

Abstract
Activation of ribosomal RNA (rRNA) synthesis is pivotal during cell growth and proliferation, but its aberrant upregulation may promote tumorigenesis. Here, we demonstrate that the candidate oncoprotein, LYAR, enhances ribosomal DNA (rDNA) transcription. Our data reveal that LYAR binds the histone-associated protein BRD2 without involvement of acetyl-lysine–binding bromodomains and recruits BRD2 to the rDNA promoter and transcribed regions via association with upstream binding factor. We show that BRD2 is required for the recruitment of the MYST-type acetyltransferase KAT7 to rDNA loci, resulting in enhanced local acetylation of histone H4. In addition, LYAR binds a complex of BRD4 and KAT7, which is then recruited to rDNA independently of the BRD2-KAT7 complex to accelerate the local acetylation of both H4 and H3. BRD2 also helps recruit BRD4 to rDNA. By contrast, LYAR has no effect on rDNA methylation or the binding of RNA polymerase I subunits to rDNA. These data suggest that LYAR promotes the association of the BRD2-KAT7 and BRD4-KAT7 complexes with transcription-competent rDNA loci but not to transcriptionally silent rDNA loci, thereby increasing rRNA synthesis by altering the local acetylation status of histone H3 and H4.
INTRODUCTION
The ribosome is the essential cellular machinery for protein synthesis. The eukaryotic ribosome consists of a large 60S subunit and a small 40S subunit, the biogenesis of which requires transcription of a large ribosomal RNA precursor (47S pre-rRNA) by RNA polymerase (RNAP) I, a 5S rRNA by RNAP III, and translation of ribosomal proteins from mRNAs transcribed by RNAP II (1–6). In addition, numerous small nucleolar RNAs and non-ribosomal proteins (called trans-acting factors) participate in ribosome biogenesis (5,6). Among these factors, many contribute to the synthesis of 47S pre-rRNA, which alone constitutes ∼80% of the total transcription of proliferating cells (1,2). Because of its predominance, rDNA transcription is regulated by ≥30 trans-acting factors, including proto-oncogene products (MYC, etc.) and tumor suppressors (p53, retinoblastoma, etc.) to maintain appropriate ribosome numbers in normal cells (1–3,7). Upon oncogenic transformation of a cell, however, rRNA synthesis is upregulated by altered function of these types of factors, e.g. owing to their dysregulated expression and/or mutation (1). In certain tumor cells, the upregulation of rRNA synthesis occurs via phosphorylation of upstream binding factor (UBF), a rDNA promoter–binding protein that mediates binding of selectivity factor 1 (SL1) to the rDNA promoter, resulting in the recruitment of RNAP I. The increased activity of casein kinase II or of complexes formed between the mitotic cyclins and the cyclin-dependent kinases (CDK4-cyclin D1, CDK2-cyclin E) or the mitogen-activated protein kinase ERK (extracellular signal–regulated kinase) is responsible for this upregulation in certain cell types (1,2,7). For instance, casein kinase II phosphorylates the serine-rich carboxyl-terminal acidic tail of UBF, thereby promoting its interaction with SL1 (8). Failure to interrupt the binding between SL1 and UBF is another mechanism underlying the upregulation of rDNA transcription in cancer cells, which is often caused by inactivating mutations of the tumor-suppressor protein retinoblastoma or p53 (7,8). Another mechanism of rDNA upregulation occurs through increased expression of MYC along with the sequence-specific DNA-binding protein Max, which associates with CACGTG in the rDNA promoter and activates RNAP I–dependent transcription via recruitment of transformation/transcription domain–associated protein that bridges between MYC and the histone acetyltransferase GCN5, resulting in local acetylation of nucleosomal histones in rDNA loci (9–11). In addition, rRNA synthesis is accelerated at the level of transcriptional elongation by the actions of chromatin-remodeling and elongation complexes including the FACT and PAF complexes (12,13) and/or histone chaperones including nucleolin (14).
LYAR, the human ortholog of the mouse nucleolar protein LYAR (Ly-1 antibody-reactive clone), is a 45-kDa protein with 379 amino acid residues including a zinc-finger motif and three nuclear localization signals (15). LYAR is conserved across many species, including mammals. Mouse LYAR mRNA is detectable in immature spermatocytes and testes in early embryos and to a lesser extent in fetal liver and thymus tissue (15–17). In adult mice, the mRNA is detectable at low levels in kidney and spleen but not in other differentiated tissues including brain; however, the mRNA is expressed at very high levels in a number of mouse B- and T-cell leukemia lines (15). Mouse fibroblasts overexpressing mouse LYAR cDNA contribute to tumor formation in nude mice; thus, LYAR is believed to be a nucleolar oncoprotein that regulates the growth and proliferation of cancer cells (15). In support of this idea, LYAR is overexpressed in human medulloblastoma, the most common brain tumor in children (18), and it is also highly expressed in human metastatic colorectal cancer cells (19). Moreover, when overexpressed in vitro, LYAR increases the already potent proliferative capacity of HeLa cells (human uterine cervical cancer) and MCF cells (human breast cancer) (20).
Moreover, mouse LYAR is necessary for the expression of the fetal globin gene via its ability to associate with protein arginine methyl transferase 5 (21), and it is indispensable for cell proliferation and development of female mouse embryos in which TP53 (encoding p53) is experimentally disrupted (16,22). Mouse LYAR also associates with nucleolin, a trans-acting factor involved in various stages of ribosome biogenesis, and together these proteins help maintain the pluripotency of embryonic stem cells (23). In addition, LYAR can bind to immature ribosome particles as well as a number of nucleolar proteins, including nucleophosmin (also known as B23), DDX21, UBF, and treacle, which is the product of TCOF1, the causative gene for Treacher Collins syndrome (24–32). LYAR also participates in pre-rRNA processing (20). Although modulation of rRNA synthesis is linked directly to differentiation of various stem-cell types including human embryonic stem cells (33–35), the involvement of LYAR in rRNA synthesis remains to be determined.
To address this shortcoming, we investigated LYAR function in human cells and found that LYAR binds rDNA via UBF and enhances pre-rRNA synthesis via its ability to mobilize the histone-associated proteins, the BRDs, to rDNA. UBF binds to the active nucleolar organizer region of genes, interacts directly with RNAP I (36), and has many functions during rDNA transcription, including the pause and release of RNAP I from the promoter (37). BRD2 enhances the recruitment of the acetyltransferase KAT7 and BRD4 to rDNA. Both BRD2 and BRD4 belong to the BET family of proteins and contain two tandem bromodomains and an extra-terminal domain (38). They are involved in RNAP II–dependent transcription and are recruited to euchromatin regions via their interaction with acetylated histones H3 and H4 (38,39). BRD2, which was originally identified as a Ser/Thr kinase (40,41), is necessary for the acetylation of lysine (K) residues of H4 (42), and BRD2 has histone chaperone activity that is required for RNAP II–dependent transcription of certain genes including cyclin A (42). BRD4 is a global regulator of p-TEFb (positive transcription elongation factor b)-dependent phosphorylation of RNAP II (43) and was recently found to be a histone acetyltransferase specific for histones H3 and H4, including H3K122 (44). Despite the roles of BRD family proteins in RNAP II–dependent transcription, their involvement in RNAP I–dependent transcription is unknown. On the other hand, KAT7, a member of a family of the MYST-type histone acetyltransferases, is required for acetylation of histones H3 and H4 depending on which co-binding partner is involved (JADE1, etc.) and for expression of development-specific genes in mammalian embryos (45). Our results suggest a mechanism by which RNAP I–dependent rRNA synthesis is regulated by LYAR, BRD proteins, and KAT7 in human cells.
MATERIALS AND METHODS
Metabolic labeling of newly synthesized 47S/45S pre-rRNA
The incorporation of 4-thiouridine and subsequent biotinylation were performed as described (46) with some modifications, noted as follows. HeLa or 293T cells in 35-mm Petri dishes were cultured in the presence of 10 μM 4-thiouridine (Sigma) for 30 min. Cells were collected with ice-cold PBS, and total RNA was isolated with TRIzol Reagent (Invitrogen). For the biotinylation of 4-thiouridine–labeled RNA, 20 μg of total RNA was incubated for 3 h at 25°C with 0.2 mg/ml biotin-coupled N-[6-(biotinamide)hexyl]-3’-(2’-pyridyldithio)propionamide (Pierce, 1 mg/ml in dimethylformamide) in 200 μl of 10 mM Tris–HCl, pH 7.4, containing 1 mM EDTA. An equal volume of chloroform was added, mixed, and incubated for 5 min. The mixture was subjected to centrifugation at 20,000 × g for 5 min, and the biotinylated RNA was precipitated from solution with isopropanol. After washing with 75% ethanol, each biotinylated RNA sample was dried and dissolved in formamide. For the detection of biotinylated 47S/45S pre-rRNA, 4 μg total RNA was electrophoresed on a 0.9% agarose/formaldehyde gel in MOPS running buffer at 3.5 V/cm for 3 h. Separated RNAs were transferred to a Hybond N+ membrane, which was subsequently dried, UV crosslinked using the CX-2000 UV Crosslinker (UVP, CA, USA) at 120 mJ/cm2, soaked in 10% acetic acid, and stained with methylene blue. Biotinylated 47/45S pre-rRNA was detected with horseradish peroxidase (HRP)–conjugated streptavidin using the Chemiluminescent Nucleic Acid Detection Module (Thermo Scientific). Signals were detected with the LAS-4000 system. Signal intensities of RNA bands were measured using NIH ImageJ software. To ensure equal loading, control RNA in each gel was stained using SYBR GOLD (Invitrogen) and quantified after the detection with the LAS-4000 system.
All other materials and methods are provided in supplementary materials including the list of antibodies (Supplementary Table S1) and that of DNA primers and small interference RNAs (Supplementary Table S2) used in this study.
RESULTS
LYAR is overexpressed in many human cancer cells
LYAR is a candidate oncoprotein (15), so we assessed the intracellular levels of LYAR or LYAR mRNA in tissues from normal/control subjects and cancer patients. In many of the tumor samples from cancer patients, including those with colorectal carcinoma or small-cell lung carcinoma, LYAR mRNA levels were higher than in normal tissues (Supplementary Figure S1A). In tumor samples from patients with colorectal carcinoma, LYAR mRNA levels, as determined by GeneChip, were higher than those in normal cells from those patients as well as in cells from healthy subjects (Supplementary Figure S1B). In addition, we examined LYAR mRNA levels in the low-metastasis colorectal cancer cell line SW480 and the highly metastatic line SW620 in comparison with HeLa cells. These cell lines were established from the primary lesion (SW480) and lymph-node metastases (SW620) of a single colorectal cancer patient (47,48). SW620 cells expressed a much higher level of both LYAR protein and mRNA than did SW480 cells (Supplementary Figure S1C).
