkyl bromides reacted efficiently under the Rh-catalyzed conditions (171a–f), whereas activated primary alkyl bromides required Ir-catalyzed conditions for the same coupling (171g–k). Later, Cho employed ethyl 2-bromo-2,2-difluoro-acetate (173) in a photoinduced Ir-catalyzed Heck reaction (Scheme 32).69 Besides styrene derivatives (products 174a–c), unactivated aliphatic alkenes (products 174d–f) were also suitable partners in reactions with alkyl bromide 173, which is quite rare for alkyl Heck reactions.
Scheme 32.

Rh- and Ir-catalyzed Heck reactions
In 2015, the Fe-catalyzed Heck reaction of benzyl chlorides 175 under UV irradiation was reported by Mankad (Scheme 33).70 The authors favored a classical Heck reaction mechanism for this transformation, where UV irradiation promoted the CO dissociation to reveal reactive, coordinatively unsaturated Fe-containing intermediates. A radical-type process was considered to be an unproductive pathway under these conditions, competing with the Heck reaction and leading to decomposition. Therefore, the reaction suffered from moderate yields and regioselectivity due to alkene isomerization (products 176a–g). In 2017, Bao reported another Fe-catalyzed Heck reaction using peresters 177 as the source of alkyl radicals (Scheme 33).71 This reaction was proposed to occur via a radical-polar crossover mechanism initiated by the Fe(II)-catalyst. Methylated and ethylated styrene derivatives 178a–g, enyne 178h, and diene 178i were obtained in reasonable yields under these conditions.
Scheme 33.

Fe-catalyzed Heck reactions
In 2014, decarbonylative Heck-type reactions employing tert-butyl peroxide and a catalytic amount of MnBr2 were reported by Li (Scheme 34).72 Peroxide-induced H-atom abstraction from aldehyde 179 gives the carbonyl radical 181, which upon releasing CO fragments into an alkyl radical 182 (Scheme 35). Subsequent radical addition of the latter to the alkene, followed by oxidation and deprotonation furnishes the Heck-type product 180. The reaction proceeds well with primary, secondary, and tertiary aldehydes, however, it is limited to 1,1-disubstituted styrenes (products 180a–f).
Scheme 34.

Mn-catalyzed Heck reaction
Scheme 35.

Li’s mechanism for the Mn-catalyzed Heck reaction
In 2016, Hashmi’s group showed that dinuclear gold complex 186 could catalyze the Heck reaction of alkyl bromides under UVA irradiation (315–400 nm) (Scheme 36).73 In this reaction, electron-rich vinyl arenes were efficiently alkylated with primary, secondary, and tertiary alkyl bromides to afford Heck products 185a–f in good yields. The method was also applicable for the late-stage functionalization of a complex molecule, e.g., pregnenolone derivative 185c. Mechanistic studies supported an Au-catalyzed radical polar-crossover mechanism induced by SET from the photoexcited Au(I)–Au(I) complex to an alkyl bromide.
Scheme 36.

Au-catalyzed Heck reaction
7. Conclusion
Although the first alkyl Heck reaction was reported by Richard Heck in his original work,1d this transformation has remained underdeveloped until recently, especially for unactivated hindered alkyl halides possessing eliminable β-hydrogens. However, a merger of radical- and transition-metal-catalyzed approaches has significantly driven this area. Nowadays, in addition to Pd, other transition metals such as Ni, Co, Cu, Fe, and others have been shown to efficiently catalyze Heck-type reactions, generally following a hybrid-organometallic radical mechanism. Unactivated alkyl electrophiles possessing eliminable β-hydrogens appeared to be non-problematic for this mechanism. Moreover, the employment of mild photoinduced conditions has further broadened the scope of the alkyl Heck reaction. Although, significant effort has been made to expand the scope with regards to the alkyl component, the scope of the alkenes remains mostly limited to good radical acceptors such as styrenes and acrylate derivatives. Examples of the alkylation of unactivated aliphatic alkenes are rare. Therefore, the future direction of this transformation may rely on the development of new systems that enable unactivated alkenes to undergo selective and efficient Heck-type reactions. Obviously, detailed mechanistic studies are warranted for better understanding of alkyl Heck-type reactions.
