


Abstract
Interstrand crosslinks (ICLs) are highly toxic DNA lesions that are repaired via a complex process requiring the coordination of several DNA repair pathways. Defects in ICL repair result in Fanconi anemia, which is characterized by bone marrow failure, developmental abnormalities, and a high incidence of malignancies. SLX4, also known as FANCP, acts as a scaffold protein and coordinates multiple endonucleases that unhook ICLs, resolve homologous recombination intermediates, and perhaps remove unhooked ICLs. In this study, we explored the role of SLX4IP, a constitutive factor in the SLX4 complex, in ICL repair. We found that SLX4IP is a novel regulatory factor; its depletion sensitized cells to treatment with ICL-inducing agents and led to accumulation of cells in the G2/M phase. We further discovered that SLX4IP binds to SLX4 and XPF–ERCC1 simultaneously and that disruption of one interaction also disrupts the other. The binding of SLX4IP to both SLX4 and XPF–ERCC1 not only is vital for maintaining the stability of SLX4IP protein, but also promotes the interaction between SLX4 and XPF–ERCC1, especially after DNA damage. Collectively, these results demonstrate a new regulatory role for SLX4IP in maintaining an efficient SLX4–XPF–ERCC1 complex in ICL repair.
INTRODUCTION
Genomic DNA is constantly challenged by various endogenous and exogenous lesions, such as interstrand crosslinks (ICLs), which are DNA lesions that covalently link two opposite DNA strands together. ICLs are highly toxic, as they can physically block DNA replication, transcription, and any other type of DNA transaction requiring the separation of DNA strands (1–3). Chemicals that induce ICLs, such as cisplatin and mitomycin C (MMC), are widely used as anti-cancer chemotherapeutic agents because they are very effective in preventing tumor growth (3–5). However, ICLs can also be induced by byproducts of cellular metabolism, such as reactive aldehydes (6,7). In either case, the ICLs must be removed to permit normal cellular proliferation (6–8). Cells have developed complex processes to remove ICLs and repair the DNA.
The importance of efficient ICL repair was underscored by the finding that defective ICL repair is associated with Fanconi anemia (FA) (9). FA is characterized by bone marrow failure, developmental abnormalities, and a high incidence of malignancies (10–12). At least 22 FA-associated genes have been identified, and investigations into the functions of these FA gene products have provided tremendous insights into ICL repair (13–16). We now know that ICL repair is a complex process that requires the coordination of several different DNA repair pathways. It is believed that, in G1-phase cells, ICLs are recognized and repaired by nucleotide excision repair (NER) machinery and bypassed by the translesion DNA synthesis pathway (17–22). In S-phase cells, ICLs cause the stalling of DNA replication forks, which leads to the recruitment of FA pathway proteins (23–26). Resolution of ICLs results in the formation of DNA double-strand breaks (DSBs), which can be further repaired by the homologous recombination (HR) pathway (27–29).
A number of nucleases, including XPF/-ERCC1, SLX4/FANCP-SLX1, MUS81-EME1 and FAN1, are thought to be involved in the ICL repair process (1,30–39). They are thought to act at different stages of ICL repair, such as the initial ICL unhooking, resolution of holiday junctions during HR, and/or removal of residual unhooked products. XPF–ERCC1 was first identified as a critical 3′-flap endonuclease involved in the NER pathway (40–42). Investigators later recognized that defects in XPF–ERCC1 resulted in cell hypersensitivity to ICL-inducing agents and the persistence of ICL lesions (27,31,43,44). Thus, it was thought that XPF–ERCC1 was the critical enzyme involved in ICL unhooking and that it might also participate in the resolution of HR intermediates during the subsequent ICL repair process (27,30,45–47). MUS81-EME1, another 3′-flap endonuclease, has been implicated in the conversion of ICLs to DSBs (33,48). However, cells in which MUS81 has been depleted exhibit only mild sensitivity to treatment with ICL-inducing agents, suggesting that MUS81 may act at a specific cell-cycle phase and/or function redundantly with other structure-specific nucleases in ICL repair (32,48). In addition, MUS81 is involved in the processing of stalled replication forks and HR intermediates, which can also contribute to ICL repair (49–51). Previous studies by our group and others led to the discovery that FAN1 is an FANCD2/FANCI-associated nuclease, i.e., it is a 3′-flap structure-specific endonuclease as well as a 5′ to 3′ exonuclease (36–39). FAN1 deficiency results in ICL repair defects, but FAN1 can also be involved in the resolution of ICLs independent of the FA pathway (52–54). FAN1 may have additional functions in maintaining stalled replication forks that depend on the FA pathway (55). Besides these endonucleases, exonucleases such as SNM1A also confer resistance to ICL-inducing agents and may function with XPF–ERCC1 in unhooking ICLs (48,56–58).
SLX4 was first discovered as a gene exhibiting synthetic lethality with yeast Sgs1-Top3 (a homolog of human BLM-TOP3α) (59). SLX4 functions as a scaffold protein that associates with multiple partners, including XPF–ERCC1, MUS81-EME1, SLX1, TERF2IP-TRF2, SLX4IP, PLK1 and MSH2-MSH3 (60–63). Researchers have speculated that it acts as a mediator and brings its interacting proteins to different types of DNA lesions to facilitate DNA repair (43,64,65). Therefore, SLX4 is considered the central module of the structure-specific endonucleases required for different repair processes. It has been suggested that several SLX4-associated nucleases, such as XPF–ERCC1, MUS81-EME1 and SLX1, participate in ICL repair (43,66). Although cells with deficiency in MUS81 or SLX1 exhibit only mild sensitivity to treatment with ICL-inducing agents, SLX4-deficient cells are hypersensitive to these agents, which may be mainly due to the association of SLX4 with XPF–ERCC1 (43,66,67). The importance of SLX4 in ICL repair was further demonstrated when its mutations were identified in patients with FA (68,69). Thus, SLX4 is also called FANCP. Researchers confirmed the involvement of SLX4 in the FA pathway in a mouse genetic study, which demonstrated that SLX4-deleted mice had many FA-associated phenotypes (32). However, it remains to be determined how SLX4 coordinates several endonucleases and participates in different steps of ICL repair.
SLX4IP, a SLX4-interacting protein, was discovered many years ago (60–63). However, its function in ICL repair has not yet been documented. In this study, we investigated the roles of SLX4IP in ICL repair. We showed that SLX4IP interacts with SLX4–XPF–ERCC1 complex and plays an accessory role in facilitating SLX4–XPF function in DNA repair.
MATERIALS AND METHODS
Cell culture and transfection
HEK293A and HEK293T cells were purchased from the ATCC (Manassas, VA) and maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal calf serum (FCS) at 37°C with 5% CO2. Transient plasmid transfections were conducted using polyethylenimine (PEI). In brief, cells were seeded into six-well plates 1 day before transfection, and 6 μg of PEI and 2 μg of plasmid were diluted separately in 100 μl of Opti-MEM Reduced Serum Media (Life Technologies, Carlsbad, CA, USA). Diluted PEI and plasmid were mixed together and incubated at room temperature for 15 min. The mixture was then carefully added to cells.
To generate cell lines stably expressing a given protein, HEK293T cells were transfected with PEI with an expression construct. Cells were diluted into 100-mm plates 48 h after transfection and were selected with 2 μg/ml of puromycin. After 10 days of selection, single colonies were picked and subjected to western blotting to determine protein expression; immunofluorescent staining was performed to determine protein localization.
Antibodies
In this study, antibodies against the following proteins were used for western blotting and/or immunostaining: SLX4 (A302-270A; Bethyl Laboratories, Montgomery, TX, USA) or an antibody developed in-house against the antigen comprising SLX4231-460, XPF (A301-315A; Bethyl Laboratories), MUS81 (sc-53382; Santa Cruz Biotechnology, Dallas, TX, USA), SLX1 (ab182501; Abcam, Cambridge, UK), MSH2 (2017S; Cell Signaling Technology, Danvers, MA), SLX4IP (an antibody developed in-house against the antigen comprising SLX4IP205-408), γH2AX (2577S; Cell Signaling Technology), Flag M2 (F3165-5MG; Sigma-Aldrich, St. Louis, MO, USA), α-tubulin (T6199-200UL; Sigma-Aldrich), vinculin (V9131; Sigma-Aldrich), MBP (sc-13564; Santa Cruz Biotechnology), GST (sc-138; Santa Cruz Biotechnology), ERCC1 (sc-17809; Santa Cruz Biotechnology), phospho-Chk2 (Thr68, 2661S; Cell Signaling Technology), Chk2 (2662S; Cell Signaling Technology), phospho-Chk1 (Ser345, 2348S; Cell Signaling Technology), and Chk1 (2360S; Cell Signaling Technology).
CRISPR/Cas9-mediated gene knockout
Guide RNAs (gRNAs) targeting specific genes were designed using online service tools and ligated into a BsmBI-lineated LentiCRISPR v2 plasmid according to a protocol described previously (70,71). Correct insertion of the gRNA sequence was confirmed via sequencing. HEK293A cells were transfected with PEI and constructs as described above. Cells were then selected with puromycin for 2 days and then divided and placed in 96-well plates; 10 days later, single clones were picked up. Western blotting was used to measure the expression level of the targeted genes. Finally, sequencing of gRNA-targeting sites was used to verify the correct knockout (KO) of a gene.
The gRNAs used to generate KO clones in this study were (i) SLX4IP-KO gRNA: GCAAGATATGAAGATCCACG AGG; (ii) XPF-KO gRNA1: CTATATCACTCTTGGAGCGG AGG; (iii) XPF-KO gRNA2: TTGTCATCGGATGCTGCTTT CGG and (iv) MUS81-KO gRNA: TCTGAAATACGAAGCGCGTG CGG.
CRISPR/Cas9-mediated gene knock-in
We knocked CMV-OsTIR1 into the genomic AAVS1 loci by co-transfecting pMK232 (AAVS1-CMV-OsTIR1-PURO-AAVS1) with AAVS1-T2 CRISPR/Cas into HEK293A cells (72). Polymerase chain reaction (PCR) was used to confirm the correct knock-in (KI) of CMV-OsTIR1. We designed gRNAs targeting the C-terminus of SLX4 and ligated them into a BsmBI-lineated lentiGuide-Puro (#219854; Addgene, Watertown, MA). The gRNA sequences used in this study were (i) SLX4-CKI-1: AAGGTGGAGCGGAACTGATG GGG and (ii) SLX4-CKI-2: AGAAGGTGGAGCGGAACTGA TGG.
To obtain the template plasmid, PCRs were used to obtain the homologous sequence of SLX4 from genomic DNA, the mini Auxin-inducible degron (mAID) sequence from plasmid pMK290 (# 72828; Addgene) (72), and SFB-P2A-hygo from a plasmid in the laboratory. All PCR fragments were purified and assembled into linear pUC19 via a Gibson assembly. The template plasmid was then mutated by mutagenesis PCR at the NGG sites corresponding to the gRNA used. HEK293A AAVS-OsTIR1 cells were co-transfected with lentiGuide-Puro-gRNA and a mutated template plasmid with the help of PEI. Single colonies were picked up and analyzed by PCR and the target sites were then sequenced.
SLX4 knockdown with inducible shRNA
SLX4 shRNA sequences were inserted into a pTRIPZ lentiviral vector (Dharmacon, Lafayette, CO, USA). HEK293A cells were infected with a supernatant containing the virus and selected with 2 μg/ml puromycin. To induce knockdown (KD) of SLX4 protein, we added 1 μg/ml doxycycline (DOX) to cells for 48 h.
ishSLX4-84: AGGAGAAAGGAAGACACAA
ishSLX4-85: TGGAGCTAGAACAAACCAA
Colony formation assay
A day before treatment, HEK293A cells were seeded into 60-mm plates (200 cells/plate). Cells were exposed to ionizing (IR) or ultraviolet (UV) radiation or were treated with increasing concentrations of DNA damage-inducing agents (MMC and camptothecin [CPT]) for 24 h. Cells were washed to remove the drugs and incubated for 10 more days before being stained with a crystal violet solution for visualization of colonies. Colonies of more than 50 cells were counted.
