

Abstract
Motivation
Metagenomic contig binning is an important computational problem in metagenomic research, which aims to cluster contigs from the same genome into the same group. Unlike classical clustering problem, contig binning can utilize known relationships among some of the contigs or the taxonomic identity of some contigs. However, the current state-of-the-art contig binning methods do not make full use of the additional biological information except the coverage and sequence composition of the contigs.
Results
We developed a novel contig binning method, Semi-supervised Spectral Normalized Cut for Binning (SolidBin), based on semi-supervised spectral clustering. Using sequence feature similarity and/or additional biological information, such as the reliable taxonomy assignments of some contigs, SolidBin constructs two types of prior information: must-link and cannot-link constraints. Must-link constraints mean that the pair of contigs should be clustered into the same group, while cannot-link constraints mean that the pair of contigs should be clustered in different groups. These constraints are then integrated into a classical spectral clustering approach, normalized cut, for improved contig binning. The performance of SolidBin is compared with five state-of-the-art genome binners, CONCOCT, COCACOLA, MaxBin, MetaBAT and BMC3C on five next-generation sequencing benchmark datasets including simulated multi- and single-sample datasets and real multi-sample datasets. The experimental results show that, SolidBin has achieved the best performance in terms of F-score, Adjusted Rand Index and Normalized Mutual Information, especially while using the real datasets and the single-sample dataset.
Availability and implementation
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Microbial communities consist of mixtures of large number of microorganisms. In metagenomic studies, genetic material is directly extracted from the microbial communities and is sequenced using high-throughput sequencing technologies resulting in large quantity of sequencing reads of different read lengths depending on the sequencing technologies and platforms. During sequencing, the information on what reads come from the same genome or closely related genomes is lost. Usually the first step in metagenomic data analysis is ‘reads assembly’ aiming to assemble the reads into relative longer genomic fragments called contigs based on the overlapping information of the reads. The second step is ‘contig binning’ aiming to cluster the contigs from the same genome or closely related genomes into the same bin. In the third step, reads are mapped to known genomic sequences or the contigs in the bins and the relative abundance profiles of genomes and genomic bins are estimated in each sample. Finally, statistical methods are used to associate the abundance profiles of genomes/bins with the environmental factors or phenotypes of the individuals (Bahram et al., 2018; Huttenhower et al., 2012; Qin et al., 2010; Sunagawa et al., 2015). Metagenomic studies have shown that microbial communities in human gut have been shown to be significantly associated with several diseases (Jostins et al., 2012; Khor et al., 2011) and the effect of immunotherapy (Chen et al., 2017; Wilck et al., 2017). In this paper, we concentrate on the usual second step of metagenomic data analysis, contig binning.
The available contig binning methods can be divided into two major categories: taxonomy dependent methods (also called supervised methods) that map the contigs to known reference databases, such as MEGAN (Huson et al., 2007), and taxonomy independent methods (also called unsupervised methods), such as MaxBin ( Wu et al., 2014, 2016 ), CONCOCT (Alneberg et al., 2014), MetaBAT (Kang et al., 2015), COCACOLA (Lu et al., 2017a) and BMC3C (Yu et al., 2018) that cluster the contigs ‘de novo’ without mapping the contigs to known genomes. Both categories of methods have their own limitations. Since the known genomes are limited and a large number of genomes are still unknown, many contigs cannot be mapped to known genomes. In addition, alignment is computationally expensive and thus alignment based contig binning methods are generally slow. Without relying on reference genomes, unsupervised binning methods can partially overcome some of the above-mentioned shortcomings. On the other hand, existing unsupervised binning methods have not made full use of additional available biological information such as the alignment of some the contigs to known genomes, which can improve the binning performance. Therefore, it is imperative to develop effective contig binning approaches that can not only be used for the datasets that contain a large amount of contigs from unknown genomes, but also make use of the available biological information from alignment for better binning performance.
To tackle this issue, we develop a novel contig binning method, Semi-supervised Spectral Normalized Cut for Binning (SolidBin), which is inspired by various studies in semi-supervised clustering (Gu et al., 2013; Wagstaff et al., 2001) that can improve the clustering performance by incorporating pairwise constraints. There are two types of pairwise constraints: must-link (ML) constraints and cannot-link (CL) constraints. Specifically, SolidBin is based on semi-supervised spectral clustering. Compared with other clustering methods, spectral clustering algorithm can relatively easily incorporate additional information on pairwise relationships of contigs. SolidBin mainly utilizes two kinds of sequence features: contig coverage information and tetranucleotides frequencies. The pairwise constraints can be generated from both internal (contig features) and external information (additional biological information such as co-alignment). With respect to different kinds of information used for generating constraints, SolidBin has several different modes, SolidBin-sequence feature similarity (SFS), SolidBin-coalign, SolidBin-CL and SolidBin-SFS-CL. SolidBin-SFS generates ML constraints between contig pairs with high similarity based on internal information of sequence features. On the other hand, SolidBin-coalign and SolidBin-CL generate constraints using external co-alignment information. By aligning contigs to known genomes, TAXAassign (https://github.com/umerijaz/taxaassign) can assign some contigs belonging to the known genomes to the corresponding genomes/species. The contig pairs with the same species assignment by TAXAassign have a ML constraint in SolidBin-coalign, while the contig pairs with different genus assignment have a CL constraint in SolidBin-CL. SolidBin-SFS-CL removes the CL pairs from the constraints set on the basis of SolidBin-SFS. The performance of SolidBin has been extensively investigated on five next-generation sequencing benchmark datasets including simulated multi- and single-sample datasets, as well as real multi-sample datasets. Compared with five state-of-the-art genome binners, CONCOCT, COCACOLA, MaxBin, MetaBAT and BMC3C, SolidBin has achieved the best performance in terms of F-score, Adjusted Rand Index (ARI) and Normalized Mutual Information (NMI), especially while using the real datasets and the single-sample dataset. Specifically, SolidBin-SFS achieves better performance when there are sufficient samples. SolidBin-coalign also works very well except the case that the dataset contains many genomes on the strain level.
2 Related work
Unsupervised contig binning methods can be roughly divided into three categories based on the sequence characteristics used for binning: (i) Nucleotide Composition-based, (ii) Differential Abundance-based and (iii) both Nucleotide Composition and Abundance-based (Sangwan et al., 2016). Nucleotide Composition-based methods (Dick et al., 2009; Laczny et al., 2015) take oligonucleotide frequency as features, based on the assumption that the oligonucleotide composition of fragments from the same genome are much more similar than fragments from different genomes. Differential Abundance-based methods [Abundancebin (Wu and Ye, 2011)] cluster contigs mainly according to the differential coverage of average nucleotide coverage of each contig, based on the assumption that the contigs that belong to the same genome should have similar abundance in the same sample. Nucleotide Composition and Abundance-based methods (MaxBin, MetaBAT, CONCOCT and COCACOLA) combine the two kinds of information and have been shown to outperform methods using only one kind of information (Sedlar et al., 2017).
