


Abstract
The association of reverse transcriptases (RTs) with CRISPR–Cas system has recently attracted interest because the RT activity appears to facilitate the RT-dependent acquisition of spacers from RNA molecules. However, our understanding of this spacer acquisition process remains limited. We characterized the in vivo acquisition of spacers mediated by an RT-Cas1 fusion protein linked to a type III-D system from Vibrio vulnificus strain YJ016, and showed that the adaptation module, consisting of the RT-Cas1 fusion, two different Cas2 proteins (A and B) and one of the two CRISPR arrays, was completely functional in a heterologous host. We found that mutations of the active site of the RT domain significantly decreased the acquisition of new spacers and showed that this RT-Cas1-associated adaptation module was able to incorporate spacers from RNA molecules into the CRISPR array. We demonstrated that the two Cas2 proteins of the adaptation module were required for spacer acquisition. Furthermore, we found that several sequence-specific features were required for the acquisition and integration of spacers derived from any region of the genome, with no bias along the 5′and 3′ends of coding sequences. This study provides new insight into the RT-Cas1 fusion protein-mediated acquisition of spacers from RNA molecules.
INTRODUCTION
CRISPR–Cas (clustered regularly interspaced short palindromic repeats and CRISPR-associated proteins) systems are adaptive defense systems that provide acquired immunity against foreign nucleic acids (1). They are present in almost all archaea and about 50% of all bacterial genomes (2,3). CRISPR–Cas systems provide immunity through three steps: adaptation, expression and interference (4,5). During the adaptation stage, an integrase complex incorporates short DNA or RNA sequences, known as spacers, into the CRISPR array (6–8). The CRISPR array consists of repeated sequences (direct repeats) separated by variable sequences corresponding to the nucleic acid of the invading agent (spacers). This array is transcribed to generate a precursor transcript (precursor crRNA) that is processed into short (mature crRNA) structured RNAs during the expression stage, leading to the formation of crRNA-Cas effector complexes that recognize and bind complementary nucleic acids, resulting in degradation of the target molecule, during the interference step (9–11). These immunogenic systems are highly diverse and are currently classified into two broad functional classes, six types and 33 subtypes (12).
The integration complex generally consists of Cas1 and Cas2 proteins and is largely conserved in all CRISPR–Cas systems (12). Cas1 seems to originate from a family of transposons called casposons, and it interacts with other proteins involved in the adaption stage (12–14). One of these ancillary proteins is the reverse transcriptase (RT) responsible for converting RNA into cDNA (15,16). There are several groups of prokaryotic RTs. Those closely related to group II intron-encoded RTs predominate among the RTs associated with CRISPR–Cas systems, either separately or frequently via the generation of a natural fusion, at the C-terminus, with Cas1. These RTs are usually linked to type III CRISPR–Cas systems (17,18), which have multisubunit effector complexes capable of targeting single-stranded RNA and/or DNA, which must be transcriptionally active (19–22).
The association of RTs with CRISPR–Cas systems has recently attracted considerable attention because, in a type III-B system present in the marine bacterium Marinomonas mediterranea (MMB-1), the associated Cas6-RT-Cas1 protein fusion has been shown to facilitate the RT-dependent acquisition of RNA spacers in vivo through a mechanism displaying several similarities to group II intron retrohoming (18). An RT-Cas1 fusion associated with a type III-D system from Fusicatenibacter saccharivorans was recently shown to acquire RNA spacers efficiently in a heterologous host (Escherichia coli) and has been used as a transcriptional recorder, describing both continuous and transient complex cellular behaviors (23). The RTs associated with CRISPR–Cas systems evolving from group II intron RTs, which co-evolve with their associated Cas1 protein, form 13 distinct clades (24,25). The two RT-associated CRISPR-cas loci studied to date, from M. mediterranea and F. sacharivorans, belong to clades 8 and 12, respectively. Thus, our knowledge of the function and mechanisms of the CRISPR–Cas adaptation modules encoding these RTs remains limited and further studies are required. Exploration of the diversity of RTs associated with different CRISPR–Cas systems could also reveal novel properties and potential biotechnological applications.
We characterized the in vivo acquisition of spacers mediated by a RT-Cas1 fusion protein from clade 6 linked to a type III-D system from Vibrio vulnificus strain YJ016. Acquisition efficiency was high in only one of the two arrays present at this locus, and was inversely correlated with the level of transcription of the CRISPR array. This RT-Cas1-associated adaptation module was able to incorporate spacers from RNA molecules into the CRISPR array. Furthermore, we found that spacers could be acquired from any region of the genome, and from within coding sequences, with no bias along the length of the gene. The nucleotide sequences of the spacers acquired collectively displayed particular features potentially related to recognition of the RT-linked CRISPR adaptation machinery. Our results indicate that the V. vulnificus type III-D system is a good model for studying the mechanism of spacer acquisition from RNA molecules with potential for use as a biotechnological tool.
MATERIALS AND METHODS
Strain and culture conditions
The E. coli strains used in this study were DH5α (Bethesda Research Lab) for cloning purposes, Rosetta 2 (DE3) (Novagen) for protein production and HMS 174 (DE3) for spacer acquisition assays. Bacteria were grown in LB medium (10 g/l tryptone, 5 g/l yeast extract, 5 g/l NaCl). Antibiotics were added as required (ampicillin, and tetracycline).
Construction of expression vectors
Plasmids for protein production and purification were based on the pMal-Flag backbone (Supplementary Table S1) for RT or RT-Cas1. Plasmids for inducible overexpression of the adaptation operon of V. vulnificus YJ016 (RT-Cas1–Cas2A–Cas2B) were built with the pGEM-T Easy backbone (Promega). The two CRISPR arrays associated with the two systems, complete or with only the first DR and the first spacer, were inserted into the pMP220 backbone (26). RT-Cas1 mutants with mutations of Cas1 (E517A and E597A) or RT (YADD to YAAA at amino acid positions 220–223) and deletion mutants of Cas2A and Cas2B were generated, using the pMal-Flag and pGEMT-Easy backbones for in vitro and in vivo analyses, respectively. The plasmids and oligonucleotides used in this study are listed in Supplementary Tables S1–S3. All plasmids were verified by sequencing. Plasmids are available upon request.
