

Abstract
Motivation
Detection of somatic mutations from tumor and matched normal sequencing data has become among the most important analysis methods in cancer research. Some existing mutation callers have focused on additional information, e.g. heterozygous single-nucleotide polymorphisms (SNPs) nearby mutation candidates or overlapping paired-end read information. However, existing methods cannot take multiple information sources into account simultaneously. Existing Bayesian hierarchical model-based methods construct two generative models, the tumor model and error model, and limited information sources have been modeled.
Results
We proposed a Bayesian model integration framework named as partitioning-based model integration. In this framework, through introducing partitions for paired-end reads based on given information sources, we integrate existing generative models and utilize multiple information sources. Based on that, we constructed a novel Bayesian hierarchical model-based method named as OHVarfinDer. In both the tumor model and error model, we introduced partitions for a set of paired-end reads that cover a mutation candidate position, and applied a different generative model for each category of paired-end reads. We demonstrated that our method can utilize both heterozygous SNP information and overlapping paired-end read information effectively in simulation datasets and real datasets.
Availability and implementation
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Cancer is driven by genomic alterations. Acquired somatic mutations, together with individual germ line variations, are important factors in cancer evolution. Together with decreasing massively parallel sequencing costs, mutation calling from tumor and matched normal sequence data has become fundamental analysis methods in cancer research (Meyerson et al., 2010).
Previous statistical mutation callers can be mainly categorized into two types. The first type of mutation caller does not assume any probability distribution that is specific for sequence data (Koboldt et al., 2012; Yoshida et al., 2011), and mutation calling is conducted based on Fisher’s exact test (Fisher, 1925). For this type, the numbers of reference supporting reads and variant supporting reads are counted in tumor and normal samples, and a P-value is computed based on a 2 × 2 contingency table. These methods only consider differences in variant allele frequencies between tumor and normal samples and ignore the biases found in sequence errors and mapping errors.
The second type of mutation caller constructs generative models that are specific for sequence data. This type of method first prepares generative models for sequence data, and then computes statistical scores based on techniques, e.g. maximum a posteriori inference of genotypes (Roth et al., 2012) and Bayes factor-based model selection (Cibulskis et al., 2013; Moriyama et al., 2017; Usuyama et al., 2014). The most important advantage of the second approach is that we can construct generative models based on sequence data-specific information sources, and then utilize the given information sources. Some methods are known to perform well by using some characteristic information of heterozygous single-nucleotide polymorphisms (SNPs) nearby mutation candidates (Usuyama et al., 2014) or overlapping paired-end reads (Moriyama et al., 2017).
Simultaneous usage of multiple characteristic information sources, e.g. heterozygous SNPs nearby mutation candidates and overlapping paired-end reads, is preferable for improving performance for the second type of method. However, existing mutation callers do not consider various information sources simultaneously. To utilize multiple information sources, we proposed a Bayesian model integration framework, named as partitioning-based model integration, and then we developed a novel mutation calling method named as OHVarfinDer based on the framework.
In Section 2, we explain the partitioning-based model integration framework, and then describe details of OHVarfinDer.
In Section 3, we first show that our method can utilize both heterozygous SNPs information and overlapping paired-end read information effectively in simulation datasets. In this experiment, we demonstrate the comparable performance of our method with other methods when only one of the two information sources is available; we also demonstrate the superior performance of our method compared to the other methods when both information sources are available. Second, we demonstrate the better performance of our method for real datasets.
In Section 4, we discuss the advantages and limitations of the proposed method.
2 Materials and methods
In this section, we first explain multiple characteristic information sources for mutation calling. Second, we elucidate our proposed framework of partitioning-based model integration in a general form. Third, we describe how these multiple information sources are incorporated in OHVarfinDer based on the partitioning-based model integration.
2.1 Characteristic information sources for mutation calling
2.1.1. Heterozygous SNPs covered by paired-end reads
The first additional information source in somatic mutation calling is heterozygous SNPs near somatic mutation candidates. The human genome is a diploid set of haplotypes, i.e. the maternal haplotype and paternal haplotype. Each somatic mutation is known to occur typically only on one side of the haplotypes, i.e. heterozygous mutation. Therefore, variant supporting reads that cover heterozygous SNPs are generated from only one side of the haplotypes as shown in the left side of Figure 1a. However, when sequence errors occur on the mutation candidate position, variant supporting reads covering heterozygous SNPs probably have both heterozygous SNPs as in the right side of Figure 1a. This information source was used in HapMuC (Usuyama et al., 2014).