LYAR associates with rDNA and increases the expression of 47S/45S pre-rRNA
We previously reported that cancer cells, such as HeLa and MCF7, have reduced ribosome production and proliferation when the level of LYAR is low; when the level of LYAR is elevated, however, cell proliferation increases (20). Because LYAR accelerates the processing of pre-rRNA in cultured cells including HeLa cells, we postulated that LYAR also enhances ribosome production by accelerating rRNA synthesis in tumor cells to help maintain their rapid proliferation. To test this idea, we first examined the association of LYAR with rDNA in cultured cells including HeLa, MCF7, and 293T cells by ChIP in combination with quantitative PCR using primer sets specific for regions corresponding to the rDNA loci H0, H8, H13 and H27 that had been defined by O’Sullivan et al. (49) (Figure 1A and B, Supplementary Figure S2A and B). We found that LYAR binds to the promoter region (H0) and transcribed region (H8 to H13) of rDNA but not to the intergenic spacer encompassing H27 in HeLa (Figure 1B), MCF7 (Supplementary Figure S2A), and 293T cells (Supplementary Figure S2B). This association of LYAR with rDNA was reduced upon LYAR knockdown in HeLa (Figure 1B) and MCF7 (Supplementary Figure S2C) cells without affecting UBF binding to rDNA, indicating that LYAR is present at the promoter and transcribed regions of rDNA. To minimize the effect of pre-rRNA processing (20), we next examined a 30-min incorporation of 4-thiouridine in 47S/45S pre-rRNA prepared from cells treated with one of two different short interfering RNAs (siRNA), and showed that it was reduced to 40–60% compared with that measured in cells treated with negative-control RNA (ncRNA) (Figure 1C).
Figure 1.

LYAR is involved in rDNA transcription. (A) Schematic diagram of a single human rDNA genic region (rDNA repeats are separated by an intergenic spacer). Primer sets (short bars: subregions H0, H8, H13, and H27) used for ChIP analysis are indicated along with their approximate positions relative to the transcription start site (0 kb). (B) ChIP analysis of UBF and LYAR binding to rDNA loci in HeLa cells transfected with scRNA (control) or siRNA specific for LYAR. Nonspecific mouse IgG or rabbit IgG was used as the antibody control. The chromatin-immunoprecipitated (ChIPed) DNA was quantified by qPCR using the primer sets indicated in A. The graph shows the amount of ChIPed DNA (% of input). Data represent the mean ± SEM of three independent experiments. The efficiency of LYAR knockdown was assessed with immunoblotting. (C) Metabolic labeling (4-thiouridine) of newly synthesized 47/45S pre-rRNA in HeLa cells upon LYAR knockdown. The RNA extracted for the cells treated with siRNA #1, siRNA #2 or ncRNA specific for LYAR (or ncRNA, control), was biotinylated and then subjected to agarose gel electrophoresis under denaturing condition and northern blotting. Signals for 47/45S pre-rRNA were detected by chemiluminescence. 28S and 18S rRNAs were used as loading controls (stained with methylene blue). The graph shows the relative band intensities of biotin-labeled 47/45S pre-rRNA normalized to that of 18S rRNA. Data reflect the mean ± SEM of three independent experiments. *P < 0.05 (paired t-test). LYAR mRNA levels, normalized to GAPDH mRNA, were assessed by reverse transcription-qPCR. (D) ChIP analysis of LYAR binding to rDNA loci (H0, H8, H13, H27) in HBF-LYAR-TO cells with or without HBF-LYAR induction via Dox. Nonspecific rabbit IgG was used as the antibody control. The graph shows the amount of ChIPed DNA (% of input). Data reflect the mean ± SEM of three independent experiments. (E) Metabolic labeling (4-thiouridine) of newly synthesized 47/45S pre-rRNA in HBF-LYAR-TO cells with or without Dox treatment. The pre-rRNA was biotinylated and then subjected to agarose gel electrophoresis under denaturing condition and northern blotting. Signals for 47/45S pre-rRNA were detected by chemiluminescence. 18S rRNAs were used as loading controls (stained with methylene blue). The graph shows the relative band intensities of 2 biotin-labeled 47/45S pre-rRNA normalized to that of 18S rRNA. Data reflect the mean ± SEM of six independent experiments. **P < 0.01 (paired t-test). (F) prHu3-Luc assay showing the effect of LYAR on RNAP I–dependent transcription. 293T cells were co-transfected with the prHu3-Luc reporter gene (firefly luciferase) and internal control vector pRL-TK (Renilla luciferase) along with the indicated amount of HBF and/or HBF-LYAR expression vectors, and luciferase activities were measured. The graph shows the ratio of the luciferase activities (firefly/Renilla) along with the corresponding HBF-LYAR expression levels or levels of HBF only. Data represent the mean ± SEM of three independent experiments. *P < 0.05, **P < 0.01 (paired t-test). (G) Methylation of rDNA in HBF-LYAR-TO cells was assessed with the HpaII resistance assay. HBF-LYAR-TO cells were treated with Dox for 24 h and then subjected to HpaII resistance assay. ChIP-CHOP experiment, in which the rDNA promoter-proximal DNA that had been subjected to ChIP with anti-LYAR or anti-UBF was subjected to HpaII digestion to determine whether LYAR associates with transcriptionally active, unmethylated rDNA repeats.
To examine the effects of LYAR overexpression on its binding to rDNA and on 47S/45S pre-rRNA synthesis, we produced a doxycycline (Dox)-inducible Flp-In T-REx 293 cell line expressing HBF (His6-biotinylation sequence-FLAG tag)-LYAR (HBF-LYAR-TO cells). The expression of HBF-LYAR increased gradually over time after the addition of Dox, reaching a steady state at a level 10-fold greater than that of endogenous LYAR (Supplementary Figure S2D). The induced HBF-LYAR localized to the nucleolus, as did endogenous LYAR, based on immunocytochemistry (Supplementary Figure S2E). The upregulated expression of HBF-LYAR led to an increase in HBF-LYAR association with the promoter and transcribed region of rDNA (Figure 1D) and promoted the synthesis of 47S/45S pre-rRNA compared with noninduced cells (Figure 1E). In addition, the increased transcription of rDNA upon HBF-LYAR overexpression was demonstrated with a luciferase assay using the prHu3-Luc reporter gene fused in tandem with a rDNA promoter (Figure 1F). Notably, overexpression of HBF-LYAR did not alter the DNA methylation status at rDNA loci, although HBF-LYAR indeed localized to regions of activated, unmethylated rDNA, as did UBF (Figure 1G). Collectively, these data suggested that LYAR promotes rDNA transcription without activating silent rDNA.
LYAR associates with a number of proteins involved in transcription
To gain insight into the role of LYAR in rDNA transcription, we isolated LYAR-associated proteins present in the nucleus. Because exogenously expressed HBF-LYAR was indistinguishable from endogenous LYAR in terms of the elution profile of the sucrose gradient ultracentrifugation of the nuclear extract (Supplementary Figure S3A), we prepared a nuclear fraction containing UBF (Nuc in Supplementary Figure S3B) from HBF-LYAR-TO cells and isolated HBF-LYAR-associated complexes from the Nuc fraction with a two-step purification using His and FLAG tags after RNase A treatment (Figure 2A), which did not affect the interaction between LYAR and nucleolin (Supplementary Figure S3C) as reported (23). The purified HBF-LYAR-associated complexes were separated by SDS-PAGE. The gel was then cut into four pieces, each of which was subjected to in-gel digestion with trypsin and endopeptidase Lys-C. The resulting peptides were analyzed with a nanoscale liquid chromatography–coupled tandem mass spectrometry (LC-MS/MS) system (50,51). These proteomic analyses assigned 148 proteins in total, including those known to associate with LYAR, i.e., nucleophosmin/B23, nucleolin, DDX21, and treacle (Table 1, Supplementary Tables S3 and S4). Among those, seven proteins, namely SSRP1, SPT16, BRD4, BRD2, SPT5, CTR9 and treacle, were also detected by immunoblotting (Figure 2B). Although UBF was identified only occasionally during our MS analyses (not shown in Supplementary Tables S3 and S4, and Table 1), it was detected unambiguously by immunoblotting (Figure 2B). In part, this may have been a consequence of the lower sensitivity of the MS-based method compared with immunoblotting and partly because of the tight binding of UBF to rDNA, which results in low extraction efficiency. As a negative control, lamin B was not detected by MS or immunoblotting (Figure 2B). The identified proteins were classified in Gene Ontology terms that were assigned using DAVID (http://www.geneontology.org/). The top three most abundant proteins had Gene Ontology terms for transcription (under Biological process) and those related to protein-binding, poly(A) RNA binding, and DNA binding (under Molecular function) (Supplementary Figure S3D and Supplementary Table S5). Among the identified proteins, nine are components of three elongation complexes involved in rDNA transcription, i.e., the PAF1 complex (RTF1, CTR9, LEO1, WDR61, PAF1) (13,52), FACT complex (SPT16, SSRP1) (12,53,54), and DSIF complex (SPT4, SPT5) (55,56) (Table 1). The other proteins, namely CD11B, CSK22, CSK2B, PELP1, MAX, treacle, TOP1, TOP2 and XRCC5, also participate in rDNA transcription (3) (Table 1). In addition, nucleophosmin and nucleolin participate in rDNA transcription as histone chaperones (Table 1) (14,57). Collectively, these analyses suggested that LYAR is a component of certain complexes that mediate rDNA transcription.
Figure 2.