Protein purification
We used pMal-Flag derivatives with the different RTs to transform E. coli strain Rosetta2 (DE3), and single transformed colonies were then grown overnight in LB medium supplemented with ampicillin, chloramphenicol and 0.2% glucose, at 37°C, with shaking. A flask containing 50 ml LB was inoculated with 1% of the overnight culture, and the bacteria were grown to exponential growth phase at 37°C, with shaking. When the culture reached an optical density of ∼0.6, IPTG was added to a final concentration of 0.3 mM and the cultures were incubated overnight at 20°C. Cells were harvested by centrifugation, and the pellet was resuspended in column buffer (CB: 20 mM Tris–HCl (pH 7.5), 200 mM NaCl, 1 mM EDTA, 1 mM DTT and 1x EDTA-free protease inhibitor (Roche)) at 4°C. Cells were lysed by three freeze-thaw cycles and subjected to sonication (Sonifier® Cell Disrupters, Branson Ultrasonics). The lysate was cleared by centrifugation (16,000 x g, 15 min, 4°C).
Proteins were purified with a liquid chromatography system (BioRad). Different RT or RT-Cas1 fusion proteins with maltose binding protein (MBP) were purified by loading the filtered crude protein onto an amylose column (30 ml; NEB Amylose High Flow Resin), incubating for 2 h at 4°C and then washing the column five times with 2 ml CB. Bound proteins were eluted in CB supplemented with 10 mM maltose. The proteins were concentrated with an Amicon ultracentrifugation filter (Ultracel 50-K), and dialyzed against storage buffer (SB: 10 mM Tris–HCl (pH 7.5), 1 mM DTT, 50% glycerol). The various proteins were stable in SB for several months at −20°C.
All protein concentrations were determined by the Bradford method (BioRad) (27), according to the kit manufacturer's protocol.
RT assay
RT activities were assessed with poly(rA)/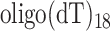 , prepared by boiling in RT buffer (10 mM KCl, 25 mM
, prepared by boiling in RT buffer (10 mM KCl, 25 mM 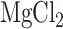 , 50 mM Tris–HCl (pH 7.5), 5 mM DTT) for 2 min and then placed on ice. The substrate was incubated with 1mM unlabeled deoxythimidine triphosphate (dTTP) and 5 μCi [α-32P]dTTP (800 Ci/mmol; GE healthcare) at 37°C, and the reaction was initiated by adding the RT or RT-Cas1 protein (final concentration 0.1–0.5 μM) in a final volume of 10 μl and incubating for 10 min. The reaction was stopped by spotting 8 μl of the reaction mixture onto Whatman DE81 paper. The paper was dried and washed in 250 ml of 2× SSC to eliminate unincorporated labeled dTTP. Radioactivity was quantified with a scintillation counter (Liquid Scintillation Analyzer Tri-Carb 1500, ΔPackard).
, 50 mM Tris–HCl (pH 7.5), 5 mM DTT) for 2 min and then placed on ice. The substrate was incubated with 1mM unlabeled deoxythimidine triphosphate (dTTP) and 5 μCi [α-32P]dTTP (800 Ci/mmol; GE healthcare) at 37°C, and the reaction was initiated by adding the RT or RT-Cas1 protein (final concentration 0.1–0.5 μM) in a final volume of 10 μl and incubating for 10 min. The reaction was stopped by spotting 8 μl of the reaction mixture onto Whatman DE81 paper. The paper was dried and washed in 250 ml of 2× SSC to eliminate unincorporated labeled dTTP. Radioactivity was quantified with a scintillation counter (Liquid Scintillation Analyzer Tri-Carb 1500, ΔPackard).
β-Galactosidase assay
β-Galactosidase assays were performed as described by Miller (28). Briefly, E. coli DH5α cells were transformed with the various pCA constructs. Individual transformed colonies were picked and cultured overnight at 37°C. Cultures were diluted 1:50 in fresh LB medium and incubated until the exponential growth phase was reached (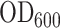 ∼0.6). They were then cooled on ice for 20 min and bacterial density was recorded by measuring
∼0.6). They were then cooled on ice for 20 min and bacterial density was recorded by measuring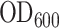 . We then mixed 100 μl of culture with 900 μl Z buffer (60 mM
. We then mixed 100 μl of culture with 900 μl Z buffer (60 mM 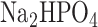 and 40 mM
and 40 mM 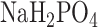 (pH 7.0), 10 mM KCl, 1 mM Mg
(pH 7.0), 10 mM KCl, 1 mM Mg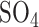 and 50 mM β-mercaptoethanol), 50 μl chloroform and 25 μl 0.1% SDS) and vortexed the mixture for 30 seconds. The samples were incubated at 28°C for 5 min and the reaction was initiated by adding 0.2 ml o-nitrophenyl-β-d-galactopyranoside (ONPG). The reaction mixture was incubated at 28°C for 10 min and the reaction was then stopped by adding 0.3 M
and 50 mM β-mercaptoethanol), 50 μl chloroform and 25 μl 0.1% SDS) and vortexed the mixture for 30 seconds. The samples were incubated at 28°C for 5 min and the reaction was initiated by adding 0.2 ml o-nitrophenyl-β-d-galactopyranoside (ONPG). The reaction mixture was incubated at 28°C for 10 min and the reaction was then stopped by adding 0.3 M 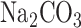 . Samples were centrifuged for 2 min to eliminate cell debris and absorbance was measured at 420 nm.
. Samples were centrifuged for 2 min to eliminate cell debris and absorbance was measured at 420 nm.
Spacer acquisition assay
Escherichia coli strain HMS174 (DE3) was cotransformed by electroporation with pAGDt plasmids harboring RT-Cas1–Cas2A–Cas2B and derivatives, and pCA plasmids harboring the CRISPR array with only the first DR and the first spacer (Supplementary Table S1). Individual colonies were cultured overnight at 37°C in LB medium supplemented with ampicillin and tetracycline. The culture was diluted 1:500 in LB medium, and split into triplicates, which were cultured with the same antibiotics and 0.1 mM IPTG for 14–18 h. The bacterial cells were harvested by centrifugation and plasmids containing CRISPR arrays were isolated by standard plasmid mini-prep procedures to serve as a template for PCR amplification and the preparation of NGS samples.
