


Abstract
Mitochondrial dysfunction plays critical roles in cancer development and related therapeutic response; however, exact molecular mechanisms remain unclear. Recently, alongside the discovery of mitochondrial-specific DNA methyltransferases, global and site-specific methylation of the mitochondrial genome has been described. Investigation of any functional consequences however remains unclear and debated due to insufficient evidence of the quantitative degree and frequency of mitochondrial DNA (mtDNA) methylation. This study uses WGBS to provide the first quantitative report of mtDNA methylation at single base pair resolution. The data show that mitochondrial genomes are extensively methylated predominantly at non-CpG sites. Importantly, these methylation patterns display notable differences between normal and cancer cells. Furthermore, knockdown of DNA methyltransferase enzymes resulted in a marked global reduction of mtDNA methylation levels, indicating these enzymes may be associated with the establishment and/or maintenance of mtDNA methylation. DNMT3B knockdown cells displayed a comparatively pronounced global reduction in mtDNA methylation with concomitant increases in gene expression, suggesting a potential functional link between methylation and gene expression. Together these results demonstrate reproducible, non-random methylation patterns of mtDNA and challenge the notion that mtDNA is lowly methylated. This study discusses key differences in methodology that suggest future investigations must allow for techniques that assess both CpG and non-CpG methylation.
INTRODUCTION
Mitochondria maintain cellular homeostasis by generating metabolic energy via oxidative phosphorylation (OXPHOS) as well as regulating apoptotic pathways. Mitochondrial dysfunction in a wide range of human malignancies is well documented and targeting mitochondria remains a promising avenue for the development of novel strategies for cancer treatment (1).
Mitochondrial DNA (mtDNA) is a double helical molecule composed of a heavy and light strand that encodes for 13 protein subunits that make up the complexes required for OXPHOS, transfer RNAs that carry specific amino acids for protein synthesis as well as ribosomal RNAs also involved in protein synthesis (2). A 1200-bp non-coding region of the mitochondrial genome, called the Displacement Loop (D-Loop), controls mitochondrial replication and transcription of its encoded genes (2) through a number of different start sites and promoter regions (3–5). However, exact molecular mechanisms involved in transcriptional control of the mitochondrial genome remain unclear.
The existence of mtDNA methylation has been reported previously via a large array of techniques, while some studies have reported an absence of mtDNA methylation (6,7), the majority have detected it (8–11). Older studies reported human fibroblast cultures and cell lines to have 2–5% of all mtDNA molecules fully methylated at specific CpG regions analysed via the method involving restriction digestion (12). Similar findings were reported using radioactive labelling in mouse fibroblast and hamster kidney cell cultures (13). Later studies confirmed the presence of mtDNA methylation in mouse brain tissue using ELISA (14), in lymphoblastoid cells of Down Syndrome children using mass spectrometry (15) as well as in human colorectal cancer cell lines (10) and human brain tissue (16) using immunoprecipitation. Pyrosequencing and bisulfite sequencing have also detected site specific mtDNA methylation in human blood cells (17–19). In addition, methylation-specific PCR techniques have detected substantial mtDNA methylation in liver tumour samples (11). Furthermore, the biological relevance of mtDNA methylation was suggested by a study showing that DNMT1 contains a targeted sequence upstream of its TSS that allows translocation into the mitochondria where it binds to the D-Loop (10). Elevated levels of DNMT1 correlated with increased expression of some mitochondrial genes adding weight to the notion that DNMT1 may have functional capacity in the mitochondria (10). Recently, DNMTIso3 (an isoform of DNMT1) was shown to be more concentrated in the mitochondria than DNMT1; suggesting that certain isoforms of DNA methyltransferases are mitochondria specific (20). Two independent studies reported the presence of DNMT1, DNMT3A and DNMT3B enzymes in the mitochondrial protein fraction of mouse skeletal muscle (21) and mouse embryonic stem cells (17). Finally, a more recent study utilized CpG and GpC methyltransferases to target mitochondrial DNA and concluded that increased GpC methylation decreased the abundance of mitochondrial encoded transcripts (22). Collectively these findings suggest the existence of mtDNA methylation, which may have a biological relevance, however mtDNA methylation remains debated largely due to contradictory reports from bisulfite sequencing studies (6,23). Since a genome-wide quantitative approach has not been demonstrated successfully for comprehensive mapping of mtDNA methylation patterns, exploration of the biological relevance of mtDNA methylation remains impeded.
Recently, several reports in the field aimed at improving techniques to be used for identification of mtDNA methylation however these studies focused primarily on CpG methylation (23–26). The present study builds on those findings by addressing key methodological adaptations which are required for investigating mtDNA methylation via whole genome bisulfite sequencing. This study presents the first report of genome-wide mitochondrial DNA methylation across different cell lines and tissue samples at single nucleotide resolution. The results show non-random, reproducible patterns of mtDNA methylation predominantly in a non-CpG context. Differences in global methylation for normal versus cancer cells derived from both liver and breast cancer are observed. Finally, the data implicates DNMT1, DNMT3A and DNMT3B to be associated with mtDNA methylation.
MATERIALS AND METHODS
Cell line selection
Normal breast epithelial cells MCF10A were maintained in DMEM (Invitrogen No. 11039-047) with 10% FBS, 1% penicillin-streptomycin, 20 ng/ml epidermal growth factor (Peprotech 1 mg), 0.5 μg/ml hydrocortisone (Sigma No. H-0888), 100 ng/ml cholera toxin (Sigma No. C-8052) and 10 μg/ml insulin (Sigma No. I-1882). Human breast cancer cells MCF7 were maintained in DMEM (Invitrogen No. 11039-047) with 10% FBS, 1% penicillin-streptomycin, sodium pyruvate (1 mM) and glutamine (2 mM). Human hepatocarcinoma cells HepG2 were maintained in DMEM (Invitrogen No. 11039-047) with 10% FBS and 1% penicillin-streptomycin. All cells were maintained in an atmosphere of 5% CO2 at 37°C. Frozen pellets of primary derived human breast cancer cells (HMEC) were a gift from the Jiri Zavadil’s lab and primary human hepatocytes were a gift from Prof. Michel Rivoire - Centre Léon Bérard (CLB), INSERM U1032, Lyon, France.
Mitochondrial DNA extraction
Frozen cell pellets (1 × 107 cells) were resuspended in 500 μl ice-cold homogenization buffer (100 mM Tris-HCl pH 7.4, 250 mM sucrose, 10 mmM EDTA) on ice. The cell pellet was homogenized with a 30G syringe and centrifuged (Eppendorf Centrifuge 5424 R) at 4°C, 1500 g for 10 min to collect debris. The supernatant was then transferred to a new tube and centrifuged again at 4°C, 10 000 g for 10 min. The pellet containing mitochondria was resuspended in 50 μl high-salt buffer (10 mM Tris-HCl pH 7.6, 10 mM KCl, 10 mM MgCl2, 0.4M NaCl, 2 mM EDTA). About 8 μl of 10% SDS was then added to the homogenate and the homogenate was incubated for 7 min at 55°C to denature and solubilize proteins. Proteins were then removed by addition of 20 μl of 5 M NaCl and centrifugation at 11 300 g for 20 min at room temperature. The supernatant was collected into a new 1.5 ml Eppendorf tube. Twice the volume of 100% ethanol was added and the supernatant was centrifuged for 10 min at 10 000 g for precipitation of the mtDNA pellet. The pellet was resuspended in 50 μl of milliQ H20 and immediately processed with phenol-chloroform method for clean-up. An equal volume of phenol/chloroform/isoamyl alcohol (25:24:1) was added and sample was mixed gently by inverting the tube. The tube was then centrifuged at room temperature for 5 min at 12 000 g. The top aqueous layer was carefully collected and the same process was repeated with chloroform/isoamyl alcohol (24:1). To the upper aqueous layer, collected in a fresh tube, 1/10th volume of 3 M NaOAc (pH 5.2), 2 μl glycogen (15 mg/ml Ambion) and 2.5× volume of absolute ethanol was added and mixed by gently inverting the tube. The mixture was incubated overnight at −20°C or for 2 h at −80°C. The crude mitochondrial pellet was obtained by centrifugation at 4°C for 15 min, 12 000 g. The pellet was resuspended in 20 μl MilliQ grade H20 and concentration of mtDNA was assessed using Qubit fluorometer. Multiple pellets of the same cell type were processed this way and mtDNA was pooled until a minimum 2 μg concentration was achieved for bisulfite modification.