Fig. 1.
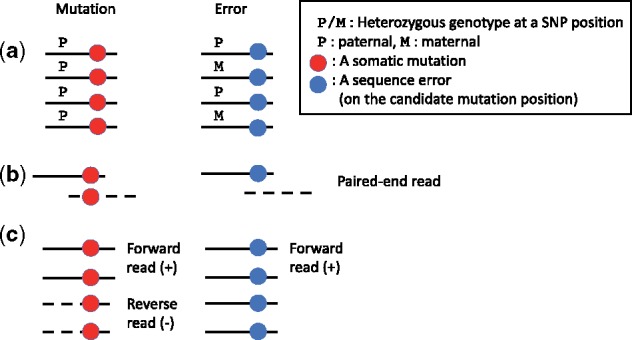
(a) The typical pattern of reads when heterozygous SNPs near the mutation candidate appear. (b) The typical pattern of paired-end reads when overlapping paired-end reads cover the mutation candidate. (c) The typical pattern of reads when both strand bias of variant supporting reads appear
2.1.2. Overlaps of paired-end reads
The second additional information source is overlaps of paired-end reads. Through Illumina’s sequencing, a pair of paired-end reads, i.e. forward and reverse reads, is sequenced from both sides of the same DNA fragment. If the DNA fragment is shorter than 2-fold the read length, the pair of reads has an overlapping region where sequence process is conducted twice from different directions independently.
If the both forward and reverse reads show the same alteration in the overlapping region as in the left side of Figure 1b, it is likely that the change is because of a mutation and not because of errors, as the occurrence probability of two errors at the same site in the overlapping region is expected to be very low, except for PCR errors in the sample preparation phase (Chen-Harris et al., 2013). In contrast, an error case is probable when only one of the reads contains an alteration in the overlapping region as in the right side of Figure 1b. This information source has been used in OVarCall (Moriyama et al., 2017).
2.1.3. Strand biases of paired-end reads
The third additional information source we considered is strand biases in variant supporting reads that cover a mutation candidate. If only forward (or reverse) reads contain a mutation candidate despite sufficient numbers of both forward and reverse reads, this phenomenon is known as strand bias as in the right side of Figure 1c. If a true somatic mutation exists, strand bias rarely occurs, and the proportion of variant supporting forward/reverse reads should be ideally similar as in the left side of Figure 1c. This information source is used for filtering in MuTect (Cibulskis et al., 2013).
2.1.4. Representative examples in real datasets
We show the examples from real datasets, in which we can find that given mutation candidates are only errors. Figure 2 shows screenshots of IGV (http://software.broadinstitute.org/software/igv/).
Fig. 2.

Typical cases of error are shown in the IGV screenshot. (a) In this case, both heterozygous SNPs near the mutation candidate appear in the variant supporting reads. See the erroneous case in Figure 1a. (b) One corresponding paired-end read is highlighted in red line. In this case, inconsistent bases in a paired-end read occur at a mutation candidate position. See the erroneous case in Figure 1b. Our method successfully evaluates these errors with low Bayes factor scores, i.e. 0.000059 in (a) and 0.0000011 in (b)
The first erroneous case shown in Figure 2a represents the variant supporting reads with both heterozygous SNPs. In this case, variant supporting reads have both heterozygous SNPs, as indicated by red and blue circles. This case corresponds with the erroneous case in Figure 1a.
The second erroneous case shown in Figure 2b represents a paired-end reads with inconsistent bases at a mutation candidate position. In this case, reads in a paired-end reads that are highlighted in red line have different bases at the mutation candidate position. This case corresponds with the erroneous case in Figure 1b.
Simpler methods, e.g. a Fisher’s exact test-based method of VarScan2, evaluate these two types of errors as somatic mutations. In the case of Figure 2a, VarScan2 showed a low P-value of 0.043 and in Figure 2a, VarScan2 also showed a low P-value of 0.0050. The main purpose of this paper is to construct a Bayesian method which discriminates these errors from somatic mutations.