CellTiter-Glo assay
Cells were diluted in DMEM containing 10% FCS at a concentration of 104 cells/ml, and 100 μl (1000 cells) of the diluted cells was seeded into 96-well plates. After 24 h, 20 μl of serially diluted concentrations of MMC, or CPT was added to the cells. Cells were then incubated for 3 days before being analyzed using CellTiter-Glo (G7572; Promega, Madison, WI) reagents.
For CellTiter-Glo analysis, cell culture media were removed from 96-well plates and CellTiter-Glo reagents were added to the wells to induce cell lysis. After incubation at room temperature for 15 min, cell lysates were transferred into opaque-walled, 96-well plates and subjected to luminescence detection using a BioTek Synergy™ 2 Multi-Mode Microplate Reader.
Cell lysis and immunoprecipitation
Cells were lysed with ice-cold NETN lysis buffer (50 mM Tris–HCl, pH 7.4, 100 mM sodium chloride [NaCl], 0.4% NP-40, 1 mM EDTA), and protease inhibitor cocktail (Roche, Basel, Switzerland) for 30 min on ice. Cell lysates were centrifuged at 13 200 rpm for 10 min in a cold room. Supernatants were subjected to immunoprecipitation (IP) with S beads or protein A/G beads conjugated with anti-SLX4. All IPs were conducted in a cold room for 2 h.
Tandem affinity purification (TAP) and mass spectrometry analysis of SLX4- and SLX4IP-interacting proteins
To identify SLX4- and SLX4IP-interacting proteins, HEK293T cells stably expressing S protein, 2 × Flag, and streptavidin-binding peptide (SFB)-tagged SLX4 or SLX4IP were seeded into 150-mm plates. Four plates of nearly 100% confluent cells were used for each purification. For treatment with MMC, 100 ng/ml of MMC was added into the cell culture media 24 h before cell collection. Cells were then scratched down and collected via centrifugation. Next, the cells were lysed with ice-cold NETN lysis buffer with rotation for 30 min in a cold room. Cell lysis was clarified at 13 000 rpm for 30 min at 4°C. Supernatants from centrifugation were mixed with 200 μl of pre-equilibrated streptavidin beads for 2 h in a cold room. After being washed with NETN lysis buffer three times, 2 mg/ml of biotin was used to elute all of the binding proteins from the beads. The biotin-eluted samples were mixed with 50 μl of pre-equilibrated S beads for 2 h in a cold room. S beads were washed with NETN lysis buffer for three times and boiled at 95°C for 10 min in 2× Laemmli buffer.
For mass spectrometry (MS) analysis, a standard procedure was used as previously reported (73,74). In general, the boiled samples were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and stained with Coomassie Blue. The gel was excised into pieces. Sliced gels were completely destained in tubes before being digested into peptides with trypsin. Digested peptides were extracted with acetonitrile and dried by vacuum. Dried samples were resolved with a solution of 5% methanol and 0.1% formic acid and subjected to nanoscale liquid chromatography coupled with tandem mass spectrometry (nanoLC-MS/MS) analysis with an EASY-nLC 1000 liquid chromatography system (Thermo Fisher Scientific, Waltham, MA, USA). The resolved samples were separated on nanoscale reverse-phase, high-performance liquid chromatography (HPLC) that was prepared by packing Reprosil-Pur Basic C18 silica into a fused silica capillary (100-μm inner diameter × ∼20-cm length) with a flame-drawn tip, with a 75-min discontinuous gradient of 4–26% acetonitrile/0.1% formic acid at a flow rate of 800 nl/min. Separated sample fractions were electro-sprayed into a Q Exactive Plus Orbitrap mass spectrometer (Thermo Fisher Scientific). For nanoLC–MS/MS analysis, the mass spectrometer was set to data-dependent mode, and the precursor MS spectrum was scanned at 375–1300 m/z with 140k resolution at 400 m/z (3 × 106 AGC target). The MS/MS resolution was 17 500. The 35 strongest ions were fragmented via collision-induced dissociation with a normalized collision energy of 27, a 3.0 m/z isolation width with a dynamic exclusion time of 30 s, 2 × 104 AGC target, and 60 ms of maximum injection time.
For the MS data analysis, raw data were searched in Proteome Discoverer 1.4 (Thermo Fisher Scientific) using Mascot algorithm 2.4 (Matrix Science, Boston, MA, USA). The Human UniProt FASTA database (October 2015) that containing 70 097 entries was searched. Acetylation of protein N-terminus and oxidation of methionine were set as variable modification. The precursor mass tolerance was set within 20 ppm with a fragment mass tolerance of 0.02 Da, and a maximum of two missed cleavages were allowed. Assigned peptides were filtered with a 1% false discovery rate using percolator validation. Further protein–protein interaction analysis was processed using a previously described two-step method (74,75). Briefly, CRAPome methodology was firstly used to assess the identified proteins and corresponding peptide spectrum matches (PSMs). Next, tandem affinity purification (TAP)-MS data for randomly selected baits were selected as negative controls and protein enrichment after TAP-MS was compared with HEK293T whole-cell lysis. Proteins with enrichment above the average fold enrichment after TAP-MS were considered high-confidence candidate interacting proteins (HCIP).
Analysis of protein localization to UV laser-induced sites of DNA damage
In brief, cells were seeded into 35-mm, glass-bottomed dishes. The next day, UV laser-induced damage of cells was conducted under a Nikon (Tokyo, Japan) TE200 inverted microscope coupled with a 365-nm UV laser MicroPoint system. After irradiation, cells were immediately fixed with 3% paraformaldehyde and processed for immunostaining.
Cell-cycle analysis using flow cytometry
To analyze the effects of treatment on cell cycle distribution, we seeded 106 of HEK293A-WT, SLX4IP-KO, and XPF-KO cells as well as cells from other cell lines as indicated into 100-mm plates. The next day, 10 or 20 ng/ml MMC was added to cells for 6, 12, 24 or 48 h. Next, cells were trypsinized, washed with phosphate-buffered saline (PBS), and fixed in 70% ethanol. After fixation, cells were washed with PBS and stained with a propidium iodide staining solution (50 μg/ml propidium iodide and 100 μg/ml RNase A) at 37°C for at least 30 min. Cell-cycle analysis was performed using a Gallios Flow Cytometer system (Beckman Coulter, Brea, CA, USA).
Immunofluorescence staining
HEK293A-WT, SLX4IP-KO, XPF-KO and MUS81-KO cells were grown on coverslips and were exposed to 0.5 μg/ml MMC for 1 h and allowed to recover after removal of the drug. The cells were then washed with PBS and fixed in 3% paraformaldehyde at room temperature for 10 min. Cells were permeabilized with 0.5% Triton X-100 for 5 min and incubated overnight with a corresponding primary antibody solution with 3% bovine serum albumin and 0.1% Triton X-100. Following three washes with PBS, cells were incubated with fluorescein isothiocyanate- or rhodamine-conjugated secondary antibodies. Finally, cells were washed with PBS and coverslips were mounted using ProLong Diamond Antifade Mountant with DAPI (P36971; Thermo Fisher Scientific). Images were obtained under a Nikon 90i microscope.
Gel filtration
For our gel filtration experiment, HEK293A-wild-type (WT) and SLX4IP-KO cells were mock-treated, treated with 1 μm MMC for 24 h, or irradiated with 100 J/m2 UV and released for 4 h. 2.5 × 107 cells were lysed in 1 ml buffer containing 50 mM Tris–HCl 7.4, 1 mM EDTA, 0.15 M sodium chloride, 0.1% (v/v) 2-mercaptoethanol, 1% NP-40, 750 units of TurboNuclease (Biovision, Milpitas, CA, #9207), and protease inhibitor cocktail for 30 min on ice. Cell lysates were centrifuged at 13,200 rpm for 30 min in a cold room and the supernatant was filtered through a 0.22 μm filter and loaded into a Superdex 200 10/300 GL preparative-grade column (GE Healthcare, Chicago, IL) using the same buffer used for lysis. The flow rate was 0.25 ml/min. 96 fractions were collected in volumes of 200 μl. Fractions as indicated were analyzed with antibodies against SLX4, XPF, MUS81 and SLX4IP.
Whole-cell extract and chromatin extract preparation
For preparation of whole-cell extract, cells were collected, washed with PBS, and boiled at 95°C for 10 min in 1× Laemmli buffer. For preparation of chromatin extract, cells were lysed with ice-cold NETN lysis buffer (50 mM Tris–HCl, pH 7.4, 100 mM NaCl, 0.4% NP-40, 1 mM EDTA) and protease inhibitor cocktail for 10 min on ice. Cell lysates were centrifuged at 13 200 rpm for 10 min in a cold room. Pellets were washed with NETN lysis buffer two more times and boiled at 95°C for 10 min in 1× Laemmli buffer.
Modified alkaline comet assay
To detect the unhooking of ICL lesions, HEK293A-WT, XPF-KO and SLX4IP-KO cells were treated with 1 μm MMC for 1 h. Cells were washed and incubated in fresh medium for 9, 12, 24 or 48 h. The unhooking of ICL lesions was detected via a modified alkaline comet assay (76). In brief, cells were digested with trypsin and diluted to a density of 2.5 × 104 cells/ml. Diluted cells were transferred into 24-well plates and irradiated with 8 Gy of X-irradiation while on ice. Next, 500 μl of irradiated cells or unirradiated control cells were mixed with 1 ml of 1% (wt/vol) low-gelling-temperature agarose (A4018; Sigma-Aldrich) and spread over slides precoated with 1% (wt/vol) agarose LE (Thomas Scientific, Swedesboro, NJ, USA). After the agarose solidified, slides were placed into ice-cold lysis buffer (100 mM disodium EDTA, 2.5 M NaCl, 10 mM Tris–HCl, pH 10.5) containing 1% Triton X-100 and were incubated on ice for 1 h in the dark. Slides were then washed three times with ice-cold water for 15 min and were transferred into an electrophoresis tank containing ice-cold alkaline solution (50 mM NaOH, 1 mM disodium EDTA, pH 12.5). After 45 min incubation in the dark, slides were subjected to electrophoresis at 18 V for 25 min. After electrophoresis, slides were carefully removed and neutralized with 0.5 M Tris–HCl (pH 7.5) for 10 min and then were washed with PBS for 10 min. Slides were dried overnight and stained with 2.5 μg/ml propidium iodide solution on the second day. Comet assay results were analyzed using a Nikon 90i microscope at ×20 magnification. At least 50 cells were analyzed for each sample by using the OpenComet plug-in in ImageJ. The degree of DNA interstand cross-linking was defined as the percentage decrease in olive tail movement and calculated by the formula:
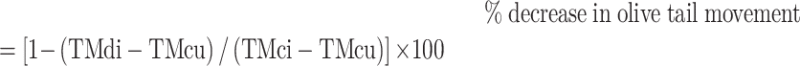 |
where TMdi = tail movement of MMC-treated irradiated samples; TMcu = tail movement of untreated, unirradiated controls; and TMci = tail movement of untreated irradiated controls.
Statistical analysis
All the experiments, such as the fluorescence-activated cell sorting, comet assay, and immunofluorescence staining experiments, were performed at least three times. Differences between groups were analyzed using Student's t-tests. P values <0.05 were considered statistically significant.