Amplification of CRISPR arrays and preparation of NGS samples
Leader proximal spacers were amplified by PCR from 3–4 ng of plasmid DNA per μl of PCR mix, with a forward primer binding to the leader sequence of the corresponding CRISPR array and a reverse primer binding to the first native spacer (Supplementary Tables S2 and S3). For each biological replicate, a 25 μl PCR mixture was subjected to the following cycling sequence: 94°C for 4 min; 30 cycles of 94°C for 30 s and 62°C for 30 s. The dominant amplicon contained the first native spacer from the unexpanded CRISPR array. Electrophoresis was performed in a 2% agarose gel for the excision of gel slices corresponding to a molecular weight of ∼300 bp (70 bp above the 233-bp band, consistent with the expected size of an amplicon from the expanded CRISR array). The slices were purified with the Illustra GFX PCR DNA and Gel Band Purification Kit (GE Healthcare) and eluted in 30 μl of buffer. We then used 2 μl of the eluted product for a second round of a semi-nested PCR in a 50 μl reaction mixture, with barcoded Illumina sequencing adaptors annealing to the leader region (closer to the first repeat) and to the first native spacer (see Supplementary Table S3), as follows: 94°C for 4 min; 35 cycles of 94°C for 30 s and 62°C for 30 s. Expanded CRISPR array amplicons were separated from unexpanded arrays by an additional round of purification by electrophoresis in a 2% agarose gel, and the final product was eluted in 10 μl of buffer. We increased the percentage of expanded amplicons by performing a third round of amplification by gel electrophoresis, for the tdI spacer acquisition assays. The resulting samples were quantified with Qubit (Life Technologies) and analyzed on a 2100 Bioanalyzer (Agilent Technologies). Libraries were sequenced on an Illumina Miseq at the Genome Sequencing Unit of the IPBLN-CSIC (Granada, Spain).
Data processing pipeline
FASTQ files were mate-paired with fastq-join (https://github.com/brwnj/fastq-join), with a minimum overlap of 40 nt. They were then converted to FASTA format with FASTX-Toolkit v0.0.14 (fastq-to-fasta) (htpp://hannolab.cshl.edu/fastx_toolkit) and trimmed with Cutadapt (29). In all samples, ∼90% of total read pairs were successfully merged, and ∼80% of the merged read pairs had the correct primer-encoded barcodes located exactly at the ends of the amplicon. Using a custom script written in Python v2.7, spacers were identified, grouped on the basis of unique start and end coordinates (unique spacers), and mapped on the plasmid and genome with Bowtie2.0, with two mismatches allowed. This approach preserves strand information.
Construction and validation of td intron constructs
The 393-bp intron sequence and its native exons (CTTGGGT│CT) were inserted in the SalI restriction site of the pAGDt-439 plasmid, just downstream from the adaptation operon, as a SalI/XhoI insertion. The intron was introduced at this site because we had previously noted that this was one of the regions of the plasmid into which new acquisition were preferentially inserted (Supplementary Figure S5A). In this context, the splicing product was easy to distinguish from non-spliced transcripts and DNA. In vivo splicing efficiency assessed by extracting total RNA and reverse transcribing 1.5 μg (SuperScriptII; Life Technologies) with random hexamers in a 20 μl reaction mixture. We then subjected 1 μl of cDNA to amplification in a 25 μl PCR mixture with the Accuprime Polymerase and the Cas2.2-439f and SP6 primers (Supplementary Table S2), which bind on either side of the splice site. The cycling conditions were as follows: 94°C for 4 min; 35 cycles of 94°C for 30 s, 62°C for 30 s, 72°C for 40 s; 72°C for 4 min. The PCR products were analyzed by electrophoresis in a 0.8% agarose gel, to check that splicing rates were close to 100%.
td intron spacer acquisition assay
For the detection of spacers originating from the exon junction (RNA) after tdI splicing, the spacer acquisition assay was optimized by searching for the maximum number of new different spacers after Illumina-Miseq sequencing. After cotransformation with pAGDt-439-tdI and pCA2s-1DR, two different sets corresponding with 80 and 200 individual colonies were selected for the standard spacer acquisition assay (Supplementary Table S6). Once the plasmid DNA had been extracted, individual PCRs were performed, as previously described. The first purification step was carried out by mixing the PCR products in groups of 10 different colonies, and then performing the second PCR step. The PCR mixtures were then combined and two band purification steps were performed to increase the proportion of expanded arrays. With this method, 50–70% of the reads after Illumina-Miseq sequencing corresponded to the expanded array, corresponding to >10,000 newly acquired spacers per assay performed with the tdI construct.
RESULTS
A unique type III-D CRISPR–Cas locus encoding an RT-Cas1 fusion from V. vulnificus
In our search for functional RT genes linked to CRISPR–Cas systems, we focused on a unique type III-D CRISPR–Cas locus from Vibrio vulnificus strain YJ016. The genomic neighborhood of the RT-Cas1 locus from V. vulnificus strain YJ016 encoding genes was retrieved, as previously described (24,25) and spans 21.6 kb on chromosome II (Figure 1A). This CRISPR–Cas system has an associated RT-Cas1 fusion protein (690 aa) from CRISPR-RT clade 6 (25) that displays exogenous RT activity in vitro (Supplementary Figure S1). A blast search within V. vulnificus showed this locus to be located downstream from a highly conserved operon encoding two peptide-methionine-S-oxide reductase genes (msrA and msrB) representing an island (30) within the genome probably acquired by lateral transfer (Figure 1A). This RT-Cas1 type III-D locus has a canonical gene-cassette encoding the type-III-D Csm effector complex including csx19 and cas6 at its 5′ end, transcribed in the opposite direction to the adaptation unit complex constituted by the RT-Cas1 fusion protein and two different Cas2 (A and B) proteins displaying ∼70% identity. This adaptation module is flanked by two CRISPR arrays: CRISPR01, with only two spacers, and CRISPR02, with nine spacers. These two arrays contain identical direct repeats (DRs; Figure 1B), and a set of ancillary genes is located downstream from the larger CRISPR array. We assessed the promoter activity of the presumed leader region of the two arrays, by constructing transcriptional fusions of CRISPR01 and CRISPR02 with a LacZ reporter gene in E. coli (Figure 1C). Constitutive promoter activity was detected only for the leader sequence of the CRISPR01 array, consistent with the transcriptional RNAseq data for this V. vulnificus strain showing that transcription rates for the CRISPR01 array are several times higher than those for the CRISPR02 array in various environmental conditions (31; Supplementary Figure S2).
Figure 1.

Characteristics of the VvYJ016 type III-D CRISPR operon. (A) Schematic diagram of the type III-D CRISPR–Cas loci in VvYJ016. The operon consists of a canonical five-gene cassette putatively encoding the type III-B Cmr effector complex (indicated by a beige background) followed by the gene encoding Cas6, which is involved in crRNA maturation. The opposite strand carries the adaptation module, which consists of an operon of three genes encoding RT-Cas1 and two Cas2 proteins, located between two CRISPR arrays containing three and nine spacers (CRISPR02). Four ancillary genes (two csx1 and two csx16) and two genes of unknown function (VVA1542 and VVA1551) complete this CRISPR-island. The black arrows indicate the array promoters identified. (B) Sequence alignment of the leader sequence and first direct repeat of the VvYJ016 CRISPR01 and CRISPR02 arrays. The nucleotide sequence of the first repeat is framed by a gray box. The putative promoter of the two sequences is framed by a blue box. The red letters represent the bases differing between the two sequences. (C) Determination of the level of transcription of the leader sequence of CRISPR01 and CRISPR02. β-Galactosidase activity was measured for the empty plasmid (pMP220) and the two complete arrays, in both orientations with respect to the lacZ gene (sense: pCA1s; pCA2s for CRISPR01 and CRISPR02, respectively; antisense: pCA1as; pCA2as for CRISPR01 and CRISPR02, respectively). Errors bars show the standard deviation for three biological replicates.