2.2 Bayes factor for finding mutations
We denote a dataset as , where xn is the n-th string consisting of and d is the depth on the mutation candidate position. We denote tumor and error models as , and corresponding parameters as . Next, the Bayes factor (Kass and Raftery, 1995) is written as follows:
where .
2.3 Partitioning-based model integration
First, we assume models in each tumor and error model, and denote these models as , where . We denote corresponding parameters as , where . We also assume that we can observe indicator variable with each data xn. We assume that the original dataset is partitioned into K subsets and tn indicates the subset of data to which xn belongs. We also assume that the k-th subset of data is generated through the k-th model of or . We denote this augmented dataset as .
We assume the graphical model of Figure 3 and that the distribution of each parameter is dependent on the k-th model of .
| (1) |
| (2) |
Fig. 3.
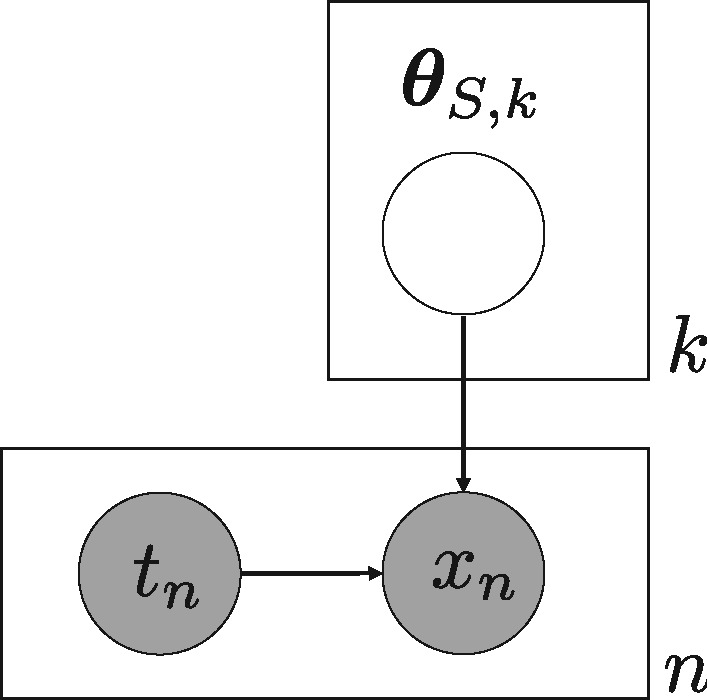
Graphical model for partitioning-based integration of generative models. Where states the hypothesis
Our purpose here is to compute the following Bayes factor:
From the graphical model in Figure 3 and above assumptions of
Equations (1) and (2), the joint probability can be computed as follows:
where .
From this joint probability, the marginal likelihood can be computed as follows:
where
If we can assume for any , we do not need to set for computation of Bayes factor, because
This manner of model integration requires two conditions. The first condition is a partition rule on the dataset and we can construct a corresponding generative model for each partitioned dataset. The second condition is that partition probabilities should be the same among the tumor and error model (). The merit of this manner is that partition probabilities do not affect the Bayes factor and thus careful and explicit settings of these probabilities are not necessary.
2.4 Notations in practical models of OHVarfinDer
The graphical model of OHVarfinder is shown in Figure 4a. rn is the n-th paired-end read, i.e. tuple of two forward/reverse reads of , and each is a string sequence of . tn is the n-th partition indicator variable. is an set of template paired-end reads and contains paired-end reads like Figure 1. zn (if ) is the n-th categorical latent variable indicating the template paired-end read for the n-th paired-end read rn. For the generation process of rn as a whole, we assume that n-th paired-end read rn is generated from zn-th paired-end read of with sequence errors and mapping errors added randomly.
Fig. 4.