Funding Information
We thank the National Institutes of Health (GM120281) and National Science Foundation (CHE-1663779) for the financial support of this work.
Biography

Daria Kurandina received her BS degree from St. Petersburg State University, Russia. In 2014, she joined Prof. Gevorgyan’s group at the University of Illinois at Chicago as a PhD student. Her work focuses on the development of novel transition-metal-catalyzed synthetic methodologies.
Padon Chuentragool obtained his BS and MS degrees from Chulalongkorn University, Bangkok, Thailand. He received his PhD in 2018 under the guidance of Prof. Gevorgyan at the University of Illinois at Chicago, where his work focused on the development of selective methods for C(sp3)–H functionalizations via photoexcited Pd catalysis.
Vladimir Gevorgyan received his PhD in 1984 from the Latvian Institute of Organic Synthesis, where he then worked as a group leader. After postdoctoral research at Tohoku University with Prof. Y. Yamamoto as a JSPS Postdoctoral Fellow and then as a Ciba-Geigy International Postdoctoral Fellow, he joined the faculty at the same institute in 1996. In 1999, he moved to the University of Illinois at Chicago (UIC) as an associate professor and was promoted to full professor in 2003. In 2012, he received the LAS Distinguished Professor Award. His group is interested in the development of novel synthetic methodologies.
References
- 1.(a) Oestreich M The Mizoroki-Heck Reaction; John Wiley & Sons: Chichester, 2009. [Google Scholar]; (b) Gharpure SJ; Shelke YG; Reddy SR B. RSC Adv 2014, 4, 46962. [Google Scholar]; (c) Heck RF J. Am. Chem. Soc 1969, 91, 6707. [Google Scholar]; (d) Heck RF; Nolley JP J. Org. Chem 1972, 37, 2320. [Google Scholar]; (e) Dieck HA; Heck RF J. Am. Chem. Soc 1974, 96, 1133. [Google Scholar]; (f) Mizoroki T; Mori K; Ozaki A Bull. Chem. Soc. Jpn 1971, 44, 581. [Google Scholar]
- 2.de Meijere A; Bräse S; Oestreich M Metal-Catalyzed Cross-Coupling Reactions and More; Wiley-VCH: Weinheim, 2014. [Google Scholar]
- 3.(a) Biajoli AFP; Schwalm CS; Limberger J; Claudino TS; Monteiro AL J. Braz. Chem. Soc 2014, 25, 2186. [Google Scholar]; (b) Torborg C; Beller M Adv. Synth. Catal 2009, 351, 3027. [Google Scholar]; (c) de Vries JG Can. J. Chem 2001, 79, 1086. [Google Scholar]; (d) Majid MH; Razieh M; Masoumeh M Curr. Org. Chem 2018, 22, 165. [Google Scholar]
- 4.Roy D; Uozumi Y Adv. Synth. Catal 2018, 360, 602. [Google Scholar]
- 5.Wang S-S; Yang G-Y Catal. Sci. Technol 2016, 6, 2862. [Google Scholar]
- 6.McCartney D; Guiry PJ Chem. Soc. Rev 2011, 40, 5122. [DOI] [PubMed] [Google Scholar]
- 7.(a) Beletskaya IP; Cheprakov AV Chem. Rev 2000, 100, 3009. [DOI] [PubMed] [Google Scholar]; (b) Cabri W; Candiani I Acc. Chem. Res 1995, 28, 2. [Google Scholar]; (c) Whitcombe NJ; Hii KK; Gibson SE Tetrahedron 2001, 57, 7449. [Google Scholar]; (d) de Meijere A; Meyer FE Angew. Chem. Int. Ed 1995, 33, 2379. [Google Scholar]