RESULTS
SLX4-interacting proteins are identified by Tandem affinity purification (TAP)
As described above, SLX4 functions as a platform for recruiting its coordinated proteins to different sites of DNA damage. However, how DNA damage regulates the recruitment of SLX4 and its function at distinct DNA damage sites, especially at ICLs, has yet to be fully elucidated. To explore the specific regulation of SLX4 recruitment and its function in DNA damage repair, we initiated this study by identifying SLX4-interacting proteins before and after DNA damage. We fused S protein, 2× Flag, and streptavidin-binding peptide (SFB tags to the C-terminus of SLX4 and generated stable HEK293T-derivative cell lines expressing these fusion proteins in a manner similar to that done in our previous studies (77,78). We performed TAP with streptavidin and S beads and analyzed SLX4 coprecipitated proteins using MS (Figure 1A). As expected, we successfully recovered several reported SLX4-interacting proteins at the top of our HCIP list, including XPF–ERCC1, PLK1, MSH2, SLX1A, TERF2-TERF2IP, SLX4IP and MUS81 (Figure 1B; Supplemental Table S1). Meanwhile, we also fused SFB tags to the N-terminus of SLX4 and performed TAP-MS experiments similar to those performed for the C-terminus tag. In the N-terminus SFB-SLX4 purification, some of the reported SLX4-interacting proteins, including PLK1, SLX1A, TERF2 and MUS81, did not exhibit strong interaction with SLX4 (Supplemental Figure S1). It has been reported that these proteins interact with the C-terminus or the middle region of SLX4; for example, SLX1 interacts with SLX4 residues 1751–1811 and MUS81 with SLX4 residues 1540–1623 (60–63). We suspected that N-terminal-tagged SLX4 might not be stable during purification and that it might lose its C-terminus-interacting proteins. Additionally, we identified PGAM5, RTEL1, TOPBP1 and KEAP1 as potential SLX4-interacting proteins. RTEL1 is an ATP-dependent DNA helicase implicated in counteracting SLX4 for the maintenance of telomere length (79–81); the roles of PGAM5, TOPBP1 and KEAP1 in SLX4-mediated functions have yet to be determined.
Figure 1.

SLX4-interacting proteins are identified by Tandem affinity purification (TAP). (A) Schematic of approaches to profiling SLX4-interacting proteins via tandem affinity purification (TAP) with streptavidin and S beads. (B) List of high-confidence candidate interacting proteins from mass spectrometry (MS) analysis of C-terminal, SFB-tagged SLX4 TAP results with and without mitomycin C (MMC)-based treatment. PSM, peptide spectrum match. SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis.
To identify ICL-related, SLX4-interacting proteins, we treated HEK293T cells stably expressing SLX4-SFB with 100 ng/ml of MMC before TAP-MS. We did not observe any obvious enhanced interactions between SLX4 and several reported SLX4-interacting proteins, such as XPF–ERCC1, MUS81-EME1, SLX1A and SLX4IP, following MMC exposure. One explanation is that the TAP protocol obscured any subtle differences that might have occurred following DNA damage. Nevertheless, these results demonstrated that the associations of these proteins with SLX4 do not change dramatically before or after DNA damage. Among these proteins, SLX4IP binds strongly to SLX4 and appears to be a constitutive member of the SLX4 complexes. SLX4IP (also called C20orf94) was first identified as part of the SLX4 complexes (60–63). The relationship between SLX4IP and SLX4 complexes and the roles of SLX4IP in DNA repair have yet to be explored and are the foci of this study.
SLX4IP is involved in ICL repair
To investigate the functions of SLX4IP in SLX4 complexes, we generated SLX4IP-KO cells and other repair gene-KO cells using CRISPR/Cas9 gene editing technology (Supplemental Figure S2A) and determined the sensitivity of these KO cells to treatment with different DNA-damaging agents. We failed to create HEK293A cells with complete XPF-KO. However, we obtained an XPF-KO cell line in which one allele has a frameshift mutation and stops at codon (residue) 431 and the other allele has a deletion of 32 residues (from residue 405 to residue 436). The XPF expression level in this cell line is extremely low and barely detectable. We decided to use this XPF-KO cell line (clone #12) for subsequent experiments. As shown in Figure 2A, SLX4IP-KO cells were highly sensitive to treatment with the ICL-inducing agent MMC but not to that with UV radiation, IR, or CPT in clonogenic assays. These results strongly suggested that SLX4IP mainly functions in ICL repair. Because SLX4 coordinates at least two nucleases (XPF–ERCC1 and MUS81-EME1) in ICL repair, we further compared the sensitivities of SLX4IP-KO, XPF-KO, and MUS81-KO cells to treatment with MMC. Specifically, we treated these KO cells with MMC at a series of concentrations for 3 days and monitored cell viability using a CellTiter-Glo luminescent assay. In this assay, SLX4IP-KO cells exhibited moderate sensitivity to MMC, with about a twofold lower half-maximal inhibitory concentration (IC50) than that seen in WT cells (Supplemental Figure S2B). Reconstitution of SLX4IP-KO cells with the SLX4IP-SFB construct fully rescued this phenotype (Supplemental Figure S2B). In addition, whereas XPF-KO cells were hypersensitive to MMC, MUS81-KO cells were only slightly sensitive to it (Supplemental Figure S2C). However, MUS81-KO cells exhibited hypersensitivity to treatment with CPT, whereas SLX4IP-KO and XPF-KO cells did not (Supplemental Figure S2D).
Figure 2.

SLX4IP is involved in ICL repair. (A) Results of clonogenic survival assays conducted with HEK293A-WT and SLX4IP-KO cells exposed to DNA damage-inducing agents (mitomycin C [MMC], campothecin [CPT], ultraviolet [UV] radiation, or ionizing radiation [IR]). For each cell line, the viability of untreated cells was defined as 100%. Data are presented as the mean ± standard error of the mean (SEM; n = 3). (B and C) HEK293A-WT, SLX4IP-KO, XPF-KO and MUS81-KO cells were treated with the indicated concentrations of MMC for 24 h or were nontreated (NT). Cells were collected and fixed with 70% ethanol before fluorescence-activated cell sorting (FACS) analysis of the cell-cycle distribution under different treatment conditions. The mean percentages of cells in the G2/M phase from three independent repeats of FACS are shown. The results were compared statistically with those for untreated cells. Statistical analysis was performed using the Student's t-test. **P < 0.01, ***P < 0.001, ns: not significant. P values <0.05 were considered statistically significant. (D) Immunostainings of SLX4, SLX4IP and γH2AX proteins performed following UV laser-induced DNA damage with the indicated antibodies. KO, knockout; WT, wild type.
We next examined other hallmarks of defective ICL repair in SLX4IP-KO cells. The presence of DNA damage in a cell triggers a complex DNA damage response, resulting in activation of the DNA damage checkpoint and leading to cell-cycle arrest. Because cells from FA patients accumulate in G2/M phase after exposure to low doses of ICL-inducing agents (82,83), we treated SLX4IP-KO, XPF-KO, and MUS81-KO cells with 10 or 20 ng/ml MMC for 24 h and determined the cell-cycle distributions (Figure 2B and C). As expected, WT cells nearly bypassed the G2/M arrest induced by treatment with low doses of MMC. Positive control XPF-KO cells exhibited dramatic accumulation of cells in the G2/M phase after MMC-based treatment. We also observed significant accumulation of SLX4IP-KO cells in the G2/M phase, demonstrating that these cells are defective in ICL repair. On the other hand, MUS81-KO cells did not undergo obvious G2/M arrest after MMC-based treatment. These results are consistent with those of DNA-damage sensitivity assays and support a role for SLX4IP in ICL repair.
We next employed a UV laser-induced damage system to investigate the recruitment of SLX4IP to sites of DNA damage. UV laser-induced stripes formed on exogenously expressed SLX4IP after damage, just like SLX4 (Figure 2D). Taken together, these data suggested that SLX4IP is involved in ICL repair.
The interaction between SLX4IP and SLX4 is coordinated with XPF
To reveal the underlying mechanism of the involvement of SLX4IP in ICL repair, we tagged SFB to the C-terminus of SLX4IP and performed TAP-MS as described above. We found SLX4 and XPF–ERCC1 at the top of the list of SLX4IP-binding proteins (Figure 3A; Supplemental Table S1). We also identified other reported SLX4-interacting proteins, such as MUS81, PLK1, and MSH2, but only limited numbers of peptides were found (Supplemental Table S1). Meanwhile, we identified some other potential SLX4IP-interacting proteins, such as PASK, AJUBA, LRP4 and TRIM26, although no studies have demonstrated that they participate in DNA repair. SLX4 and XPF–ERCC1 are the major SLX4IP-interacting proteins, with or without MMC-based treatment (Figure 3A). Studies have demonstrated that SLX4 coordinates with XPF–ERCC1 and participates in the unhooking of ICLs during the repair process (31,43,67,84). The binding of SLX4IP to both SLX4 and XPF–ERCC1 inspired us to further investigate whether SLX4IP plays a role in regulating SLX4–XPF–ERCC1 interaction.
Figure 3.

The interaction between SLX4IP and SLX4 is coordinated with XPF. (A) Tandem affinity purification and mass spectrometry (TAP-MS) was conducted to identify SLX4IP-interacting proteins. Lists of high-confidence candidate interacting proteins from mass spectrometry analysis of cells treated with or without mitomycin C (MMC) are presented. (B) HEK293A cells were transfected with constructs encoding SLX4-SFB or its deletion variants and were subjected to immunoprecipitation (IP) with S beads. Western blotting was conducted with antibodies as indicated. (C) Experiments were done as in B, except that SLX4-SFB wild type and SLX4m-SFB (L530A, F545A, Y546A and L550A) variants were subjected to precipitation with S beads. FL, full length (controls); PSM, peptide spectrum match.
We first examined the interaction between SLX4 and SLX4IP in HEK293A cells. We generated a series of SLX4 deletion mutants based on SLX4’s known interaction domain or functional motif. We found that deletion of the MEI9XPF interaction like region (MLR) of SLX4 (residues 409–555), which is considered the binding site for XPF–ERCC1 (84,85), abolished its interaction with SLX4IP (Figure 3B). We generated further small-deletion mutants in the MLR domain and found that deletion of residues 501–555 of SLX4 not only abolished its interaction with XPF but also impaired its interaction with SLX4IP (Supplemental Figure S3A). Meanwhile, these SLX4 deletion mutants could still bind to MUS81 and SLX1 (Supplemental Figure S3B). We also examined the four-point SLX4 mutation SLX4L530A, F545A, Y546A, L550A, which has been shown to be defective in its binding to XPF , and observed that this mutant also had a significant reduction in its binding to both XPF and SLX4IP, although it still had some residual interaction with SLX4IP (Figure 3C). Collectively, these data strongly suggested that the binding of XPF and SLX4IP to SLX4 are coordinated. We envisioned at least three possibilities to explain these interactions: (i) SLX4IP may bridge the interaction between SLX4 and XPF, (ii) XPF may bridge the interaction between SLX4 and SLX4IP and (iii) SLX4IP may bind to the SLX4–XPF–ERCC1 complex.
The conserved N-terminus of SLX4IP is responsible for its interaction with SLX4–XPF–ERCC1
To test the possibility that SLX4IP bridges the interaction between SLX4 and XPF–ERCC1, we monitored the interaction sites on SLX4IP for SLX4 or XPF. We generated a series of SLX4IP deletion mutants based on its predicted secondary structures (Figure 4A) and examined the mutant's interactions with SLX4 and XPF–ERCC1. Deletion of residues 1–27, 28–67 and 68–140 of SLX4IP abolished the interaction of SLX4IP with SLX4 but also impaired SLX4IP’s interaction with XPF–ERCC1 (Figure 4A). Because SLX4IP is a relatively small protein (45 kDa), we reasoned that disruption of any part of its N-terminus may disrupt the structure of the N-terminus and potentially affect SLX4IP’s binding to its associated proteins.