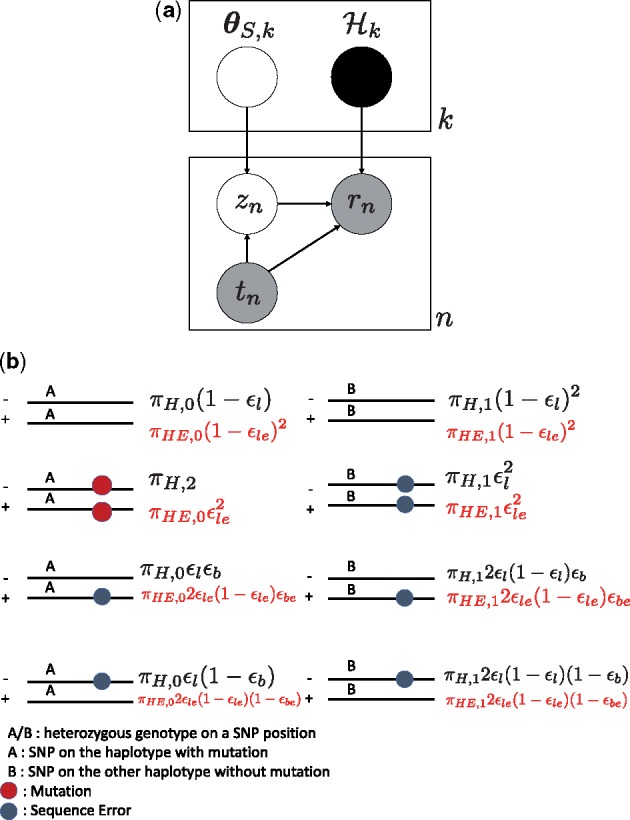
(a) Graphical model of OHVarfinDer. (b) Ideal paired-end reads set in and corresponding proportion and for the tumor model and error model. and are error rate for overlapping reads, strand bias rate and haplotype frequency used in the error model of O(+)H(+). Characteristic information of heterozygous SNPs, overlapping paired-end reads and strand bias can be considered by setting the proportions and . Occurrence probabilities shown in black (red) letters are for (). The black colored formulations in the left hand side are based on the tumor model of O(+)H(−) category. The black colored formulations in the right hand side and all the red colored formulations are based on the error model of O(+)H(−), cf. the Supplementary Material A.2 and A.4
2.5 Partition rules for each paired-end read in OHVarfinDer
In our method, we split paired-end reads into five types. is determined for each paired-end read by the following partitioning rule.
2.5.1. O(+)H(−) category
A paired-end read in this category (tn = 0) is overlapping between the forward read and reverse read at the mutation candidate position and covers no heterozygous SNPs nearby the candidate position.
2.5.2. O(−)H(+) category
A paired-end read in this category (tn = 1) is not overlapping between the forward read and reverse read at the mutation candidate position and covers heterozygous SNPs nearby the candidate position.
Note that global haplotype phasing is not necessary and we only conduct haplotype phasing locally around the mutation candidate positions as previously conducted in (Usuyama et al., 2014). The genotype A and B as in Figure 4b is determined from the number of variant supporting reads for each SNP.
2.5.3. O(+)H(+) category
A paired-end read in this category (tn = 2) is overlapping between the forward read and reverse read at the mutation candidate position and covers heterozygous SNPs nearby the candidate position.
2.5.4. O(−)H(−)S(+) category
A paired-end read in this category (tn = 3) is not overlapping between the forward read and reverse read at the mutation candidate position and covers no heterozygous SNPs nearby the candidate position. The mutation candidate position is covered by the forward read. (Forward/reverse is determined by the mapping direction compared to the reference sequence.)
2.5.5. O(−)H(−)S(−) category
A paired-end read in this category (tn = 4) is not overlapping between the forward read and reverse read at the mutation candidate position and covers no heterozygous SNPs nearby the candidate position. The mutation candidate position is covered by the reverse read.
2.5.6. Suitability of partition based integration
We should note that partitioning-based integration is suited for this problem setting for two reasons. The first reason is that we can set partition rules on paired-end reads and construct generative models for each dataset by referring to existing methods. The second reason is that partitioning probabilities are thought to be the same, e.g. the existence of a mutation does not affect whether a paired-end read will cover a heterozygous SNP.
2.6 Details of tumor generative model for O(+)H(+) type of paired-end read
Here, we only show the details of the tumor generative model for O(+)H(+) type (tn = 3) due to the limitation of the space. See the Supplementary Material A.1–A.8 for the details of our models. zn is the one of eight expression vector indicating an idealized paired-end read. For the parameters, we used (that is ). is the error rate when the paired-end read is overlapping at the mutation candidate position, is the strand bias rate. is a 3-dimensional non-negative simplex, indicating the proportion of paired-end reads from a maternal haplotype, paternal haplotype and haplotype with somatic mutation. Let the hyperparameters for . The tumor generative model for the O(+)H(+) type of paired-end read is defined as follows:
where are the probability density function of beta, Dirichlet, and multinomial distributions respectively. is the alignment probability which is formulated by profile hidden Markov model (HMM) (Albers et al., 2011; Usuyama et al., 2014). is a non-negative simplex defined by . We only show the case for in Figure 4b. is a function that returns the index where the value is 1 from a given one-hot encoding vector.