DNA quality check
Relative enrichment of mtDNA to nuclear DNA of all extractions was assessed with qPCR to ensure the extractions were not contaminated with nuclear DNA. Primers specific to the mitochondrial genome, Forward Primer (5′-GCTCAACACCCACTACCTAAA-3′), Reverse Primer (5′-TGATCTGACGCAGGCTTATG-3′) as well as primers specific to nuclear encoded gene GAPDH, Forward Primer (5′-CCCTTCATACCCTCACGTATTC-3′), Reverse Primer (5′-CCATTCTGTCTTCCACTCACTC-3′) were used to amplify the mtDNA extraction. The quality of the extraction was assessed by comparing the fold change of mitochondrial genome versus nuclear genome using the ΔCt method.
Mitochondrial DNA pre-processing
Linearization of 700 ng of HepG2 mtDNA was achieved via a 10-min incubation of mtDNA at 65 degrees to encourage uncoiling followed by a 9-h incubation with 2U of BamHI restriction enzyme and NEB Buffer. Linearization was confirmed by visualization on 1% Agarose gel. Subsequently, sonication was selected as a better method since this has been shown to improve library preparation quality specifically for NGS in the context of plasmid DNA (27). Since mtDNA is of a similar structure to plasmid DNA, all mtDNA samples were sonicated with the Diagenode bioruptor Next Gen until fragments between 100 and 10 000 bp was achieved. MtDNA from each cell type was diluted to an appropriate concentration determined experimentally and sonicated with different conditions ranging from 3 cycles on the HIGH setting to 25 cycles on HIGH setting with a sonication time of 30 s per cycle. Fragment sizes of HepG2 cells were initially assessed on the BioAnalayser as well as 1% Agarose gel to confirm the method works. All other samples were subsequently checked on 1% Agarose gel to ensure no visible secondary structure remained. The mtDNA was then bisulfite modified using the EZ DNA Methylation Gold Kit (Zymo Research, USA), and the bisulfite modified DNA was quantified on Nano Drop using the ssDNA setting.
Pyrosequencing
Bisulfite modified DNA was amplified with previously published pyrosequencing primers ((19); Supplementary Table S1) in a 50 μl reaction. Following visualization of amplicon via gel agarose, 45 μl of PCR product was used with the PyroMark Kit (QIAGEN Cat. No. 972804) for each sequencing reaction as per manufacturer instructions. Briefly, DNA was immobilized on to streptavidin-coated beads in binding buffer for 10 min. The biotin-labeled template was purified using the pyrosequencing vacuum prep tool and incubated with 10 pmol/reaction sequencing primer in annealing buffer (20 mM Tris-acetate, 2 mM MgAc2; pH 7.6). The DNA strands were denatured at 80°C for 2 min and reannealed at room temperature for 10 min. Sequencing was performed according to manufacturer instructions.
Library preparation and next-generation sequencing
Library preparation of the extracted, phenol-chloroform cleaned, sonicated and bisulfite modified mtDNA was then performed using the TruSeq DNA Methylation Kit (Illumina, EGMK81312) according to manufacturer instructions. Briefly, 200 ng of bisulfite-converted mtDNA was ligated to adapters and amplification indexes from the TruSeq Indexes Kit (Illumina) were introduced during PCR amplification. Adapter-Tagged libraries were purified using AMPure beads (Beckman Coulter). Libraries were assessed on Bioanalyser for quality assurance. Libraries were then subjected to 150 bp paired-end sequencing using the Illumina MiSeq instrument.
Bioinformatics—data filtering and alignment
For all WGBS runs, internal controls showed greater than 95% of all reads were detected by the MiSeq to be ≥ Q30. Just over 45 million reads passed filter for all samples. All reads that passed filter from the MiSeq were then further assessed through FastQC v0.11.3 for standard quality control. Reads were trimmed with Trimgalore version 0.4.4 with a minimum quality cut off of 28 bp and a minimum length cut off of 30 bp. Following adapter trimming, all reads below a QC threshold of 30 were discarded. Mapping and methylation calls were done using BSMAP v2.42 (28).
For MeDIP runs, TrimGalore software was used to http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) (29) to remove the custom adapters of the paired-end sequences, and 20 additional nucleotides from each 3-prime end, due to the specific requirements of the sequencing kit. The custom adapters were GCCAAT, CAGATC, CTTGTA and AGTCAA, for both read files of sample ‘HepG2’, ‘HepG2-replicate’, ‘Hepatocyte’ and ‘Hepatocyte-replicate’, respectively. The NC_012920.1 mitochondrial genome was used as the reference genome for the alignment that was done by bowtie2 software (30). The same software was used to perform the paired-end alignment and obtained SAM files. After replacing ‘NC_012920.1’ by ‘chrM’, sorted BAM files were generated with samtools (31) and the indexes were obtained of each file using the command index. Next, the BAM files were uploaded into the Integrative Genome Viewer (IGV) software to obtain the read profile of each sample and their plots and calculated the differential methylation across the mitochondrial chromosome by using the default parameters of the MEDIPS R package (32). Briefly, we used chromosome M of the ‘BSgenome.Hsapiens.UCSC.hg38’ library as a reference genome and fixed a P value = 1e-3 as a maximum of stacked reads per genomic position, extending all reads to a length of 300 nucleotides according to the given strand information and dividing the genome into adjacent windows of length = 100 nucleotides.
Noise filtering of mitochondrial DNA methylation sequencing data
Conventional methods used for assessing bisulfite conversion efficiency of nuclear DNA (33), which require spiking samples with λ DNA, were unsuccessful for sequencing of mtDNA (data not shown). Hence, in order to assess the quality of the sequencing reads, apart from standard read filtering methods described above, a 100% unmethylated plasmid DNA (ThermoFisher Scientific Cat. No SD0021) sample was processed and sequenced alongside the samples of interest. The amount of potential incorrect methylation calls was assessed from these data to determine the threshold of sequencing reads that need to be considered for avoiding potential false positive methylation calls. The following formula was used:
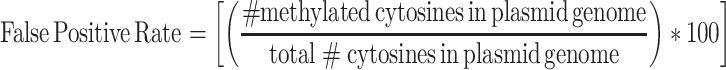 |
From this analysis, we found that including data points under the threshold of 10 reads and all cytosines with methylation levels under 9% considerably increased the false positive detection of methylation, decreasing the accuracy to <97%. Therefore, for all datasets, all sites with coverage of <10 reads as well as all sites with methylation levels under 9% were removed. This ensured that the methylation calls from the data are at least 98.95% accurate.
Methylation calculations
Descriptive Statistics on the global patterns of methylation across the four cell lines was conducted in order to compare between cell lines. The following formulas were used to determine the methylation ratio at individual cytosines, methylation index (MI) and proportion of methylation across the mitochondrial genome:
 |
Non-parametric, paired and unpaired Wilcox T-test was used to calculate significant differences between mean methylation of functionally relevant genomic regions between cell types.