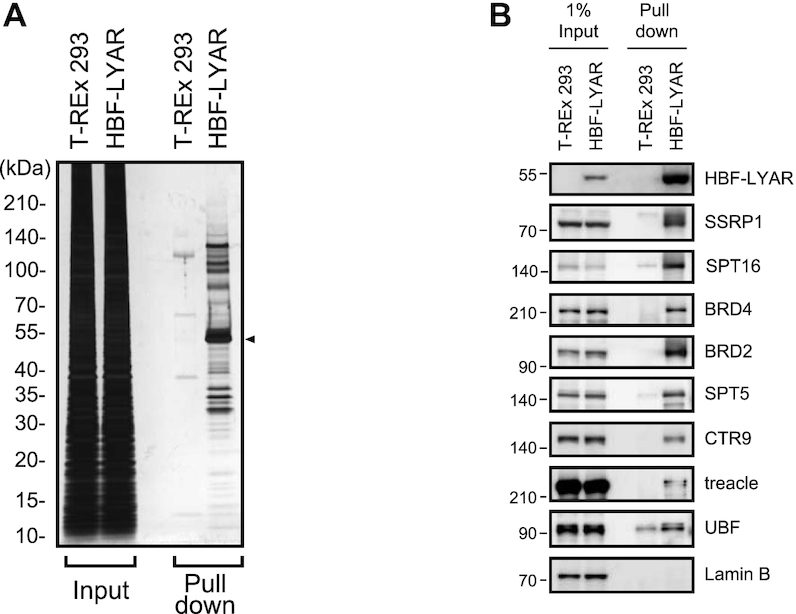
Pulldown of LYAR and identification of LYAR-associated proteins (A) Silver staining of HBF-LYAR-associated proteins. HBF-LYAR-associated complexes were isolated via sequential two-step pulldown (Ni-NTA pulldown, RNase A treatment, and pulldown of FLAG-tagged HBF-LYAR) from nuclear extract of HBF-LYAR-TO cells or T-REx 293 cells (control) treated with Dox for 24 h. Proteins were subjected to SDS-PAGE and visualized with silver staining. The arrowhead represents HBF-LYAR, as the bait protein. Molecular mass markers (kDa) are indicated to the left. Input: nuclear extract (10 μg). (B) Immunoblotting of HBF-LYAR-associated proteins using antibodies indicated to the right of each panel. 1% Input: 1% of the nuclear extract used for pull down of HBF-LYAR complexes. HBF-LYAR was detected by Stabilized Streptavidin-HRP Conjugate.
Table 1.
LYAR-associating proteins involved or expected to be involved in transcription
| Entry name | Protein name | Gene symbol | Gene ID | Note |
|---|---|---|---|---|
| Transcription-FACT complex | ||||
| SP16H_HUMAN | FACT complex subunit SPT16 | SUPT16H | 11198 | involved in rDNA transcription |
| SSRP1_HUMAN | FACT complex subunit SSRP1 | SSRP1 | 6749 | involved in rDNA transcription |
| Transcription-DSIF complex | ||||
| SPT5H_HUMAN | Transcription elongation factor SPT5 | SUPT5H | 6829 | involved in rDNA transcription |
| SPT4H_HUMAN | Transcription elongation factor SPT4 | SUPT4H1 | 6827 | involved in rDNA transcription |
| Transcription-PAF1 complex | ||||
| RTF1_HUMAN | RNA polymerase-associated protein RTF1 homolog | RTF1 | 23168 | involved in rDNA transcription |
| CTR9_HUMAN | RNA polymerase-associated protein CTR9 homolog | CTR9 | 9646 | involved in rDNA transcription |
| LEO1_HUMAN | RNA polymerase-associated protein LEO1 | LEO1 | 123169 | involved in rDNA transcription |
| WDR61_HUMAN | WD repeat-containing protein 61 | WDR61 | 80349 | involved in rDNA transcription |
| PAF1_HUMAN | RNA polymerase II-associated factor 1 homolog | PAF1 | 54623 | involved in rDNA transcription |
| Transcription-Super elongation complex (SEC) | ||||
| AFF4_HUMAN | AF4/FMR2 family member 4 | AFF4 | 27125 | |
| ENL_HUMAN | Protein ENL | MLLT1 | 4298 | |
| Kinase | ||||
| CD11B_HUMAN | Cyclin-dependent kinase 11B | CDK11B | 984 | |
| CSK22_HUMAN | Casein kinase II subunit alpha | CSNK2A2 | 1459 | |
| CSK2B_HUMAN | Casein kinase II subunit beta | CSNK2B | 1460 | |
| Topoisomerase & DNA damage | ||||
| TOP1_HUMAN | DNA topoisomerase 1 | TOP1 | 7150 | involved in rDNA transcription |
| DDB1_HUMAN | DNA damage-binding protein 1 | DDB1 | 1642 | |
| TOP2A_HUMAN | DNA topoisomerase 2-alpha | TOP2A | 7153 | involved in rDNA transcription |
| XRCC5_HUMAN | X-ray repair cross-complementing protein 5 | XRCC5 | 7520 | involved in rDNA transcription |
| Transcription-Other | ||||
| SAFB2_HUMAN | Scaffold attachment factor B2 | SAFB2 | 9667 | |
| SAFB1_HUMAN | Scaffold attachment factor B1 | SAFB | 6294 | |
| BRD4_HUMAN | Bromodomain-containing protein 4 | BRD4 | 23476 | |
| BRD2_HUMAN | Bromodomain-containing protein 2 | BRD2 | 6046 | |
| BRD3_HUMAN | Bromodomain-containing protein 3 | BRD3 | 8019 | |
| CN166_HUMAN | UPF0568 protein C14orf166 | C14orf166 | 51637 | |
| PELP1_HUMAN | Proline-, glutamic acid- and leucine-rich protein 1 | PELP1 | 27043 | involved in rDNA transcription |
| MATR3_HUMAN | Matrin-3 | MATR3 | 9782 | |
| RBP56_HUMAN | TATA-binding protein-associated factor 2N | TAF15 | 8148 | |
| T2FA_HUMAN | General transcription factor IIF subunit 1 | GTF2F1 | 2962 | |
| BCLF1_HUMAN | Bcl-2-associated transcription factor 1 | BCLAF1 | 9774 | |
| YLPM1_HUMAN | YLP motif-containing protein 1 | YLPM1 | 56252 | |
| TCOF_HUMAN | Treacle protein | TCOF1 | 6949 | involved in rDNA transcription |
| IWS1_HUMAN | Protein IWS1 homolog | IWS1 | 55677 | |
| SPT6H_HUMAN | Transcription elongation factor SPT6 | SUPT6H | 6830 | |
| MTA1_HUMAN | Metastasis-associated protein MTA1 | MTA1 | 9112 | |
| SATB2_HUMAN | DNA-binding protein SATB2 | SATB2 | 23314 | |
| MAX_HUMAN | Protein max | MAX | 4149 | involved in rDNA transcription |
| SLTM_HUMAN | SAFB-like transcription modulator | SLTM | 79811 | |
| TCF20_HUMAN | Transcription factor 20 | TCF20 | 6942 | |
| ILF3_HUMAN | Interleukin enhancer-binding factor 3 | ILF3 | 3609 | |
| S30BP_HUMAN | SAP30-binding protein | SAP30BP | 29115 | |
UBF is involved in recruitment of LYAR to rDNA
Given that LYAR binds the DNA sequence GGTTAT through its zinc-finger domain that is similar to the one found in the globin gene (21), we first examined whether LYAR interacts directly with rDNA via the GGTTAT sequence that is located in five different regions of rDNA (GenBank, U13369.1): nucleotides 42992–42997 (promoter), 3955–3960 (complementary strand, transcribed region), 32632–32637 and 34176–34181 (complementary strand, intergenic spacer), and 37256–37261 (intergenic spacer). However, the locations of these LYAR-binding sequences were not consistent with the regions of rDNA bound to LYAR observed by ChIP analysis; i.e., LYAR was not present in intergenic spacer. In fact, an H0-specific oligonucleotide containing the 42992–42997 sequence present in the promoter region did not bind LYAR as determined with electrophoretic mobility shift assay (Supplementary Figure S4A). To further examine the involvement, if any, of the LYAR zinc-finger domain in rDNA binding, we constructed domain mutants of LYAR, including ones that lack the N-terminal zinc-finger domain (LYAR168–379 and LYAR168–260; Supplementary Figure S4B) and found that LYAR168–379 could minimally bind rDNA (Supplementary Figure S4C). Collectively, these data suggested that the zinc-finger domain of LYAR and LYAR-binding sequences in rDNA are not involved in LYAR binding to rDNA. Because a LYAR mutant lacking the zinc-finger domain has a dominant-negative effect on the processing of pre-rRNAs (20), the zinc-finger domain of LYAR probably participates in pre-rRNAs processing rather than rDNA transcription.
We next considered whether LYAR binds rDNA loci via the basal pre-transcription structural proteins UBF and treacle (58–67), each of which can associate with LYAR; this analysis was carried out in concert with knockdown of UBF, treacle or SPT5 (as a control). We found that UBF is required for the recruitment of LYAR to regions H0, H8, and H13 of rDNA as shown by ChIP analysis of UBF-knockdown cells (Figure 3A), whereas treacle seems to contribute to the recruitment of LYAR to other regions in rDNA (H0 and H8) (Supplementary Figure S4D). In addition, UBF knockdown reduced the localization of LYAR in the nucleolus and shifted to the nucleolar cap (Figure 3B). Given our previous report that LYAR knockdown did not alter the nucleolar localization of UBF (20), these data suggested that LYAR is recruited to rDNA via UBF. We expected that the knockdown of SPT5 would not affect LYAR binding to rDNA; however, it significantly increased LYAR binding to region H0 of rDNA (Figure 3A). This increase coincided with the increase of UBF binding to the H0 region upon SPT5 knockdown (Figure 3A). These data suggested that LYAR is recruited to rDNA via UBF and that SPT5 can control LYAR binding to region H0. On the other hand, the knockdown of UBF or SPT5 reduced the binding of LYAR to SPT5 or UBF, respectively (Figure 3C), suggesting that LYAR can form a complex with UBF and SPT5. These results could not definitively determine whether the LYAR-UBF-SPT5 complex forms upon binding to rDNA or forms prior to binding, i.e. as a pre-formed unit that might help maintain the equilibrium among the three protein monomers. Therefore, the mechanism by which UBF and SPT5 regulate LYAR recruitment to rDNA may be more complicated than previously suspected.
Figure 3.

LYAR recruitment to rDNA loci depends on UBF (A) ChIP analysis of LYAR (left) or UBF binding (right) to rDNA loci in 293T cells upon knockdown of UBF or SPT5. 293T cells were treated with an siRNA specific for UBF or SPT5 (ncRNA as a control) for 72 h, and whole-cell extract was subjected to immunoblotting using the antibodies indicated to the right of the panels. β-actin was used as the loading control. Molecular mass markers (kDa) are indicated to the left. The graphs show the amount of ChIPed DNA (% of input) with respect to particular rDNA loci. Data represent the mean ± SEM of three independent experiments. *P < 0.05 (unpaired t-test). (B) Immunocytostaining of LYAR and UBF upon UBF knockdown. 293T cells were treated with ncRNA or an siRNA specific for UBF for 72 h and subjected to immunocytostaining with anti-LYAR (rabbit) and anti-UBF (mouse). FITC-conjugated anti-rabbit IgG and Cy3-conjugated anti-mouse IgG were used as the secondary antibodies. DAPI staining indicates the nucleus. Bar: 10 μm. (C) Immunoblotting for HBF-LYAR-associated proteins upon siRNA-mediated knockdown of treacle, UBF or SPT5 in HBF-LYAR-TO cells for 72 h. A two-step pulldown was carried out with His6- or FLAG-tag of HBF-LYAR. The antibodies used for immunoblotting are indicated to the right.