2.7 Bayes factor in OHVarfinDer
Here, we show the Bayes factor in OHVarfinDer and explain that our method is truly based on the partitioning-based integration and that setting of and are not necessary. Let is the set of paired-end reads for both tumor and normal sample data which cover a mutation candidate position, and the marginal likelihoods can be computed as follows:
where
Therefore, if , it is not necessary to set these distributions in the Bayes factor of OHVarfinDer as shown in the previous section.
2.8 Computation of marginal likelihoods
We applied the variational Bayes procedures for computing . We can obtain a lower bound for from the convexity of log function (Jensen, 1906).
| (3) |
where we denote all latent variables and parameters of as . is the variational distribution for which is formulated in the independent form as follows:
In the above inequality of Equation (3), the equality holds true when is equal to the posterior distribution of the . In the variational Bayes procedure (Beal, 2003), we maximize the lower bound for each variational distribution of and iteratively until the updated lower bound converges, and approximate the log marginal likelihood using this maximized lower bound. We described the full procedures for variational Bayes in the Supplementary Material A.9–A.16.
3 Results
3.1 Performance evaluation of OHVarfinDer using simulation data
3.1.1. Simulation data generation procedure
We tested OHVarfinDer using simulation datasets. The simulation procedure is described as follows. In the following procedure, we prepared two types of errors. The first type of errors are position-specific ones, and known as error prone sites (Moriyama et al., 2017; Shiraishi et al., 2013). The second type of errors are non-position-specific ones.
Generate a random reference DNA sequence.
Generate a heterozygous germ line variant in a random location, as well as two haplotypes (h1 and h2)
Generate a somatic mutation randomly around a heterozygous germ line variant, according to an empirical distribution of whole genome data, as well as two haplotypes (h3 and h4)
- Randomly generate paired-end reads around 900 somatic mutations and 2100 error prone sites randomly.
- Determine the number of paired-end reads covering the position, by generating a random value d from a norm distribution of N(50, 2), and round d to the nearest integer value.
- Randomly determine the haplotype of the original DNA fragment. We set the frequency of haplotypes as h1: 50-v%, h2: 50%, h3: v%, h4: 0% if a somatic mutation truly exists. We set the frequency of haplotypes as h1: 50%, h2: 50%, h3: 0%, h4: 0% otherwise.
- For each paired-end read, determine the DNA fragment size by generating a random value l from , and round l to the nearest integer value.
- Generate the 100-bp length read sequence on forward strand. Each observed base flips with the sequence error probability of perror. If the position of each observed base is the error prone site, perror is generated from a beta distribution of Beta(2, 30). If the position of each observed base is not the error prone site, perror is generated from Beta(10, 1000).
- Generate the read sequence on the reverse strand like (d).
3.1.2. Performance evaluation of OHVarfinDer using simulation data
As a counterpart method, we prepared OVarCall, HapMuC, and a simple Fisher’s exact test (Fisher, 1925) method, which uses a 2 × 2 contingency table of read counts, tumor and normal samples/variant and reference alleles. We calculated the area under the curve (AUC) values from the plotted receiver-operating characteristic curve (ROC) (Bradley, 1997) for each simulation condition as shown in Table 1. We described the filter conditions in the Supplementary Material B.1.
Table 1.