The MaxBin algorithm utilizes the single-copy marker genes to determine the number of bins and expectation–maximization algorithm is performed while binning. MaxBin 2.0 can be applied to the datasets that contain multiple samples based on the assumption that all samples are independently sequenced. The MetaBAT algorithm utilizes two different genomic features to calculate the distance of contig pairs, and then converts the distance to the probability of them being in the same genome. Finally, the modified K-medoids algorithm is used for binning. CONCOCT takes sequence composition and coverage across multiple samples as sequence feature vectors, and then uses principal component analysis to reduce the dimension of the feature vectors. Finally, it obtains the binning results using the Gaussian mixture model. It determines the number of bins by the variational Bayesian approach. COCACOLA employs the similar sequence feature vectors as CONCOCT and it takes advantage of both the soft and hard clustering by employing non-negative matrix factorization with sparse regularization. The performances of COCACOLA and CONCOCT depend largely on the number of samples, partially due to the way to construct feature vectors.
Several investigators realized the importance of incorporating additional biological information for binning, such as co-alignment and pair-end read linkage. However, the co-alignment information was not considered by CONCOCT, MaxBin and MetaBAT, while the pair-end reads linkage information was not considered in MaxBin and MetaBAT. CONCOCT utilizes the pair-end reads linkage information only in the post-processing stage of binning. It merges the clusters with sufficient pair-end links and similar coverage to perform a hierarchical clustering (https://concoct.readthedocs.io/en/latest/), which may improve the recall of the binning result but reduce the precision. On the other hand, COCACOLA uses non-negative matrix factorization to factorize the feature matrix of contigs, and integrates co-alignment information and paired-end read linkage across multiple samples into a cost function. It was shown that the additional information can improve the binning performance, especially when the number of samples is small. Hetero-RP (Lu et al., 2017b) automatically adjusts the weights of contig features according to the additional co-alignment information before using other binning methods, and the method is integrated into the COCACOLA-python version. However, the performance of Hetero-RP depends heavily on the accuracy of the additional information and the fraction of contig pairs having such information. These studies have inspired us to develop a new binning method with a semi-supervised spectral clustering approach, SolidBin, where pairwise constraints among contigs can be effectively integrated into the spectral clustering framework of contig graph partition.
Recently, several ensemble clustering methods have been developed for metagenomic contig binning. BMC3C (Yu et al., 2018) takes sequence composition, coverage information and codon usage as sequence feature vectors and repeatedly applying the K-means clustering with different initialization to form a weight graph of contigs for partitioning. Additionally, ensemble models combing results from multiple binning algorithms have been proposed. Binning refiner (Song and Thomas, 2017) performs a pairwise comparison of contigs between the output bins of two binners using blastn to obtain shared contigs between two sets of bins. Each set of shared contigs is treated as a refined bin if its total length is longer than a predefined threshold. DAS Tool (Sieber et al., 2018) proposes a scoring function based on single-copy genes and uses it to rank bins generated by different binners. It picks up high-scoring bins iteratively and updates the scores of the remaining candidate bins. MetaWRAP (Uritskiy et al., 2018) uses the output bin sets of MetaBAT2, MaxBin2 and CONCOCT to generate hybrid bin sets, in which every pair of contigs in different bins from any original sets are separated. MetaWRAP then selects the best bin based on the estimated completion and contamination scores. In contrast to these ensemble approaches, SolidBin is a stand-alone method that can be integrated into these ensemble approaches as a component for better binning performance.
3 Materials and methods
3.1 Semi-supervised clustering and spectral clustering
Semi-supervised clustering methods can use prior information to improve clustering performance. One important type of prior information is pairwise constraints that can be divided into two categories: ML constraints and CL constraints. A ML constraint means that two instances should be grouped in the same cluster, while a CL constraint indicates that two instances should appear in different clusters. For the contig binning problem, instances correspond to contigs and affinity between instances correspond to similarity between contigs.
Spectral clustering methods are based on the spectral graph theory (Ng et al., 2002). A given dataset can be mapped to a graph whose nodes represent the instances of the dataset and the edges reflect the affinity of the instances. The essence of spectral clustering methods is to transform the clustering problem into an optimal partition problem of the graph. Spectral clustering has several variants according to different criteria for graph partitioning and the normalized cut (Ncut) criterion is a reliable one that measures both the total inter-cluster dissimilarity and the total intra-cluster similarity (Shi and Malik, 2000). Compared with other clustering methods, the combination of spectral clustering algorithm and the semi-supervised method has obvious advantages. Spectral clustering algorithm utilizes the similarity among instances, and thus the pairwise constraints among instances can be easily incorporated into the algorithm to improve clustering performance.
3.2 The SolidBin algorithm
In this paper, we developed a contig binning method, SolidBin, based on the semi-supervised spectral clustering method that can utilize additional biological information and make full use of both sequence composition and coverage information. The method has five major steps. First, the contigs are mapped into a graph after obtaining the feature representations of contigs. Then, the bin number K is initialized according to the single-copy marker genes, and the final bin number will be determined by the iterative binning results of K-means. Next, constraints are generated differently according to the modes of SolidBin, and then the constraints will be used for semi-supervised NCut with the final bin number. Finally, the method will evaluate the clustering performance with different parameter values without labels and keep the optimal one as the final result. The general pipeline of SolidBin is shown in Figure 1 and more details of the steps are as follows.
Fig. 1.