Methylated DNA immunoprecipitation
A total of 1.2 μg of mtDNA per reaction was extracted as outlined in the ‘Mitochondrial DNA Extraction’ section above from HepG2 and hepatocyte cells. Cell lines were processed in technical duplicate using the Auto MagMeDIP Kit (Diagenode #C02010013) as per instructions. Briefly, extracted mtDNA was sonicated, quantified and assessed for fragment size and quality via Qubit and BioAnalyser. Immunoprecipitation was conducted by pre-set protocols on the IP-Star Compact robot (Diagenode) that performed the necessary antibody incubation and washing steps required. Library preparation for sequencing of the MeDIP output mtDNA was then performed using the ACCEL-NGS Methyl-Seq DNA Library Kit (Swift Bioscience #30024) as per manufacturer instructions.
RNA interference-mediated knockdown of DNMT enzymes
Small interfering RNAs (siRNAs) were introduced into MCF10A cells by transfection using the Lipofectamine® RNAiMAX Reagent (Invitrogen) according to the manufacturer instructions. Cells were transfected with 100 nM of the following human ON-TARGETplus SMARTpools (Dharmacon): DNMT1 (ON-TARGET plus SMARTpool siDNMT1, L-004605-00-0005), DNMT3a (ON-TARGET plus SMARTpool siDNMT3a, L-006672-01-0005), DNMT3b (ON-TARGET plus SMARTpool siDNMT3b, L-006395-00-0005) or ON-TARGET plus non-targeting pool (D-001810-10-05) that was used as a negative control. Cells were harvested 96 h post-transfection and protein was extracted to validate the silencing with western blot.
Western blot analysis of DNMT enzymes
Whole cell protein lysates were extracted using ice-cold RIPA buffer supplemented with protease inhibitors. Protein (50 μg) was subjected to SDS-PAGE followed by immunoblotting. Primary antibodies against DNMT3a (ab13888, 1:500) and DNMT3b (ab13604, 1:500) were obtained from Abcam. Monoclonal antibodies against DNMT1 (MAB0079, 1:1000) and β-actin (Clone C4, 1:1000) were obtained from Abnova and MP Biomedicals, respectively.
Relative protein levels were quantified by ImageJ (https://imagej.nih.gov/ij/). The relative intensity was calculated for each DNMT band with respect to the siControl and normalized to the relative intensity of actin in each sample.
Mitochondrial DNA content assessment
Whole cell DNA and RNA extractions were performed using the All Prep DNA/RNA Mini Kit (Qiagen, REF80204) according to manufacturer instructions. The mitochondrial DNA content in each cell type was determined using qPCR on whole cell DNA extractions. Primers specific to the mitochondrial genome, Forward Primer (5′-GCTCAACACCCACTACCTAAA-3′), Reverse Primer (5′-TGATCTGACGCAGGCTTATG-3′) as well as primers specific to nuclear encoded gene GAPDH, Forward Primer (5′-CCCTTCATACCCTCACGTATTC-3′), Reverse Primer (5′-CCATTCTGTCTTCCACTCACTC-3′) were used to amplify the whole cell DNA extraction. The mtDNA content of the extraction was assessed by comparing the fold change in expression of mitochondrial genome versus nuclear genome using the ΔCt method.
mRNA expression analysis
Following cDNA synthesis from the above RNA extractions, relative enrichment of mRNA expression of mitochondrial encoded genes was assessed with qPCR using previously published primers (see Supplementary Table S1). GAPDH, Forward Primer (5′AACGGGAAGCTTGTCATCAA-3′), Reverse Primer (5′TGGACTCCCACGACGTACTCA-3′), was used as housekeeping gene for breast cell types. SFRS4, Forward Primer(5′-GGCTACGGGAAGATCCTGGA-3′), Reverse Primer (5′-TGCATCACGCAGATCATCAA-3′), was used as housekeeping gene for liver cell types (34). In order to compare mRNA expression between cell lines, mRNA expression was normalized to the mtDNA content in each cell in order to account for vastly differing mitochondrial DNA content between cell types:
 |
RESULTS
Development of NGS methodology for high-resolution DNA methylation analysis specifically for the mitochondrial genome
To investigate genome-wide mtDNA methylation patterns at single base pair resolution in human normal and cancer cell lines and tissues, we aimed to optimize the method that combines bisulfite conversion with NGS. To this end, the mtDNA enrichment achieved from the extraction protocol was confirmed by qPCR analysis using mitochondrial and nuclear specific primers (Figure 1A). Based on previous studies, mtDNA was initially extracted from HepG2 cells and linearized with restriction enzyme BamHI in order to avoid bisulfite conversion inefficiency as previously described (24). This was then followed by deep sequencing using the MiSeq platform. The degree of methylation detected at each cytosine position was compared to the sequencing depth achieved for that position (Figure 1B). In line with previous reports, the data showed that only small methylation values had high sequencing depth and that reads exhibiting higher methylation values had low sequencing depth (23). Moreover, the ratio of sequencing reads (not shown) which aligned to the H-strand compared to the L-strand was also uneven (0.08), suggesting some amplification bias. However, mtDNA that was fragmented mechanically prior to bisulfite modification and sequencing, eliminated this strand bias (ratio = 1.16) (Figure 1C). Furthermore, the cells that were sonicated prior to sequencing also produced far better coverage (Figure 1E). Hence, all further samples were fragmented before bisulfite modification and subsequent sequencing.
Figure 1.

Mitochondrial DNA sequencing requires method adjustments. (A) Raw amplification cycles of mtDNA enrichment assessed with qPCR of a mitochondrial specific genomic region compared to nuclear specific GAPDH genomic region (left panel). Relative fold enrichment of mtDNA to nuclear DNA (2−ΔCt) for HepG2 mtDNA extractions is indicated (right panel). The lack of nuclear DNA enrichment confirms the quality of mtDNA extraction. (B) Cytosine methylation compared to sequencing read depth (left panel) of HepG2 mtDNA which was digested with 2U of BamHI overnight at 37°C and visualised on agarose gel (right panel). (C) Cytosine methylation compared to sequencing read depth (left panel) of HepG2 mtDNA that was fragmented via sonication (100–7000 bp) and visualized on agarose gel (right panel). (D) HepG2 sonicated mtDNA (left panel) and bisulfite modified mtDNA (right panel) was assessed on the BioAnalyser to ensure sonication was efficient and that modified mtDNA produced the standard expected curve. (E) Average number of reads aligning to the mitochondrial genome per 1000 bp. HepG2 sample that was sonicated prior to sequencing showed an improvement in coverage compared to the BamHI-treated HepG2 sample. (F) A 100% unmethylated plasmid DNA control was processed and sequenced alongside mtDNA samples to ensure an appropriate false positive threshold can be used for data analysis. The X-axis represents the genomic position of the plasmid genome (48 kb). The Y-axis represents the methylation value detected at each cytosine. The dots represent the 1.56% false detection rate.
To ensure quality methylation calls, 100% unmethylated plasmid DNA was processed and sequenced simultaneously alongside the mtDNA samples (Figure 1F). When all methylation calls with <10× coverage and <9% methylation were excluded, 378 cytosines remained false positive out of a potential 24 182 cytosines. Hence 98.44% accuracy in methylation calls can be achieved with minimum 10× coverage and a detection threshold of 9% methylation. All reads not matching this threshold criteria were excluded as background noise (see ‘Materials and Methods’ section).
To further confirm that these results are not the consequence of bisulfite conversion-related false positives, mitochondrial DNA of HepG2 cells and primary hepatocytes were processed via a bisulfite-free method to confirm that the mtDNA is methylated. MeDIP-seq assay performed on technical replicates of two cell lines also showed that the mitochondrial genome was globally methylated, hence complementing the WGBS data (Supplementary Figure S1A).
The results indicate this stringent approach allows adequate investigation of mtDNA methylation at single base pair resolution that accounts for both CpG and non-CpG methylation.