Given that the small molecule CX-5461 inhibits RNAP I transcription initiation via exclusion of SL1 from the rDNA promoter and that actinomycin D (actD) inhibits the elongation of rDNA transcription (68,69), we examined the effects of these compounds on the binding of UBF and LYAR to rDNA. Although both compounds inhibited rRNA synthesis (Supplementary Figure S4E), they did not affect the binding of LYAR to UBF at 6 h treatment (Supplementary Figure S4E and F), suggesting that these two proteins form a complex independently of transcription. However, actD reduced the binding between LYAR and UBF at 2 h treatment and increased the binding of LYAR to the transcribed region of rDNA but not the promoter or the intergenic spacer (Supplementary Figure S4G). Thus, the inhibition of rDNA transcriptional elongation seems to rearrange the binding between LYAR and UBF and to trap LYAR on the transcribed region of rDNA.
LYAR recruits BRD2 to rDNA loci
Given that we identified a number of transcription regulatory factors as LYAR-binding proteins (Table 1), we investigated which transcription regulatory factors have to be recruited to rDNA to increase the transcription of 47S/45S pre-rRNA via binding to LYAR and looked for transcription regulatory factors for which the binding to rDNA is reduced upon siRNA-mediated knockdown of LYAR. Among the transcription regulatory factors we examined (Supplementary Figure S5A), BRD2 exhibited reduced binding to rDNA (regions H0, H8 and H13) upon LYAR knockdown, although the overall expression of BRD2 did not change (Figure 4A). Knockdown of BRD2 itself reduced its binding to rDNA, including the region corresponding to H27 (Supplementary Figure S5B), implying that BRD2 is recruited at a low basal level to rDNA loci in the absence of LYAR. The binding of LYAR to BRD2 was not affected by the inhibition of RNAP I transcription with actD or CX-5461 (Supplementary Figure S4F), suggesting that the binding of LYAR to BRD2 is independent on the RNAP I transcription. By contrast, knockdown of UBF reduced the binding of BRD2 in regions H0, H18, and H13 but not H27, as did LYAR (Figure 4A and B). Collectively, these data strongly suggested that LYAR enhances the recruitment of BRD2 to the promoter and transcribed regions of rDNA via UBF.
Figure 4.

LYAR recruits BRD2 to rDNA transcription sites (A) ChIP analysis of BRD2 binding to rDNA loci in 293T cells upon LYAR knockdown (KD). 293T cells were treated with an siRNA specific for LYAR (or ncRNA, control) and then subjected to ChIP analysis with an antibody against LYAR or BRD2. LYAR KD was confirmed by immunoblotting with antibodies indicated to the right of the panels. β-actin was used as the loading control. Molecular mass markers (kDa) are indicated to the left of the panels. The graphs show the amount of ChIPed DNA (% of input) relative to the number of rDNA loci with the antibody indicated under each graph. Data represent the mean ± SEM of three independent experiments. *P < 0.05, **P < 0.01 (unpaired t-test). (B) ChIP analysis of BRD2 binding to rDNA loci in 293T cells upon UBF knockdown. The cells were treated with an siRNA specific for UBF (or ncRNA) and then subjected to ChIP analysis with an antibody against BRD2. The graph shows the amount of ChIPed DNA (% of input). (C) Re-ChIP analysis showing that LYAR-BRD2-associated complexes bind rDNA loci. The first ChIP of HBF-LYAR, with anti-FLAG, was performed using HBF-LYAR-TO cells with or without Dox treatment (1st ChIP; left graph). The second ChIP, with anti-BRD2, was performed using the first ChIPed HBF-LYAR-associated complexes (2nd ChIP; right graph). As an antibody control for anti-BRD2, a nonspecific rabbit IgG was used. The graphs show the amount of ChIPed DNA (% of input). Data represent the mean ± SEM of three independent experiments. *P < 0.05, **P < 0.01 (unpaired t-test). (D) Metabolic labeling (4-thiouridine) of newly synthesized 47/45S pre-rRNA in 293T cells upon BRD2 knockdown (siRNA). The pre-rRNA was biotinylated and then subjected to agarose gel electrophoresis under denaturing condition and northern blotting. The signals for 47/45S pre-rRNA were detected by chemiluminescence. 28S and 18S rRNAs were used as loading controls (stained with SYBR gold). The graph shows the relative band intensities of biotin-labeled 47/45S pre-rRNA normalized to that of 18S rRNA. Data represent the mean ± SEM of four independent experiments. *P < 0.05, **P < 0.01 (paired t-test). Knockdown of BRD2 was confirmed by immunoblotting with anti-BRD2. β-actin was used as the loading control.
Given our result that the peptide LYAR168–260, which is rich in lysine residues, could interact with BRD2 (Supplementary Figure S4B), we examined in vitro binding between LYAR168–260 and each of the recombinant domain mutants of BRD2 (Supplementary Figure S5C). LYAR168–260 could interact with BRD2458–720 or BRD2458–801, each of which lacks the two canonical bromodomains (Supplementary Figure S5D). Isothermal titration calorimetry also revealed that peptide BRD2489–540 could interact with peptide LYAR175–219 (Kd = 12.3 µM) with 1:1 stoichiometry (Supplementary Figure S5E). These results were consistent with recent reports by Luna-Pelaez et al. showing that the motif B region of BRD2 interacts with the C-terminal region of LYAR (70). In addition, overexpression of LYAR increased the binding of BRD2 to rDNA loci without affecting the overall cellular level of BRD2 (Supplementary Figure S5F). Sequential re-ChIP analysis revealed co-occupancy of LYAR and BRD2 in rDNA regions H0, H8 and H13 (Figure 4C), supporting the idea that LYAR forms a complex with BRD2 via direct interaction and then the complex associates with rDNA loci via UBF. The fact that BRD2 overexpression did not affect the binding of LYAR or UBF to rDNA (Supplementary Figure S5G) excluded the possibility that BRD2 could increase the binding of LYAR or UBF to rDNA loci. Because BRD proteins had not previously been shown to be involved in rDNA transcription, we then knocked down BRD2 and found that 47S/45S pre-rRNA synthesis was reduced to the level of ∼60% compared with the mock-knockdown control, as detected by a 30-min incorporation of 4-thiouridine (Figure 4D). Collectively, these data suggested that BRD2 is required for LYAR-dependent rRNA synthesis.
BRD2 recruits KAT7 to rDNA loci
We further addressed the issue of how BRD2 can upregulate LYAR-dependent rDNA transcription. Given that BRD2 recruits the histone H4–specific acetyltransferase to the RNAP II transcription start site (43,71,72), we first examined whether LYAR-recruited BRD2 could induce acetylation of local histones at rDNA loci by using ChIP with antibodies that recognize acetylated H3 (at K9, K14, K18, K23 and K27) or acetylated H4 (at K5, K8, K12 and K16). Similar to the report by Sinha et al. (43), BRD2 knockdown mainly reduced H4 acetylation at rDNA loci (Figure 5A). Based on our MS-based identification of two LYAR-associated histone acetyltransferases, namely KAT7 (45) and BRD4 (44) (Table 1 and Supplementary Table S3), we first examined whether KAT7 is responsible for the acetylation of histones at rDNA loci. We found that LYAR overexpression increased the binding of KAT7 to rDNA, including the intergenic spacer region corresponding to the probe for region H27 (Figure 5B). By contrast, LYAR knockdown reduced KAT7 binding to rDNA (Figure 5C). These data demonstrated that LYAR has an effect on KAT7 binding to rDNA beyond the LYAR-binding region. Moreover, knockdown of BRD2 reduced the amount of rDNA-associated KAT7 at regions H8, H13 an H27 (Figure 5D). This BRD2 knockdown also reduced the binding of LYAR to KAT7 and JADE3 that was identified as one of the LYAR-binding proteins, a possible scaffold partner of KAT7 (Figure 5E), suggesting that BRD2 bridges between LYAR and KAT7 (or more likely between LYAR and KAT7-JADE3). In addition, KAT7 knockdown mainly reduced the acetylation of histone H4 at rDNA loci (Figure 5F). Collectively, these data suggested that LYAR recruits BRD2-KAT7 and possibly JADE3 to acetylate histone H4 within nucleosomes at rDNA loci.
Figure 5.

BRD2 recruits KAT7 to rDNA loci (A) ChIP analysis of the extent to which H3 or H4 is acetylated. 293T cells were treated with an siRNA specific for BRD2 or ncRNA (control) and then subjected to ChIP analysis with an antibody against H3ac or H4ac, as indicated. The graphs show the amount of ChIPed DNA (% of input) relative to the number of rDNA loci indicated under each graph. Data represent the mean ± SEM of three independent experiments. *P < 0.05, **P < 0.01 (unpaired t-test). B–D) ChIP analysis of the binding of KAT7 to rDNA loci. HBF-LYAR-TO cells were treated with (+Dox) or without (–Dox) Dox (B). 293T cells were treated with ncRNA (control) or an siRNA specific for LYAR (C) or BRD2 (D) for 72 h. These cells were subjected to ChIP analysis with an antibody against KAT7. The graphs show the amount of ChIPed DNA (% of input) relative to the number of rDNA loci indicated under each graph. Data represent the mean ± SEM of three independent experiments. *P < 0.05, **P < 0.01 (unpaired t-test). (E) Immunoblotting for HBF-LYAR-associated proteins upon the knockdown of BRD2. HBF-LYAR-TO cells were treated with an siRNA specific for BRD2 for 72 h; after induction with Dox, the nuclear extract was subjected to the pulldown with two-step pull down with His6- and FLAG-tag of HBF-LYAR. HBF-LYAR-associated proteins were detected by immunoblotting with antibodies indicated to the right of the panels. (F) ChIP analysis of the extent to which H3 or H4 is acetylated. 293T cells were treated with an siRNA specific for KAT7. These cells were subjected to ChIP analysis with an antibody against H3ac or H4ac, as indicated. 293T cells were treated with ncRNA (control) or an siRNA specific for KAT7. The graphs show the amount of ChIPed DNA (% of input) relative to the number of rDNA loci indicated under each graph. Data represent the mean ± SEM of three independent experiments. *P < 0.05, **P < 0.01 (unpaired t-test).