Figure 4.

The conserved N-terminus of SLX4IP is responsible for its interaction with SLX4–XPF–ERCC1. (A) HEK293A cells were transfected with constructs encoding SLX4IP-SFB or its deletion variants and subjected to immunoprecipitation (IP) pulldown with S beads. Western blotting conducted with antibodies as indicated. A schematic overview of the deletion strategy and protein-protein interaction results is presented. (B) Conservation of sequences at the N-terminus of SLX4IP. The alignment of the N-terminal sequences of SLX4IP in humans, mice, zebrafish and Xenopus, established using the Clustal Omega sequence alignment program, is shown. (C) HEK293A cells were transfected with constructs encoding SLX4IP-SFB or its point mutation variants and subjected to IP with S beads. Western blotting was conducted with antibodies as indicated. (D) SLX4 was subjected to IP with anti-SLX4 serum from HEK293A-WT or SLX4IP-KO cells (clone #6 and clone #13). Western blotting was conducted with antibodies as indicated. (E) HEK293A-WT or SLX4IP-KO cells were transfected with SFB-XPF and subjected to IP with S beads. Western blotting was conducted with antibodies as indicated. (F) Western blots detecting the interaction between XPF and SLX4 in HEK293A-WT and SLX4IP-KO cells were analyzed by ImageJ. The mean relative interaction from five experiments is shown. FL, full length (controls); KO, knockout; WT, wild type.
We then asked whether we could identify any point mutations that could differentiate the interactions between SLX4IP and SLX4 or XPF–ERCC1. Because SLX4IP is conserved in organisms ranging from Xenopus to humans, especially at its N-terminus (Figure 4B), we generated a number of point mutations in these conserved amino acids and examined the ability of the mutants to interact with SLX4 and/or XPF–ERCC1. Notably, we found that four-point mutants, L16K/V17K, W32A/F33A, K91R92A and V115K/V116K, were defective in binding to both SLX4 and XPF–ERCC1 (Figure 4C).
The inability to separate the interaction of SLX4IP with SLX4 from its interaction with XPF suggests that it is unlikely that SLX4IP would bridge the interaction between SLX4 and XPF–ERCC1. Thus, we directly tested the interaction between SLX4 and XPF in SLX4IP-KO cells. We first used anti-SLX4 serum to immunoprecipitate endogenous SLX4 from cells and found that the absence of SLX4IP did not abolish the interaction between SLX4 and XPF–ERCC1 (Figure 4D). Similarly, we used overexpressed, SFB-tagged XPF and found that XPF was able to bind to SLX4 in the absence of SLX4IP (Figure 4E). When we used ImageJ to quantify Western blots from five experiments detecting the relative interaction between XPF and SLX4 in WT control and SLX4IP-KO cells, we found no significant change in SLX4–XPF interaction in the SLX4IP-KO cells (Figure 4F), although there was a trend toward a reduction in this interaction. Therefore, we conclude that SLX4IP is not essential for the interaction between SLX4 and XPF–ERCC1.
XPF’s helicase-like domain is responsible for its interaction with SLX4 and SLX4IP
Next, we tested the possibility that XPF may bridge the interaction between SLX4 and SLX4IP. We overexpressed SFB-tagged SLX4IP in XPF-KO cells and found that the interaction between SLX4 and SLX4IP was dramatically impaired in these KO cells (Figure 5A). These data suggest that, while XPF may not bridge the interaction between SLX4 and SLX4IP, it is nevertheless important for this interaction. We also performed IP of endogenous SLX4 using anti-SLX4 serum in WT and XPF-KO cells and showed that, without XPF, the interaction between SLX4 and SLX4IP was greatly diminished or abolished (Supplemental Figure S3C). However, the caveat is that endogenous SLX4IP expression level was greatly reduced in XPF-KO cells (Supplemental Figure S3C).
Figure 5.

The helicase-like domain of XPF is responsible for its interaction with SLX4 and SLX4IP. (A) HEK293A-WT or XPF-KO cells were transfected with SLX4IP-SFB and subjected to immunoprecipitation (IP) with S beads. Western blotting was conducted with antibodies as indicated. (B) A schematic overview of the XPF domains, XPF deletion mutants, and results of protein-protein interactions is presented. (C and D) XPF-KO cells were transfected with a construct encoding SFB-XPF, a deletion mutant of SFB-XPF, or a point mutation variant of SFB-XPF along with a construct encoding GFP-SLX4 (C) or GFP-SLX4IP (D). Cell lysates were subjected to IP with S beads. Western blotting was conducted with antibodies as indicated. (E) A schematic model shows the interaction domain of SLX4–XPF–ERCC1-SLX4IP. (F) To induce degradation of SLX4-mAID-SFB protein, 1 mM auxin (IAA) was added to cells for 48 h. Western blotting was conducted with antibodies as indicated. (G) HEK293A SLX4-mAID-SFB knock-in (KI) cells were infected with a virus encoding HA-SLX4IP and treated with or without auxin for 48 h. Cells were collected, lysed, and subjected to IP with hemagglutinin (HA) beads. Western blotting was conducted with antibodies as indicated. (H) HEK293A cells were infected with a virus encoding inducible control shRNA or shRNA for SLX4 (#84 and #85). To induce knockdown of SLX4 protein, 1 μg/ml of doxycycline (DOX) was added to cells for 48 h. Western blotting was conducted to detect the levels of the indicated proteins in whole-cell extract and chromatin extracts. (I) The expression levels of proteins in HEK293A-WT, SLX4IP-KO, XPF-KO and XPF/SLX4IP-DKO cells were detected with antibodies as indicated. HLD, helicase-like domain; KO, knockout; WT, wild type.
We again attempted to identify XPF mutants that can separate the interactions of XPF with SLX4 or SLX4IP. We generated a series of XPF deletion mutants and monitored their interactions with SLX4 and SLX4IP (Figure 5B). We co-transfected cells with constructs encoding GFP-tagged SLX4 or SLX4IP and SFB-tagged WT or XPF mutants (Figure 5C and D). We found that deletion of any part of the N-terminus of XPF’s helicase-like domain (HLD) simultaneously impaired its interaction with SLX4 and SLX4IP (Figure 5C and D).
Previous studies have shown that XPF mutations can cause various human diseases, such as progeria, Cockayne syndrome, and/or FA (86–89). Accordingly, we generated several disease-associated XPF point mutations and examined their interactions with SLX4 and SLX4IP. We observed decreased interaction of XPFR153P or XPFL230P mutants with both SLX4 and SLX4IP, but not that of XPFR689S or other XPF mutants (Figure 5C and D; Supplemental Figure S3D). The XPFR153P and XPFL230P mutations are mapped to XPF’s HLD, which is mainly responsible for mediating protein–protein interactions, while the XPFR689S mutation is located in the nuclease domain of XPF. The XPFR153P mutation was found in a patient who was diagnosed with an extreme progeroid syndrome and with both NER and ICL repair defects with cellular sensitivity to UV and ICLs (90). XPFL230P and XPFR689S mutations were identified in FA patients (89). We reconstituted XPF-KO cells with WT or mutant XPF (XPFR153P, XPFL230P or XPFR689S) and examined WT or mutant XPF’s ability to rescue MMC sensitivity in XPF-KO cells. Whereas XPF-WT fully rescued MMC sensitivity, XPFR153P, XPFL230P or XPFR689S only partially suppressed MMC sensitivity (Supplemental Figure S3E). We reasoned that the inability of XPFR153P and XPFL230P mutants to interact with SLX4 and SLX4IP may be responsible, at least in part, for their defects in ICL repair. The XPFR689S mutant was able to interact with SLX4 and SLX4IP. However, this mutation is located within the XPF nuclease domain, suggesting that the enzymatic function of XPF is important for ICL repair. Taken together, these data revealed that, while XPF is important for the interaction between SLX4 and SLX4IP, it does not act as a protein that bridges the SLX4-SLX4IP interaction. Instead, XPF, SLX4 and SLX4IP interactions appear to be coordinated (Figure 5E).
The stability of SLX4IP depends on its binding to the SLX4–XPF–ERCC1 complex
The data described above showed that, whereas SLX4IP is not essential for the interaction between SLX4 and XPF–ERCC1, loss of XPF significantly reduced the binding between SLX4 and SLX4IP. We therefore asked what would happen to the interaction between XPF and SLX4IP in the absence of SLX4. We used auxin to induce the degradation of endogenously expressed SLX4-mAID-SFB in HEK293A cells. In this system, we firstly introduced a construct encoding the auxin-responsive F-box protein TIR1 from Oryza sativa (OsTIR1) into the genomic AAVS1 locus (72). We then tagged endogenous SLX4 with mAID using a CRISPR/Cas9-mediated HR approach (Supplemental Figure S4A). We confirmed single clones with insertions at both alleles using genomic PCR and sequencing (Supplemental Figure S4B). We induced degradation of SLX4 protein by adding auxin to cell culture media, and we checked SLX4 protein expression with Western blotting (Figure 5F). We then infected SLX4-mAID-SFB cells with a virus encoding for HA-SLX4IP and obtained cells stably expressing HA-SLX4IP. We immunoprecipitated SLX4IP with hemagglutinin (HA) beads before and after induction of SLX4 protein degradation. Of note, induction of SLX4 downregulation led to a reduction in SLX4IP protein expression, but not of XPF protein expression (Figure 5G). Although the SLX4IP protein level decreased, the residual HA-SLX4IP could still interact with XPF (Figure 5G). We could not draw any reliable conclusion about SLX4IP-XPF interaction based on these experiments because we could not completely abolish SLX4 expression using this auxin-inducible system. However, the results described above (Figure 5G) suggested that the SLX4IP protein level may depend on SLX4. We further confirmed these results by KD SLX4 protein expression via inducible shRNAs targeting SLX4. We found that SLX4IP protein expression was dramatically decreased upon induction of SLX4 KD, while the levels of XPF and MUS81 were not changed in these SLX4-KD cells (Figure 5H).
We also found that, in XPF-KO cells, the expression level of SLX4IP protein also decreased dramatically (Figure 5I). It is known that depletion of XPF causes significant reduction of its heterodimer partner, ERCC1 (85,91). Indeed, we observed the same extent of reduction of SLX4IP and ERCC1 protein levels in XPF-KO cells. With reintroduction of XPF into these KO cells, the expression level of SLX4IP protein and the interaction of SLX4IP with SLX4 were recovered (Supplemental Figure S4C). These data further confirmed the formation of a stable SLX4IP-SLX4–XPF–ERCC1 complex and suggested that binding to the SLX4–XPF–ERCC1 complex helps stabilize SLX4IP protein. On the other hand, we did not observe any change of SLX4IP expression level in MUS81-KO cells (Supplemental Figure S4D).
Meanwhile, the chromatin binding of residual SLX4IP also decreased in XPF-KO and SLX4 KD cells, which we determined by comparing the ratio of SLX4IP in KO/KD cells to WT in whole-cell extracts versus those in chromatin extracts (Figure 5H and Supplemental Figure S4D). These data suggest that the binding of SLX4IP to chromatin also depends on SLX4 and XPF. We also observed that the amount of XPF and MUS81 binding to chromatin decreased considerably in the absence of SLX4 (Figure 5H), which demonstrated the important role of SLX4 in mediating the functions of these proteins on chromatin DNA.