- 8.Ariafard A; Lin Z Organometallics 2006, 25, 4030. [Google Scholar]
- 9.(a) Frisch AC; Beller M Angew. Chem. Int. Ed 2005, 44, 674. [DOI] [PubMed] [Google Scholar]; (b) Tang S; Liu K; Liu C; Lei A Chem. Soc. Rev 2015, 44, 1070. [DOI] [PubMed] [Google Scholar]; (c) Rudolph A; Lautens M Angew. Chem. Int. Ed 2009, 48, 2656. [DOI] [PubMed] [Google Scholar]
- 10.(a) Phapale VB; Buñuel E; García-Iglesias M; Cárdenas DJ Angew. Chem. Int. Ed 2007, 46, 8790. [DOI] [PubMed] [Google Scholar]; (b) Schley ND; Fu GC J. Am. Chem. Soc 2014, 136, 16588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Newcomb M; Toy PH Acc. Chem. Res 2000, 33, 449. [DOI] [PubMed] [Google Scholar]; (b) Baldwin JE Chem. Rev 2003, 103, 1197. [DOI] [PubMed] [Google Scholar]
- 12.(a) Wang C; Lei Y; Guo M; Shang Q; Liu H; Xu Z; Wang R Org. Lett 2017, 19, 6412. [DOI] [PubMed] [Google Scholar]; (b) Fan J-H; Wei W-T; Zhou M-B; Song R-J; Li J-H Angew. Chem. Int. Ed 2014, 53, 6650. [DOI] [PubMed] [Google Scholar]; (c) Kancherla R; Muralirajan K; Maity B; Zhu C; Krach PE; Cavallo L; Rueping M Angew. Chem. Int. Ed 2019, in press; DOI: 10.1002/anie.201811439. [DOI] [PubMed] [Google Scholar]
- 13.Yi P; Zhuangyu Z; Hongwen H Synthesis 1995, 245. [Google Scholar]
- 14.Higuchi K; Sawada K; Nambu H; Shogaki T; Kita Y Org. Lett 2003, 5, 3703. [DOI] [PubMed] [Google Scholar]
- 15.Wang L; Pan Y; Jiang X; Hu H Tetrahedron Lett. 2000, 41, 725. [Google Scholar]
- 16.Hu Y.-m.; Zhou J; Long X.-t.; Han J.-l.; Zhu C.-j.; Pan Y Tetrahedron Lett. 2003, 44, 5009. [Google Scholar]
- 17.Nagayama K; Shimizu I; Yamamoto A Bull. Chem. Soc. Jpn 1999, 72, 799. [Google Scholar]
- 18.Narahashi H; Yamamoto A; Shimizu I Chem. Lett 2004, 33, 348. [Google Scholar]
- 19.Yang Z; Zhou J J. Am. Chem. Soc 2012, 134, 11833. [DOI] [PubMed] [Google Scholar]
- 20.Wu GZ; Lamaty F; Negishi E J. Org. Chem 1989, 54, 2507. [Google Scholar]
- 21.Liu Z; Shi C; Chen Y Synlett 2008, 1734. [Google Scholar]
- 22.Zhou W; An G; Zhang G; Han J; Pan Y Org. Biomol. Chem 2011, 9, 5833. [DOI] [PubMed] [Google Scholar]
- 23.(a) Mori M; Oda I; Ban Y Tetrahedron Lett. 1982, 23, 5315. [Google Scholar]; (b) Mori M; Kanda N; Oda I; Ban Y Tetrahedron 1985, 41, 5465. [Google Scholar]
- 24.Glorius F Tetrahedron Lett. 2003, 44, 5751. [Google Scholar]
- 25.Kurandina D; Rivas M; Radzhabov M; Gevorgyan V Org. Lett 2018, 20, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Q-Y; Yang Z-Y; Zhao C-X; Qiu Z-M J. Chem. Soc., Perkin Trans 1 1988, 563. [Google Scholar]
- 27.Surapanich N; Kuhakarn C; Pohmakotr M; Reutrakul V Eur. J. Org. Chem 2012, 5943. [Google Scholar]
- 28.Feng Z; Min Q-Q; Zhao H-Y; Gu J-W; Zhang X Angew. Chem. Int. Ed 2015, 54, 1270. [DOI] [PubMed] [Google Scholar]
