

Abstract
Motivation
High performance computing (HPC) clusters play a pivotal role in large-scale bioinformatics analysis and modeling. For the statistical computing language R, packages exist to enable a user to submit their analyses as jobs on HPC schedulers. However, these packages do not scale well to high numbers of tasks, and their processing overhead quickly becomes a prohibitive bottleneck.
Results
Here we present clustermq, an R package that can process analyses up to three orders of magnitude faster than previously published alternatives. We show this for investigating genomic associations of drug sensitivity in cancer cell lines, but it can be applied to any kind of parallelizable workflow.
Availability and implementation
The package is available on CRAN and https://github.com/mschubert/clustermq. Code for performance testing is available at https://github.com/mschubert/clustermq-performance.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
The volume of data produced in the biological sciences has recently increased by orders of magnitude across many disciplines, most apparent in single cell sequencing (Svensson et al., 2018). In order to analyze this data, there is a need not only for efficient algorithms, but also for efficient and user-friendly utilization of high performance computing (HPC). Having reached a limit in the speed of single processors, the focus has shifted to distributing computing power to multiple processors or indeed multiple machines. HPC clusters have played and are continuing to play an integral role in bioinformatic data analysis and modelling. However, efficient parallelization using low-level systems such as MPI, or submitting jobs that later communicate via network sockets, requires specialist knowledge.
For the popular statistical computing language R (Ihaka and Gentleman, 1996) several packages have been developed that are able to automate parallel workflows on HPC without the need for low-level programming. The best-known packages for this are BatchJobs (Bischl et al., 2015) and batchtools (Lang et al., 2017). They provide a consistent interface for distributing tasks over multiple workers by automatically creating the files required for processing each individual computation, and collect the results back to the main session upon completion.
However, these packages write arguments and results of individual function calls to a networked file system. This is highly inefficient for a large number of calls and effectively limits these packages at about 106 function evaluations (cf. Fig. 1 and Supplementary Methods). In addition, it hinders load balancing between computing nodes (as it requires a file-system based lock mechanism) and the use of remote compute facilities without shared storage systems.
Fig. 1.
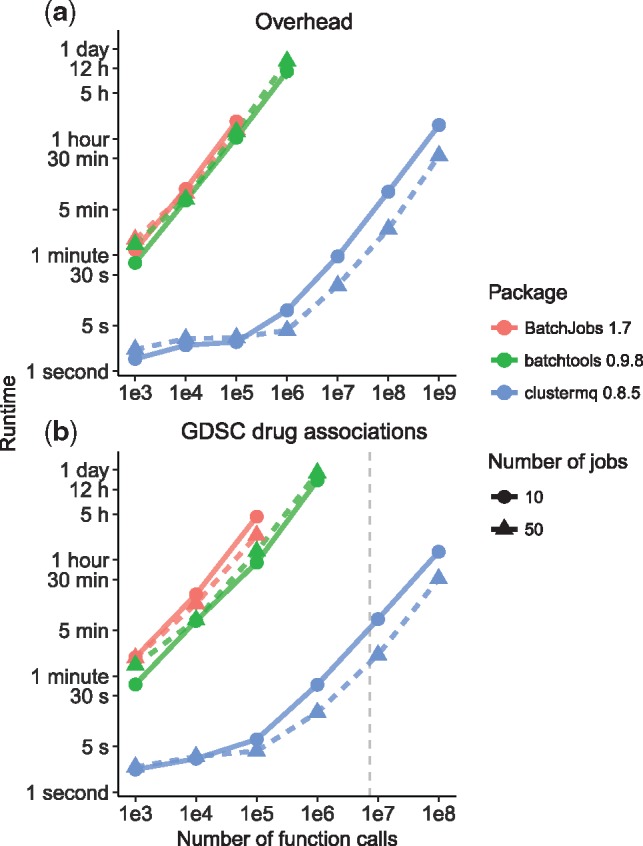
Performance evaluation of HPC packages for (a) processing overhead and (b) application to GDSC data. Along the range of tested number of function calls, clustermq requires substantially less time for processing in both scenarios. Indicated measurements are averages of two runs with range shown as vertical bars. (b) The dashed grey line indicates the actual number of calls required for all GDSC associations
Here we present the R package clustermq that overcomes these limitations and provides a minimal interface to submit jobs on a range of different schedulers (LSF, SGE, Slurm, PBS, Torque). It distributes data over the network without involvement of network-mounted storage, monitors the progress of up to 109 function evaluations and collects back the results.