Reduction of SLX4IP protein expression in XPF-deficient cells is partially responsible for the hypersensitivity of these cells to treatment with MMC
The above experiments demonstrated that SLX4IP forms a stable complex with SLX4–XPF–ERCC1. To determine whether the function of SLX4IP is closely associated with that of XPF, we generated XPF/SLX4IP-double knockout (DKO) cells and examined the epistatic relationship between SLX4IP and XPF. We found that XPF/SLX4IP-DKO cells were slightly more sensitive to treatment with MMC than XPF-KO cells (Figure 6A and Supplemental Figure S5A). Also, XPF/SLX4IP-DKO cells grew much more slowly than WT, XPF-KO or SLX4IP-KO cells (Supplemental Figure S5B). Because the expression level of SLX4IP is already reduced to a relatively low level in XPF-KO cells, the effects of further deletion of SLX4IP may be due to an XPF-independent function of SLX4IP or to further reduction of residual XPF activity that depends on SLX4IP. We did not observe any increased sensitivity of XPF/SLX4IP-DKO cells to treatment with CPT (Supplemental Figure S5C), demonstrating that SLX4IP’s function may not be important for other repair pathways.
Figure 6.

SLX4IP functions in the same pathway as XPF but not as MUS81. (A–C) Proliferation of HEK293A-WT, SLX4IP-KO, XPF-KO, and other cell lines as indicated was measured using a CellTiter-Glo assay after 3 days in the presence of the indicated concentrations of mitomycin C (MMC). Data are presented as mean ± the standard error of the mean (n = 3). (D) Experiments were done as presented in A, B, and C. Proliferation of the indicated cell lines was measured using a CellTiter-Glo assay after 3 days in the presence of the indicated concentrations of camptothecin (CPT). DKO, double knockout; IC50, half-maximal inhibitory concentration; KO, knockout; WT, wild type.
Next, we examined whether the increased sensitivity to treatment with MMC and growth defects observed in XPF/SLX4IP-DKO cells are due to further reduction of SLX4IP protein expression. When we reintroduced XPF into XPF/SLX4IP-DKO cells, the MMC sensitivity of these reconstituted cells was comparable with that of SLX4IP-KO cells, as expected (IC50 of 28.43 compared with 30.9 in SLX4IP-KO cells) (Figure 6A). However, when we reintroduced SLX4IP into XPF/SLX4IP-DKO cells, the MMC IC50 in these cells increased to 21.47, which was much higher than that in XPF-KO cells (3.999) (Figure 6A). We further confirmed this observation by introducing SLX4IP into XPF-KO cells and also observed a reduction in MMC sensitivity in them (Supplemental Figure S5D). We suspected that MMC hypersensitivity in XPF-KO cells may partially result from the reduction of SLX4IP protein expression and that overexpression of SLX4IP somehow restores the ICL repair efficiency of these cells.
We also examined the epistatic relationship between SLX4IP and MUS81 by generating SLX4IP/MUS81-DKO cells. Although MUS81-KO cells were hypersensitive to treatment with CPT, they were not very sensitive to treatment with MMC (Supplemental Figure S2C and D). SLX4IP/MUS81-DKO cells exhibited slightly increased MMC sensitivity (Figure 6B), which can be explained by the fact that they function in two different pathways. Similarly, XPF/MUS81-DKO cells had only very modest increases in their sensitivity to treatment with MMC (Figure 6C). Of note, XPF/MUS81-DKO and SLX4IP/MUS81-DKO cells exhibited a slight reduction, not an increase, in CPT sensitivity compared to MUS81-KO cells (Figure 6D), which indicates that XPF and SLX4IP are likely to act very differently from MUS81 in CPT-based damage repair. We also treated these KO cells with 20 ng/ml MMC and determined the cell-cycle distributions at different time points (Supplemental Figure S6A). XPF/SLX4IP-DKO and XPF/MUS81-DKO cells exhibited dramatic accumulation of cells in the G2/M phase after MMC-based treatment for 48 h (Supplemental Figure S6B). However, we did not observe any statistically significant differences in these DKO cells compared to XPF-KO cells or in SLX4IP/MUS81-DKO cells compared to SLX4IP-KO cells (Supplemental Figure S6B).
SLX4IP promotes the interaction between SLX4 and XPF–ERCC1
We observed ICL repair defects in SLX4IP-KO cells and hypersensitivity of XPF-KO cells to treatment with MMC, which may be due in part to the reduction of SLX4IP expression in these cells. We next sought to reveal the mechanism of ICL repair defects in SLX4IP-KO cells. Previous studies have revealed that cells have two pools of XPF–ERCC1: one is co-eluted with SLX4, and the other is eluted at a lower molecular weight (61,92). We performed gel filtration experiments using HEK293A whole-cell extract prepared under normal conditions or after MMC and UV radiation treatment and confirmed the co-elution of SLX4IP with SLX4, XPF, and MUS81 in the higher molecular weight fractions (Figure 7A). Of note, we found a slight decrease in the amount of XPF co-eluted with SLX4 in SLX4IP-KO cells, as well as a corresponding increase in the amount of XPF in lower molecular weight fractions, after treatment with MMC (Figure 7A and Supplemental Figure S7); these findings indicated a very modest decrease of SLX4–XPF interaction in MMC-treated SLX4IP-KO cells. No differences were found under normal conditions or after UV radiation treatment.
Figure 7.

SLX4IP stabilizes the interaction between SLX4 and XPF–ERCC1, especially after DNA damage. (A) HEK293A-WT and SLX4IP-KO cells were nontreated (NT) or treated with 1 μm mitomycin C (MMC) for 24 h, and radiated with 100 J/m2 ultraviolet (UV) radiation and released for 4 h. Whole-cell extracts were analyzed by gel filtration on a Superdex 200 10/300 GL preparative-grade column. 96 fractions were collected in volumes of 200 μl. Fractions as indicated were analyzed with antibodies as indicated. (B) SLX4 was subjected to immunoprecipitation (IP) with anti-SLX4 serum using lysates of HEK293A-WT cells, XPF-KO cells, and other indicated cell lines. (C) The same amount of GFP or GFP-SLX4293-638 was mixed with GFP beads. After rotation in a cold room for 1 h, protein-bound beads were washed with NETN lysis buffer. The indicated amounts of XPF and/or SLX4IP were mixed with NETN lysis buffer to the same volume, and the mixture was allowed to bind to protein bound on GFP beads. After mixing in a cold room for 1 h, beads were thoroughly washed and boiled at 95°C in Laemmli buffer. GFP and GFP-SLX4293-638 were detected using Coomassie Blue staining. XPF was detected using an anti-GST antibody, and SLX4IP was detected using an anti-MBP antibody. Quantification analysis was done with ImageJ and the amount of relative XPF protein bound to the beads is shown. DKO, double knockout; KO, knockout; WT, wild type.
In our XPF-KO cells, the level of expression of the truncated form of XPF was very low (Supplemental Figure S8A), and we usually could not detect its interaction with SLX4 (Figure 7B, lane 3). However, when we overexpressed SLX4IP in XPF-KO cells, the interaction between the truncated form of XPF and SLX4 increased (Figure 7B, lanes 7 and 8). This was not due to a change in the expression level of this truncated XPF (Supplemental Figure S8A). The increased interaction of SLX4 with the truncated mutant of XPF may explain why reconstitution of XPF/SLX4IP-DKO and XPF-KO cells with SLX4IP strongly suppressed the MMC sensitivity in these cells (Figure 6A and Supplemental Figure S5D); in other words, SLX4IP promoted the interaction between SLX4 and truncated XPF. To further confirm SLX4IP’s ability to promote interaction between SLX4 and XPF, we purified GFP-SLX4293-638, GST-XPF1-655, and MBP-SLX4IP1-300 from Escherichia coli. We loaded GFP-SLX4 into beads and added XPF, together with an increasing amount of SLX4IP protein. We found that, with increased addition of SLX4IP protein, the interaction between SLX4 and XPF also increased (Supplemental Figure S8B). Although we observed some nonspecific binding of SLX4IP protein to the beads, the binding did not induce an increase in binding of XPF to GFP (the control) (Supplemental Figure S8B). When repeated these experiments with the inclusion of another negative control—i.e., purified MBP-SLX4IP205-408, which does not interact with SLX4 and XPF—we obtained the same results (Figure 7C). We also tried to monitor the interaction between SLX4 and XPF in SLX4IP-KO cells under normal and DNA damage conditions but failed to detect obvious changes (Supplemental Figure S8C and S8D). We reasoned that the technical limitations of our immunoprecipitation experiments may have prevented the detection of any subtle changes.
Loss of SLX4IP reduces ICL repair efficiency in vivo
Based on the results above, we hypothesized that, whereas a fraction of XPF proteins always form a complex with SLX4 under normal conditions, this interaction must be further enhanced or stabilized for SLX4-coordinated XPF function after DNA damage. SLX4IP may stabilize the interaction between SLX4 and XPF to achieve a highly efficient SLX4–XPF complex for ICL repair. Therefore, SLX4IP may act as a unique ICL repair factor that assists the XPF-SLX4/ICL pathway following DNA damage (Figure 8A). To test our hypothesis, we monitored the ICL repair process in SLX4IP-KO cells in vivo. We performed a modified comet assay to measure the unhooking process of ICL (76). We found that, after cells had been treated with 0.5 μg/ml of MMC for 1 h and released for 48 h, ICLs formed in WT cells were nearly fully unhooked, whereas some ICLs remained in XPF-KO and SLX4IP-KO cells (Figure 8B). These data suggest that, as was the case with the loss of XPF, the loss of SLX4IP reduced unhooking efficiency.
Figure 8.

Loss of SLX4IP reduces the repair efficiency of interstrand crosslink repair in vivo. (A) Working model of SLX4–XPF–ERCC1-SLX4IP interaction and function. Our current working hypothesis is that SLX4IP acts to promote the interaction between SLX4 and XPF–ERCC1, especially following DNA damage. This function of SLX4IP is critical for interstrand crosslink (ICL) repair. In our XPF-KO cells, truncated XPF was expressed only at low levels and SLX4IP protein was mostly degraded. Therefore, a very limited SLX4–XPF–ERCC1-SLX4IP complex formed and we observed severe ICL repair defects. When we overexpressed SLX4IP in these cells, the interaction between truncated XPF and SLX4 was enhanced, which suppressed the hypersensitivity of XPF-KO or XPF/SLX4IP-DKO cells to mitomycin C (MMC). We found that mAID-mediated downregulation of SLX4 also caused downregulation of SLX4IP protein, which may result in the failure of XPF–ERCC1 recruitment and diminished cell survival. As for SLX4IP-KO cells, SLX4 could still interact with XPF–ERCC1, but the complex was not stable; this was especially true following DNA damage, which resulted in reduced ICL repair efficiency. (B) Cells were treated with 1 μm MMC for 1 h and released for the indicated times. Samples were taken to detect the unhooking of ICL using a modified comet assay (please see the Material and Methods section for detailed description). DNA ICL is expressed as the percentage of decrease in Olive tail movement. Data are presented as the mean ± the standard error of the mean (n = 3). (C) HEK293A cells were mock-treated (Mock) or treated with MMC (either 1# 0.5 μg/ml MMC for 1 h and release for 24 h, or 2# 0.1 μg/ml MMC for 24 h). After treatment, whole-cell extracts were prepared and subjected to Western blotting with the indicated antibodies. (D) HEK293A-WT, SLX4IP-KO, XPF-KO, and MUS81-KO cells were exposed to 0.5 μg/ml MMC for 1 h and allowed to recover after removal of the drug. Cells were then fixed with 3% paraformaldehyde and stained with an anti-γH2AX antibody. The percentages of cells with more than five γH2AX foci are shown. Data are presented as the mean ± the standard error of the mean (n = 3). Statistical analysis was performed using the Student's t-test. *P < 0.05, **P < 0.01, ***P < 0.001, ns: not significant. P values less than 0.05 were considered statistically significant. KO, knockout; WT, wild type.