Spacer acquisition by the V. vulnificus RT-Cas1-associated CRISPR–Cas adaptation module
We investigated whether the adaptation operon of strain YJ016 could acquire new spacers in a heterologous host (E. coli), with expression vectors carrying the genes encoding the RT-Cas1 fusion and the Cas2A and Cas2B proteins and vectors carrying the CRISPR01 or CRISPR02 arrays containing the leader sequence, 1DR and 1 spacer (Figure 2A). After cotransformation with the two plasmids, we assessed spacer acquisition in E. coli by overexpressing the RT-Cas1, Cas2A and Cas2B operon. The acquisition of new spacers by the two arrays was evident after two rounds of PCR/purification of the expanded array band, followed by Illumina-Miseq sequencing. Spacers were acquired from the E. coli genome (∼95%) with the rest being derived from plasmids DNA (∼5%) (Supplementary Table S4). Using this assay, we demonstrated that the strain YJ016 CRISPR–Cas adaptation module could acquire new spacers in E. coli. Spacer acquisition by the CRISPR02 array occurred at a rate about 20 times higher than that for the CRISPR01 array (Figure 2B; Supplementary Table S5). Based on these findings, we used CRISPR02 for further spacer acquisition assays.
Figure 2.

Spacer acquisition by the VvYJ016 adaptation operon in the heterologous E. coli system. (A) Schematic diagram of the high-throughput spacer acquisition assay. Overexpression of the adaptation operon in E. coli HMS 174 (DE3) followed by the extraction of plasmid DNA, two rounds of PCR/purification of the expanded CRISPR array, and deep sequencing, analysis and characterization of the spacers identified. (B) Frequency of new spacer detection per million reads for the wildtype RT-Cas1, RT active site mutant (YAAA), Cas1 domain mutants E517A and E597A and the ΔCas2A, ΔCas2B and ΔCas2A-B mutants. The bars indicate the range for three biological replicates.
For evaluation of the role of the different domains of the RT-Cas1 fusion protein in spacer acquisition, we constructed two single mutants: one with a YAAA mutation of the catalytic RT (RT5) and another with an E597A mutation of the Cas1 active site. RT in vitro assays performed (see method section) revealed that the YAAA mutant lacked RT activity, whereas the E597A mutant had a level of RT activity similar to that of the wild-type protein (Supplementary Figure S3A). The mutants and the wild-type yield similar amount of protein fusion (Supplementary Figure S3B). The mutation affecting the Cas1 domain abolished spacer acquisition, whereas the point mutation in the RT active site decreased the acquisition of new spacers by ∼90% (Figure 2B; Supplementary Figure S3C). These findings reveal the importance of the functional RT catalytic site in the acquisition of spacers in vivo in the heterologous E. coli host. We investigated the requirement for Cas2A and/or Cas2B for the formation of an active acquisition complex in vivo, by construction deletion mutants of Cas2A, Cas2B or both. Each deletion completely abolished the acquisition of new spacers, clearly demonstrating that both Cas2 proteins were required for the adaptation stage (Figure 2B), and raising the possibility that the Cas2 dimer of the functional integration complex consists of two different Cas2 proteins. In fact, both proteins present the characteristic conserved structure consisting in 2 alpha helix and 4/5 beta strands in which a critical aspartic of the catalytic site at the end of the first beta strand is conserved in both subunits (Supplementary Figure S4).
Features of the acquired spacers
We investigated the features of the V. vulnificus RT-Cas1-associated acquisition system, by characterizing the pool of newly acquired spacers. The spacers matched sequences throughout the genome of the host bacteria (Figure 3A) with the rRNA genes as the most abundant in our spacer libraries. However, we detected a bias of acquired spacers with a specific antisense orientation of coding sequences, reflecting the complementary nature of the newly acquired spacers relative to the predicted messenger RNA (Figure 3A and B). These findings suggest a relationship between the orientation of the spacer and transcription, even in the absence of the effector module.
Figure 3.

Characterization of the spacers acquired by the VvYJ016 adaptation operon. (A) Coverage of spacers aligning with the E. coli HMS174 (DE3) genome and a representative locus. Identical alignments represent recurrent spacers acquired in independent biological samples (n = 11). (B) Strand bias in pools of newly acquired spacers relative to the source transcript. Proportion of newly acquired spacers with the Wild-type RT-Cas1 in the sense or antisense strand of coding genes or in intergenic regions of the E. coli genome (n = 11). (C) Histogram showing normalized counts of E. coli genome or pAGDt-Op439 plasmid spacers, by length. (D) GC content distribution of genome- and plasmid-aligned spacers. The dotted lines represent the GC content of the plasmid (light gray) and the genome (dark gray). For C and D, the bars indicate the range for the three assays in which the largest numbers of spacers were detected (>10,000 newly acquired spacers per experiment).
The spacers were mostly between 34 and 38 base pairs (bp) long, consistent with the length distribution of the natural spacers present in these arrays (Figure 3C). Nevertheless, spacers originating from plasmids were slightly longer (1 bp longer on average) than those of genomic origin. Consistent with their origin, the median ‘GC’ content of the spacers was correlated with the ‘GC’ content of the ‘template’ used, either the E. coli genome or the plasmid (Figure 3D). As in other type III CRISPR–Cas systems (32), no conserved protospacer-adjacent motif (PAM) was observed (Figure 4A), even if the analysis was based on dinucleotide frequency (33; data not shown).
Figure 4.