Mitochondrial DNA within normal and cancer liver cells exhibit significantly different methylation patterns that are primarily in a non-CpG context
In order to establish baseline methylation patterns within the mitochondrial genome, normal liver cells derived from primary human liver (hepatocytes) and liver cancer cells (HepG2) were assessed first. Globally, in both cases, the L-strand displayed a higher degree and frequency of methylation compared to the H-strand (Figure 2A and B). Hepatocytes had 37% of all cytosines were methylated, whereas HepG2 had 44% of their mitochondria methylated (Figure 2C). Interestingly, in contrast to nuclear DNA, a substantial amount of methylation in the hepatocyte and HepG2 mitochondrial genome was detected in CpT, CpC and CpA contexts. The highest frequency of methylation (around 30%) was detected within a CpT and CpC dinucleotide context, whereas methylation of CpG sites had the lowest methylation frequency (12% and 5%, respectively) (Figure 2C). A striking difference in methylation was observed across tRNA encoding regions. Specifically, hepatocytes are completely devoid of methylation on the L-strand within tRNAs which carry the amino acids arginine and lysine (Arg and Lys MI = 0%) (Figure 2D); however, these same regions are methylated in HepG2 cells (MI = 11.4%) (Figure 2D). Significant differences in mean methylation across almost all gene encoding regions in both strands of HepG2 was detected compared to hepatocytes (P value < 0.001; Figure 2E). In addition, the distribution of methylation values across the displacement loop in each cell type was compared since this region contains multiple promoters and functionally relevant transcriptional start sites. Hepatocytes showed a bimodal distribution of methylation values indicating both highly and lowly methylated regions across the D-Loop, whereas HepG2 showed a right skewed distribution indicating the D-Loop is mostly highly methylated (Figure 2F). Methylation across the L-strand promoter region (392–435 bp) was compared to the level of ND6 mRNA expression in each cell type. Finally, the higher methylation across the L-strand promoter region detected in HepG2 was associated with lower expression of ND6 mRNA compared to hepatocytes (Figure 2G).
Figure 2.

Baseline patterns of the mitochondrial methylome in normal and cancer liver cells. (A andB) MtDNA isolated from hepatocytes and liver cancer cells (HepG2) that was sequenced using the NSG MiSeq platform as described in Figure 1. The circular plot represents genomic position (1–16 kb) of all methylated cytosines with respect to sequence order. Each segment of the circle represents a separate functionally relevant region, tRNA, rRNA, gene or D-Loop. The Y-axis indicating that methylation level is represented on the left-hand side of the D-Loop segment. The large outer ring displays methylation at each cytosine within the H-Strand, whereas the large inner ring displays methylation at each cytosine on the L-Strand. Thin inner bands indicate the genomic position of all cytosines within the H-Strand and L-Strand sequence. Global mtDNA methylation patterns differ between hepatocytes and cancer cells and are also strand specific. (C) Summary statistics of the frequency of mitochondrial mCpN context in liver cells. (D) Methylation Index across tRNA encoding regions in liver cancer versus normal cells. Each horizontal segment compares the MI within tRNAs that have been grouped according to the amino acid they carry (acidic, basic, aromatic or hydrophobic). The left panel indicates MI across H-Strand and right panel indicates MI across L-Strand. (E) Comparative boxplots indicating significant (P < 0.001) difference of mean methylation across gene encoding regions of normal versus cancer cells in each strand. (F) Density plot of the distribution of methylation values along the D-Loop region. (G) Relative ND6 mRNA expression normalized to mtDNA amount compared with methylation index across the LSP. High LSP methylation associates with low ND6 expression.
To ensure biological relevance of mtDNA methylation in the disease context, primary tissue resections of normal liver and hepatocellular carcinoma samples were also examined (Supplementary Figure S2). Similar to the cell line samples, significant differences (P value < 0.001) in mean methylation of both global and some gene-encoding regions across both strands was observed (Supplementary Figure S2A and C). Both primary samples showed a bimodal distribution of methylation values across the D-Loop, however in line with methylation patterns observed in cell line samples (Figure 2D), this distribution was slightly skewed toward the right in tumour cells (Supplementary Figure S2B).
Together these results show that specific mtDNA methylation profiles across biologically relevant genomic regions can be observed among different normal and cancer cell types.
Baseline mitochondrial methylation patterns of normal and cancer breast cell lines differ significantly
To test the generality of mitochondrial genome methylation, baseline mtDNA methylation patterns were next examined in an independent cell type. MtDNA isolated from normal mammary epithelial cells (MCF10A) and breast cancer cells (MCF7) were assessed and compared. Similar to the liver context, globally in both cases, the L-strand displayed a higher degree and frequency of methylation compared to the H-strand (Figure 3A and B). MCF10A had 43% of the entire mitochondrial genome methylated, whereas MCF7 had 38% of their mitochondria methylated (Figure 3C). In contrast to nuclear DNA, and much like what was observed in liver cells, a substantial amount of methylation in the MCF10A and MCF7 mitochondrial genome was detected in CpT, CpC and CpA contexts. Again, the highest frequency of methylation was detected within a CpT or CpC dinucleotide context, whereas methylation of CpG sites was the least frequently methylated dinucleotides (12% and 10%, respectively) (Figure 3C). In breast cells a different pattern of tRNA methylation was observed compared to liver cells. On the H-strand of breast cancer cells (MCF7) (Figure 3D), methylation in tRNA encoding regions, which carry the amino acids arginine, lysine, asparagine and glutamine, is almost completely lost (MI = 0.29%) compared to normal breast (MCF10A) cells (MI = 8.84%) (Figure 3D). The distribution of methylation values across the displacement loop in each cell type was compared since this region contains multiple promoters and functionally relevant transcriptional start sites. MCF7 exhibited a bimodal distribution of methylation values indicating the presence of both highly and lowly methylated regions across the D-Loop, whereas MCF10A showed a right skewed distribution, consistent with high level methylation in the D-Loop (Figure 3F). Methylation levels across the L-strand promoter region (392–435 bp) were then compared with the level of ND6 mRNA expression in each cell type. Higher methylation across the region detected in MCF10A coincided with lower expression of ND6 compared to MCF7 (Figure 3G), these findings were consistent with those observed in liver cells.
Figure 3.

Baseline patterns of the mitochondrial methylome in normal and cancer breast cells. MtDNA was sequenced using the NSG MiSeq platform. The circular plot represents genomic position (1–16 kb) of all methylated cytosines with respect to sequence order. Each segment of the circle represents a separate functionally relevant region, tRNA, rRNA, gene or DLoop. The Y-axis indicating methylation level is represented on the left-hand side of the D-Loop segment. The large outer ring displays methylation at each cytosine within the H-Strand whereas the large inner ring displays methylation at each cytosine on the L-Strand. Thin inner bands indicate the genomic position of all cytosines within the H-Strand, L-Strand sequence. Global mtDNA methylation patterns differ between these cell types and are also strand specific. (A) Normal breast epithelial cells (MCF10A); (B) Cancer epithelial cells (MCF7); (C) Summary statistics of the frequency of mitochondrial mCpN context in liver cells and (D) Methylation Index across tRNA encoding regions in breast cancer versus normal. Each horizontal segment compares the MI within tRNAs that have been grouped according to the amino acid they carry (acidic, basic, aromatic or hydrophobic). The left panel indicates MI across H-Strand and right panel indicates MI across L-Strand. (E) Comparative boxplot indicating significant (P < 0.001) difference of mean methylation across gene encoding regions of normal versus cancer cells in each strand. (F) Density plot of the distribution of methylation values along the D-Loop region. (G) Relative ND6 mRNA expression normalized to mtDNA amount compared with methylation index across the LSP. High LSP methylation associates with low ND6 expression
To investigate concordance of baseline methylation patterns among cell types, additional normal liver (HepaRG) and normal breast (HMEC) cell lines were subjected to mtDNA methylation profiling. Significant differences in mean methylation of both global as well as some gene encoding regions across both strands was observed between HMEC and MCF10A cells (Supplementary Figure S2D–F) as well as hepatocytes and HepaRG cells (Supplementary Figure S2G–I).