Simulation results summary (AUC)
| v (%) | Heterozygous SNPs | Overlap | Distance to SNP | μl | σl | OHVarfinDer | OVarCall | HapMuC | Fisher | #SNV | #Error | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A | 5 | − | − | 500–5000 | 300 | 30 | 0.828 | 0.750 | 0.828 | 0.810 | 341 | 822 |
| 10 | − | − | 0.891 | 0.867 | 0.880 | 0.891 | 713 | 871 | ||||
| 20 | − | − | 0.967 | 0.978 | 0.950 | 0.983 | 896 | 872 | ||||
| B | 5 | − | + | 500–5000 | 180 | 30 | 0.938 | 0.917 | 0.786 | 0.817 | 407 | 1394 |
| 10 | − | + | 0.958 | 0.954 | 0.843 | 0.899 | 763 | 1413 | ||||
| 20 | − | + | 0.989 | 0.991 | 0.947 | 0.988 | 897 | 1411 | ||||
| C | 5 | + | − | 1–100 | 300 | 30 | 0.880 | 0.765 | 0.882 | 0.825 | 301 | 851 |
| 10 | + | − | 0.916 | 0.877 | 0.907 | 0.886 | 733 | 871 | ||||
| 20 | + | − | 0.986 | 0.984 | 0.977 | 0.983 | 896 | 925 | ||||
| D | 5 | + | + | 1–100 | 180 | 30 | 0.943 | 0.923 | 0.838 | 0.803 | 388 | 1356 |
| 10 | + | + | 0.975 | 0.952 | 0.918 | 0.914 | 757 | 1398 | ||||
| 20 | + | + | 0.994 | 0.991 | 0.977 | 0.990 | 896 | 1354 |
The highest AUC values are written in italic letters.
In the simulation dataset under the condition of B, only overlapping paired-end read information was available. In this case, our method performs comparable with OVarCall. In the simulation dataset in the condition of C, only heterozygous SNP information was available. In this case, our method performed comparably well with HapMuC that can utilize this information source. In the simulation dataset under the condition of A, neither of the above types of information was available. In this case, our method performed comparably well with Fisher’s exact test. In the simulation dataset under the condition of D, both overlapping paired-end read information and heterozygous SNP information were available. In this case, our method outperformed both OVarCall and HapMuC. We summarized the ROC curves in the Supplementary Material B.10.1–B.10.3.
3.2 Performance evaluation of OHVarfinDer using real data
3.2.1. SNVs in exome sequence dataset
We confirmed whether the performance of our method could be improved by using overlapping information using real exome datasets, as shown in
Table 2 for the real datasets, we used exome sequence data from renal clear-cell carcinoma, which has already been used for performance evaluation of OVarCall (Moriyama et al., 2017). In these datasets, ∼40% of paired-end reads overlapped, and thus the use of overlapping paired-end reads is expected to affect the performance. In this dataset, true somatic SNVs were validated by deep sequencing (Shiraishi et al., 2013). In both the case of lower variant allele frequency of 2–7% and the case of moderate variant allele frequency above 7%, OHVarfinDer performed comparably well with OVarCall and outperformed HapMuC. Furthermore, we observed that our method returned low Bayes factor of 0.0000011 in the false positive case in Figure 2b. Therefore, we confirmed that our method can incorporate overlapping information and improve its performance. For the details of this experiment, see the Supplementary Material B.2, B.8 and B.10.4.
Table 2.
Exome datasets summary (AUC)
| SNV/InDel | VAF | OVarCall | OHVarfinDer | HapMuC | Strelka | MuTect | VarScan2 | #SNV | #Error |
|---|---|---|---|---|---|---|---|---|---|
| SNV | 2–7% | 0.982 | 0.990 | 0.965 | 0.933 | 0.875 | 0.625 | 52 | 2422 |
| SNV | ≥7% | 0.991 | 0.988 | 0.955 | 0.995 | 0.994 | 0.900 | 184 | 1982 |
The highest AUC values are written in italic letters. VAF, represents variant allele frequency; SNV, represents single nucleotide variant.
3.2.2. SNVs and InDels in whole genome dataset
We examined whether we could improve the performance of our method by using heterozygous SNP and strand bias information using whole genome sequence data. The results are summarized in
Table 3 for the dataset, we used whole genome sequence datasets from breast cancer cell lines, which are publicly available as a part of The Cancer Genome Atlas (TCGA) Mutation Calling Benchmark 4 datasets (These datasets can be downloaded from https://gdc.cancer.gov/resources-tcga-users/tcga-mutation-calling-benchmark-4-files) and have been used for performance evaluation of HapMuC.
Table 3.