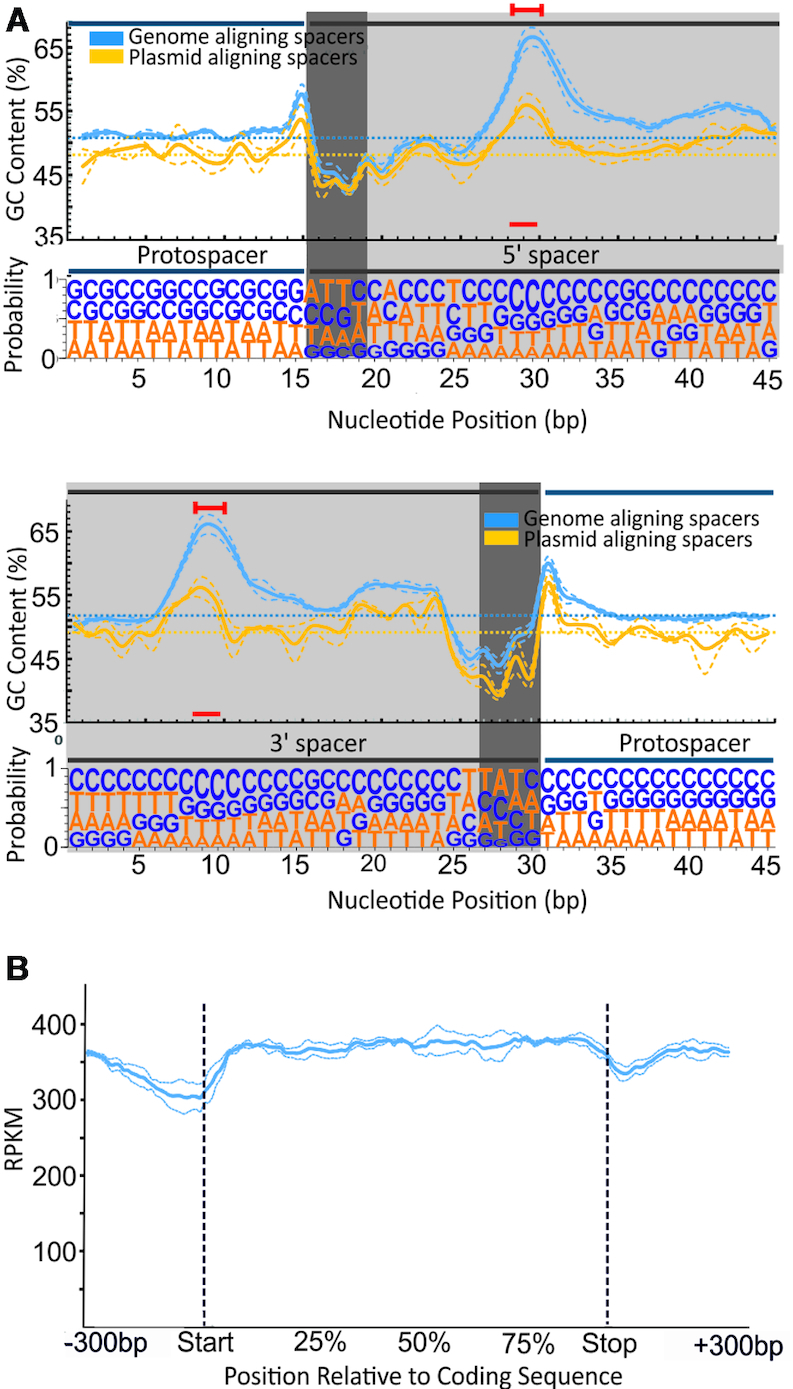
Spacer composition and position relative to coding sequences. (A) GC content (above) and nucleotide probabilities (below) at each position along the wild-type RT-Cas1–acquired protospacers. Given the variation of protospacer length, two panels are shown, with the spacer anchored 5′and 3′ at positions 15 and 35, respectively. Spacer (gray background) and flanking (white background) nucleotides are shown. The dark gray background indicates asymmetry at the two ends of the spacers, with a stretch of positions rich in ‘AT’ (see text). The particular bias towards ‘C’ within the spacer observed is indicated by a red line. The ‘GC’ content is shown for spacers aligning with the genome (blue) and plasmid (yellow). (B) Gene body coverage of spacer alignments along the length of transcripts. The relative position corresponds to the percentile of coding sequence length ± 300 bp of the adjacent genomic regions. Dotted lines in A and B represent the mean error of alignment for the three assays in which the largest numbers of spacers were detected (>10,000 newly acquired spacers per experiment).
Given the large numbers of different spacers obtained, a significant deviation of the expected ‘GC’ content was observed at different positions in the spacer unit. Thus, a symmetric bias emerged at both ends of the spacers, at which a stretch of four to five positions rich in ‘AT’ residues was observed. These ‘AT’-rich positions contrast clearly with the bias towards ‘GC’ enrichment observed at the first nucleotide of the protospacer flanking the spacer (Figure 4A). A strong bias towards ‘GC’ was observed at positions +14 and +15 of the spacer (present at ∼67% of the spacers acquired from the genome), and the high frequency of C at these positions was particularly marked (∼40% of the total). These specific biases were observed not only for spacers originating from the genome, but also for those originating from plasmids, suggesting a probable preference of the acquisition complex. An analysis of the position of the spacer relative to a coding sequence from the E. coli genome revealed no bias at the beginning or end of the gene (Figure 4B), by contrast observations for the spacer acquisition machinery of Fusicatenibacter saccharivorans (23). Taken together, these findings indicate that several sequence-specific requirements for acquisition and integration are characteristic of these spacers, regardless of their origin (plasmid or genome) or relative position in a gene.
The V. vulnificus RT-Cas1-associated CRISPR–Cas adaptation module acquires spacers directly from RNA
We investigated whether the spacers acquired via the RT-Cas1 system originated from RNA molecules, with an acquisition assay in which we looked for spacers harboring the ligated exon junction of the self-splicing td group I intron, a ribozyme that catalyzes its own excision from the original transcript and is not present in DNA (18,23), We designed a construct containing the td intron just downstream from the adaptation operon, expressed under the control of the T7 promoter (Figure 5A). The tdI cloning site corresponds to a region in which a large number of spacers were observed in the antisense orientation (bias source transcription; Supplementary Figure S5A), making it more likely that the spacers detected originated from RNA. We checked that the exon-junction was not present elsewhere in the genome. Thus, the detection of the exon junction as a DNA spacer demonstrates that the system can acquire spacers directly from RNA. We confirmed that efficient self-splicing occurred in vivo (almost 100%; Supplementary Figure S5B). We then performed assays for newly integrated spacers in plasmid copies of CRISPR02, and we recovered more than 100,000 new spacers mapping to plasmids or to the E. coli genome. We found three unique spacers spanning the splice junction (Figure 5B), confirming that the V. vulnificus RT-Cas1-associated CRISPR–Cas adaption module can acquire spacers from RNA molecules in E. coli.
Figure 5.