Together these results provide further evidence to the generality of previously mentioned methylation patterns and confirm the methylation patterns are cell type specific.
DNMT3B, DNMT3A and DNMT1 are involved in modulating mtDNA methylation
Since multiple DNMTs have previously been detected in the mitochondria by various independent studies (10,17,20,21), it was important to assess the dependence of mtDNA methylation patterns on different DNMT enzymes. Thus, the impact of knockdown of DNMT1, DNMT3A and DMT3B on mtDNA methylation was assessed using a siRNA approach. Global cellular protein levels of each enzyme were depleted individually in MCF10A cells (Supplementary Figure S3) and was followed by mtDNA methylation analysis. Silencing of all three DNMT enzymes resulted in markedly reduced levels of cytosine methylation globally across the MCF10A mitochondrial genome compared to the control cells (Supplementary Figure S4).
The degree and frequency of methylation were especially affected in the DNMT3B silenced cells (Figure 4A and B) with the L-strand most heavily affected (Figure 4A and B), therefore the impact of DNMT3B knockdown on mtDNA methylation was analysed in further detail. Methylation between DNMT3B knockdown and control cells showed the L-strand in both cases displayed a higher degree and frequency of methylation compared to the H-strand (Figure 4A). Significant decreases (P value < 0.001) were observed in global methylation in the knockdown cells as well as within gene encoding regions on both strands (Figure 4C and E). The distribution of methylation values across the displacement loop in the knockdown cells was also observed to be markedly lower than in control cells (Figure 4D). To assess potential functional impact of the observed mtDNA methylation loss, mRNA expression of a set of mitochondrial genes was analysed in DNMT3B knockdown cells. A drop in methylation in the D-Loop region in DNMT3B knockdown cells, which accounts for a drop in methylation across all promoter regions, resulted in higher mRNA expression of the genes COX1, COX3, ND4, ND4L and ND5 (Figure 4G). Furthermore, similar to previous observations, the drop in global methylation induced by DNMT3B knockdown resulted in reduced methylation of the LSP and this was found to be concomitant with an increase in mRNA expression of the ND6 gene (Figure 4H).
Figure 4.

Silencing DNMT3B modulates mitochondrial DNA methylation concomitant to expression changes of mitochondrial transcripts. (A) MtDNA methylation in MCF10A cells exposed to a 96-h treatment of DNMT3B siRNA compared to control. A global drop in methylation ratio is observed for H and L strand methylation in DNMT3Bsi treated cells. (B) Whole cell protein was assayed for relative amounts of DNMT3B via western blot to confirm assay efficiency. (C) Comparative boxplot indicating significant (P < 0.001) difference of mean methylation across gene encoding regions of control versus knockdown treated cells in each strand. (D) Paired Wilcox t-test showing significance of the global drop in methylation between control and treated cells (P < 0.001). (E) Density plot of the distribution of methylation values along the D-Loop region. (F) Whole cell protein of repeat experiment assayed for relative amounts of DNMT3B via western blot to confirm efficiency. (G) Relative mRNA expression normalized to mtDNA amount compared of genes encoded on the H-strand. Treated cells show an increase in gene expression compared to control. (H) Relative ND6 mRNA expression normalized to mtDNA amount compared with methylation index across the LSP. High LSP methylation associates with low ND6 expression.
Since knockdown experiments implicated a potential role for DNMTs in mtDNA methylation, whether DNMT enzymes are present in mitochondria was further explored with confocal microscopy. Analysis of 2D images suggest small amounts of all three DNMT1, DNMT3A and DNMT3B may be co-localized in both MCF7 and HepG2 cells (Supplementary Figure S4); however, additional assays such as z-stack imaging are needed to confirm and consolidate these data.
Together these results suggest that DNMT1, DNMT3A and DNMT3B enzymes may be associated with the observed baseline methylation patterns of the mitochondrial genome and there may be a link between mtDNA methylation and gene expression as is the case in the nuclear genome.
DISCUSSION
The present study for the first time combines appropriate stepwise methodology for quantitatively assessing genome-wide mtDNA methylation at both CpG and non-CpG methylation sites with base-pair resolution. This methodology has revealed human mitochondrial genomes can be extensively methylated. Results show that the methylation is cell type specific occurs predominantly in a non-CpG context and is more heavily present on the L-strand than the H-Strand. MtDNA methylation can be modulated by decreasing global protein levels of DNMT1, DNMT3A and DNMT3B enzymes, suggesting an association of these enzymes with mtDNA methylation. A potential functional relevance of this methylation is suggested by the observed association between mtDNA methylation levels within the displacement loop region and mRNA of relevant genes encoded on the mitochondrial DNA. Considering the different mitochondrial methylome patterns observed in the normal and cancer cells examined, this study suggests important implications for the role of mtDNA methylation in tumorigenesis.
There are three crucial steps in methodology that is unique and specific for WGBS of mtDNA in this study (Supplementary Figure S5). These are (i) the separation of mtDNA from nuclear DNA, (ii) removal of excess protein via phenol-chloroform and (iii) finally shearing of the mtDNA to ensure no coiled or secondary structures remain prior to bisulfite sequencing. The following describes how methodology may be a key contributor to underestimation of mtDNA in previous reports.
One of the key issues pertaining to sequencing mtDNA for methylation analysis is in understanding the intrinsic differences of mtDNA function and structure as well as how these differences require adaptation of existing methodologies which have evolved with a primary focus on nuclear DNA and CpG methylation. Liu et al., for instance, showed that the mtDNA needs to be linearized prior to methylation analysis with pyrosequencing to avoid overestimation of methylation values owing to inefficient bisulfite conversion (24) caused by the coiled, circular structures of mtDNA. Results from this study concur with previous findings since in all cell lines analysed via pyrosequencing, linearized mtDNA via sonication showed lower methylation levels compared to unfragmented, circular mtDNA (data not shown). In addition, pyrosequencing detected lower amounts of mtDNA methylation than those detected by NGS (Supplementary Figure S1C). Since global methylation was observed in both methylation results from NGS and MeDIP-seq data of two different cell lines (Supplementary Figure S1A), it is likely that pyrosequencing may underestimate cytosine methylation of mtDNA due to its CpG nucleotide bias (Supplementary Figure S1C). One explanation for the discrepancy between pyrosequencing and NGS results can be attributed to the previously established notion that standard pyrosequencing assays are biased towards CpG methylation (35,36). This is especially relevant to the mitochondrial genome where the sequence is heavily dispersed with repetitive cytosine nucleotides (n > 3) throughout the genome. Hence methodology that adequately allows assessment of both CpG and non-CpG methylation is essential for investigating mtDNA methylation; this may be a possible explanation for previous reports that have concluded low or no mitochondrial methylation (6).
The complimentary bisulfite methylation data (Figure 2A and B) and MeDIP data (Supplementary Figure S1A) observed in this study is in line with previous reports that comprehensively showed cytosine methylation within mitochondrial genomes to be present across 39 pre-existing datasets spanning different human cell and tissue types (37). Although our results concur with conclusions made by Ghosh et al., it should be noted that since the paper was first published, a series of methodological processing required to address bisulfite conversion efficiency that is unique for mtDNA has been reported (23,24). This may be another potential factor that has lead to the idea that mtDNA is generally lowly methylated. Pre-existing WGBS datasets were not produced with experimental considerations unique to mtDNA in mind. Hence assessing mtDNA methylation in these databases, although a viable option for exploratory analysis, may underestimate mtDNA methylation levels and this should be considered for future studies (37).