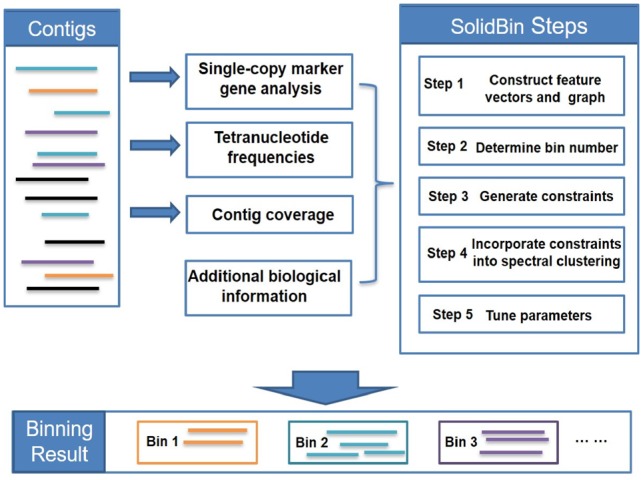
The general workflow of SolidBin
3.2.1. Construct feature vectors and graph
Similar to CONCOCT and COCACOLA, we employ the combination of an M dimensional coverage vector and a T dimensional composition vector to represent each contig, where M is the number of the samples and T is the number of distinct tetramers. T=136 in our method considering one tetramer and its reverse-complement are combined. After adding a pseudo-count, the composition vectors are normalized over contigs to account for different contig lengths. The coverage vectors are normalized over contigs and over samples, so that different read numbers from a sample are accounted for Alneberg et al. (2014). Therefore, the feature matrix of the contigs is denoted as , where N denotes the number of contigs.
For graph construction, we use L1-distance to measure the dissimilarity between each pair of contigs (Lu et al., 2017a) and convert it to the affinity using the following formula, where A denotes the affinity matrix, Dist denotes the dissimilarity matrix of the contigs and max(.) and min(.) denotes the maximum and minimum of all the elements of the matrix, respectively.
| (1) |
3.2.2. Bin number determination
Similar to the method used in COCACOLA, we utilize single-copy marker genes to estimate the initial bin number k0, and then, we try a list of numbers larger than k0 as the bin numbers sequentially and use the K-means algorithm for binning. Then we calculate the silhouette coefficient (Rousseeuw, 1987) of the binning result, which is an index to evaluate the clustering performance without labels by measuring the cohesion and the separation of the clusters. Our methods will find two local maxima of the silhouette coefficient values, and the larger one corresponds to a bin number, which is our final bin number.
3.2.3. Generate constraints
Generate ML constraints
ML constraints are mainly generated in three ways as three modes of SolidBin:
- SolidBin-SFS mode: constraints are generated according to SFS, which is a taxonomy-independent method. In this mode, we underline the reliable information from sequence features themselves without using reference genomes. The contig pairs with high similarity are chosen as ML constraints. Because the affinity graphs of different datasets have different connection structures, the ratio of the pairwise constraints we selected from whole similarity matrix may vary widely. Therefore, we determine the ratio of the pairwise constraints adaptively as follows. We assume that the connection relationship obtained from the binning results is close to the real connection relationship and varies among different datasets. The assumption will be examined in the experiments. We run the NCut method and obtain the initial binning results for the connection graphs. The connection ratio is calculated as follows, where K represents the number of the bins used by NCut method and Numk represents the number of contigs contained in the k-th bin.
(2) (3) SolidBin-coalign mode: we take the contig pairs with the same assignment on species level using NCBI taxonomy by TAXAassign as ML constraints. In this mode, we take advantage of some contigs that belong to the known reference genomes although the reference genomes are incomplete.
SolidBin-SFS-CL: constraints are first generated according to the SFS as in SolidBin-SFS. We next remove the contig pairs assigned to different genera by TAXAassign.
Generate CL constraints
SolidBin-CL: to obtain CL constraints with high accuracy, the contigs assigned to different genus by TAXAassign are regarded as CL pairs.
3.2.4. Incorporate constraints into spectral clustering
Spectral clustering with NCut
A popular criterion in spectral clustering is the NCut, for which the cost function of given K clusters is defined as follows (Gu et al., 2013; Ji et al., 2006):
| (4) |
where V denotes the vertex set, and W denotes the affinity matrix. Let be the binary indicator vector of cluster Pi, and hni = 1 means the n-th contig belongs to Pi. Let D be a diagonal matrix with
FNcut can be rewritten as follows:
| (5) |
where , and Y TY = I.
| (6) |
where are the largest K eigenvalues of matrix , and when are their corresponding eigenvectors, the equality sign of in Equation (6) holds.
Integrate ML constraints
If the i-th contig and the j-th contig belong to the same species, the i-th and j-th row of the indicator matrix H should be the same. Therefore, If there are C ML constraints, the c-th ML constraints can be represented as a constraint row vector , where uic = 1, ujc = –1, and the rest of all elements are zero. We can encode all ML constraints by matrix such that:
| (7) |
Integrate CL constraints
If the i-th contig and the j-th contig belong to different genomes, the i-th and j-th rows of the indicator matrix H should be orthogonal to each other. So, we can use an N × N non-negative matrix to represent the CL information, where Zij = 1 means that the i-th contig and the j-th contig belong to different genomes, while Zij = 0 means that the relationship between the i-th contig and the j-th contig is unknown. The CL constraints can be represented as follows:
| (8) |
ML and CL constraints can be incorporated into the framework of the NCut as follows (Gu et al., 2013):
| (9) |
3.2.5. Tune parameters (α or β)
For parameter tuning, we use the Calinski–Harabasz index (Caliński and Harabasz, 1974) to evaluate the clustering performance without labels by measuring the cohesion and the separation of the clusters. There is only one parameter to be tuned in all the modes, and a one-dimensional search is conducted for the optimal parameter in a range of candidate α or β.
3.2.6. Different SolidBin modes
There are several SolidBin modes according to the prior adopted information. The summary of different modes and the summary of performance of different modes compared in the experiments are shown in Table 1.
Table 1.
Summary of different SolidBin modes
| SolidBin mode | Constraints | Type | Parameter | Description | Performance profiles |
|---|---|---|---|---|---|
| SolidBin-naive | None | Taxonomy-independent | None | The NCut mode | Comparable performance to other binners |
| SolidBin-SFS | ML | Taxonomy-independent | α | Take the contig pairs with high similarity as ML constraints | Good performance when the number of samples is large. It can obtain good results with high similarity quality, and more samples could bring more useful coverage information |
| SolidBin-coalign | ML | Taxonomy-dependent | α | Take the contig pairs with the same assignment by TAXAassign as ML constraints | Good performance when the genomes contained in the datasets are on the species level, while using the results from TAXAassign. It can obtain good results if the pairwise ML constraints have high accuracy |
| SolidBin-CL | CL | Taxonomy-dependent | β | Take the contig pairs assigned to different genera by TAXAassign as CL constraints | Good performance when the genomes contained in the datasets are on the species level, often worse than SolidBin-coalign, while using the results from TAXAassign. It can obtain good results if the pairwise CL constraints have high accuracy |
| SolidBin-SFS-CL | ML | Taxonomy-dependent | α | Remove the contig pairs assigned to different genera by TAXAassign from the constraints set on the basis of SolidBin-SFS | Often a little better than SolidBin-SFS at a cost of spending much time on taxonomy alignment |
4 Experiments
4.1 Data
As shown in Table 2, we compared SolidBin with the-state-of-art binners on five benchmark datasets.