ICLs induce complex DNA damage responses in cells. A replication fork stalls when encountering an ICL, since it cannot unwind double-stranded DNA in the ICL’s presence. This stalled replication fork may activate a replication stress-induced checkpoint. Additionally, several steps in the processing of an ICL can potentially produce DSBs (3,25). For example, if a replication fork is encountered during nuclease-mediated unhooking of an ICL, the replication fork may collapse and cause DSBs. Alternatively, stalled replication forks may regress into ‘chicken foot’ structures with double-stranded ends (93). Replication-associated DSBs may not only activate the ATR-CHK1 pathway, but also activate the ATM-CHK2 pathway. We thus examined several established DNA-damage response markers and found that, after MMC-based treatment at either (i) 0.5 μg/ml for 1 h and release for 24 h or (ii) 0.1 μg/ml for 24 h, the levels of γH2AX, P-Chk2 (T68), and P-Chk1 (S345) increased in SLX4IP-KO cells (Figure 8C), indicating persistent DNA damage and delayed DNA repair without SLX4IP. After treating cells with 0.5 μg/ml MMC for 1 h and releasing them at different time points, we further detected γH2AX foci and confirmed delayed repair in SLX4IP-KO cells compared with WT cells (Figure 8D). These results further demonstrate that SLX4IP is important for efficient ICL repair in vivo.
DISCUSSION
SLX4 is a multi-domain protein that functions as a platform for recruiting other proteins to various sites of DNA damage to facilitate DNA repair. SLX4-coordinated protein complexes are reported to participate in ICL repair, HR, and telomere-length maintenance (63–65,80,94,95). In the present study, we demonstrated the regulatory roles of SLX4IP in SLX4-containing protein complexes. We observed binding of SLX4IP to both SLX4 and XPF–ERCC1 and showed that these interactions are highly coordinated; disrupting one interaction also affects the other, making SLX4IP a bona fide candidate for regulating the stability and activity of the SLX4–XPF–ERCC1 complex. In addition, the binding of SLX4IP to the SLX4–XPF–ERCC1 complex is important for the stability of the SLX4IP protein, suggesting that SLX4IP is a key component of the SLX4–XPF–ERCC1 complex. We further showed that SLX4IP promotes the interaction between SLX4 and XPF in vitro and enhances the interaction between truncated XPF and SLX4 in vivo. The finding that fractions of SLX4-co-eluted XPF may decrease, albeit only slightly, in SLX4IP-KO cells after MMC-based treatment suggests that SLX4IP stabilizes SLX4–XPF interaction following DNA damage and therefore promotes ICL repair (Figure 7A). We further examined the epistatic relationship between SLX4IP and XPF and found that XPF/SLX4IP-DKO cells had slightly greater MMC sensitivity than did XPF-KO cells, which may be due to the need for residual SLX4IP in the promotion of interaction between truncated XPF and SLX4. Our current working hypothesis also explains the growth defect in XPF/SLX4IP-DKO cells and the suppression of MMC sensitivity of XPF-KO or XPF/SLX4IP-DKO cells by SLX4IP overexpression.
Our data suggested that the major function of SLX4IP is to participate in ICL repair, as the loss of SLX4IP sensitized cells to treatment with the ICL-inducing agent MMC but not to treatment with IR, UV or CPT. In addition, we observed accumulation of SLX4IP-KO cells in the G2/M phase after low-dose MMC treatment, which is a typical indication of an ICL repair defect. On the other hand, others have reported that SLX4 deficiency also sensitized cells to CPT-based treatment (64) and that cells with defective XPF protein are hypersensitive to treatment with UV radiation. However, it is known that SLX4’s CPT-related DNA repair function depends on its interaction with MUS81 endonuclease and that XPF’s function in the repairing of UV radiation-induced DNA damage is linked with the known contributions of XPF in NER via its association with XPA, but not its interaction with SLX4. The fact that SLX4IP simultaneously binds to SLX4 and XPF limits its function to SLX4–XPF–ERCC1 complex-involved repair pathways, such as ICL repair. This implies that the main function of SLX4IP is to augment the SLX4–XPF–ERCC1 complex following DNA damage.
XPF–ERCC1 is known to act in both the NER and FA pathways. Mutations of the XPF gene can cause progeria, Cockayne syndrome, and FA (86–90,96). Researchers have speculated that cells have two distinct XPF pools: one associates with XPA and functions in the NER pathway, whereas the other associates with SLX4 and functions in the FA pathway (61,92). We did not identify any NER-related proteins in our TAP-MS purification of SLX4 or SLX4IP, further indicating the exclusive nature of these two XPF pools. However, it is likely that both NER and FA pathways are required for ICL repair. As Supplemental Figure S3E shows, the SLX4 interaction-deficient mutation XPFL230P partially suppresses the MMC hypersensitivity of XPF-KO cells, suggesting that other SLX4-independent functions of XPF may also contribute to ICL repair.
Although an in vitro assay with a recombinant SLX4 complex without SLX4IP can cleave a broad range of DNA substrates (97), the ICL repair processes in vivo may be considerably more complex. SLX4IP is not essential for ICL repair, nor is it essential for the interaction between SLX4 and XPF–ERCC1. However, the results of our in vivo studies implied that SLX4IP may stabilize the SLX4–XPF–ERCC1 complex after DNA damage, which is important for maintaining highly efficient, SLX4-coordinated XPF activity during ICL repair. ICL repair is a complex process that requires the coordination of several different DNA repair pathways. Our findings indicate the existence of a novel regulatory mechanism involved in the interplay between the different repair pathways in ICL repair.
Structural analysis of SLX4IP-SLX4–XPF complex with DNA substrates will be very helpful in elucidating the details of SLX4IP function in promoting SLX4–XPF interaction. Although the phenotypes of SLX4IP-KO cells are relatively mild, we cannot exclude the possibilities that SLX4IP may affect the biochemical activities and/or other functions of SLX4–XPF–ERCC1 complex. Thus, we should determine whether SLX4IP may have any additional roles in regulating the enzymatic activity and/or substrate preference of SLX4–XPF–ERCC1 complex. We would like to point out that while this manuscript was under review, Dr. Simon Boulton and colleagues from the Francis Crick Institute showed that SLX4IP also functions in alternative lengthening of telomere (98), suggesting that SLX4IP-SLX4–XPF complex has roles beyond ICL repair.
Although depletion of XPF or SLX4 is rare in cancers, several studies have reported deletion of SLX4IP by illegitimate V/(D)/J-mediated recombination in about 30% of patients with childhood acute lymphoblastic leukemia (99–101). Whether patients with this cancer have ICL repair defects due to SLX4IP deletion must be further investigated. To better understand the relationship between SLX4IP and cancer, and to explore the potential of targeting SLX4IP deficiency in cancer treatment, we searched several public databases (e.g. the Broad Institute Cancer Cell Line Encyclopedia, the cBioPortal for Cancer Genomics, and The Human Protein Atlas) for information regarding SLX4IP. We found that SLX4IP expression level closely correlates with survival probability in pancreatic cancer (Supplemental Figure S9A). In addition, SLX4IP expression is relatively low in some cases of renal cancer (Supplemental Figure S9B). Furthermore, SLX4IP’s mRNA level is significantly reduced in a subset of Hodgkin lymphoma cell lines (Supplemental Figure S9C). Whether SLX4IP can be used as a biomarker for these cancers remains to be determined. If so, ICL-inducing agents may be effective treatment options for acute lymphoblastic leukemia, pancreatic cancer, and Hodgkin lymphoma in patients with SLX4IP deletions or downregulation.
CONCLUSION
In this study, we showed that the loss of SLX4IP caused cells to become sensitive to treatment with ICL-inducing agents and led to accumulation of cells at the G2/M phase after low-dose treatment with MMC. We further demonstrated that SLX4IP localizes to ultraviolet laser-induced sites of DNA damage, suggesting that SLX4IP is directly involved in DNA damage repair. Moreover, we showed that SLX4IP interacts with both SLX4 and XPF–ERCC1 and that this interaction is important for SLX4IP protein stability. Finally, we observed that SLX4IP is important for promotion of the interaction between SLX4 and XPF–ERCC1 after DNA damage. Collectively, these results demonstrate a novel regulatory role for SLX4IP in SLX4-coordinated ICL repair.
DATA AND SOFTWARE AVAILABILITY
The MS proteomics data (dataset identifier PXD011727) have been deposited with the ProteomeXchange Consortium via the PRIDE partner repository.
Supplementary Material
ACKNOWLEDGEMENTS
We thank all the members of the Chen laboratory for their help and constructive discussions. We also thank Laura Russell in the Department of Scientific Publications from The University of Texas MD Anderson for help in the scientific editing of the manuscript.
Author contributions: H.Z. and J.C. conceived the project. H.Z., Z.C., Y.Y., Z.Y., D.C., Y.X., M.S., X.F., M.T. and C.W. performed the experiments. J.A.T. helped with gel filtration experiments. H.Z. and J.C. wrote the manuscript with input from all authors.
Notes
Present address: Dan Cao, Department of Gastroenterology, The First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, Anhui 230001, P.R. China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health/National Cancer Institute (NIH/NCI) [P01CA193124 to J.C]; J.C. also received support from Cancer Prevention & Research Institute of Texas [RP160667 to J.C.]; NIH [CA157448, CA210929, CA216911, CA216437 to J.C.]; and the Pamela and Wayne Garrison Distinguished Chair in Cancer Research; J.A.T. is supported by NIH [P01 CA092584. NIH R35 CA220430], a Robert A. Welch Chemistry Chair, and the Cancer Prevention and Research Institute of Texas; University of Texas MD Anderson Cancer Center's Flow Cytometry and Cellular Imaging Facility (supported by the NIH/NCI [P30CA016672]; MD Anderson's Functional Genomics Core, which is also supported by NIH/NCI [P30CA016672], for help with SLX4 shRNAs. Funding for open access charge: NIH [P01 CA193124].
Conflict of interest statement. None declared.