Accurate fragmentation is a key technical adaption that needs to be optimized in order to ensure high quality, reliable methylation reads from sequencing data. Recently, Mechta et al. concluded that secondary structures of mitochondrial DNA may have caused hindrance in the bisulfite conversion step leading to false positive methylation values (23). In concordance, results from this study further demonstrate that fragmentation of mtDNA is required for optimal mtDNA accessibility for accurate bisulfite conversion and sequencing (Figure 1). This was further consolidated by the high correlation (R2 = 0.97) observed between biological replicate data (Supplementary Figure S1B). Accessibility needs to be addressed when considering mtDNA for bisulfite sequencing due to the highly coiled form it exists within cells (38). When cells were treated with BamHI and sequenced (Figure 1B), the data showed an almost exact match to the ‘read depth versus methylation value’ patterns obtained by Mechta et al. (23) and we concur this was most likely due to hindrance toward bisulfite conversion from secondary structures post linearisation (23). However, when mtDNA was fragmented through sonication a more even distribution of methylation values and sequencing depth was achieved (Figure 1C). One of the pitfalls of using sonication for fragmentation is the lack of control in obtaining fragment sizes. Each cell line had to be individually optimized (Figure 1D) through a rigorous process of trial and error until an ideal range of fragments were observed on agarose gel (∼100–7000 bp) and these size ranges were then confirmed with BioAnalyser. Since these fragments then underwent a further strenuous process of bisulfite conversion, it is possible that a number of fragments (especially fragments within 100–200 bp size) would have been more vulnerable to degradation, making them impossible to be incorporated in the library preparation stage. Indeed those mitochondrial DNA samples which were sonicated to the point where all fragments were within 100–200 bp range did not produce successful libraries (data not shown). Hence although not perfected, fragmentation via sonication can overcome secondary structure obstructions to bisulfite conversion. The data presented in this study have been validated via a secondary non-bisulfite dependent method (Supplementary Figure S1A) and shown to be reproducible (R2 = 0.97) through cell line biological replicates (Supplementary Figure S1B). This line of evidence provides support that the data is unlikely to be false positive results.
Another key step in the methodology that is crucial for consideration is the phenol-chloroform extraction that immediately followed extraction of the crude mitochondrial DNA pellet (see ‘Materials and Methods’ section). Using crystal structure imaging, it has been previously shown that the mtDNA transcription factor TFAM binds to both H-Strand promoter, as well as non-specific mtDNA regions, and imposes U-turn shapes on the mtDNA. Hence, transcription factor binding encourages compaction and packaging of mtDNA molecules (38). This combined with the difficulty observed in HepG2 mtDNA double digestion with restriction enzymes BamHI and EcoRI (data not shown), we hypothesized that some protein–DNA interactions may have remained intact during the crude mtDNA extraction protocol. Since phenol-chloroform is a technique used to separate nucleic acids from proteins and lipids (39,40), it was added as a final step in the extraction method. After this step was added to the protocol, complete fragmentation of the mtDNA in subsequent steps was easier as observed by number of sonication cycles required and visualization on agarose gel (data not shown). Hence there is a chance mtDNA bound transcription factors, and/or any restriction enzymes used for linearization, remain and encourage compaction of mtDNA following extraction methods that have not undergone phenol-chloroform clean up. Hence further investigation is required to confirm results from any reports (26,41) of mtDNA methylation that have relied on restriction enzyme digestion alone as this may be a point of hindrance in efficient bisulfite conversion of mtDNA. These findings show that methodology for bisulfite sequencing needs to be specifically adapted for mitochondrial genomes.
Since the D-Loop consists of promoter regions and is responsible for controlling mtDNA replication, we considered whether the frequency distribution of methylation within this region differs in normal and cancer contexts. A striking shift from the bimodal distribution of methylation frequency in normal liver cells was observed in liver cancer cells, which was strongly right skewed (Figure 2D). A similar but less pronounced pattern was also observed in primary liver and liver tumour resections (Supplementary Figure S2A–C). However, a different distribution of D-Loop methylation was observed in breast cancer and normal cells (Figure 3F). MtDNA content and cancer have been previously implicated in disease prognosis. For example, low mtDNA content was found to be associated with better survival (42) and treatment outcomes (43) for breast cancer patients, whereas low mtDNA content in liver has been associated with higher risk of developing non-alcoholic fatty liver disease, a precursor to cancer development (44). Since the D-Loop region is involved in mtDNA replication, whether D-Loop methylation determined by this study (Figures 2F, 3F and 4D) has direct regulatory consequences for mitochondrial replication should be investigated further. The functional consequences of this could mean a cell type specific layer of regulation concerning mtDNA replication that has remained unexplored.
Because MtDNA is known to insert into the nuclear genome, resulting in ‘nuclear DNA sequences of mitochondrial origin’ (NUMTs), the presence of NUMTs in some analytical procedures may generate misleading results (6,23). It is estimated that there are between 1000 and 10 000 copies of mtDNA in each mammalian cell (45); hence, the probability of identical sequences in the nuclear genome being amplified when mtDNA is the target sequence is extremely low due to the vast excess of mtDNA (6). Although mtDNA enrichment was confirmed using qPCR, to avoid any complication, data from this study was aligned to the human nuclear genome (Human Genome Assembly GRCh38.p12, June 2018) as well as to the mitochondrial genome alone (revised Cambridge Reference Sequence NC_012920, June 2018). Any reads that aligned to both were excluded to ensure only those reads that uniquely mapped to mtDNA were considered in the analysis. Therefore, the presence of NUMT sequences is highly unlikely to contribute to mtDNA methylation levels and patterns observed in this study.
Previous work investigating non-CpG methylation in the nuclear genome has shown a conservation of methylation context. The trend observed in nuclear genome, i.e. mCpG > mCpA > mCpT > mCpC (36), has been attributed to the observed patterns being non-random and hence biologically significant. Similarly in this study, distinct trends in the frequency of methylation at each dinucleotide, i.e. mCpC>mCpA /mCpT>mCpG, is largely conserved in all cell types. Further studies are warranted to examine the potential biological significance of the consistent occurrence of non-CpG methylation detected in mtDNA. In the nuclear genome, unlike CpG methylation, non-CpG methylation largely occurs asymmetrically and this has been attributed to its acute regulation in response to environmental stimuli/stressors. It has also been shown to regulate the expression of specific genes, in particular PGC-1a (35) which is involved in mitochondrial function and biogenesis. In the nuclear genome, characteristic areas of the genome, such as gene promoter regions, are often aberrantly hypo- or hyper-methylated in cancer versus normal cells and this has been directly linked to transcriptional regulation of gene expression (46). Likewise, this study has shown a similar association where higher average promoter methylation of the L-strand has been concomitant with lower expression of ND6 between different cell types (Figures 2E, 3E and 4E). This suggests the detected methylation may play a role in strand specific regulation of gene expression. These results concur with previous investigations where gene expression was associated with induced promoter methylation within GpC dinucleotides (22). It should be noted that GpC methylation from the previous study has been reported as part of the CpN methylation in the present study. It is also noteworthy that in mtDNA, unlike in the nuclear genome, one promoter region such as the Origin of replication (100–441 bp) has been identified to regulate multiple genes (3,5). Previous reports have shown a link between the expression of specific genes, such as ND1, to be associated with induced GpC methylation within specific promoters such as HSP2 (22). To further emphasize this, strand specific methylation of CpG and adjacent CpN sites has been shown across a known transcription factor binding site (523–550 bp) of the mitochondrial genome in Supplementary Figure S4 (47). The complex relationship between multiple H-strand promoters and origins of replication in the D-Loop as well as varying distributions of methylation in the D-Loop between cell types observed in this study should be investigated further to determine any functional links to transcription of genes encoded on the H-strand. The asymmetry of non-CpG methylation has previously been attributed to its high response to environmental stimuli (36). In addition, mitochondria are known to be highly responsive to changing cellular conditions as part of many of their physiological functions such as maintaining cellular energy demands. Hence, it can be postulated that the detected mtDNA methylation could play a role in regulation of specific genes in a transient manner with respect to immediate cellular demand.