2 Implementation
In order to provide efficient distribution of data as well as compute instructions, we use the ZeroMQ library (Hintjens, 2013), which provides a level of abstraction of simple network sockets and handles low-level operations such as message envelopes and timeouts. The main function used to distribute tasks on compute nodes and subsequently collect the results is the Q function. It takes named iterated arguments, and a list of const (objects that do not change their value between function calls) and export objects (which will be made available in the worker environment). The Q function will check which schedulers are available, and is hence often usable without any additional required setup (cf. Supplementary User Guide).
# load the library and create a simple function
library(clustermq)
fx= function(x, y) x * 2 + y
# queue the function call on your scheduler
Q(fx, x = 1: 3,const=list(y = 1),n_jobs = 1)
# list(3, 5, 7)
Another way of parallelization is to register clustermq as a parallel foreach backend. This is particularly useful if a third-party package uses foreach loops internally, like all Bioconductor (Gentleman et al., 2004) packages that make use of BiocParallel.
library(foreach)
register_dopar_cmq(n_jobs = 2, memory = 1024)
# this will be executed as jobs
foreach(i = 1: 3)
# also for Bioconductor packages using this
BiocParallel:: register(BiocParallel::DoparParam())
BiocParallel::bplapply(1: 3,sqrt)
In addition, the package provides a documented worker API that can be used to build tools that need fine-grained control over the calls sent out instead of the normal scatter-gather approach (cf. Supplementary Technical Documentation).
3 Evaluation
In order to evaluate the performance of clustermq compared to the BatchJobs and batchtools packages (cf. Supplementary Methods), we first tested the overhead cost for each of these tools by evaluating a function of negligible runtime and repeating this between 1000 and 109 times. We found that clustermq has about 1000× less overhead cost compared to the other packages when processing 105 or more calls, although across the whole range a clear speedup is apparent (Fig. 1a). The maximum number of calls that BatchJobs could successfully process was 105, while batchtools increased this limit to 106. By contrast, clustermq was able to process 109 calls in about one hour.
For our second evaluation, we chose a realistic scenario with application to biological data. The Genomics of Drug Sensitivity in Cancer (GDSC) project published molecular data of approximately 1000 cell lines and their response (IC50) to 265 drugs (Iorio et al., 2016). We ask the question if any one of 1073 genomic or epigenomic events (mutation/copy number aberration of a gene and differential promoter methylation, respectively) is correlated with a significant difference in drug sensitivity across all cell lines or for 25 specific cancer types (n = 7 392 970 associations). We found that for this setup, clustermq is able to process the associations in about one hour with 10% lost to overhead (Fig. 1b; dashed line). The other packages produced too many small temporary files for our networked file system to handle, and by extrapolation processing all associations would have taken over a week.
To achieve similar results using the previously published packages one would need to adapt the analysis code to chunk together related associations and explicitly loop through different subsets of data. clustermq lifts this requirement and lets the analyst focus on the biological question they are trying to address instead of manually optimizing code parallelization for execution time (cf. Supplementary Discussion).
4 Conclusion
The clustermq R package enables computational analysts to efficiently distribute a large number of function calls via HPC schedulers, while reducing the need to adapt code between different systems. We have shown its utility for drug screening data, but it can be used a broad range of analyses. This includes Bioconductor packages that make use of BiocParallel.
Conflict of Interest: none declared.
Supplementary Material
References
- Bischl B. et al. (2015) BatchJobs and BatchExperiments: abstraction mechanisms for using R in batch environments. J. Stat. Softw., 64, 1–25. [Google Scholar]
- Gentleman R.C. et al. (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol., 5, R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hintjens P. (2013) ZeroMQ: Messaging for Many Applications. O’Reilly Media, Inc, Sebastopol, California. [Google Scholar]
- Ihaka R., Gentleman R. (1996) R: a language for data analysis and graphics. J. Comput. Graph. Stat., 5, 299–314. [Google Scholar]
- Iorio F. et al. (2016) A landscape of pharmacogenomic interactions in cancer. Cell, 166, 740–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang M. et al. (2017) batchtools: tools for R to work on batch systems. J. Open Source Softw., 2, 135. [Google Scholar]
- Svensson V. et al. (2018) Exponential scaling of single-cell RNA-seq in the past decade. Nat. Protoc., 13, 599–604. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.