Real datasets summary whole genome (AUC)
| Sample | SNV/InDel | OVarCall | OHVarfinDer | HapMuC | Strelka | MuTect | VarScan2 | #SNV/InDel | #Error |
|---|---|---|---|---|---|---|---|---|---|
| HCC1143_n20t80 | SNV | 0.869 | 0.906 | 0.827 | 0.873 | 0.848 | 0.801 | 10 618 | 2327 |
| HCC1143_n40t60 | 0.870 | 0.901 | 0.824 | 0.877 | 0.855 | 0.799 | 8517 | 2049 | |
| HCC1143_n60t40 | 0.884 | 0.912 | 0.843 | 0.901 | 0.876 | 0.814 | 5450 | 1684 | |
| HCC1143_n80t20 | 0.901 | 0.941 | 0.870 | 0.938 | 0.918 | 0.830 | 1874 | 1451 | |
| HCC1954_n20t80 | 0.882 | 0.934 | 0.852 | 0.903 | 0.869 | 0.862 | 10 653 | 2854 | |
| HCC1954_n40t60 | 0.893 | 0.941 | 0.852 | 0.917 | 0.880 | 0.858 | 7969 | 2327 | |
| HCC1954_n60t40 | 0.917 | 0.949 | 0.865 | 0.937 | 0.905 | 0.852 | 4638 | 1770 | |
| HCC1954_n80t20 | 0.941 | 0.970 | 0.880 | 0.972 | 0.942 | 0.848 | 1389 | 1404 | |
| Total | 0.895 | 0.935 | 0.860 | 0.913 | 0.886 | 0.852 | 51 108 | 15 866 | |
| HCC1143_n20t80 | InDel | 0.707 | 0.796 | 0.678 | 0.713 | — | 0.722 | 926 | 4951 |
| HCC1143_n40t60 | 0.733 | 0.814 | 0.700 | 0.755 | — | 0.748 | 617 | 4761 | |
| HCC1143_n60t40 | 0.760 | 0.834 | 0.723 | 0.784 | — | 0.778 | 328 | 4563 | |
| HCC1143_n80t20 | 0.809 | 0.855 | 0.770 | 0.816 | — | 0.800 | 94 | 4899 | |
| HCC1954_n20t80 | 0.800 | 0.860 | 0.771 | 0.822 | — | 0.825 | 1771 | 5219 | |
| HCC1954_n40t60 | 0.821 | 0.866 | 0.778 | 0.843 | — | 0.835 | 1172 | 5215 | |
| HCC1954_n60t40 | 0.819 | 0.863 | 0.770 | 0.848 | — | 0.831 | 607 | 5200 | |
| HCC1954_n80t20 | 0.815 | 0.887 | 0.777 | 0.864 | — | 0.823 | 159 | 5053 | |
| Total | 0.777 | 0.838 | 0.774 | 0.794 | — | 0.792 | 5674 | 39 861 |
The highest AUC values are written in italic letters.
In these datasets, pure cell line sequence datasets of normal and tumor cell line and computational mixtures of these sequence datasets are prepared, e.g. HCC1143_n40t60 represents that 40% of pure normal and 60% of pure tumor sequence data are mixed. In this experiment, we obtained answers of true mutations from these pure cell line datasets, and we conducted performance evaluations for tumor sequence datasets with several mixture rates, i.e. n20t80, n40t60, n60t40, n80t20. For these datasets, the use of heterozygous SNPs information and strand bias information is important for improving performance because the average proportion of overlapping paired-end reads was ∼3% within these datasets.
For the performance of OHVarfinDer, OHVarfinDer performed better than any other mutation caller, except for HCC1954_n80t20. We also observed that our method returned low Bayes factor of 0.000059 in the false positive case in Figure 2a. Therefore, we confirmed that our method can incorporate heterozygous SNP and strand bias information and improve its performance. For the details of this experiment, see the Supplementary Material B.3, B.9 and B.10.5.
4 Discussion
Some mutation calling methods, e.g. HapMuC and OVarCall, can incorporate a characteristic information source, e.g. heterozygous SNPs and overlapped paired-end reads, in their mutation calling process. However, no existing methods utilize multiple types of such characteristic information sources simultaneously.