REFERENCES
- 1. Hashimoto S., Anai H., Hanada K.. Mechanisms of interstrand DNA crosslink repair and human disorders. Genes Environ. 2016; 38:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Muniandy P.A., Liu J., Majumdar A., Liu S.T., Seidman M.M.. DNA interstrand crosslink repair in mammalian cells: step by step. Crit. Rev. Biochem. Mol. Biol. 2010; 45:23–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Deans A.J., West S.C.. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer. 2011; 11:467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bradner W.T. Mitomycin C: a clinical update. Cancer Treat. Rev. 2001; 27:35–50. [DOI] [PubMed] [Google Scholar]
- 5. Crooke S.T., Bradner W.T.. Mitomycin C: a review. Cancer Treat. Rev. 1976; 3:121–139. [DOI] [PubMed] [Google Scholar]
- 6. Van Wassenhove L.D., Mochly-Rosen D., Weinberg K.I.. Aldehyde dehydrogenase 2 in aplastic anemia, Fanconi anemia and hematopoietic stem cells. Mol. Genet. Metab. 2016; 119:28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pontel L.B., Rosado I.V., Burgos-Barragan G., Garaycoechea J.I., Yu R., Arends M.J., Chandrasekaran G., Broecker V., Wei W., Liu L. et al.. Endogenous formaldehyde is a hematopoietic stem cell genotoxin and metabolic carcinogen. Mol. Cell. 2015; 60:177–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Duxin J.P., Walter J.C.. What is the DNA repair defect underlying Fanconi anemia. Curr. Opin. Cell Biol. 2015; 37:49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang L.C., Gautier J.. The Fanconi anemia pathway and ICL repair: implications for cancer therapy. Crit. Rev. Biochem. Mol. Biol. 2010; 45:424–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Auerbach A.D. Fanconi anemia and its diagnosis. Mutat. Res. 2009; 668:4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim H., D’Andrea A.D.. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev. 2012; 26:1393–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deakyne J.S., Mazin A.V.. Fanconi anemia: at the crossroads of DNA repair. Biochemistry. 2011; 76:36–48. [DOI] [PubMed] [Google Scholar]
- 13. Soulier J., Leblanc T., Larghero J., Dastot H., Shimamura A., Guardiola P., Esperou H., Ferry C., Jubert C., Feugeas J.P. et al.. Detection of somatic mosaicism and classification of Fanconi anemia patients by analysis of the FA/BRCA pathway. Blood. 2005; 105:1329–1336. [DOI] [PubMed] [Google Scholar]
- 14. Joenje H., Levitus M., Waisfisz Q., D’Andrea A., Garcia-Higuera I., Pearson T., van Berkel C.G., Rooimans M.A., Morgan N., Mathew C.G. et al.. Complementation analysis in Fanconi anemia: assignment of the reference FA-H patient to group A. Am. J. Hum. Genet. 2000; 67:759–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Genuardi M., Chiurazzi P., Capelli A., Neri G.. X-linked VACTERL with hydrocephalus: the VACTERL-H syndrome. Birth Defects Orig. Artic. Ser. 1993; 29:235–241. [PubMed] [Google Scholar]
- 16. Meetei A.R., Medhurst A.L., Ling C., Xue Y., Singh T.R., Bier P., Steltenpool J., Stone S., Dokal I., Mathew C.G. et al.. A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat. Genet. 2005; 37:958–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sarkar S., Davies A.A., Ulrich H.D., McHugh P.J.. DNA interstrand crosslink repair during G1 involves nucleotide excision repair and DNA polymerase zeta. EMBO J. 2006; 25:1285–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wood R.D. Mammalian nucleotide excision repair proteins and interstrand crosslink repair. Environ. Mol. Mutagen. 2010; 51:520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McHugh P.J., Sarkar S.. DNA interstrand cross-link repair in the cell cycle: a critical role for polymerase zeta in G1 phase. Cell Cycle. 2006; 5:1044–1047. [DOI] [PubMed] [Google Scholar]
- 20. Williams H.L., Gottesman M.E., Gautier J.. Replication-independent repair of DNA interstrand crosslinks. Mol. Cell. 2012; 47:140–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Klug A.R., Harbut M.B., Lloyd R.S., Minko I.G.. Replication bypass of N2-deoxyguanosine interstrand cross-links by human DNA polymerases eta and iota. Chem. Res. Toxicol. 2012; 25:755–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Minko I.G., Harbut M.B., Kozekov I.D., Kozekova A., Jakobs P.M., Olson S.B., Moses R.E., Harris T.M., Rizzo C.J., Lloyd R.S.. Role for DNA polymerase kappa in the processing of N2-N2-guanine interstrand cross-links. J. Biol. Chem. 2008; 283:17075–17082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ben-Yehoyada M., Wang L.C., Kozekov I.D., Rizzo C.J., Gottesman M.E., Gautier J.. Checkpoint signaling from a single DNA interstrand crosslink. Mol. Cell. 2009; 35:704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Knipscheer P., Raschle M., Smogorzewska A., Enoiu M., Ho T.V., Scharer O.D., Elledge S.J., Walter J.C.. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science. 2009; 326:1698–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Raschle M., Knipscheer P., Enoiu M., Angelov T., Sun J., Griffith J.D., Ellenberger T.E., Scharer O.D., Walter J.C.. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell. 2008; 134:969–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rothfuss A., Grompe M.. Repair kinetics of genomic interstrand DNA cross-links: evidence for DNA double-strand break-dependent activation of the Fanconi anemia/BRCA pathway. Mol. Cell Biol. 2004; 24:123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. De Silva I.U., McHugh P.J., Clingen P.H., Hartley J.A.. Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol. Cell Biol. 2000; 20:7980–7990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hinz J.M. Role of homologous recombination in DNA interstrand crosslink repair. Environ. Mol. Mutagen. 2010; 51:582–603. [DOI] [PubMed] [Google Scholar]
- 29. Michl J., Zimmer J., Tarsounas M.. Interplay between Fanconi anemia and homologous recombination pathways in genome integrity. EMBO J. 2016; 35:909–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Niedernhofer L.J., Odijk H., Budzowska M., van Drunen E., Maas A., Theil A.F., de Wit J., Jaspers N.G., Beverloo H.B., Hoeijmakers J.H. et al.. The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol. Cell Biol. 2004; 24:5776–5787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kuraoka I., Kobertz W.R., Ariza R.R., Biggerstaff M., Essigmann J.M., Wood R.D.. Repair of an interstrand DNA cross-link initiated by ERCC1-XPF repair/recombination nuclease. J. Biol. Chem. 2000; 275:26632–26636. [DOI] [PubMed] [Google Scholar]
- 32. Crossan G.P., van der Weyden L., Rosado I.V., Langevin F., Gaillard P.H., McIntyre R.E., Gallagher F., Kettunen M.I., Lewis D.Y., Brindle K. et al.. Disruption of mouse Slx4, a regulator of structure-specific nucleases, phenocopies Fanconi anemia. Nat. Genet. 2011; 43:147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hanada K., Budzowska M., Modesti M., Maas A., Wyman C., Essers J., Kanaar R.. The structure-specific endonuclease Mus81-Eme1 promotes conversion of interstrand DNA crosslinks into double-strands breaks. EMBO J. 2006; 25:4921–4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim Y., Lach F.P., Desetty R., Hanenberg H., Auerbach A.D., Smogorzewska A.. Mutations of the SLX4 gene in Fanconi anemia. Nat. Genet. 2011; 43:142–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Stoepker C., Hain K., Schuster B., Hilhorst-Hofstee Y., Rooimans M.A., Steltenpool J., Oostra A.B., Eirich K., Korthof E.T., Nieuwint A.W. et al.. SLX4, a coordinator of structure-specific endonucleases, is mutated in a new Fanconi anemia subtype. Nat. Genet. 2011; 43:138–141. [DOI] [PubMed] [Google Scholar]
- 36. Kratz K., Schopf B., Kaden S., Sendoel A., Eberhard R., Lademann C., Cannavo E., Sartori A.A., Hengartner M.O., Jiricny J.. Deficiency of FANCD2-associated nuclease KIAA1018/FAN1 sensitizes cells to interstrand crosslinking agents. Cell. 2010; 142:77–88. [DOI] [PubMed] [Google Scholar]
- 37. Liu T., Ghosal G., Yuan J., Chen J., Huang J.. FAN1 acts with FANCI-FANCD2 to promote DNA interstrand cross-link repair. Science. 2010; 329:693–696. [DOI] [PubMed] [Google Scholar]
- 38. MacKay C., Declais A.C., Lundin C., Agostinho A., Deans A.J., MacArtney T.J., Hofmann K., Gartner A., West S.C., Helleday T. et al.. Identification of KIAA1018/FAN1, a DNA repair nuclease recruited to DNA damage by monoubiquitinated FANCD2. Cell. 2010; 142:65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Smogorzewska A., Desetty R., Saito T.T., Schlabach M., Lach F.P., Sowa M.E., Clark A.B., Kunkel T.A., Harper J.W., Colaiacovo M.P. et al.. A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol. Cell. 2010; 39:36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Graf N., Ang W.H., Zhu G., Myint M., Lippard S.J.. Role of endonucleases XPF and XPG in nucleotide excision repair of platinated DNA and cisplatin/oxaliplatin cytotoxicity. Chembiochem. 2011; 12:1115–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fisher L.A., Bessho M., Wakasugi M., Matsunaga T., Bessho T.. Role of interaction of XPF with RPA in nucleotide excision repair. J. Mol. Biol. 2011; 413:337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ciccia A., McDonald N., West S.C.. Structural and functional relationships of the XPF/MUS81 family of proteins. Annu. Rev. Biochem. 2008; 77:259–287. [DOI] [PubMed] [Google Scholar]
- 43. Klein Douwel D., Boonen R.A., Long D.T., Szypowska A.A., Raschle M., Walter J.C., Knipscheer P.. XPF–ERCC1 acts in Unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol. Cell. 2014; 54:460–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Niedernhofer L.J., Odijk H., Budzowska M., van Drunen E., Maas A., Theil A.F., de Wit J., Jaspers N.G.J., Beverloo H.B., Hoeijmakers J.H.J. et al.. The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol. Cell Biol. 2004; 24:5776–5787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bergstralh D.T., Sekelsky J.. Interstrand crosslink repair: can XPF–ERCC1 be let off the hook. Trends Genet. 2008; 24:70–76. [DOI] [PubMed] [Google Scholar]
- 46. Adair G.M., Rolig R.L., Moore-Faver D., Zabelshansky M., Wilson J.H., Nairn R.S.. Role of ERCC1 in removal of long non-homologous tails during targeted homologous recombination. EMBO J. 2000; 19:5552–5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fisher L.A., Bessho M., Bessho T.. Processing of a psoralen DNA interstrand cross-link by XPF–ERCC1 complex in vitro. J. Biol. Chem. 2008; 283:1275–1281. [DOI] [PubMed] [Google Scholar]
- 48. Wang A.T., Sengerova B., Cattell E., Inagawa T., Hartley J.M., Kiakos K., Burgess-Brown N.A., Swift L.P., Enzlin J.H., Schofield C.J. et al.. Human SNM1A and XPF–ERCC1 collaborate to initiate DNA interstrand cross-link repair. Genes Dev. 2011; 25:1859–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen X.B., Melchionna R., Denis C.M., Gaillard P.H., Blasina A., Van de Weyer I., Boddy M.N., Russell P., Vialard J., McGowan C.H.. Human Mus81-associated endonuclease cleaves Holliday junctions in vitro. Mol. Cell. 2001; 8:1117–1127. [DOI] [PubMed] [Google Scholar]
- 50. Regairaz M., Zhang Y.W., Fu H.Q., Agama K.K., Tata N., Agrawal S., Aladjem M.I., Pommier Y.. Mus81-mediated DNA cleavage resolves replication forks stalled by topoisomerase I-DNA complexes. J. Cell Biol. 2011; 195:739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Osman F., Whitby M.C.. Exploring the roles of Mus81-Eme1/Mms4 at perturbed replication forks. DNA Repair (Amst.). 2007; 6:1004–1017. [DOI] [PubMed] [Google Scholar]