Table 2.
Datasets used in the experiments
| Datasets | Data type | Sample num | Contig num | Labeled contig num |
|---|---|---|---|---|
| SpeciesMock | Simulated | 96 | 37 628 | 37 628 |
| StrainMock | Simulated | 64 | 9417 | 9417 |
| SimHC | Simulated | 1 | 29 535 | 13 918 |
| Sharon | Real | 18 | 5579 | 2614 |
| MetaHIT | Real | 264 | 192 673 | 17 136 |
Multi-sample dataset on the species level: ‘SpeciesMock’ (Alneberg et al., 2014)
The dataset was constructed based on the analysis of 16S rRNA samples originated from the Human Microbiome Project (Huttenhower et al., 2012) and consisted of 101 different species across 96 samples. A total of 37 628 contigs remain for binning after co-assembly and filtering. The dataset is used for evaluating the influence of sample numbers on different SolidBin modes.
Multi-sample dataset on the strain level: ‘StrainMock’ (Alneberg et al., 2014)
The dataset was constructed to test the performance of the binners at different levels (Huttenhower et al., 2012). It consisted of 20 different species or strains across 64 samples. A total of 9417 contigs remain for binning after co-assembly and filtering.
Single-sample dataset: ‘SimHC’ (Wu et al., 2014)
The single-sample dataset was simulated by Wu et al. (2014) and 29 535 contigs remain for binning after assembling and filtering. SimHC simulated high-complexity communities lacking dominant populations and it contains 100 genomes. A total of 13 918 out of 29 535 co-assembled contigs are unambiguously labeled on the species level for evaluation by Wang (Wang et al., 2017) and we binned all the contigs. The dataset is used for evaluating the performance of different binners on single-sample datasets.
Real dataset: ‘MetaHIT’
The dataset from MetaHIT consortium (Qin et al., 2010) contains 264 samples, is the same dataset used in COCACOLA (Lu et al., 2017a) and MetaBAT (Kang et al., 2015). A total of 17 136 out of 192 673 co-assembled contigs are unambiguously labeled on the species level for evaluation (Lu et al., 2017a). We binned the contigs with unambiguously labels for evaluation as in Lu et al. (2017a) and Yu et al. (2018). The dataset is used for evaluating the performance of different binners on high-complexity real datasets.
Real dataset: ‘Sharon’ (Sharon et al., 2013)
The dataset from a time-series study of microbiome samples from a premature infant contains 18 samples. All the contigs are binned, and 2614 of 5579 contigs are unambiguously labeled on the species level for evaluation (Lu et al., 2017a). The dataset is used for evaluating the performance of different binners on low complexity real datasets.
4.2 Evaluation metric
For the unambiguously labeled contigs belonging to the datasets, the measures including precision, recall, F-score, Normalized Mutual Information and Adjusted Rand Index are used to evaluate the binning results and their definitions are shown in Supplementary Material.
4.3 Experimental procedures
We compared SolidBin with different modes to five advanced binners: CONCOCT-0.4.0, COCACOLA-python, MaxBin 2.2.4, MetaBAT 2.12.1 and BMC3C, respectively.
First, we conducted some preliminary experiments to show the necessity and effectiveness of determining the ratio of the pairwise constraints adaptively in SolidBin-SFS, and the accuracy of different constraints. We then compared the performance SolidBin with different modes with other binners. Finally, we investigated the effect of the number of samples on the performance of SolidBin-SFS and SolidBin-coalign based on sub-samples of the speciesmock dataset.
4.4 Experimental results
To make a fair comparison, the input bin numbers are determined by the respective binners. Tables 5–9 show the results of SolidBin-SFS and SolidBin-SFS-CL when the ratio of pairwise constraints =40% of the estimated ratio. The values of ‘α*’ and ‘β*’ are the estimated parameters chosen by the Calinski–Harabasz index. COCACOLA-coalign means incorporating co-alignment information into COCACOLA.
Table 5.
The performance of COCACOLA, CONCOCT, MaxBin, MetaBAT, BMC3C and SolidBin on the StrainMock dataset
| StrainMock | Recall (%) | Precision (%) | F-score (%) | NMI (%) | ARI (%) |
|---|---|---|---|---|---|
| COCACOLA | 99.20 | 99.26 | 99.23 | 98.63 | 99.03 |
| COCACOLA-coalign | 95.87 | 93.53 | 94.69 | 95.46 | 94.57 |
| MaxBin | 96.35 | 91.00 | 93.60 | 98.36 | 86.69 |
| CONCOCT | 98.21 | 93.85 | 95.98 | 96.24 | 93.99 |
| MetaBAT | 81.24 | 59.87 | 68.94 | 69.76 | 33.50 |
| BMC3C(9401/9417) | 99.01 | 99.01 | 99.01 | 98.42 | 98.73 |
| SolidBin-naive | 98.92 | 98.92 | 98.92 | 98.49 | 98.91 |
| SolidBin-SFS(α*=20) | 99.29 | 99.29 | 99.29 | 98.75 | 99.13 |
| SolidBin-coalign(α*=10) | 99.46 | 92.59 | 95.90 | 95.93 | 87.74 |
| SolidBin-CL(β*=0.1) | 99.29 | 99.29 | 99.29 | 98.74 | 99.12 |
| SolidBin-SFS-CL(α*= 30) | 99.29 | 99.29 | 99.29 | 98.75 | 99.12 |
Note: The optimal values of the results are in bold.
4.4.1. Preliminary experiments
Determine the ratio of the pairwise constraints adaptively.
Table 3 shows the results of our method for determining the ratio of the pairwise constraints adaptively. The ‘real link ratio’ and ‘estimated link ratio’ are computed using Equation (2) according to the real labels and the binning result of SolidBin-naive, respectively. The ‘real link ratio’ varies greatly among different datasets and the estimated link ratios are close to their corresponding real link ratio on most datasets. For the Sharon dataset, the cluster numbers are underestimated by SolidBin using the contigs unambiguously labeled on species level. Overall, the consistency between real and estimated link ratios shows the validity of our estimation method.
Table 3.