- 29.Fan T; Meng W-D; Zhang X Beilstein J. Org. Chem 2017, 13, 2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parasram M; Iaroshenko VO; Gevorgyan V J. Am. Chem. Soc 2014, 136, 17926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kurandina D; Parasram M; Gevorgyan V Angew. Chem. Int. Ed 2017, 56, 14212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.(a) Bräse S; Waegell B; de Meijere A Synthesis 1998, 148. [Google Scholar]; (b) Firmansjah L; Fu GC J. Am. Chem. Soc 2007, 129, 11340. [DOI] [PubMed] [Google Scholar]
- 33.Bloome KS; McMahen RL; Alexanian EJ J. Am. Chem. Soc 2011, 133, 20146. [DOI] [PubMed] [Google Scholar]
- 34.Bloome KS; Alexanian EJ J. Am. Chem. Soc 2010, 132, 12823. [DOI] [PubMed] [Google Scholar]
- 35.Venning ARO; Kwiatkowski MR; Roque Peña JE; Lainhart BC; Guruparan AA; Alexanian EJ J. Am. Chem. Soc 2017, 139, 11595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dong X; Han Y; Yan F; Liu Q; Wang P; Chen K; Li Y; Zhao Z; Dong Y; Liu H Org. Lett 2016, 18, 3774. [DOI] [PubMed] [Google Scholar]
- 37.McMahon CM; Alexanian EJ Angew. Chem. Int. Ed 2014, 53, 5974. [DOI] [PubMed] [Google Scholar]
- 38.Zou YJ; Zhou JR Chem. Commun 2014, 50, 3725. [DOI] [PubMed] [Google Scholar]
- 39.Wang G-Z; Shang R; Cheng W-M; Fu Y J. Am. Chem. Soc 2017, 139, 18307. [DOI] [PubMed] [Google Scholar]
- 40.Gevorgyan V; Chuentragool P; Yadagiri D; Morita T; Sarkar S; Parasram M; Wang Y Angew. Chem. Int. Ed 2019, in press; DOI: 10.1002/anie.201812398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Teng S; Tessensohn ME; Webster RD; Zhou JS ACS Catal. 2018, 8, 7439. [Google Scholar]
- 42.(a) Wang G-Z; Shang R; Fu Y Org. Lett 2018, 20, 888. [DOI] [PubMed] [Google Scholar]; (b) Koy M; Sandfort F; Tlahuext-Aca A; Quach L; Daniliuc CG; Glorius F Eur. J. Chem 2018, 24, 4552. [DOI] [PubMed] [Google Scholar]
- 43.Matsubara R; Jamison TF J. Am. Chem. Soc 2010, 132, 6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matsubara R; Gutierrez AC; Jamison TF J. Am. Chem. Soc 2011, 133, 19020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Standley EA; Jamison TF J. Am. Chem. Soc 2013, 135, 1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harris MR; Konev MO; Jarvo ER J. Am. Chem. Soc. 2014, 136, 7825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu H; Hu B; Huang H J. Org. Chem 2018, 83, 13922. [DOI] [PubMed] [Google Scholar]
- 48.Liu C; Tang S; Liu D; Yuan J; Zheng L; Meng L; Lei A Angew. Chem. Int. Ed 2012, 51, 3638. [DOI] [PubMed] [Google Scholar]
- 49.Lebedev SA; Lopatina VS; Petrov ES; Beletskaya IP J. Organomet. Chem 1988, 344, 253. [Google Scholar]
- 50.Millán A; Álvarez de Cienfuegos L; Miguel D; Campaña AG; Cuerva JM Org. Lett 2012, 14, 5984. [DOI] [PubMed] [Google Scholar]
- 51.Kwiatkowski MR; Alexanian EJ Angew. Chem. Int. Ed 2018, 57, 16857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Okabe M; Abe M; Tada M J. Org. Chem 1982, 47, 1775. [Google Scholar]
- 53.Busato S; Tinembart O; Zhang Z-D; Scheffold R Tetrahedron 1990, 46, 3155. [Google Scholar]