BRD2 assists the recruitment of BRD4 to rDNA loci
We next examined the contribution of another LYAR-binding acetyltransferase, namely BRD4, to histone acetylation at rDNA loci. The overexpression of LYAR increased the binding of BRD4 to rDNA loci without affecting the overall expression level of BRD4 (Supplementary Figure S5F). In addition, the knockdown of LYAR or BRD2 reduced the binding of BRD4 to rDNA (Figure 6A and B); as expected, knockdown of BRD4 reduced its own association with rDNA loci (Supplementary Figure S6A). BRD4 knockdown reduced the acetylation of both histones H3 and H4 at rDNA loci, except for region H27 (Figure 6C). Moreover, LYAR knockdown reduced the overall acetylation of both H3 and H4 at the rDNA promoter as well as the transcribed and intergenic spacer regions of rDNA loci (Figure 6D), whereas LYAR overexpression increased the overall acetylation of both H3 and H4 (Figure 6E). Given that BRD4 catalyzes the acetylation of H3K122 (H3acK122), which is sufficient to stimulate transcription of rDNA (73), we also tested whether acetylation at H3K122 is increased by overexpression of LYAR via the ability to recruit BRD4 to rDNA. ChIP analysis with anti-H3acK122 revealed that LYAR overexpression caused a significant increase in H3acK122 at rDNA loci including region H27 (Figure 6F), suggesting that LYAR-recruited BRD4 is also responsible for H3K122 acetylation at rDNA loci. Consistent with these data, BRD4 knockdown reduced 47S/45S pre-rRNA synthesis to ∼35% compared with the mock-knockdown control, as detected by a 30-min incorporation of 4-thiouridine (Figure 6G). However, the histone acetyltransferases GCN5 and p300 were not identified as LYAR-associated proteins (Supplementary Tables S3 and S4), and indeed LYAR did not affect the binding of either GCN5 or p300 to rDNA (Supplementary Figure S6B) (11,74). Collectively, these data suggested that LYAR-mediated recruitment of BRD2, KAT7 and BRD4 to rDNA controls the acetylation of local histones H3 and H4 to cause the relaxation of these histones from rDNA loci and thereby promote rDNA transcription.
Figure 6.

LYAR and BRD2 assist the recruitment of BRD4 to rDNA (A, B) ChIP analysis of the binding of BRD4 to rDNA upon the knockdown of LYAR (A) or BRD2 (B). 293T cells were treated with ncRNA or an siRNA specific for LYAR (A) or BRD2 (B) for 72 h. These cells were subjected to ChIP analysis with an antibody against BRD4. The graphs show the amount of ChIPed DNA (% of input) relative to the number of rDNA loci indicated under each graph. Data represent the mean ± SEM of three independent experiments. *P < 0.05, **P < 0.01 (unpaired t-test). (C, D) ChIP analysis of the extent to which H3 or H4 is acetylated in 293T cells. The cells were treated with ncRNA or an siRNA specific for BRD4 (C) or LYAR (D) for 72 h. These cells were subjected to ChIP analysis with an antibody against H3ac or H4ac, as indicated. (E, F) ChIP analysis of the extent to which H3 or H4 is acetylated in HBF-LYAR-TO cells. The cells were treated with (Dox+) or without Dox (Dox–) for 24 h. These cells were subjected to ChIP analysis with an antibody against H3ac or H4ac (E), or H3K122ac (F). (G) Metabolic labeling (4-thiouridine) of newly synthesized 47/45S pre-rRNA in 293T cells upon BRD4 knockdown (siRNA) for 72 h. The pre-rRNA was biotinylated and then subjected to agarose gel electrophoresis and northern blotting. The signals for 47/45S pre-rRNA were detected by chemiluminescence. 28S and 18S rRNAs were used as loading controls (stained with SYBR gold). The graph shows the relative band intensities of biotin-labeled 47/45S pre-rRNA normalized to that of 18S rRNA. Data represent the mean ± SEM of four independent experiments. *P < 0.05, **P < 0.01 (paired t-test). Knockdown of BRD4 was confirmed by immunoblotting with anti-BRD4. β-actin was used as the loading control.
BRD2 and BRD4 form complexes with LYAR and KAT7-JADE3 independently of each other
To examine the relationship among complexes formed by LYAR, UBF, BRD2/4, KAT7-JADE3 and SPT5, we carried out a reverse pulldown assay using each of those proteins, individually, as the affinity bait. We initially performed a two-step pulldown using HBF-LYAR as the first affinity bait and endogenous SPT5 or UBF as the second affinity bait. Although LYAR-UBF could associate with BRD2 or BRD4 even at basal cellular levels (Figure 7A), LYAR-SPT5 did not associate with BRD2 or BRD4 at all (Figure 7B). Conversely, a pulldown assay using BRD2 or BRD4 as bait showed that neither protein could associate with SPT5 or UBF; however, BRD2 and BRD4—independently of each other—could form a complex containing both LYAR and KAT7-JADE3 (Figure 7C). In addition, knockdown of BRD2 did not affect the binding of BRD4 to LYAR, and vice versa (Figures 5E and 7D). Collectively, these data support the previous notice that LYAR forms a complex with BRD2 or BRD4, after which either complex can be recruited to rDNA via UBF. Furthermore, the association of LYAR with KAT7-JADE3 is dependent on BRD2 (Figure 5E) but not on BRD4 (Figure 7D), suggesting that BRD2 and BRD4 differentially promote the recruitment of KAT7-JADE3 to rDNA.
Figure 7.
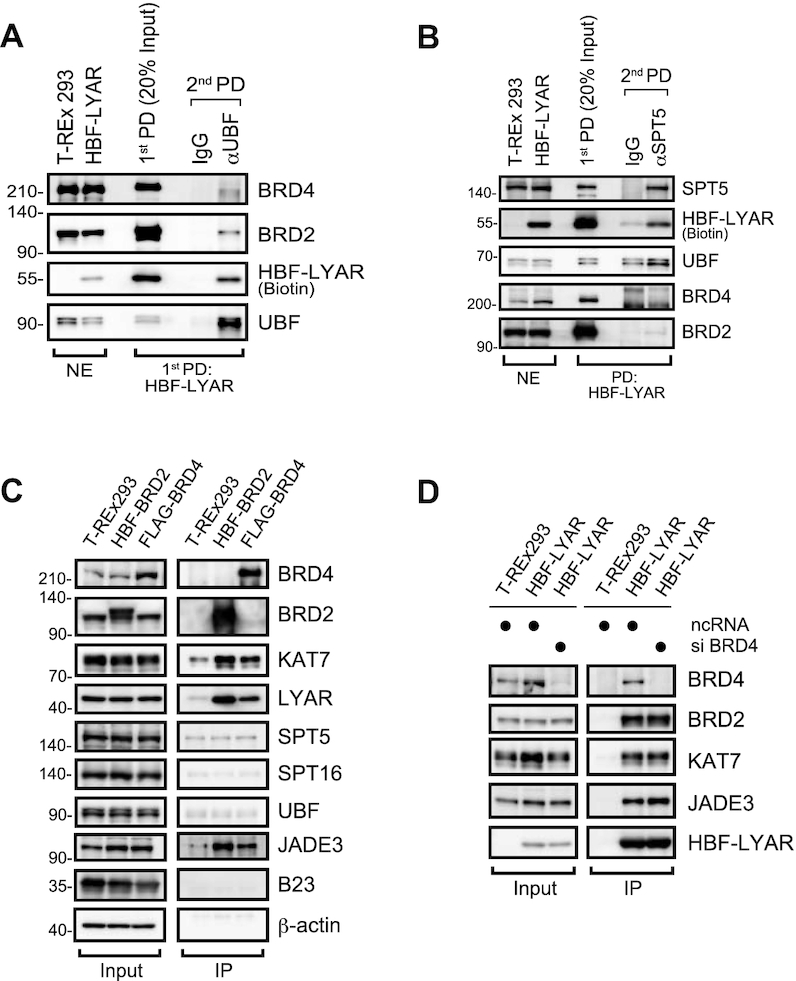
BRD2, BRD4, and SPT5 bind LYAR independently (A, B) Reverse pulldown (PD) of UBF (A) or SPT5 (B) from HBF-LYAR complexes, assessed with immunoblotting. HBF-LYAR-TO cells were treated with Dox for 24 h, and then nuclear extracts (NE) were subjected to first pulldown (first PD) by two-step immunoprecipitation using His6- and FLAG-tag of HBF-LYAR. The purified HBF-LYAR complexes were immunoprecipitated with anti-UBF (αUBF) (A) or anti-SPT5 (αSPT5) (B) as the second affinity bait (second PD). T-REx 293 cells treated with Dox were used as the control. Proteins were detected by immunoblotting using the antibodies indicated to the right of the panels. Molecular mass markers (kDa) are indicated to the left of the panels. The nuclear extract (NE, 10 μg) was used as a loading control. (C) Immunoblotting for proteins that associated with BRD2 or BRD4. HBF-BRD2-TO cells and FLAG-BRD4-TO cells were treated with Dox for 24 h, and then the nuclear extracts were subjected to a two-step immunoprecipitation (IP) with anti-His6 and anti-FLAG. T-REx 293 cells treated with Dox were used as the control. Proteins that associated with HBF-BRD2 or FLAG-BRD4 were detected by immunoblotting using the antibodies indicated to the right of the panels. β-actin was used as the loading control. (D) Immunoblotting for HBF-LYAR-associated proteins upon the knockdown of BRD4. HBF-LYAR-TO cells were treated with an siRNA specific for BRD4 for 72 h. After induction with Dox, the nuclear extracts were subjected to two-step immunoprecipitation (IP) using His6- and FLAG-tag of HBF-LYAR.
DISCUSSION
Our results demonstrate that LYAR has a role in rDNA transcription. Given that LYAR overexpression does not affect the cellular levels of MYC, PTEN, retinoblastoma, and p53 (20), we hypothesized that LYAR promotes rDNA transcription via a pathway independent of those proteins, although we could not exclude the possibility that LYAR-induced proteins other than those listed above may be involved in rDNA transcription. Among several transcription regulatory factors that associate with LYAR (Table 1), we identified BRD2, KAT7, and BRD4 as being recruited by LYAR to rDNA. Although BET family proteins and MYST-type acetyltransferases are essential mediators of RNAP II–dependent transcription (75,76), their involvement in RNAP I–dependent transcription is unknown. Thus, to our knowledge, this is the first report that provides evidence of a role for BRD proteins and KAT7 in RNAP I–dependent transcription.