Another important finding from this study is that the methylation may be modulated by global cellular DNMT enzymes (Supplementary Figure S3). It is also noteworthy that DNMT3B was the enzyme that severely impacted the global frequency and amount of mtDNA methylation in normal breast cells (Figure 4). It should be noted that a wide variety of nuclear encoded genes are known to have direct, functional effects on mitochondrial processes as well as mitochondrial DNA compaction and transcription (48). The effects of DNMT knockdown on these nuclear encoded genes and any potential consequential effects on the observed changes in mtDNA methylation cannot be ruled out. Co-operative maintenance of DNA methylation by the DNMT family of enzymes in the nuclear genome has been previously well characterized (49). However, little is known about the consequences of aberrant overexpression of DNMT3B in particular that has been reported for a number of malignancies including breast cancer (50) and colorectal cancer (51,52). Due to its high relevance in disease, there is a need for a better understanding of the complex interplay between the DNMT family of enzymes and how they may be involved in modulating mitochondrial DNA methylation. The complex nature of the nuclear-mitochondrial crosstalk, and how methylation of the two genomes plays a role (48) is another area of future investigation.
In summary, the present study demonstrates, for the first time via NGS-based bisulfite sequencing, that the mitochondrial genome is heavily methylated with predominant non-CpG methylation and that this methylation is associated with multiple DNMT enzymes. Furthermore, the results show that mtDNA methylation patterns are cell type specific and that they may be functionally linked to mitochondrial gene expression. Importantly, this study highlights key methodological requirements for adequate assessment of methylation within mitochondrial DNA. This study implores the importance of adapting methodology to allow assessment of both CpG and non-CpG methylation for future investigations. Future efforts aimed at deciphering the potential functional consequences and biological variances of mtDNA methylation may prove to be highly useful.
DATA AVAILABILITY
Available upon request.
Supplementary Material
ACKNOWLEDGEMENTS
HMEC cells were a gift from Jiri Zavadil’s lab (Lyon, France), we would like to thank Dr Mauraj Pandey for optimizing growth conditions for the HMEC cells. Human primary hepatocytes were a gift from Prof. Michel Rivoire - Centre Léon Bérard (CLB), INSERM U1032, Lyon, France. We would like to thank Maud Michelet for technical assistance and isolation of PHHs; F. Zoulim and M. Rivoire for access to liver resections.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Institut National du Cancer (INCa, France); European Commission (EC) Seventh Framework Programme (FP7) Translational Cancer Research (TRANSCAN) Framework; Foundation ARC pour la Recherche sur le Cancer (France); Plan Cancer-Eva-Inserm research grant (to Z.H.); Postdoctoral Fellowship from International Agency for Research on Cancer (to V.P.), partially supported by the EC FP7 Marie Curie Actions – People – Co-funding of regional, national and international programmes (COFUND)’. Funding for open access charge: Institut National du Cancer (INCa, France); European Commission (EC) Seventh Framework Programme (FP7) Translational Cancer Research (TRANSCAN) Framework; Foundation ARC pour la Recherche sur le Cancer (France); Plan Cancer-Eva-Inserm research grant (to Z.H.).
Conflict of interest statement. The authors alone are responsible for the views expressed in this article and they do not necessarily represent the views, decisions or policies of the institutions with which they are affiliated.
REFERENCES
- 1. Porporato P.E., Filigheddu N., Pedro J.M.B., Kroemer G., Galluzzi L.. Mitochondrial metabolism and cancer. Cell Res. 2018; 28:265–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chinnery P.F., Hudson G.. Mitochondrial genetics. Br. Med. Bull. 2013; 106:135–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tapper D.P., Clayton D.A.. Mechanism of replication of human mitochondrial DNA. Localization of the 5′ ends of nascent daughter strands. J. Biol. Chem. 1981; 256:5109–5115. [PubMed] [Google Scholar]
- 4. Crews S., Ojala D., Posakony J., Nishiguchi J., Attardi G.. Nucleotide sequence of a region of human mitochondrial DNA containing the precisely identified origin of replication. Nature. 1979; 277:192–198. [DOI] [PubMed] [Google Scholar]
- 5. Fish J., Raule N., Attardi G.. Discovery of a major D-loop replication origin reveals two modes of human mtDNA synthesis. Science. 2004; 306:2098–2101. [DOI] [PubMed] [Google Scholar]
- 6. Hong E.E., Okitsu C.Y., Smith A.D., Hsieh C.L.. Regionally specific and genome-wide analyses conclusively demonstrate the absence of CpG methylation in human mitochondrial DNA. Mol. Cell Biol. 2013; 33:2683–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Maekawa M., Taniguchi T., Higashi H., Sugimura H., Sugano K., Kanno T.. Methylation of mitochondrial DNA is not a useful marker for cancer detection. Clin. Chem. 2004; 50:1480–1481. [DOI] [PubMed] [Google Scholar]
- 8. Gao J., Wen S., Zhou H., Feng S.. De-methylation of displacement loop of mitochondrial DNA is associated with increased mitochondrial copy number and nicotinamide adenine dinucleotide subunit 2 expression in colorectal cancer. Mol. Med. Rep. 2015; 12:7033–7038. [DOI] [PubMed] [Google Scholar]
- 9. Feng S., Xiong L., Ji Z., Cheng W., Yang H.. Correlation between increased ND2 expression and demethylated displacement loop of mtDNA in colorectal cancer. Mol. Med. Rep. 2012; 6:125–130. [DOI] [PubMed] [Google Scholar]
- 10. Shock L.S., Thakkar P.V., Peterson E.J., Moran R.G., Taylor S.M.. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:3630–3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pirola C.J., Gianotti T.F., Burgueño A.L., Rey-Funes M., Loidl C.F., Mallardi P., Martino J.S., Castaño G.O., Sookoian S.. Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut. 2013; 62:1356–1363. [DOI] [PubMed] [Google Scholar]
- 12. Shmookler Reis R.J., Goldstein S.. Mitochondrial DNA in mortal and immortal human cells. Genome number, integrity, and methylation. J. Biol. Chem. 1983; 258:9078–9085. [PubMed] [Google Scholar]
- 13. Nass M.M. Differential methylation of mitochondrial and nuclear DNA in cultured mouse, hamster and virus-transformed hamster cells. In vivo and in vitro methylation. J. Mol. Biol. 1973; 80:155–175. [DOI] [PubMed] [Google Scholar]
- 14. Dzitoyeva S., Chen H., Manev H.. Effect of aging on 5-hydroxymethylcytosine in brain mitochondria. Neurobiol. Aging. 2012; 33:2881–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Infantino V., Castegna A., Iacobazzi F., Spera I., Scala I., Andria G., Iacobazzi V.. Impairment of methyl cycle affects mitochondrial methyl availability and glutathione level in Down’s syndrome. Mol. Genet. Metab. 2011; 102:378–382. [DOI] [PubMed] [Google Scholar]