The adjacency relationship between contigs in different datasets
| Dataset | Estimated link ratio (%) | Real link ratio (%) |
|---|---|---|
| SpeciesMock | 1.54 | 1.55 |
| StrainMock | 9.30 | 9.67 |
| SimHC | 1.58 | 1.74 |
| Sharon | 34.65 | 19.19 |
| MetaHIT | 3.71 | 3.44 |
Note: For Sharon and MetaHIT datasets, only unambiguously labeled contigs are considered.
Accuracy of constraints.
The accuracy of constraints defined as the fraction of true relationships among all the constrained contig pairs on different datasets is shown in Table 4. In order to obtain adequate constraints with high accuracy on the SolidBin-SFS mode, we take the contig pairs with high similarity according to the estimated ratio as constraints. We test the accuracy of SolidBin-SFS mode constraints with the proportion ranging from 20 to 80%, with 20% as an incremental interval, and the proportion is set to 40% in our experiments. The results show that all the multi-sample datasets can obtain constraints with high accuracy on the SolidBin-SFS mode with the proportion 20 and 40%. Single-sample dataset ‘SimHC’ cannot obtain constraints with high accuracy on the SolidBin-SFS mode, which is reasonable given that the effect of coverage information is limited in the case of a single sample. In contrast, the SolidBin-SFS-CL mode can generate constraints with higher accuracy than the corresponding SolidBin-SFS mode in the case of a single sample, for instance, from 0.7238 to 0.9436 on the SimHC dataset. Moreover, the SolidBin-coalign mode can generate relatively accurate constraints except for the strain level dataset ‘StrainMock’, because the co-alignment information is reliable on the species or higher levels other than the strain level. For example, the accuracy of SolidBin-coalign constrants on the SpeciesMock is 0.9130, while the one on the StrainMock dataset is 0.5160.
Table 4.
The accuracy of the constraints generated by different SolidBin modes
| Mode | SpeciesMock |
StrainMock |
SimHC |
Sharon_partial |
MetaHIT_partial |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Acc (%) | Num | Acc (%) | Num | Acc (%) | Num | Acc (%) | Num | Acc (%) | Num | ||
| SolidBin-SFS | ratio×0.2 | 100.00 | 4 410 072 | 100.00 | 1 658 091 | 72.38 | 1 824 430 | 99.24 | 363 286 | 99.80 | 2 190 826 |
| — | ratio×0.4 | 100.00 | 8 782 498 | 99.98 | 3 306 241 | 55.59 | 2 390 242 | 98.24 | 550 808 | 99.61 | 4 356 334 |
| — | ratio×0.6 | 100.00 | 13 154 614 | 99.89 | 4 950 081 | 45.13 | 2 633 940 | 93.86 | 769 282 | 98.83 | 6 474 940 |
| — | ratio×0.8 | 99.98 | 17 523 650 | 99.44 | 6 567 453 | 38.06 | 2 767 326 | 86.67 | 986 612 | 83.17 | 7 260 682 |
| SoliBin-coalign | — | 91.30 | 20 293 038 | 51.60 | 8 179 998 | 99.71 | 3 065 364 | 99.19 | 1 199 672 | 99.90 | 9 171 166 |
| SolidBin-CL | — | 99.99 | 1 370 250 256 | 99.62 | 67 069 672 | 100.00 | 188 819 244 | 99.96 | 3 955 520 | 99.95 | 237 793 612 |
| SolidBin-SFS-CL | ratio×0.2 | 100.00 | 4 369 002 | 100.00 | 1 646 968 | 94.36 | 1 810 484 | 99.24 | 360 192 | 99.81 | 2 172 750 |
| — | ratio×0.4 | 100.00 | 8 727 240 | 99.98 | 3 249 328 | 89.83 | 2 376 178 | 98.60 | 547 706 | 99.71 | 4 336 152 |
| — | ratio×0.6 | 100.00 | 13 071 540 | 99.89 | 4 834 258 | 87.06 | 2 619 628 | 97.43 | 766 180 | 99.14 | 6 448 034 |
| — | ratio×0.8 | 99.98 | 17 416 512 | 99.44 | 6 382 828 | 85.28 | 2 752 684 | 95.74 | 983 508 | 92.36 | 7 228 632 |
4.4.2. The binning results on the simulated datasets
For the ‘StrainMock’ dataset, COCACOLA, SolidBin and BMC3C have better performance compared with other methods as shown in Table 5, and SolidBin-SFS performs best in general. Just considering the binning results of the different SolidBin modes, SolidBin-SFS and SolidBin-CL can generate constraints with high accuracy on this dataset as shown in Table 4, and have better performance in terms of all metrics compared with SolidBin-naive. On the other hand, since the co-alignment information is not reliable on the strain level, both the COCACOLA-coalign and SolidBin-coalign have worse performance than COCACOLA and SolidBin-SFS on this strain level dataset.
To figure out whether the coalignment information is useful to the ‘StrainMock’ dataset, we evaluated the results of the StrainMock at the species level. As shown in Table 6, both the COCACOLA and SolidBin have better performance with coalignment information at the species level. SolidBin-coalign has larger improvement than COCACOLA-coalign. For example, the ARI of COCACOLA-coalign increases from 0.8667 (COCACOLA) to 0.8801, and the ARI of SolidBin-coalign increases from 0.8689 (SolidBin-naive) to 0.9883. Therefore, SolidBin-colaign can better utilize coalignment information than COCACOLA-coalign, which highlights the effectiveness of spectral clustering in incorporating pairwise constraints.
Table 6.
The performance of COCACOLA, CONCOCT, MaxBin, MetaBAT, BMC3C and SolidBin on the ‘StrainMock’ dataset (evaluated on species level)
| StrainMock | Recall (%) | Precision (%) | F-score (%) | NMI (%) | ARI (%) |
|---|---|---|---|---|---|
| COCACOLA | 91.81 | 99.96 | 95.71 | 95.90 | 86.67 |
| COCACOLA-coalign | 91.27 | 99.94 | 95.41 | 96.89 | 88.01 |
| MaxBin | 91.01 | 91.01 | 91.01 | 91.87 | 78.50 |
| CONCOCT | 91.11 | 99.63 | 95.18 | 96.82 | 89.50 |
| MetaBAT | 81.24 | 67.32 | 73.63 | 72.46 | 34.10 |
| BMC3C(9401/9417) | 91.58 | 99.97 | 95.59 | 95.94 | 86.51 |
| SolidBin-naive | 91.94 | 99.99 | 95.80 | 96.06 | 86.89 |
| SolidBin-SFS(α*=20) | 92.00 | 99.99 | 95.83 | 96.06 | 86.92 |
| SolidBin-coalign(α*=10) | 99.46 | 99.97 | 99.71 | 99.39 | 98.83 |
| SolidBin-CL(β*=0.1) | 92.01 | 99.98 | 95.83 | 96.05 | 86.92 |
| SolidBin-SFS-CL(α*=30) | 92.00 | 99.99 | 95.83 | 96.06 | 86.92 |
Note: The optimal values of the results are in bold.