BRD2 associates with the RNAP II mediator complex containing E2F-TATA-box–binding proteins that is proposed to recruit histone acetyltransferases to DNA during RNAP II–dependent transcription (71). However, it is less likely that LYAR recruits the E2F-TATA-box transcription factors to rDNA via its association with BRD2 because our MS-based analysis did not identify any of the proteins known to associate with BRD2 (Table 1). Instead of those proteins, we found that BRD2 assists the recruitment of KAT7 and BRD4 to rDNA. Based on our present findings, we propose a mechanism for LYAR-mediated enhancement of rDNA transcription (Figure 8). First, upon its increased expression, LYAR forms a complex with BRD2-KAT7 (or BRD4-KAT7); the complex probably contains JADE3. Second, LYAR-BRD2-KAT7 complex binds to the promoter and transcribed regions of rDNA loci via UBF. Third, KAT7 acetylates histone H4 in nucleosomes at rDNA. Alternatively, LYAR may recruit BRD4-KAT7 to rDNA loci regardless of whether BRD2 is already present at those loci to accelerate BRD4-KAT7 binding to acetylated H4. Finally, BRD4-KAT7 assists the acetylation of both H4 and H3 near the LYAR-binding site on rDNA and promotes BRD4-KAT7 binding to rDNA loci to relax the nucleosome structure, which enhances RNAP I–mediated transcription (Figure 8). In this mechanism, LYAR binds directly to BRD2 and recruits it to rDNA loci without involvement of acetylated histones. This is in contrast to the involvement of BRD2 in RNAP II–dependent transcription, in which it is recruited to an acetylated histone (e.g., H4K12ac) (77). By contrast, an unknown protein(s) may be required for the binding of LYAR to BRD4. This proposed mechanism may explain why BRD4 is recruited to the intergenic spacer encompassing H27 beyond region H13 (Figure 6A). A similar function of the MYST-type acetyltransferase has been reported to facilitate HIV (human immunodeficiency virus) latency in human cells; in that case, BRD4 is recruited to the HIV long, terminal repeat by interacting with acetylated H4 (acetylation catalyzed by KAT5) (78).
Figure 8.
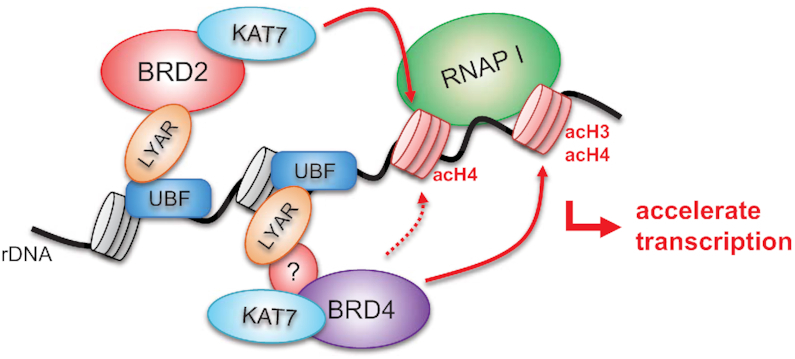
Model for LYAR function in rDNA transcription For RNAP I–dependent transcription, LYAR binds rDNA via UBF, recruits BRD2-KAT7-JADE3 through direct binding between LYAR and BRD2 (without any auxiliary factor) to rDNA transcription sites, and accelerates acetylation of histone H4. Moreover, LYAR facilitates the binding of BRD4-KAT7-JADE3 to acetylated H4 at rDNA transcription sites. BRD4-KAT7-JADE3 further accelerates the acetylation of histones H3 and H4 at rDNA loci, which further enhances BRD4-KAT7-JADE3 binding to rDNA loci, resulting in the relaxation of chromatin structure to enhance RNAP I–dependent transcription. Because the binding of LYAR to KAT7-JADE3 is dependent on BRD2 but not BRD4, an unknown factor may be involved in the binding between LYAR and KAT7-JADE3 in the LYAR-BRD4-KAT7-JADE3 complex.
One issue this mechanism does not address is whether the intrinsic histone acetylase activity of BRD4 is responsible for LYAR-induced acetylation of histones H3 and H4 or whether BRD4 recruits a different acetyltransferase. The lysine residues in H3 and H4 that are acetylated by BRD4 are distinct from those of other histone acetyltransferases, especially the acetylation of K122 in H3 (H3K112ac) (44). Given that overexpression of LYAR increased the frequency of H3acK122 (Figure 6F), it is likely that the intrinsic acetylase activity of BRD4 is involved in acetylation assisted by LYAR on rDNA loci. Although two other acetyltransferases, namely P300/CBP (79) and SIRT7 (80), can generate H3K122ac, we did not identify those acetyltransferases as LYAR-associated proteins, supporting the aforementioned hypothesis that the intrinsic histone acetylase activity of BRD4 is responsible for LYAR-induced acetylation. However, it is possible that other unknown acetyltransferases are involved in LYAR-assisted histone acetylation at rDNA loci.
Another issue with this mechanism is whether LYAR binds rDNA directly or indirectly. Our data show that LYAR binds rDNA indirectly via binding to UBF, and we serendipitously found that SPT5 regulates UBF binding to the rDNA promoter, and thus SPT5 probably regulates LYAR binding to that promoter. In addition, we demonstrated that SPT5-UBF and BRD2-KAT7-JADE3 (or BRD4-KAT7-JADE3) form complexes with LYAR independently of each other (Figure 7A–C). Given that UBF and SPT5 participate in the pause and release, respectively, of RNAP I from the rDNA promoter (35,54,81), the initiation of rDNA transcription can be regulated by a similar mechanism, i.e., RNAP I is initially paused at the promoter but is released upon LYAR-BRD2-KAT7-JADE3 binding to UBF. This RNAP I release mechanism is consistent with our result that LYAR affected neither the binding of two RNAP I subunits, RPA194 and RPA135, to rDNA (Supplementary Figure S5A) nor the state of DNA methylation (Supplementary Figure S2F). However, LYAR overexpression did not alter the binding of SPT5 to rDNA loci, including the promoter (Supplementary Figure S5A). Thus, we propose that LYAR binding to rDNA is not involved in the release of paused RNAP I from the promoter, although we cannot exclude the possibility that LYAR binding alters the association between SPT5 and UBF via histone acetylation by KAT7/BRD4 at the rDNA promoter or phosphorylation of UBF via the kinase activity of BRD2.
Based on our current results and those of our previous study (20), we propose that upregulation of LYAR in certain cancer cells promotes rDNA transcription and pre-rRNA processing, resulting in increased ribosome biogenesis and maintenance of rapid growth and proliferation. Therefore, it will be interesting to determine whether cancer cells that express LYAR at a high level can maintain their tumorigenic potential upon suppression of LYAR function in rRNA synthesis. Recent research revealed that the cell-permeable molecule JQ1 inhibits the binding of BRD proteins with acetylated histones and is a potential therapeutic agent for the treatment of acute myeloid leukemia (82,83). Our present findings concerning the potential interactions among LYAR/UBF, LYAR/BRD2-KAT7-JADE3 and/or LYAR/BRD4-KAT7-JADE3 may provide other potential targets for cancer treatment.
Finally, it has been shown that the modulation of rRNA synthesis is closely linked to cell differentiation of ovarian germline stem cells in Drosophila (35) and in human cells with differentiation potential, including embryonic stem cells (34) and promyelocytic and monocytic leukemia cells (84). Therefore, it is likely that LYAR maintains the pluripotency of embryonic stem cells by promoting rRNA synthesis. It is also possible that the physiological convergence of BRD2 and BRD4 observed in early embryogenesis in mice (85,86) may be a consequence of their roles—as mediated by LYAR—in rRNA synthesis.
CLINICAL SPECIMENS
The Ethics Committee of The University of Tokyo approved the use of human tissues, and all patients gave informed consent.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr T. Shinkawa, N. Miyazawa, I. Suzuki, M. Akiyama and K. Nagahashi for their technical assistance with the biochemical and proteomic analyses during the initial stages of this work. Vector prHu3-Luc was a kind gift from Dr Yan-Hwa Wu Lee (National Yang-Ming University, Taipei, Taiwan). This work was supported mainly by a grant for Core Research for Evolutionary Science and Technology (CREST) from the Japan Science and Technology Agency.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Scientific Research, Ministry of Education, Culture, Sports, Science & Technology of Japan (MEXT) [24241075 to N.T.]; Core Research for Evolutional Science and Technology (CREST) from Japan Science and Technology Agency (JST) [JPMJCR13M2 to I.T. and N.T.]; Naito Foundation Grant for Studying Overseas (2013) [2013-413 to H.Y.]; Uehara Memorial Foundation Postdoctoral Fellowship [201430061 to H.Y.]; European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie Individual Fellowship [657087 to H.Y]. Funding for open access charge: JST CREST.
Conflict of interest statement. None declared.