- 16. Devall M., Smith R.G., Jeffries A., Hannon E., Davies M.N., Schalkwyk L., Mill J., Weedon M., Lunnon K.. Regional differences in mitochondrial DNA methylation in human post-mortem brain tissue. Clin. Epigenetics. 2017; 9:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bellizzi D., D’Aquila P., Scafone T., Giordano M., Riso V., Riccio A., Passarino G.. The control region of mitochondrial DNA shows an unusual CpG and non-CpG methylation pattern. DNA Res. 2013; 20:537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Byun H.M., Colicino E., Trevisi L., Fan T., Christiani D.C., Baccarelli A.A.. Effects of air pollution and blood mitochondrial DNA methylation on markers of heart rate variability. J. Am. Heart Assoc. 2016; 5:e003218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Baccarelli A.A., Byun H.M.. Platelet mitochondrial DNA methylation: a potential new marker of cardiovascular disease. Clin Epigenetics. 2015; 7:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Saini S.K., Mangalhara K.C., Prakasam G., Bamezai R.N.K.. DNA Methyltransferase1 (DNMT1) Isoform3 methylates mitochondrial genome and modulates its biology. Sci. Rep. 2017; 7:1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wong M., Gertz B., Chestnut B.A., Martin L.J.. Mitochondrial DNMT3A and DNA methylation in skeletal muscle and CNS of transgenic mouse models of ALS. Front. Cell Neurosci. 2013; 7:279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van der Wijst M.G., van Tilburg A.Y., Ruiters M.H., Rots M.G.. Experimental mitochondria-targeted DNA methylation identifies GpC methylation, not CpG methylation, as potential regulator of mitochondrial gene expression. Sci. Rep. 2017; 7:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mechta M., Ingerslev L.R., Fabre O., Picard M., Barrès R.. Evidence Suggesting Absence of Mitochondrial DNA Methylation. Front. Genet. 2017; 8:166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu B., Du Q., Chen L., Fu G., Li S., Fu L., Zhang X., Ma C., Bin C.. CpG methylation patterns of human mitochondrial DNA. Sci. Rep. 2016; 6:23421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mechta M., Ingerslev L.R., Barres R.. Methodology for accurate detection of mitochondrial DNA methylation. J. Visual. Exp. 2018; 135:doi:10.3791/57772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Owa C., Poulin M., Yan L., Shioda T.. Technical adequacy of bisulfite sequencing and pyrosequencing for detection of mitochondrial DNA methylation: Sources and avoidance of false-positive detection. PLoS One. 2018; 13:e0192722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Thompson M., Aukema K., O’Bryan D., Rader S., Murray B.. Plasmid sonication improves sequencing efficiency and quality in the Beckman Coulter CEQ system. BioTechniques. 2008; 45:327–329. [DOI] [PubMed] [Google Scholar]
- 28. Xi Y., Li W.. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics. 2009; 10:232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011; 17:10–12. [Google Scholar]
- 30. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Robinson J.T., Thorvaldsdottir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P.. Integrative genomics viewer. Nat. Biotechnol. 2011; 29:24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lienhard M., Grimm C., Morkel M., Herwig R., Chavez L.. MEDIPS: genome-wide differential coverage analysis of sequencing data derived from DNA enrichment experiments. Bioinformatics. 2014; 30:284–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang Q., Gu L., Adey A., Radlwimmer B., Wang W., Hovestadt V., Bähr M., Wolf S., Shendure J., Eils R. et al.. Tagmentation-based whole-genome bisulfite sequencing. Nat. Protoc. 2013; 8:2022–2032. [DOI] [PubMed] [Google Scholar]
- 34. Waxman S., Wurmbach E.. De-regulation of common housekeeping genes in hepatocellular carcinoma. BMC Genomics. 2007; 8:243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Barrès R., Osler M.E., Yan J., Rune A., Fritz T., Caidahl K., Krook A., Zierath J.R.. Non-CpG methylation of the PGC-1alpha promoter through DNMT3B controls mitochondrial density. Cell Metab. 2009; 10:189–198. [DOI] [PubMed] [Google Scholar]
- 36. Patil V., Ward R.L., Hesson L.B.. The evidence for functional non-CpG methylation in mammalian cells. Epigenetics. 2014; 9:823–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ghosh S., Sengupta S., Scaria V.. Comparative analysis of human mitochondrial methylomes shows distinct patterns of epigenetic regulation in mitochondria. Mitochondrion. 2014; 18:58–62. [DOI] [PubMed] [Google Scholar]
- 38. Ngo H.B., Lovely G.A., Phillips R., Chan D.C.. Distinct structural features of TFAM drive mitochondrial DNA packaging versus transcriptional activation. Nat. Commun. 2014; 5:3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gedamu L., Iatrou K., Dixon G.H.. A simple procedure for the isolation and purification of protamine messenger ribonucleic acid from trout testis. Biochem. J. 1978; 171:589–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Green M.R., Sambrook J.. Isolation of high-molecular-weight DNA using organic solvents. Cold Spring Harb Protoc. 2017; 2017:doi:10.1101/pdb.prot093450.. [DOI] [PubMed] [Google Scholar]
- 41. Morris M.J., Hesson L.B., Poulos R.C., Ward R.L., Wong J.W.H., Youngson N.A.. Reduced nuclear DNA methylation and mitochondrial transcript changes in adenomas do not associate with mtDNA methylation. Biomarker research. 2018; 6:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Weerts M.J., Sieuwerts A.M., Smid M., Look M.P., Foekens J.A., Sleijfer S., Martens J.W.. Mitochondrial DNA content in breast cancer: Impact on in vitro and in vivo phenotype and patient prognosis. Oncotarget. 2016; 7:29166–29176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Weerts M.J.A., Hollestelle A., Sieuwerts A.M., Foekens J.A., Sleijfer S., Martens J.W.M.. Low tumor mitochondrial DNA content is associated with better outcome in breast cancer patients receiving Anthracycline-Based chemotherapy. Clin. Cancer Res. 2017; 23:4735–4743. [DOI] [PubMed] [Google Scholar]
- 44. Kamfar S., Alavian S.M., Houshmand M., Yadegarazari R., Seifi Zarei B., Khalaj A., Shabab N., Saidijam M.. Liver mitochondrial DNA copy number and deletion levels may contribute to nonalcoholic fatty liver disease susceptibility. Hepat Mon. 2016; 16:e40774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lightowlers R.N., Chinnery P.F., Turnbull D.M., Howell N.. Mammalian mitochondrial genetics: heredity, heteroplasmy and disease. Trends Genet. 1997; 13:450–455. [DOI] [PubMed] [Google Scholar]
- 46. Baylin S.B., Jones P.A.. Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol. 2016; 8:doi:10.1101/cshperspect.a019505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fisher R.P., Topper J.N., Clayton D.A.. Promoter selection in human mitochondria involves binding of a transcription factor to orientation-independent upstream regulatory elements. Cell. 1987; 50:247–258. [DOI] [PubMed] [Google Scholar]
- 48. Sun X., Vaghjiani V., Jayasekara W.S.N., Cain J.E., St John J.C.. The degree of mitochondrial DNA methylation in tumor models of glioblastoma and osteosarcoma. Clin Epigenetics. 2018; 10:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jin B., Robertson K.D.. DNA methyltransferases, DNA damage repair, and cancer. Adv. Exp. Med. Biol. 2013; 754:3–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Roll J.D., Rivenbark A.G., Jones W.D., Coleman W.B.. DNMT3b overexpression contributes to a hypermethylator phenotype in human breast cancer cell lines. Mol. Cancer. 2008; 7:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rhee I., Bachman K.E., Park B.H., Jair K.W., Yen R.W., Schuebel K.E., Cui H., Feinberg A.P., Lengauer C., Kinzler K.W. et al.. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature. 2002; 416:552–556. [DOI] [PubMed] [Google Scholar]
- 52. Nosho K., Shima K., Irahara N., Kure S., Baba Y., Kirkner G.J., Chen L., Gokhale S., Hazra A., Spiegelman D. et al.. DNMT3B expression might contribute to CpG island methylator phenotype in colorectal cancer. Clin. Cancer Res. 2009; 15:3663–3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Available upon request.