In this paper, we first introduced a framework for Bayesian model integration named as partitioning-based model integration, which differs from Bayesian model averaging (Hoeting et al., 1999). In this framework, we first set a partitioning rule for data and augmented the data with indicator variables which show the category of partitioning. Second, we constructed a generative model for each category of partitioned dataset. This framework requires two assumptions. The first assumption is that we can set a partitioning rule and construct corresponding generative models. The second assumption is that partitioning probabilities are common among the tumor model and error model. If the above assumptions hold true, we can compute the Bayes factor without careful setting of prior partitioning probabilities. In our problem setting of mutation calling, the above two assumptions seem natural, and thus we constructed a Bayesian mutation calling method, OHVarfinDer, based on this framework.
We conducted performance evaluations with simulation and real datasets. In the simulation datasets, we showed that our method could utilize multiple information sources, particularly overlapping paired-end read information and heterozygous SNP information. If only one information source was given, our method performed comparably well with other existing methods. If both information sources were given, our method performed better than other existing methods. In the real datasets, e.g. TCGA Mutation Calling Benchmark 4 datasets, we also demonstrated the better performance of our method compared to other existing methods.
We have demonstrated how to integrate known multiple information sources for mutation calling by our framework. We note that mapping quality and base quality of reads are also used in our method by incorporating the profile HMM modeling (Albers et al., 2011; Usuyama et al., 2014). Although our framework is practically useful for mutation calling, there is at least one limitation for this framework, i.e. our framework does not assume inference over the parameter distributions, e.g. prior distributions for the error parameters. Such inference is important if we consider using multiple sequence datasets simultaneously. For example, if we can use pooled normal sequence datasets, we can infer the error distributions depending on the genomic positions. For the future work, we plan to extend our framework to infer the form of the parameter distributions, e.g. incorporating predictive distributions for the error parameters.
Supplementary Material
Acknowledgements
The super-computing resource was provided by Human Genome Center, the Institute of Medical Science, The University of Tokyo.
Funding
This work has been supported by the Grant-in-Aid for JSPS Research Fellow (17J08884) and MEXT Grant (15H05912, hp180198, hp170227).
Conflict of Interest: none declared.
References
- Albers C.A. et al. (2011) Dindel: accurate indel calls from short-read data. Genome Res., 21, 961–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal M.J. (2003). Variational algorithms for approximate Bayesian inference. PhD Thesis, The Gatsby Computational Neuroscience Unit, University College London, London, UK.
- Bradley A.P. (1997) The use of the area under the ROC curve in the evaluation of machine learning algorithms. Pattern Recognit., 30, 1145–1159. [Google Scholar]
- Chen-Harris H. et al. (2013) Ultra-deep mutant spectrum profiling: improving sequencing accuracy using overlapping read pairs. BMC Genomics, 14, 96.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K. et al. (2013) Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol., 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher R.A. (1925). Statistical Methods for Research Workers. Edinburgh, Oliver and Boyd. [Google Scholar]
- Hoeting J.A. et al. (1999) Bayesian model averaging: a tutorial. Statist. Sci., 14, 382–417. [Google Scholar]
- Jensen J.L.W.V. (1906) Sur les fonctions convexes et les inégalités entre les valeurs moyennes. Acta Math., 30, 175–193. [Google Scholar]
- Kass R.E., Raftery A.E. (1995) Bayes Factors. J. Am. Stat. Assoc., 90, 773–795. [Google Scholar]
- Koboldt D.C. et al. (2012) VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res., 22, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerson M. et al. (2010) Advances in understanding cancer genomes through second-generation sequencing. Nat. Rev. Genet., 11, 685–696. [DOI] [PubMed] [Google Scholar]
- Moriyama T. et al. (2017) OVarCall: Bayesian mutation calling method utilizing overlapping paired-end reads. IEEE Trans. Nanobioscience, 16, 116–122. [DOI] [PubMed] [Google Scholar]
- Roth A. et al. (2012) JointSNVMix: a probabilistic model for accurate detection of somatic mutations in normal/tumour paired next-generation sequencing data. Bioinformatics (Oxford, England), 28, 907–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraishi Y. et al. (2013) An empirical Bayesian framework for somatic mutation detection from cancer genome sequencing data. Nucleic Acids Res., 41, e89.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usuyama N. et al. (2014) HapMuC: somatic mutation calling using heterozygous germ line variants near candidate mutations. Bioinformatics (Oxford, England), 30, 3302–3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K. et al. (2011) Frequent pathway mutations of splicing machinery in myelodysplasia. Nature, 478, 64–69. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.