For the only single-sample dataset ‘SimHC’, CONCOCT and SolidBin have better performance compared with other methods as shown in Table 7, and SolidBin performs better in two modes. The F-score, NMI and ARI of SolidBin-coalign reach 0.9473, 0.9732 and 0.9319, respectively. In comparison, CONCOCT achieves 0.8925 in terms of F-score, 0.9371 in terms of NMI, and 0.8217 in terms of ARI, respectively. Taking different SolidBin modes into consideration, the SolidBin-SFS mode cannot obtain constraints with high accuracy and have better binning results than SolidBin-naive on the single dataset due to insufficient sample number. In contrast, sequence alignment information is quite useful in improving the binning results of the dataset. Both SolidBin-coalign and SolidBin-CL have better performance in terms of all metrics compared with other binners and SolidBin modes. Both COCACOLA-coalign and SolidBin-coalign have better performance than COCACOLA and SolidBin-naive on this dataset, but the improvement of SolidBin-coalign is more significant, from 0.8416 to 0.9473 in terms of F-score.
Table 7.
The performance of COCACOLA, CONCOCT, MaxBin, MetaBAT, BMC3C and SolidBin on the ‘SimHC’ dataset
| SimHC | Recall (%) | Precision (%) | F-score (%) | NMI (%) | ARI (%) |
|---|---|---|---|---|---|
| COCACOLA | 92.51 | 77.09 | 84.09 | 90.17 | 76.15 |
| COCACOLA-coalign | 94.41 | 80.43 | 86.86 | 91.66 | 80.73 |
| MaxBin | 85.84 | 83.28 | 84.54 | 89.34 | 76.90 |
| CONCOCT | 96.92 | 82.70 | 89.25 | 93.71 | 82.17 |
| MetaBAT | 90.38 | 72.18 | 80.26 | 86.42 | 57.07 |
| BMC3C(13 918/13 918) | 69.18 | 93.03 | 79.35 | 90.23 | 68.58 |
| SolidBin-naive | 90.20 | 78.88 | 84.16 | 89.95 | 75.78 |
| SolidBin-SFS(α*=0) | 90.20 | 78.88 | 84.16 | 89.95 | 75.78 |
| SolidBin-coalign(α*=10) | 98.85 | 90.94 | 94.73 | 97.32 | 93.19 |
| SolidBin-CL(β*=0.2) | 97.15 | 87.22 | 91.92 | 96.30 | 87.22 |
| SolidBin-SFS-CL(α*=0) | 90.20 | 78.88 | 84.16 | 89.95 | 75.78 |
Note: The optimal values of the results are in bold.
4.4.3. The binning results of the real datasets
For the ‘MetaHIT’ dataset, the results of the 17 136 contigs with unambiguous labels are shown in Table 8. SolidBin have better performance than other tools in general. Both SolidBin-SFS and SolidBin-SFS-CL have comparable performance with COCACOLA and CONCOCT, and achieve much better performance than Maxbin and MetaBAT, but worse performance than BMC3C. Considering the performance of different SolidBin modes, all the other modes have better performance than the SolidBin-naive mode, especially the modes that use the sequence alignment information. Both COCACOLA-coalign and SolidBin-coalign have better performance than COCACOLA and SolidBin-naive on this dataset, but the improvement of SolidBin-coalign is more significant, from 0.7730 to 0.9349 in terms of ARI and from 0.8649 to 0.9710 in terms of F-score.
Table 8.
The performance of COCACOLA, CONCOCT, MaxBin, MetaBAT, BMC3C and SolidBin on the ‘MetaHIT’ (partial) dataset
| MetaHIT_partial | Recall (%) | Precision (%) | F-score (%) | NMI (%) | ARI (%) |
|---|---|---|---|---|---|
| COCACOLA | 91.87 | 83.63 | 87.56 | 84.26 | 76.38 |
| COCACOLA-coalign | 91.92 | 85.39 | 88.54 | 86.04 | 80.91 |
| MaxBin | 84.75 | 73.58 | 78.77 | 77.84 | 69.02 |
| CONCOCT | 80.41 | 90.26 | 85.05 | 85.29 | 74.33 |
| MetaBAT | 74.34 | 52.59 | 61.60 | 61.37 | 30.19 |
| BMC3C(17 122/17 136) | 87.50 | 91.45 | 89.43 | 87.90 | 82.82 |
| SolidBin-naive | 85.85 | 87.15 | 86.49 | 85.76 | 77.30 |
| SolidBin-SFS(α*=10) | 86.24 | 87.22 | 86.73 | 85.91 | 77.95 |
| SolidBin-coalign(α*=40) | 97.01 | 97.18 | 97.10 | 96.01 | 93.49 |
| SolidBin-CL(β*=0.2) | 90.47 | 91.17 | 90.82 | 89.98 | 85.34 |
| SolidBin-SFS-CL(α*=0) | 85.85 | 87.15 | 86.49 | 85.76 | 77.30 |
Note: The optimal values of the results are in bold.
For the ‘Sharon’ dataset, we binned all the contigs and the results of the 2 614 contigs with unambiguous labels are shown in Table 9 and the estimated input bin number is 7 for SolidBin. All the modes of SolidBin and BMC3C have better performance than other tools. The F-score, NMI and ARI of the best performed SolidBin-coalign reach 0.9937, 0.9771 and 0.9846, respectively. In comparison, the F-score, NMI and ARI of CONCOCT, best one among other binners on the dataset, is 0.9621, 0.9353 and 0.9453, respectively. Taking different SolidBin modes into consideration, all the other modes have better performance than the SolidBin-naive mode, which shows that both the additional biological information and the internal information are useful for the binning. On the other hand, COCACOLA-coalign achieved similar performance with COCACOLA, which indicates the inefficiency of COCACOLA in incorporating coalignment information.
Table 9.