- 52. Thongthip S., Bellani M., Gregg S.Q., Sridhar S., Conti B.A., Chen Y., Seidman M.M., Smogorzewska A.. Fan1 deficiency results in DNA interstrand cross-link repair defects, enhanced tissue karyomegaly, and organ dysfunction. Genes Dev. 2016; 30:645–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yoshikiyo K., Kratz K., Hirota K., Nishihara K., Takata M., Kurumizaka H., Horimoto S., Takeda S., Jiricny J.. KIAA1018/FAN1 nuclease protects cells against genomic instability induced by interstrand cross-linking agents. Proc. Natl Acad. Sci. U.S.A. 2010; 107:21553–21557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhou W., Otto E.A., Cluckey A., Airik R., Hurd T.W., Chaki M., Diaz K., Lach F.P., Bennett G.R., Gee H.Y. et al.. FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat. Genet. 2012; 44:910–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lachaud C., Moreno A., Marchesi F., Toth R., Blow J.J., Rouse J.. Ubiquitinated Fancd2 recruits Fan1 to stalled replication forks to prevent genome instability. Science. 2016; 351:846–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Iyama T., Lee S.Y., Berquist B.R., Gileadi O., Bohr V.A., Seidman M.M., McHugh P.J., Wilson D.M. 3rd. CSB interacts with SNM1A and promotes DNA interstrand crosslink processing. Nucleic Acids Res. 2015; 43:247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cattell E., Sengerova B., McHugh P.J.. The SNM1/Pso2 family of ICL repair nucleases: from yeast to man. Environ. Mol. Mutagen. 2010; 51:635–645. [DOI] [PubMed] [Google Scholar]
- 58. Ishiai M., Kimura M., Namikoshi K., Yamazoe M., Yamamoto K., Arakawa H., Agematsu K., Matsushita N., Takeda S., Buerstedde J.M. et al.. DNA cross-link repair protein SNM1A interacts with PIAS1 in nuclear focus formation. Mol. Cell Biol. 2004; 24:10733–10741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mullen J.R., Kaliraman V., Ibrahim S.S., Brill S.J.. Requirement for three novel protein complexes in the absence of the Sgs1 DNA helicase in Saccharomyces cerevisiae. Genetics. 2001; 157:103–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Fekairi S., Scaglione S., Chahwan C., Taylor E.R., Tissier A., Coulon S., Dong M.Q., Ruse C., Yates J.R. 3rd, Russell P. et al.. Human SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell. 2009; 138:78–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Munoz I.M., Hain K., Declais A.C., Gardiner M., Toh G.W., Sanchez-Pulido L., Heuckmann J.M., Toth R., Macartney T., Eppink B. et al.. Coordination of structure-specific nucleases by human SLX4/BTBD12 is required for DNA repair. Mol. Cell. 2009; 35:116–127. [DOI] [PubMed] [Google Scholar]
- 62. Andersen S.L., Bergstralh D.T., Kohl K.P., LaRocque J.R., Moore C.B., Sekelsky J.. Drosophila MUS312 and the vertebrate ortholog BTBD12 interact with DNA structure-specific endonucleases in DNA repair and recombination. Mol. Cell. 2009; 35:128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Svendsen J.M., Smogorzewska A., Sowa M.E., O’Connell B.C., Gygi S.P., Elledge S.J., Harper J.W.. Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell. 2009; 138:63–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kim Y., Spitz G.S., Veturi U., Lach F.P., Auerbach A.D., Smogorzewska A.. Regulation of multiple DNA repair pathways by the Fanconi anemia protein SLX4. Blood. 2013; 121:54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhang J., Walter J.C.. Mechanism and regulation of incisions during DNA interstrand cross-link repair. DNA Repair (Amst.). 2014; 19:135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Castor D., Nair N., Declais A.C., Lachaud C., Toth R., Macartney T.J., Lilley D.M., Arthur J.S., Rouse J.. Cooperative control of holliday junction resolution and DNA repair by the SLX1 and MUS81-EME1 nucleases. Mol. Cell. 2013; 52:221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hodskinson M.R., Silhan J., Crossan G.P., Garaycoechea J.I., Mukherjee S., Johnson C.M., Scharer O.D., Patel K.J.. Mouse SLX4 is a tumor suppressor that stimulates the activity of the nuclease XPF–ERCC1 in DNA crosslink repair. Mol. Cell. 2014; 54:472–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kim Y., Lach F.P., Desetty R., Hanenberg H., Auerbach A.D., Smogorzewska A.. Mutations of the SLX4 gene in Fanconi anemia. Nat. Genet. 2011; 43:142–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Stoepker C., Hain K., Schuster B., Hilhorst-Hofstee Y., Rooimans M.A., Steltenpool J., Oostra A.B., Eirich K., Korthof E.T., Nieuwint A.W. et al.. SLX4, a coordinator of structure-specific endonucleases, is mutated in a new Fanconi anemia subtype. Nat. Genet. 2011; 43:138–141. [DOI] [PubMed] [Google Scholar]
- 70. Sanjana N.E., Shalem O., Zhang F.. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014; 11:783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Shalem O., Sanjana N.E., Hartenian E., Shi X., Scott D.A., Mikkelson T., Heckl D., Ebert B.L., Root D.E., Doench J.G. et al.. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014; 343:84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Natsume T., Kiyomitsu T., Saga Y., Kanemaki M.T.. Rapid protein depletion in human cells by auxin-inducible degron tagging with short homology donors. Cell Rep. 2016; 15:210–218. [DOI] [PubMed] [Google Scholar]
- 73. Srivastava M., Chen Z., Zhang H., Tang M., Wang C., Jung S.Y., Chen J.. Replisome dynamics and their functional relevance upon DNA damage through the PCNA interactome. Cell Rep. 2018; 25:3869–3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chen Z., Tran M., Tang M., Wang W., Gong Z.. Proteomic analysis reveals a novel mutator S (MutS) partner involved in mismatch repair pathway. Mol. Cell Proteomics. 2016; 15:1299–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Li X., Han H., Zhou M.T., Yang B., Ta A.P., Li N., Chen J., Wang W.. Proteomic analysis of the human tankyrase protein interaction network reveals Its role in pexophagy. Cell reports. 2017; 20:737–749. [DOI] [PubMed] [Google Scholar]
- 76. Spanswick V.J., Hartley J.M., Ward T.H., Hartley J.A.. Measurement of drug-induced DNA interstrand crosslinking using the single-cell gel electrophoresis (comet) assay. Methods Mol. Med. 1999; 28:143–154. [DOI] [PubMed] [Google Scholar]
- 77. Huen M.S., Grant R., Manke I., Minn K., Yu X., Yaffe M.B., Chen J.. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007; 131:901–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Huang J., Huen M.S., Kim H., Leung C.C., Glover J.N., Yu X., Chen J.. RAD18 transmits DNA damage signalling to elicit homologous recombination repair. Nat. Cell Biol. 2009; 11:592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Vannier J.B., Pavicic-Kaltenbrunner V., Petalcorin M.I., Ding H., Boulton S.J.. RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell. 2012; 149:795–806. [DOI] [PubMed] [Google Scholar]
- 80. Sarkar J., Wan B., Yin J., Vallabhaneni H., Horvath K., Kulikowicz T., Bohr V.A., Zhang Y., Lei M., Liu Y.. SLX4 contributes to telomere preservation and regulated processing of telomeric joint molecule intermediates. Nucleic Acids Res. 2015; 43:5912–5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Margalef P., Kotsantis P., Borel V., Bellelli R., Panier S., Boulton S.J.. Stabilization of Reversed Replication Forks by Telomerase Drives Telomere Catastrophe. Cell. 2018; 172:439–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Heinrich M.C., Hoatlin M.E., Zigler A.J., Silvey K.V., Bakke A.C., Keeble W.W., Zhi Y., Reifsteck C.A., Grompe M., Brown M.G. et al.. DNA cross-linker-induced G2/M arrest in group C Fanconi anemia lymphoblasts reflects normal checkpoint function. Blood. 1998; 91:275–287. [PubMed] [Google Scholar]
- 83. Andreassen P.R., Ren K.. Fanconi anemia proteins, DNA interstrand crosslink repair pathways, and cancer therapy. Curr. Cancer Drug Targets. 2009; 9:101–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hashimoto K., Wada K., Matsumoto K., Moriya M.. Physical interaction between SLX4 (FANCP) and XPF (FANCQ) proteins and biological consequences of interaction-defective missense mutations. DNA Repair (Amst.). 2015; 35:48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Choi Y.J., Ryu K.S., Ko Y.M., Chae Y.K., Pelton J.G., Wemmer D.E., Choi B.S.. Biophysical characterization of the interaction domains and mapping of the contact residues in the XPF–ERCC1 complex. J. Biol. Chem. 2005; 280:28644–28652. [DOI] [PubMed] [Google Scholar]
- 86. Kashiyama K., Nakazawa Y., Pilz D.T., Guo C., Shimada M., Sasaki K., Fawcett H., Wing J.F., Lewin S.O., Carr L. et al.. Malfunction of nuclease ERCC1-XPF results in diverse clinical manifestations and causes Cockayne syndrome, xeroderma pigmentosum, and Fanconi anemia. Am. J. Hum. Genet. 2013; 92:807–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Fujiwara Y., Ichihashi M., Uehara Y., Matsumoto A., Yamamoto Y., Kano Y., Tanakura Y.. Xeroderma pigmentosum groups C and F: additional assignments and a review of the subjects in Japan. J. Radiat. Res. 1985; 26:443–449. [DOI] [PubMed] [Google Scholar]
- 88. Norris P.G., Hawk J.L., Avery J.A., Giannelli F.. Xeroderma pigmentosum complementation group F in a non-Japanese patient. J. Am. Acad. Dermatol. 1988; 18:1185–1188. [DOI] [PubMed] [Google Scholar]
- 89. Bogliolo M., Schuster B., Stoepker C., Derkunt B., Su Y., Raams A., Trujillo J.P., Minguillon J., Ramirez M.J., Pujol R. et al.. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am. J. Hum. Genet. 2013; 92:800–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Niedernhofer L.J., Garinis G.A., Raams A., Lalai A.S., Robinson A.R., Appeldoorn E., Odijk H., Oostendorp R., Ahmad A., Van Leeuwen W. et al.. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature. 2006; 444:1038–1043. [DOI] [PubMed] [Google Scholar]
- 91. Tripsianes K., Folkers G., Ab E., Das D., Odijk H., Jaspers N.G., Hoeijmakers J.H., Kaptein R., Boelens R.. The structure of the human ERCC1/XPF interaction domains reveals a complementary role for the two proteins in nucleotide excision repair. Structure. 2005; 13:1849–1858. [DOI] [PubMed] [Google Scholar]
- 92. Perez-Oliva A.B., Lachaud C., Szyniarowski P., Munoz I., Macartney T., Hickson I., Rouse J., Alessi D.R.. USP45 deubiquitylase controls ERCC1-XPF endonuclease-mediated DNA damage responses. EMBO J. 2015; 34:326–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Gari K., Decaillet C., Delannoy M., Wu L., Constantinou A.. Remodeling of DNA replication structures by the branch point translocase FANCM. Proc. Natl Acad. Sci. U.S.A. 2008; 105:16107–16112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Wan B., Yin J., Horvath K., Sarkar J., Chen Y., Wu J., Wan K., Lu J., Gu P., Yu E.Y. et al.. SLX4 assembles a telomere maintenance toolkit by bridging multiple endonucleases with telomeres. Cell Rep. 2013; 4:861–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Cybulski K.E., Howlett N.G.. FANCP/SLX4: a Swiss army knife of DNA interstrand crosslink repair. Cell Cycle. 2011; 10:1757–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Yamamura K., Ichihashi M., Hiramoto T., Ogoshi M., Nishioka K., Fujiwara Y.. Clinical and photobiological characteristics of xeroderma pigmentosum complementation group F: a review of cases from Japan. Br. J. Dermatol. 1989; 121:471–480. [DOI] [PubMed] [Google Scholar]
- 97. Wyatt H.D., Laister R.C., Martin S.R., Arrowsmith C.H., West S.C.. The SMX DNA repair tri-nuclease. Mol. Cell. 2017; 65:848–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Panier S., Maric M., Hewitt G., Mason-Osann E., Gali H., Dai A., Labadorf A., Guervilly J.H., Ruis P., Segura-Bayona S.. SLX4IP Antagonizes Promiscuous BLM Activity during ALT Maintenance. Mol. Cell. 2019; doi:10.1016/j.molcel.2019.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Meissner B., Bartram T., Eckert C., Trka J., Panzer-Grumayer R., Hermanova I., Ellinghaus E., Franke A., Moricke A., Schrauder A. et al.. Frequent and sex-biased deletion of SLX4IP by illegitimate V(D)J-mediated recombination in childhood acute lymphoblastic leukemia. Hum. Mol. Genet. 2014; 23:590–601. [DOI] [PubMed] [Google Scholar]
- 100. Mullighan C.G., Goorha S., Radtke I., Miller C.B., Coustan-Smith E., Dalton J.D., Girtman K., Mathew S., Ma J., Pounds S.B. et al.. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007; 446:758–764. [DOI] [PubMed] [Google Scholar]
- 101. Mullighan C.G., Downing J.R.. Global genomic characterization of acute lymphoblastic leukemia. Semin. Hematol. 2009; 46:3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MS proteomics data (dataset identifier PXD011727) have been deposited with the ProteomeXchange Consortium via the PRIDE partner repository.