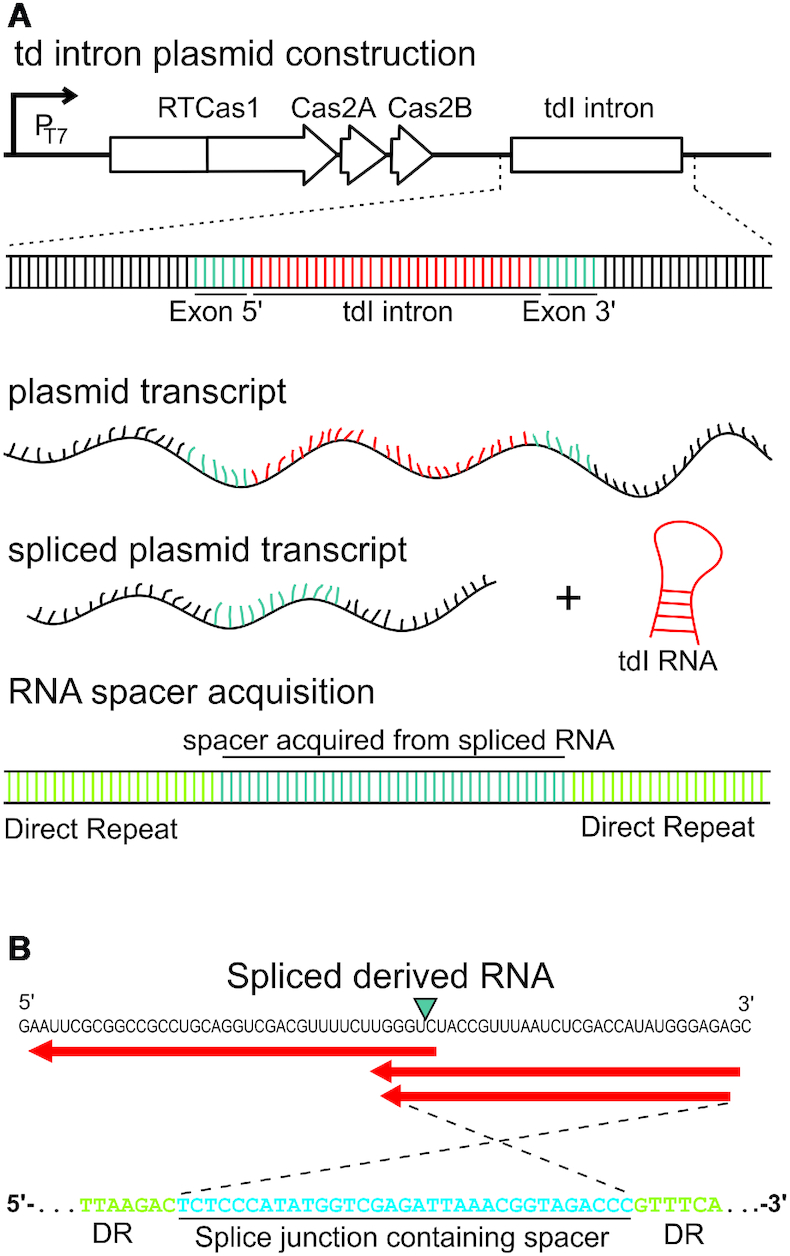
Spacer acquisition from RNA in the VVYJ016 type III-D system. (A) Schematic diagram of td intron-containing constructs. We determined whether the spacers originated from RNA, using a self-splicing transcript that produces an RNA sequence junction not encoded by DNA. Newly acquired spacers containing this exon junction may be considered to have been acquired from an RNA target. (B) RNA derived from the newly acquired exon junction-spanning spacer (blue). The splice site is indicated by a blue triangle. Red arrows indicate that the spacer are in the antisense orientation relative to the direction of transcription of the td intron. At the bottom, the highlighted sequence of one of the splice junction-containing spacers located in the CRISPR array is indicated.
DISCUSSION
We characterized the acquisition of spacers mediated by an RT-Cas1 fusion protein associated with a unique type III-D system from V. vulnificus strain YJ106. We show here that the adaptation module including the RT-Cas1 fusion, two different Cas2 proteins (A and B) and two CRISPR arrays is functional in a heterologous host (E. coli). We found that spacer acquisition differed in efficiency between the two CRISPR arrays and that it was abolished by mutations of any of the cas loci and strongly impaired by a lack of RT activity in the adaptation module. The acquired spacers throughout the genome, derived from overexpressing plasmids and within coding sequences displayed a bias for the antisense strand. The nucleotide sequence of the acquired spacers had characteristic features consistent with specific recognition by the adaptation complex. RT-Cas1 was also able to acquire spacers from RNA molecules. Overall, our findings demonstrate that the V. vulnificus type III-D system is a good model for studying the mechanism of acquisition of spacers from RNA of potential value for biotechnological applications.
Generally, when several CRISPR arrays are present in a particular CRISPR–Cas locus, only one of them has a high naïve acquisition efficiency in the systems studied (23,34). A similar situation was observed here, in the V. vulnificus YJ016 RT-Cas1 system, as only one of the arrays, CRISPR02, appears to be fully functional in acquisition (Figure 2B). Indeed, CRISPR02 contains more spacers than CRISPR01 in the V. vulnificus genome (9 and 2, respectively). An analysis of the leader region revealed only small differences in sequence more than 119 bp away from the first DR, so a difference in recognition by the RT-Cas1–Cas2A–Cas2B complex is highly unlikely. However, the two arrays displayed a significant difference in promoter activity, and this activity was inversely correlated with the ability to acquire new spacers. It is worth noting the opposite has been reported in the E. coli K12 CRISPR system that the highest expressed CRISPR locus is the most active for new spacer acquisition (35,36). Moreover, the small sequence variations between the CRISPR01 and 02 loci in V. vulnificus could be due to disruption of binding of other host factors that may be essential (i.e. IHF or other structural proteins; 37). The possible relationship between array transcription levels and spacer acquisition efficiency merits further study in other systems.
Unlike most of the RT-associated type III CRISPR–Cas systems (13 out of 14) functionally analyzed by Schmidt et al. requires of a selective amplification method (SENECA) to detect the acquisition of new spacers, the functionality of the RT- CRISPR–Cas system of V. vulnificus YJ016 can be demonstrated using previously established spacer acquisition assays (6,18).