- 54.(a) Giese B; Erdmann P; Göbel T; Springer R Tetrahedron Lett. 1992, 33, 4545. [Google Scholar]; (b) Torii S; Inokuchi T; Yukawa T J. Org. Chem 1985, 50, 5875. [Google Scholar]; (c) Ladlow M; Pattenden G Tetrahedron Lett. 1984, 25, 4317. [Google Scholar]
- 55.(a) Branchaud BP; Meier MS; Choi Y Tetrahedron Lett. 1988, 29, 167. [Google Scholar]; (b) Branchaud BP; Yu GX Organometallics 1993, 12, 4262. [Google Scholar]; (c) Branchaud BP; Detlefsen WD Tetrahedron Lett. 1991, 32, 6273. [Google Scholar]; (d) Branchaud BP; Meier MS J. Org. Chem 1989, 54, 1320. [Google Scholar]; (e) Branchaud BP; Choi YL Tetrahedron Lett. 1988, 29, 6037. [Google Scholar]; (f) Branchaud BP; Meier MS Tetrahedron Lett. 1988, 29, 3191. [Google Scholar]
- 56.(a) Ikeda Y; Nakamura T; Yorimitsu H; Oshima KJ Am. Chem. Soc 2002, 124, 6514. [DOI] [PubMed] [Google Scholar]; (b) Affo W; Ohmiya H; Fujioka T; Ikeda Y; Nakamura T; Yorimitsu H; Oshima K; Imamura Y; Mizuta T; Miyoshi KJ Am. Chem. Soc 2006, 128, 8068. [DOI] [PubMed] [Google Scholar]
- 57.Ikeda Y; Yorimitsu H; Shinokubo H; Oshima K Adv. Synth. Catal 2004, 346, 1631. [Google Scholar]
- 58.Weiss ME; Kreis LM; Lauber A; Carreira EM Angew. Chem. Int. Ed 2011, 50, 11125. [DOI] [PubMed] [Google Scholar]
- 59.Kreis LM; Krautwald S; Pfeiffer N; Martin RE; Carreira EM Org. Lett 2013, 15, 1634. [DOI] [PubMed] [Google Scholar]
- 60.Cao H; Jiang H; Feng H; Kwan JMC; Liu X; Wu J J. Am. Chem. Soc 2018, 140, 16360. [DOI] [PubMed] [Google Scholar]
- 61.Liwosz TW; Chemler SR Org. Lett 2013, 15, 3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nishikata T; Noda Y; Fujimoto R; Sakashita T J. Am. Chem. Soc 2013, 135, 16372. [DOI] [PubMed] [Google Scholar]
- 63.Nishikata T; Itonaga K; Yamaguchi N; Sumimoto M Org. Lett 2017, 19, 2686. [DOI] [PubMed] [Google Scholar]
- 64.Zhang X; Yi H; Liao Z; Zhang G; Fan C; Qin C; Liu J; Lei A Org. Biomol. Chem 2014, 12, 6790. [DOI] [PubMed] [Google Scholar]
- 65.(a) Yang F; Fu SY; Chu W; Li C; Tong DG RSC Adv. 2014, 4, 45838. [Google Scholar]; (b) Fu SY; Li YZ; Chu W; Lia C; Tong DG Catal. Sci. Technol 2015, 5, 1638. [Google Scholar]
- 66.Tang C; Zhang R; Zhu B; Fu J; Deng Y; Tian L; Guan W; Bi X J. Am. Chem. Soc 2018, 140, 16929. [DOI] [PubMed] [Google Scholar]
- 67.Terao J; Watabe H; Miyamoto M; Kambe N Bull. Chem. Soc. Jpn 2003, 76, 2209. [Google Scholar]
- 68.Liu Q; Yi H; Liu J; Yang Y; Zhang X; Zeng Z; Lei A Eur. J. Chem 2013, 19, 5120. [DOI] [PubMed] [Google Scholar]
- 69.Yu C; Iqbal N; Park S; Cho EJ Chem. Commun 2014, 50, 12884. [DOI] [PubMed] [Google Scholar]
- 70.Waldhart GW; Mankad NP J. Organomet. Chem 2015, 793, 171. [Google Scholar]
- 71.Zhu N; Zhao J; Bao H Chem. Sci 2017, 8, 2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zong Z; Wang W; Bai X; Xi H; Li Z Asian J. Org. Chem 2015, 4, 622. [Google Scholar]
- 73.Xie J; Li J; Weingand V; Rudolph M; Hashmi ASK Eur. J. Chem 2016, 22, 12646. [DOI] [PubMed] [Google Scholar]