The performance of COCACOLA, CONCOCT, MaxBin, MetaBAT, BMC3C and SolidBin on the ‘Sharon’ dataset
| Sharon | Recall (%) | Precision (%) | F-score (%) | NMI (%) | ARI (%) |
|---|---|---|---|---|---|
| COCACOLA | 91.21 | 97.98 | 94.47 | 91.58 | 93.21 |
| COCACOLA-coalign | 92.65 | 97.49 | 95.00 | 91.43 | 92.02 |
| MaxBin | 96.02 | 90.57 | 93.21 | 89.18 | 87.74 |
| CONCOCT | 93.66 | 98.90 | 96.21 | 93.53 | 94.53 |
| MetaBAT | 90.91 | 68.74 | 78.28 | 71.49 | 58.97 |
| BMC3C(2591/2614) | 99.27 | 99.02 | 99.14 | 97.16 | 97.98 |
| SolidBin-naive | 98.11 | 97.58 | 97.84 | 93.90 | 94.38 |
| SolidBin-SFS(α*=1) | 99.33 | 98.76 | 99.04 | 96.61 | 97.66 |
| SolidBin-coalign(α*=10) | 99.64 | 99.09 | 99.37 | 97.71 | 98.46 |
| SolidBin-CL(β*=0.1) | 99.44 | 98.88 | 99.16 | 97.03 | 97.96 |
| SolidBin-SFS-CL(α*=1) | 99.53 | 98.97 | 99.25 | 97.35 | 98.16 |
Note: The optimal values of the results are in bold.
4.4.4. Evaluation of the impact of incorporating constraint information on sub-samples of the ‘SpeciesMock’ dataset
In order to evaluate the impact of co-alignment information and the ML constraints generated by SolidBin-SFS mode when the number of samples is small, we investigate the performance of SolidBin-SFS and SolidBin-coalign on sub-samples without overlapping of the ‘SpeciesMock’ dataset. The sample size ranges from 10 to 90, with 10 as increment. As shown in Figure 2, the results of SolidBin-SFS vary greatly with the number of samples and SolidBin-SFS can obtain better performance when the number of samples is large. The results of ARI and NMI are in the Supplementary Material. The results of SolidBin-coalign are relatively stable and can obtain good performance even when the number of samples is relatively low.
Fig. 2.
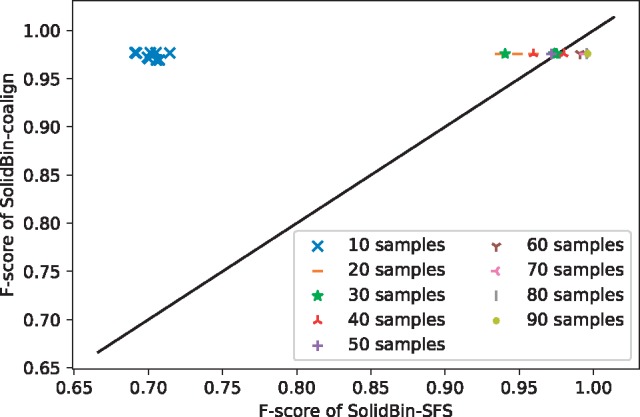
Evaluation of the result of SolidBin-coalign and SolidBin-SFS on sub-samples of the ‘SpeciesMock’ dataset
4.5 Running time and memory usage of the binners
All the experiments were done on a machine with 4-way 6-core 1.87 GHz Intel Xeon CPUs and 1 T memory. We ran all the binners with multiple threads. The running time and memory usage of the binners on different datasets are as shown in Table 10. On the one hand, SolidBin has excellent performance on the binning, but on the other hand, it takes a lot of time and memory to compute the similarity matrix for singular value decomposition, especially for the datasets with large contig numbers. However, unlike other binners, the number of the samples almost has no impact on the running time and memory of SolidBin.
Table 10.
The running time and memory usage of the binners on the different datasets
| Binners | StrainMock |
SimHC |
Sharon |
MetaHIT |
||||
|---|---|---|---|---|---|---|---|---|
| Time | Time | Memory (MiB) | Time | Memory (MiB) | Time | Memory (MiB) | Time | Memory (MiB) |
| COCACOLA | 4 min 50 s | 220 | 45 min 30 s | 403 | 2 min 01 s | 148 | 11 min 33 s | 473 |
| COCACOLA-coalign | 6 min 54 s | 2881 | 54 min 45 s | 4144 | 3 min 33 s | 1777 | 16 min 16 s | 2024 |
| MaxBin | 22 min 08 s | 49 | 63 min 10 s | 160 | 4 min 31 s | 27 | 600 min 43 s | 47 |
| CONCOCT | 2 min 07 s | 466 | 28 min 30 s | 1151 | 2 min 14 s | 359 | 88 min 05 s | 1098 |
| MetaBAT | 39 s | 112 | 6 min 16 s | 412 | 13 s | 69 | 1 min 51 s | 128 |
| BMC3C | 9 min 58 s | 1327 | 104 min 10 s | 2681 | 1 min 47 s | 477 | 25 min 23 s | 1897 |
| SolidBin-naive | 7 min 10 s | 4258 | 71 min 37 s | 35 932 | 2 min 32 s | 1549 | 26 min 31 s | 11 425 |
| SolidBin-SFS | 22 min 45 s | 5176 | 379 min 26 s | 40 631 | 6 min 51 s | 2022 | 95 min 37 s | 13 944 |
| SolidBin-coalign | 21 min 18 s | 4955 | 302 min 43 s | 40 441 | 7 min 43 s | 2639 | 73 min 43 s | 14 033 |
| SolidBin-CL | 28 min 20 s | 16 640 | 385 min 20 s | 196 770 | 6 min 43 s | 3056 | 99 min 28 s | 47 369 |
| SolidBin-SFS-CL | 37 min 48 s | 16 342 | 566 min 48 s | 197 001 | 9 min 35 s | 3155 | 143 min 02 s | 47 519 |
4.6 Analysis of the results
In summary, SolidBin with different modes have better performance than other binners in most cases. For the different SolidBin modes, the performance mainly depends on the accuracy of the constraints of different modes, which will be affected by the properties of the datasets, such as the number of samples. SolidBin-SFS is a reliable mode without any taxonomy information when the number of samples is large, and our method can determine the ratio of the pairwise constraints adaptively according to the data distribution. Sequence alignment information can improve the binning results markedly such as the SolidBin-coalign mode and the SolidBin-CL mode, especially when there are insufficient samples and the genomes contained in the dataset are at the species level, not at the strain level. The performance profiles of different SolidBin modes are shown in Table 1. Both COCACOLA and SolidBin can utilize co-alignment information, however, the improvements of SolidBin-coalign are always more significant, which shows that spectral clustering algorithm can integrate additional information better.