Moreover, our results show that the RT domain plays an essential role in the acquisition step in the heterologous E. coli host, because the abolition of RT activity greatly decreases (by >90%) the number of spacers acquired. A similar reduction in an analogous RT mutant has been observed in MMB-1 RT-CRISPR system in M. mediterranea host but not in E. coli (18). In our experimental conditions the most abundant spacers correspond to rRNA genes that is the most abundant RNA in the cell, which may reflect RNA abundance-dependent spacer acquisition. In the M. mediterranea system the RT-Cas1 mediated spacer acquisition show a bias towards highly transcribed regions, but spacers were rarely acquired from rRNA (18). Comparisons of our spacer datasets to that of Record-seq (23) by F. saccharivorans (FsRT-Cas1–Cas2) also obtained in E. coli does not show any correlation. Additionally, and in contrast to F. saccharivorans and the M. mediterranea systems we have not found a positive correlation between the frequency of spacer acquisition and overall gene expression level. Note that such correlation does not distinguish between spacer acquisition from RNA versus DNA and only indicates that the acquired spacers are preferentially derived from highly transcribed genes (23). Taken together, our findings reflect mechanistically specific features underlying the VvRT-Cas1–Cas2A/Cas2B acquisition system that need to be further elucidated. Additionally, consistent with the other two RT-Cas systems studied (23,38,39), a bias towards the antisense strand of coding sequences was observed in the newly acquired spacers in the absence of the effector unit. This property suggests that, although the RT-Cas1–Cas2A–Cas2B complex is able to acquire spacers in either orientation, presents a slight preference of integrating new spacers into the CRISPR array to generate a crRNA complementary to predicted transcripts to conduct to an effective interference in Type III-CRISPR–Cas immunity systems (40).
The presence of two different Cas2 proteins is characteristic of the RT-CRISPR systems of clade 6 and this set-up is also frequently observed in clade 13 (24,25). We show here that both Cas2 proteins are required for spacer acquisition in vivo raising the possibility that the functional integration complex carries a Cas2A/Cas2B heterodimeric unit. This may facilitate analyses of the role of these proteins in the adaptation stage and may be related with the fact that spacers with a particular sequence are selected, a finding not reported for other RT–CRISPR–Cas systems characterized by the presence of only one cas2 gene (18,23). As already described for type I CRISPR–Cas systems, Cas2 dimer plays a structural role in the formation and stabilization of the adaptation complex, binding the central area of the protospacer and acting as a bridge between two Cas1 dimers (8,41). By analogy, in our system, the bias observed at different positions within the newly acquired spacers may indicate that, within the RT-Cas1–Cas2A–Cas2B complex, the RT-Cas1 protein preferentially binds to spacers with borders rich in ‘AT’ and flanked by a ‘G’ or a ‘C’ at the derived protospacer, whereas the Cas2A–Cas2B heterodimeric unit may be responsible for the particular bias (towards ‘C’) observed at asymmetric positions (+14 and +15) within the spacer, further studies are required to validate this hypothesis.
Finally, the use of the F. saccharivorans RT-Cas1–Cas2 system as an RNA-recording tool appears to result in skewing to AT-rich regions at the ends of the transcripts (23), whereas the V. vulnificus RT-Cas1–Cas2A–Cas2B system can acquire spacers regardless of their ‘GC’ content and from any point in the coding sequence.
In summary, the RT-Cas1 system from V. vulnificus YJ016 is a new RT–CRISPR system with novel properties different from those of the systems previously studied (18,23). It is a good model for further studies not only of the role of RTs in the acquisition of spacers, but also for elucidating the particular role of heterologous Cas2 complexes and the characteristics of the spacers acquired by type III CRISPR–Cas systems.
DATA AVAILABILITY
Data has been deposited in the Sequence Read Archive under accession number PRJNA539885: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA539885
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Carmen Amaro and Colleagues for the Vibrio material.
Authors contributions: A.G.-D., F.M.-A. and N.T. defined the research objective, designed experiments and wrote the paper with input from the other authors. A.G.-D. performed all genetics experiments. A.G.-D., F.M-A. performed all biochemistry experiments. A.G.-D., M.R.M., performed all bioinformatics analyses.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Spanish Ministerio de Ciencia, Innovación y Universidades; ERDF (European Regional Development Funds) research grants [BIO2014-51953-P, BIO2017-82244-P]. A.G.-D. was supported by a FPU predoctoral fellowship grant from the Ministerio de Economía y Competitividad [FPU15/02714]. Funding for open access charge: Ministerio de Ciencia, Innovación y Universidades [BIO2017-82244-P].
Conflict of interest statement. None declared.
REFERENCES
- 1. Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D.A., Horvath P.. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007; 315:1709–1712. [DOI] [PubMed] [Google Scholar]
- 2. Grissa I., Vergnaud G., Pourcel C.. The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics. 2007; 8:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Makarova K.S., Wolf Y.I., Alkhnbashi O.S., Costa F., Shah S.A., Saunders S.J., Barrangou R., Brouns S.J., Charpentier E., Haft D.H. et al.. An updated evolutionary classification of CRISPR–Cas systems. Nat. Rev. Microbiol. 2015; 13:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. van der O.J., Westra E.R., Jackson R.N., Wiedenheft B.. Unravelling the structural and mechanistic basis of CRISPR–Cas systems. Nat. Rev. Microbiol. 2014; 12:479–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Amitai G., Sorek R.. CRISPR–Cas adaptation: insights into the mechanism of action. Nat. Rev. Microbiol. 2016; 14:67–76. [DOI] [PubMed] [Google Scholar]
- 6. Yosef I., Goren M.G., Qimron U.. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 2012; 40:5569–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nuñez J.K., Kranzusch P.J., Noeske J., Wright A.V., Davies C.W., Doudna J.A.. Cas1–Cas2 complex formation mediates spacer acquisition during CRISPR–Cas adaptive immunity. Nat. Struct. Mol. Biol. 2014; 21:528–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang J., Li J., Zhao H., Sheng G., Wang M., Yin M., Wang Y.. Structural and mechanistic basis of PAM-dependent spacer acquisition in CRISPR–Cas systems. Cell. 2015; 163:840–853. [DOI] [PubMed] [Google Scholar]
- 9. Mojica F.J., Díez-Villaseñor C., García-Martínez J., Soria E.. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 2005; 60:174–182. [DOI] [PubMed] [Google Scholar]