REFERENCES
- 1. Ruggero D., Pandolfi P.P.. Does the ribosome translate cancer. Nat. Rev. Cancer. 2003; 3:179–192. [DOI] [PubMed] [Google Scholar]
- 2. White R.J. RNA polymerases I and III, non-coding RNAs and cancer. Trends Genet. 2008; 24:622–629. [DOI] [PubMed] [Google Scholar]
- 3. Russell J., Zomerdijk J.C.. RNA-polymerase-I-directed rDNA transcription, life and works. Trends Biochem. Sci. 2005; 30:87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boisvert F.M., van Koningsbruggen S., Navascues J., Lamond A.I.. The multifunctional nucleolus. Nat. Rev. Mol. Cell Biol. 2007; 8:574–585. [DOI] [PubMed] [Google Scholar]
- 5. Henras A.K., Soudet J., Gerus M., Lebaron S., Caizergues-Ferrer M., Mougin A., Henry Y.. The post-transcriptional steps of eukaryotic ribosome biogenesis. Cell Mol. Life Sci. 2008; 65:2334–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Strunk B.S., Karbstein K.. Powering through ribosome assembly. RNA. 2009; 15:2083–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Drygin D., Rice W.G., Grummt I.. The RNA polymerase I transcription Machinery: An emerging target for the treatment of cancer. Annu. Rev. Pharmacol. 2010; 50:131–156. [DOI] [PubMed] [Google Scholar]
- 8. Grummt I. Wisely chosen paths - regulation of rRNA synthesis. FEBS J. 2010; 277:4626–4639. [DOI] [PubMed] [Google Scholar]
- 9. Arabi A., Wu S.Q., Ridderstrale K., Bierhoff H., Shiue C., Fatyol K., Fahlen S., Hydbring P., Soderberg O., Grummt I. et al.. c-Myc associates with ribosomal DNA and activates RNA polymerase I transcription. Nat. Cell Biol. 2005; 7:303–310. [DOI] [PubMed] [Google Scholar]
- 10. Grandori C., Gomez-Roman N., Felton-Edkins Z.A., Ngouenet C., Galloway D.A., Eisenman R.N., White R.J.. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat. Cell Biol. 2005; 7:311–318. [DOI] [PubMed] [Google Scholar]
- 11. McMahon S.B., Wood M.A., Cole M.D.. The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-Myc. Mol. Cell Biol. 2000; 20:556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Birch J.L., Tan B.C.M., Panov K.I., Panova T.B., Andersen J.S., Owen-Hughes T.A., Russell J., Lee S.C., Zomerdijk J.C.B.M.. FACT facilitates chromatin transcription by RNA polymerases I and III. EMBO J. 2009; 28:854–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. He N.H., Chan C.K., Sobhian B., Chou S., Xue Y.H., Liu M., Alber T., Benkirane M., Zhou Q.. Human Polymerase-Associated Factor complex (PAFc) connects the Super Elongation Complex (SEC) to RNA polymerase II on chromatin. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:E636–E645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mongelard F., Bouvet P.. Nucleolin: a multiFACeTed protein. Trends Cell Biol. 2007; 17:80–86. [DOI] [PubMed] [Google Scholar]
- 15. Su L., Hershberger R.J., Weissman I.L.. LYAR, a novel nucleolar protein with zinc finger DNA-binding motifs, is involved in cell growth regulation. Genes Dev. 1993; 7:735–748. [DOI] [PubMed] [Google Scholar]
- 16. Lee B., Jin S., Choi H., Kwon J.T., Kim J., Jeong J., Kwon Y.I., Cho C.. Expression and function of the Testis-Predominant protein LYAR in mice. Mol. Cells. 2013; 35:54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yonezawa K., Sugihara Y., Oshima K., Matsuda T., Nadano D.. Lyar, a cell growth-regulating zinc finger protein, was identified to be associated with cytoplasmic ribosomes in male germ and cancer cells. Mol. Cell Biochem. 2014; 395:221–229. [DOI] [PubMed] [Google Scholar]
- 18. Swartling F.J., Grimmer M.R., Hackett C.S., Northcott P.A., Fan Q.W., Goldenberg D.D., Lau J., Masic S., Nguyen K., Yakovenko S. et al.. Pleiotropic role for MYCN in medulloblastoma. Genes Dev. 2010; 24:1059–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wu Y., Liu M., Li Z., Wu X.B., Wang Y., Wang Y., Nie M., Huang F., Ju J., Ma C. et al.. LYAR promotes colorectal cancer cell mobility by activating galectin-1 expression. Oncotarget. 2015; 6:32890–32901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Miyazawa N., Yoshikawa H., Magae S., Ishikawa H., Izumikawa K., Terukina G., Suzuki A., Nakamura-Fujiyama S., Miura Y., Hayano T. et al.. Human cell growth regulator Ly-1 antibody reactive homologue accelerates processing of preribosomal RNA. Genes Cells. 2014; 19:273–286. [DOI] [PubMed] [Google Scholar]
- 21. Ju J., Wang Y., Liu R., Zhang Y., Xu Z., Wang Y., Wu Y., Liu M., Cerruti L., Zou F. et al.. Human fetal globin gene expression is regulated by LYAR. Nucleic Acids Res. 2014; 42:9740–9752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang G., Fulkerson C.M., Malek R., Ghassemifar S., Snyder P.W., Mendrysa S.M.. Mutations in Lyar and p53 are synergistically lethal in female mice. Birth Defects Res. A Clin. Mol. Teratol. 2012; 94:729–737. [DOI] [PubMed] [Google Scholar]
- 23. Li H., Wang B., Yang A., Lu R., Wang W., Zhou Y., Shi G., Kwon S.W., Zhao Y., Jin Y.. Ly-1 antibody reactive clone is an important nucleolar protein for control of self-renewal and differentiation in embryonic stem cells. Stem Cells. 2009; 27:1244–1254. [DOI] [PubMed] [Google Scholar]
- 24. Yanagida M. 2003; Tokyo University of Agriculture & Technology; Ph. D. Dessertation. [Google Scholar]
- 25. Fujiyama S., Yanagida M., Hayano T., Miura Y., Isobe T., Fujimori F., Uchida T., Takahashi N.. Isolation and proteomic characterization of human parvulin-associating preribosomal ribonucleoprotein complexes. J. Biol. Chem. 2002; 277:42418–42418. [DOI] [PubMed] [Google Scholar]
- 26. Yanagida M., Shimamoto A., Nishikawa K., Furuichi Y., Isobe T., Takahashi N.. Isolation and proteomic characterization of the major proteins of the nucleolin-binding ribonucleoprotein complexes. Proteomics. 2001; 1:1390–1404. [DOI] [PubMed] [Google Scholar]
- 27. Ewing R.M., Chu P., Elisma F., Li H., Taylor P., Climie S., McBroom-Cerajewski L., Robinson M.D., O’Connor L., Li M. et al.. Large-scale mapping of human protein-protein interactions by mass spectrometry. Mol. Syst. Biol. 2007; 3:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fujiyama-Nakamura S., Yoshikawa H., Homma K., Hayano T., Tsujimura-Takahashi T., Izumikawa K., Ishikawa H., Miyazawa N., Yanagida M., Miura Y. et al.. Parvulin (Par14), a Peptidyl-Prolyl cis-trans isomerase, is a novel rRNA processing factor that evolved in the metazoan lineage. Mol. Cell Proteomics. 2009; 8:1552–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hayano T., Yanagida M., Yamauchi Y., Shinkawa T., Isobe T., Takahashi N.. Proteomic analysis of human Nop56p-associated pre-ribosomal ribonucleoprotein complexes - Possible link between Nop56p and the nucleolar protein treacle responsible for Treacher Collins syndrome. J. Biol. Chem. 2003; 278:34309–34319. [DOI] [PubMed] [Google Scholar]
- 30. Lee H.K., Hsu A.K., Sajdak J., Qin J., Pavlidis P.. Coexpression analysis of human genes across many microarray data sets. Genome Res. 2004; 14:1085–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sekiguchi T., Hayano T., Yanagida M., Takahashi N., Nishimoto T.. NOP132 is required for proper nucleolus localization of DEAD-box RNA helicase DDX47. Nucleic Acids Res. 2006; 34:4593–4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yanagida M., Hayano T., Yamauchi Y., Shinkawa T., Natsume T., Isobe T., Takahashi N.. Human fibrillarin forms a sub-complex with splicing factor 2-associated p32, protein arginine methyltransferases, and tubulins alpha 3 and beta 1 that is independent of its association with preribosomal ribonucleoprotein complexes. J. Biol. Chem. 2004; 279:1607–1614. [DOI] [PubMed] [Google Scholar]
- 33. Hayashi Y., Kuroda T., Kishimoto H., Wang C., Iwama A., Kimura K.. Downregulation of rRNA transcription triggers cell differentiation. PLoS One. 2014; 9:e98586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Woolnough J.L., Atwood B.L., Liu Z., Zhao R., Giles K.E.. The regulation of rRNA gene transcription during directed differentiation of human embryonic stem cells. PLoS One. 2016; 11:e0157276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang Q., Shalaby N.A., Buszczak M.. Changes in rRNA transcription influence proliferation and cell fate within a stem cell lineage. Science. 2014; 343:298–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schnapp G., Santori F., Carles C., Riva M., Grummt I.. The HMG box-containing nucleolar transcription factor UBF interacts with a specific subunit of RNA polymerase I. EMBO J. 1994; 13:190–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Panov K.I., Friedrich J.K., Russell J., Zomerdijk J.C.. UBF activates RNA polymerase I transcription by stimulating promoter escape. EMBO J. 2006; 25:3310–3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Filippakopoulos P., Picaud S., Mangos M., Keates T., Lambert J.P., Barsyte-Lovejoy D., Felletar I., Volkmer R., Muller S., Pawson T. et al.. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012; 149:214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dey A., Chitsaz F., Abbasi A., Misteli T., Ozato K.. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:8758–8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rachie N.N., Seger R., Valentine M.A., Ostrowski J., Bomszyk K.. Identification of an inducible 85-kDanuclear protein kinase. J. Biol. Chem. 1993; 268:22143–22149. [PubMed] [Google Scholar]
- 41. Denis G.V., Green M.R.. A novel, mitogen-activated nuclear kinase is related to a Drosophila developmental regulator. Genes Dev. 1996; 10:261–271. [DOI] [PubMed] [Google Scholar]
- 42. Sinha A., Faller D.V., Denis G.V.. Bromodomain analysis of Brd2-dependent transcriptional activation of cyclin A. Biochem. J. 2005; 387:257–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. He N., Pezda A.C., Zhou Q.. Modulation of a P-TEFb functionalequilibrium for the global control of cell growth and differentiation. Mol. Cell Biol. 2006; 26:7068–7076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Devaiah B.N., Case-Borden C., Gegonne A., Hsu C.H., Chen Q.R., Meerzaman D., Dey A., Ozato K., Singer D.S.. BRD4 is a histone acetyltransferase that evicts nucleosomes from chromatin. Nat. Struct. Mol. Biol. 2016; 23:540–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Foy R.L., Song I.Y, Chitalia V.C., Choen H.T., Saksouk N., Cayrou C., Vaziri C., Cote J., Panchenko M.V.. Role of Jade-1 in the histone acetyltransferase (HAT) HBO1 complex. J. Biol. Chem. 2008; 283:28817–28826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Burger K., Muhl B., Kellner M., Rohrmoser M., Gruber-Eber A., Windhager L., Friedel C.C., Dolken L., Eick D.. 4-thiouridine inhibits rRNA synthesis and causes a nucleolar stress response. RNA Biol. 2013; 10:1623–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Leibovitz A., Stinson J.C., McCombs W.B. 3rd, McCoy C.E., Mazur K.C., Mabry N.D.. Classification of human colorectal adenocarcinoma cell lines. Cancer Res. 1976; 36:4562–4569. [PubMed] [Google Scholar]