5 Discussion and conclusion
In this paper, we developed a binning method, SolidBin, based on semi-supervised spectral clustering. To our knowledge, this is the first method to apply semi-supervised spectral clustering for contig binning. It uses two types of prior information: ML constraints and CL constraints. We examined two ways to generate ML constraints, one is taxonomy independent, named as SolidBin-SFS and the other is based on the co-alignment information, which can take advantage of some contigs that belong to the known reference genomes, named as SolidBin-coalign. In this paper, our method is compared with five advanced binning tools, CONCOCT, COCACOLA, MaxBin, MetaBAT and BMC3C, using five different types of datasets. As shown in the Section 4.4, our method, compared with the-state-of-the-art binning methods, has best performance in terms of F-score, ARI and NMI in most cases.
However, our method has its own limitations. For example, SolidBin-SFS does not perform best when the number of samples is small. But SolidBin-coalign may perform well under these circumstances if the genomes contained in the dataset are at the species level. However, how to choose α, β and bin number K is still challenging. In addition, how to extend our method to handle large-scale datasets is still worthy further investigation.
In future research, we plan to incorporate more prior information into our method, such as linkage information used in Lu et al. (2017a), gene prediction information (Sunagawa et al., 2015) and DNA methylation (Beaulaurier et al., 2018).
Funding
SZ is supported by National Natural Science Foundation of China (No. 61572139 and No. 61872094) and Shanghai Municipal Science and Technology Major Project (No. 2017SHZDZX01). ZW and ZW are supported by the 111 Project (NO. B18015), the key project of Shanghai Science & Technology (No. 16JC1420402), Shanghai Municipal Science and Technology Major Project (No. 2018SHZDZX01) and ZJLab. FS and YL are supported by US NIH grants R01GM120624 and 1R01GM131407.
Conflict of Interest: none declared.
Supplementary Material
References
- Alneberg J. et al. (2014) Binning metagenomic contigs by coverage and composition. Nat. Methods, 11, 1144–1146. [DOI] [PubMed] [Google Scholar]
- Bahram M. et al. (2018) Structure and function of the global topsoil microbiome. Nature, 560, 233–237. [DOI] [PubMed] [Google Scholar]
- Beaulaurier J. et al. (2018) Metagenomic binning and association of plasmids with bacterial host genomes using DNA methylation. Nat. Biotechnol., 36, 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caliński T., Harabasz J. (1974) A dendrite method for cluster analysis. Commun. Stat. Theory Methods, 3, 1–27. [Google Scholar]
- Chen K. et al. (2017) Towards in silico prediction of the immune-checkpoint blockade response. Trends Pharmacol. Sci., 38, 1041–1051. [DOI] [PubMed] [Google Scholar]
- Dick G.J. et al. (2009) Community-wide analysis of microbial genome sequence signatures. Genome Biol., 10, R85.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J. et al. (2013) Efficient semisupervised MEDLINE document clustering With MeSH-semantic and global-content constraints. IEEE Trans. Cybern., 43, 1265–1276. [DOI] [PubMed] [Google Scholar]
- Huson D. et al. (2007) Megan analysis of metagenomic data. Genome Res., 17, 377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenhower C. et al. (2012) Structure, function and diversity of the healthy human microbiome. Nature, 486, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X. et al. (2006) Document clustering with prior knowledge. In: Proceedings of the 29th Annual International ACM SIGIR Conference on Research and Development in Information Retrieval pp. 405–412. ACM.
- Jostins L. et al. (2012) Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature, 491, 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang D.D. et al. (2015) MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ, 3, e1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khor B. et al. (2011) Genetics and pathogenesis of inflammatory bowel disease. Nature, 474, 307.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laczny C.C. et al. (2015) VizBin - an application for reference-independent visualization and human-augmented binning of metagenomic data. Microbiome, 3, 1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.Y. et al. (2017a) COCACOLA: binning metagenomic contigs using sequence COmposition, read CoverAge, CO-alignment and paired-end read LinkAge. Bioinformatics, 33, 791–798. [DOI] [PubMed] [Google Scholar]
- Lu Y.Y. et al. (2017b) Towards enhanced and interpretable clustering/classification in integrative genomics. Nucleic Acids Res., 45, e169.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng A.Y. et al. (2002) On spectral clustering: analysis and an algorithm. In: Advances in Neural Information Processing Systems. Vol. 14, pp. 849–856.
- Qin J. et al. (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature, 464, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseeuw P.J. (1987) Silhouettes: a graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math., 20, 53–65. [Google Scholar]
- Sangwan N. et al. (2016) Recovering complete and draft population genomes from metagenome datasets. Microbiome, 4, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlar K. et al. (2017) Bioinformatics strategies for taxonomy independent binning and visualization of sequences in shotgun metagenomics. Comput. Struct. Biotechnol. J., 15, 48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon I. et al. (2013) Time series community genomics analysis reveals rapid shifts in bacterial species, strains, and phage during infant gut colonization. Genome Res., 23, 111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J., Malik J. (2000) Normalized cuts and image segmentation. IEEE Trans. Pattern Anal. Mach. Intell., 22, 888–905. [Google Scholar]
- Sieber C.M. et al. (2018) Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol., 3, 836–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W.Z., Thomas T. (2017) Binning_refiner: improving genome bins through the combination of different binning programs. Bioinformatics, 33, 1873–1875. [DOI] [PubMed] [Google Scholar]
- Sunagawa S. et al. (2015) Ocean plankton. Structure and function of the global ocean microbiome. Science, 348, 1261359.. [DOI] [PubMed] [Google Scholar]
- Uritskiy G.V. et al. (2018) MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome, 6, 158.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagstaff K. et al. (2001) Constrained k-means clustering with background knowledge. In: Eighteenth International Conference on Machine Learning (ICML) Vol. 1, pp. 577–584.
- Wang Y. et al. (2017) Improving contig binning of metagenomic data using [Formula: see text] oligonucleotide frequency dissimilarity. BMC Bioinformatics, 18, 425.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilck N. et al. (2017) Salt-responsive gut commensal modulates TH17 axis and disease. Nature, 551, 585–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.W. et al. (2014) MaxBin: an automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome, 2, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.W. et al. (2016) MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics, 32, 605–607. [DOI] [PubMed] [Google Scholar]
- Wu Y.W., Ye Y. (2011) A novel abundance-based algorithm for binning metagenomic sequences using l-tuples. J. Comput. Biol., 18, 523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G. et al. (2018) BMC3C: binning Metagenomic Contigs using Codon usage, sequence Composition and read Coverage. Bioinformatics, 34, 4172–4179. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.