- 10. Brouns S.J., Jore M.M., Lundgren M., Westra E.R., Slijkhuis R.J., Snijders A.P., Dickman M.J., Makarova K.S., Koonin E.V., van der Oost J.. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008; 321:960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hale C.R., Zhao P., Olson S., Duff M.O., Graveley B.R., Wells L., Terns R.M., Terns M.P.. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell. 2009; 139:945–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Koonin E.V., Makarova K.S.. Mobile genetic elements and evolution of CRISPR–Cas systems: all the way there and back. Genome Biol. Evol. 2017; 92:2812–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Krupovic M., Koonin E.V.. Self-synthesizing transposons: unexpected key players in the evolution of viruses and defense systems. Curr. Opin. Microbiol. 2016; 31:25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Krupovic M., Makarova K.S., Forterre P., Prangishvili D., Koonin E.V.. Casposons: a new superfamily of self-synthesizing DNA transposons at the origin of prokaryotic CRISPR–Cas immunity. BMC Biol. 2014; 12:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baltimore D. RNA-dependent DNA polymerase in virions of RNA tumour viruses. Nature. 1970; 226:1209–1211. [DOI] [PubMed] [Google Scholar]
- 16. Temin H.M., Mizutani S.. RNA-dependent DNA polymerase in virions of Rous sarcoma virus. Nature. 1970; 226:1211–1213. [DOI] [PubMed] [Google Scholar]
- 17. Toro N., Nisa-Martínez R.. Comprehensive phylogenetic analysis of bacterial reverse transcriptases. PLoS One. 2014; 9:e114083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Silas S., Mohr G., Sidote D.J., Markham L.M., Sanchez-Amat A., Bhaya D., Lambowitz A.M., Fire A.Z.. Direct CRISPR spacer acquisition from RNA by a natural reverse transcriptase-Cas1 fusion protein. Science. 2016; 351:aad4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Elmore J.R., Sheppard N.F., Ramia N., Deighan T., Li H., Terns R.M., Terns M.P.. Bipartite recognition of target RNAs activates DNA cleavage by the type III-B CRISPR–Cas system. Genes Dev. 2016; 30:447–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Estrella M.A., Kuo F.T., Bailey S.. RNA-activated DNA cleavage by the type III-B CRISPR–Cas effector complex. Genes Dev. 2016; 30:460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kazlauskiene M., Tamulaitis G., Kostiuk G., Venclovas Č., Siksnys V.. Spatiotemporal control of type III-A CRISPR–Cas immunity: coupling DNA degradation with the target RNA recognition. Mol. Cell. 2016; 62:295–306. [DOI] [PubMed] [Google Scholar]
- 22. Tamulaitis G., Venclovas Č., Siksnys V.. Type III CRISPR–Cas immunity: major differences brushed aside. Trends Microbiol. 2017; 25:49–61. [DOI] [PubMed] [Google Scholar]
- 23. Schmidt F., Cherepkova M.Y., Platt R.J.. Transcriptional recording by CRISPR spacer acquisition from RNA. Nature. 2018; 562:380–385. [DOI] [PubMed] [Google Scholar]
- 24. Toro N., Martínez-Abarca F., González-Delgado A.. The reverse transcriptases associated with CRISPR–Cas systems. Sci Rep. 2017; 7:7089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Toro N., Martínez-Abarca F., González-Delgado A., Mestre M.R.. On the origin and evolutionary relationships of the reverse transcriptases associated with type III CRISPR–Cas systems. Front Microbiol. 2018; 9:1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Spaink H.P., Okker R.J.H., Wijffelman C.A., Pees E., Lugtenberg B.. Promoters and operon structure of the nodulation region of the Rhizobium leguminosarum symbiosis plasmid pRL1JI. Recognition in Microbe-Plant Symbiotic and Pathogenic Interactions. 1986; Berlin, Heidelberg: Springer; 55–68. [Google Scholar]
- 27. Bradford M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976; 72:248–254. [DOI] [PubMed] [Google Scholar]
- 28. Miller J. Experiments in Molecular Genetics. 1972; Cold Spring Harbor Laboratory Press; 352–355. [Google Scholar]
- 29. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. 2011; 17:10–12. [Google Scholar]
- 30. Mcdonald N.D., Regmi A., Morreale D.P., Borowski D., Boyd E.F.. CRISPR–Cas systems are present predominantly on mobile genetic elements in Vibrio species. BMC Genomics. 2019; 20:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Williams T.C., Blackman E.R., Morrison S.S., Gibas C.J., Oliver J.D.. Transcriptome sequencing reveals the virulence and environmental genetic programs of Vibrio vulnificus exposed to host and estuarine conditions. PLoS One. 2014; 9:e114376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pyenson N.C., Marraffini L.A.. Type III CRISPR–Cas systems: when DNA cleavage just isn’t enough. Curr. Opin. Microbiol. 2017; 37:150–154. [DOI] [PubMed] [Google Scholar]
- 33. Kieper S.N., Almendros C., Behler J., McKenzie R.E., Nobrega F.L., Haagsma A.C., Vink J.N.A., Hess W.R., Brouns S.J.J.. Cas4 facilitates PAM-compatible spacer selection during CRISPR adaptation. Cell Rep. 2018; 22:3377–3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Staals R.H.J., Jackson S.A., Biswas A., Brouns S.J.J., Brown C.M., Fineran P.C.. Interference-driven spacer acquisition is dominant over naive and primed adaptation in a native CRISPR–Cas system. Nat. Commun. 2016; 7:12853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pougach K., Semenova E., Bogdanova E., Datsenko K.A., Djordjevic M., Wanner B.L., Severinov K.. Transcription, processing and function of CRISPR cassettes in Escherichia coli. Mol. Microbiol. 2010; 77:1367–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Datsenko K.A., Pougach K., Tikhonov A., Wanner B.L., Severinov K., Semenova E.. Molecular memory of prior infections activates the CRISPR/Cas adaptive bacterial immunity system. Nat. Commun. 2012; 3:945. [DOI] [PubMed] [Google Scholar]
- 37. Nuñez J.K., Bai L., Harrington L.B., Hinder T.L., Doudna J.A.. CRISPR immunological memory requires a host factor for specificity. Mol. Cell. 2016; 62:824–833. [DOI] [PubMed] [Google Scholar]
- 38. Mohr G., Silas S., Stamos J.L., Makarova K.S., Markham L.M., Yao J., Lucas-Elío P., Sanchez-Amat A., Fire A.Z., Koonin E.V. et al.. A reverse transcriptase-Cas1 fusion protein contains a Cas6 domain required for both CRISPR RNA biogenesis and RNA spacer acquisition. Mol. Cell. 2018; 72:700–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Silas S., Lucas-Elio P., Jackson S.A., Aroca-Crevillén A., Hansen L.L., Fineran P.C., Fire A.Z., Sánchez-Amat A.. Type III CRISPR–Cas systems can provide redundancy to counteract viral escape from type I systems. eLife. 2017; e27601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pyenson N.C., Marraffini L.A.. Type III CRISPR–Cas systems: when DNA cleavage just isn’t enough. Curr. Opin. Microbiol. 2017; 37:150–154. [DOI] [PubMed] [Google Scholar]
- 41. Wan H., Li J., Chang S., Lin S., Tian Y., Tian X., Wang M., Hu J.. Probing the behaviour of Cas1–Cas2 upon protospacer binding in CRISPR–Cas systems using Molecular Dynamics simulations. Sci. Rep. 2019; 9:3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data has been deposited in the Sequence Read Archive under accession number PRJNA539885: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA539885