- 48. Gagos S., Hopwood V.L., Iliopoulos D., Kostakis A., Karayannakos P., Yatzides H., Skalkeas G.D., Pathak S.. Chromosomal markers associated with metastasis in two colon cancer cell lines established from the same patient. Anticancer Res. 1995; 15:369–378. [PubMed] [Google Scholar]
- 49. O'Sullivan A.C., Sullivan G.J., McStay B.. UBF binding in vivo is not restricted to regulatory sequences within the vertebrate ribosomal DNA repeat. Mol. Cell Biol. 2002; 22:657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yoshikawa H., Komatsu W., Hayano T., Miura Y., Homma K., Izumikawa K., Ishikawa H., Miyazawa N., Tachikawa H., Yamauchi Y. et al.. Splicing factor 2-associated protein p32 participates in ribosome biogenesis by regulating the binding of Nop52 and fibrillarin to preribosome particles. Mol. Cell Proteomics. 2011; 10:M110 006148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Izumikawa K., Ishikawa H., Yoshikawa H., Terukina G., Miyazawa N., Nakayama H., Nobe Y., Taoka M., Yamauchi Y., Philipsen S. et al.. Friend of Prmt1, FOP is a novel component of the nuclear SMN complex isolated using biotin affinity purification. J. Proteomics Bioinformatics. 2014; S7:doi:10.4172/jpb.S7-002. [Google Scholar]
- 52. Zhang Y., Sikes M.L., Beyer A.L., Schneider D.A.. The Paf1 complex is required for efficient transcription elongation by RNA polymerase I. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:2153–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kwon S.H., Florens L., Swanson S.K., Washburn M.P., Abmayr S.M., Workman J.L.. Heterochromatin protein 1 (HP1) connects the FACT histone chaperone complex to the phosphorylated CTD of RNA polymerase II. Gene Dev. 2010; 24:2133–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Reinberg D., Sims R.J.. de FACTo nucleosome dynamics. J. Biol. Chem. 2006; 281:23297–23301. [DOI] [PubMed] [Google Scholar]
- 55. Anderson S.J., Sikes M.L., Zhang Y.F., French S.L., Salgia S., Beyer A.L., Nomura M., Schneider D.A.. The transcription elongation factor Spt5 influences transcription by RNA polymerase I positively and negatively. J. Biol. Chem. 2011; 286:18816–18824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chen Y.X., Yamaguchi Y., Tsugeno Y., Yamamoto J., Yamada T., Nakamura M., Hisatake K., Handa H.. DSIF, the Paf1 complex, and Tat-SF1 have nonredundant, cooperative roles in RNA polymerase II elongation. Gene Dev. 2009; 23:2765–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Murano K., Okuwaki M., Hisaoka M., Nagata K.. Transcription regulation of the rRNA gene by a multifunctional nucleolar protein, B23/nucleophosmin, through its histone chaperone activity. Mol. Cell Biol. 2008; 28:3114–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Valdez B.C., Henning D., So R.B., Dixon J., Dixon M.J.. The Treacher Collins syndrome (TCOF1) gene product is involved in ribosomal DNA gene transcription by interacting with upstream binding factor. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:10709–10714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gonzales B., Henning D., So R.B., Dixon J., Dixon M.J., Valdez B.C.. The Treacher Collins syndrome (TCOF1) gene product is involved in pre-rRNA methylation. Hum. Mol. Genet. 2005; 14:2035–2043. [DOI] [PubMed] [Google Scholar]
- 60. Prieto J.L., McStay B.. Recruitment of factors linking transcription and processing of pre-rRNA to NOR chromatin is UBF-dependent and occurs independent of transcription in human cells. Genes Dev. 2007; 21:2041–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lin C.I., Yeh N.H.. Treacle recruits RNA polymerase I complex to the nucleolus that is independent of UBF. Biochem. Biophys. Res. Commun. 2009; 386:396–401. [DOI] [PubMed] [Google Scholar]
- 62. Werner A., Iwasaki S., McGourty C.A., Medina-Ruiz S., Teerikorpi N., Fedrigo I., Ingolia N.T., Rape M.. Cell-fate determination by ubiquitin-dependent regulation of translation. Nature. 2015; 525:523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jordan P., Mannervik M., Tora L., CarmoFonseca M.. In vivo evidence that TATA-binding protein SL1 colocalizes with UBF and RNA polymerase I when rRNA synthesis is either active or inactive. J. Cell Biol. 1996; 133:225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cavanaugh A.H., Hempel W.M., Taylor L.J., Rogalsky V., Todorov G., Rothblum L.I.. Activity of RNA-polymerase-I transcription factor Ubf blocked by Rb gene-product. Nature. 1995; 374:177–180. [DOI] [PubMed] [Google Scholar]
- 65. Brandenburger Y., Jenkins A., Autelitano D.J., Hannan R.D.. Increased expression of UBF is a critical determinant for rRNA synthesis and hypertrophic growth of cardiac myocytes. FASEB J. 2001; 15:2051–2053. [DOI] [PubMed] [Google Scholar]
- 66. Bell S.P., Learned R.M., Jantzen H.M., Tjian R.. Functional cooperativity between transcription Factor-Ubf1 and Factor-Sl1 mediates human Ribosomal-Rna synthesis. Science. 1988; 241:1192–1197. [DOI] [PubMed] [Google Scholar]
- 67. Stefanovsky V., Langlois F., Gagnon-Kugler T., Rothblum L.I., Moss T.. Growth factor signaling regulates elongation of RNA polymerase I transcription in mammals via UBF phosphorylation and r-chromatin remodeling. Mol. Cell. 2006; 21:629–639. [DOI] [PubMed] [Google Scholar]
- 68. Drygin D., Lin A., Bliesath J., Ho C.B., O’Brien S.E., Proffitt C., Omori M., Haddach M., Schwaebe M.K., Siddiqui-Jain A. et al.. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011; 71:1418–1430. [DOI] [PubMed] [Google Scholar]
- 69. Hadjiolova K.V., Hadjiolov A.A., Bachellerie J.P.. Actinomycin D stimulates the transcription of rRNA minigenes transfected into mouse cells. Implications for the in vivo hypersensitivity of rRNA gene transcription. Eur. J. Biochem. 1995; 228:605–615. [DOI] [PubMed] [Google Scholar]
- 70. Luna-Pelaez N., Garcia-Dominguez M.. Lyar-Mediated recruitment of Brd2 to the chromatin attenuates nanog downregulation following induction of differentiation. J. Mol. Biol. 2018; 430:1084–1097. [DOI] [PubMed] [Google Scholar]
- 71. Peng J.H., Dong W., Chen L., Zou T.T., Qi Y.P., Liu Y.L.. Brd2 is a TBP-associated protein and recruits TBP into E2F-1 transcriptional complex in response to serum stimulation. Mol. Cell Biochem. 2007; 294:45–54. [DOI] [PubMed] [Google Scholar]
- 72. Galbiati L., Mendoza-Maldonado R., Gutierrez M.I., Giacca M.. Regulation of E2F-1 after DNA damage by p300-mediated acetylation and ubiquitination. Cell Cycle. 2005; 4:930–939. [DOI] [PubMed] [Google Scholar]
- 73. Tropberger P., Pott S., Keller C., Kamieniarz-Gdula K., Caron M., Richter F., Li G., Mittler G., Liu E.T., Buhler M. et al.. Regulation of transcription through acetylation of H3K122 on the lateral surface of the histone octamer. Cell. 2013; 152:859–872. [DOI] [PubMed] [Google Scholar]
- 74. Vintermist A., Bohm S., Sadeghifar F., Louvet E., Mansen A., Percipalle P., Ostlund Farrants A.K.. The chromatin remodelling complex B-WICH changes the chromatin structure and recruits histone acetyl-transferases to active rRNA genes. PLoS One. 2011; 6:e19184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yang X.J. Multisite protein modification and intramolecular signaling. Oncogene. 2005; 24:1653–1662. [DOI] [PubMed] [Google Scholar]
- 76. Yang Z.Y., Yik J.H.N., Chen R.C., He N.H., Jang M.K., Ozato K., Zhou Q.. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein brd4. Mol. Cell. 2005; 19:535–545. [DOI] [PubMed] [Google Scholar]
- 77. Nakamura Y., Umehara T., Nakano K., Jang M.K., Shirouzu M., Morita S., Uda-Tochio H., Hamana H., Terada T., Adachi N. et al.. Crystal structure of the human BRD2 bromodomain: insights into dimerization and recognition of acetylated histone H4. J. Biol. Chem. 2007; 282:4193–4201. [DOI] [PubMed] [Google Scholar]
- 78. Li Z., Mbonye U., Feng Z., Wang X., Gao X., Karn J., Zhou Q.. The KAT5-Acetyl-Histone4-Brd4 axis silences HIV-1 transcription and promotes viral latency. PLoS Pathog. 2018; 14:e1007012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hirschler-Laszkiewicz I., Cavanaugh A., Hu Q., Catania J., Avantaggiati M.L., Rothblum L.I.. The role of acetylation in rDNA transcription. Nucleic Acids Res. 2001; 29:4114–4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Li L., Shi L., Yang S., Yan R., Zhang D., Yang J., He L., Li W., Yi X., Sun L. et al.. SIRT7 is a histone desuccinylase that functionally links to chromatin compaction and genome stability. Nat. Commun. 2016; 7:12235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Qiu Y., Gilmour D.S.. Identification of regions in the Spt5 subunit of DRB sensitivity-inducing factor (DSIF) that sre involved in promoter-proximal pausing. J. Biol. Chem. 2017; 292:5555–5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Dawson M.A., Prinjha R.K., Dittmann A., Giotopoulos G., Bantscheff M., Chan W.I., Robson S.C., Chung C.W., Hopf C., Savitski M.M. et al.. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011; 478:529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Filippakopoulos P., Qi J., Picaud S., Shen Y., Smith W.B., Fedorov O., Morse E.M., Keates T., Hickman T.T., Felletar I. et al.. Selective inhibition of BET bromodomains. Nature. 2010; 468:1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hein N., Cameron D.P., Hannan K.M., Nguyen N.N., Fong C.Y., Sornkom J., Wall M., Pavy M., Cullinane C., Diesch J. et al.. Inhibition of Pol I transcription treats murine and human AML by targeting the leukemia-initiating cell population. Blood. 2017; 129:2882–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Gyuris A., Donovan D.J., Seymour K.A., Lovasco L.A., Smilowitz N.R., Halperin A.L.P., Klysik J.E., Freiman R.N.. The chromatin-targeting protein Brd2 is required for neural tube closure and embryogenesis. BBA-Gene Regul. Mech. 2009; 1789:413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Houzelstein D., Bullock S.L., Lynch D.E., Grigorieva E.F., Wilson V.A., Beddington R.S.P.. Growth and early postimplantation defects in mice deficient for the bromodomain-containing protein Brd4. Mol. Cell Biol. 2002; 22:3794–3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.