

Abstract
Background and Aims
Perennial grasses are a global resource as forage, and for alternative uses in bioenergy and as raw materials for the processing industry. Marginal lands can be valuable for perennial biomass grass production, if perennial biomass grasses can cope with adverse abiotic environmental stresses such as drought and waterlogging.
Methods
In this study, two perennial grass species, reed canary grass (Phalaris arundinacea) and cocksfoot (Dactylis glomerata) were subjected to drought and waterlogging stress to study their responses for insights to improving environmental stress tolerance. Physiological responses were recorded, reference transcriptomes established and differential gene expression investigated between control and stress conditions. We applied a robust non-parametric method, RoDEO, based on rank ordering of transcripts to investigate differential gene expression. Furthermore, we extended and validated vRoDEO for comparing samples with varying sequencing depths.
Key Results
This allowed us to identify expressed genes under drought and waterlogging whilst using only a limited number of RNA sequencing experiments. Validating the methodology, several differentially expressed candidate genes involved in the stage 3 step-wise scheme in detoxification and degradation of xenobiotics were recovered, while several novel stress-related genes classified as of unknown function were discovered.
Conclusions
Reed canary grass is a species coping particularly well with flooding conditions, but this study adds novel information on how its transcriptome reacts under drought stress. We built extensive transcriptomes for the two investigated C3 species cocksfoot and reed canary grass under both extremes of water stress to provide a clear comparison amongst the two species to broaden our horizon for comparative studies, but further confirmation of the data would be ideal to obtain a more detailed picture.
Keywords: Phalaris arundinacea, reed canary grass, Dactylis glomerata, cocksfoot, orchardgrass, transcriptome, drought, flooding, waterlogging, biomass
INTRODUCTION
Concern over the impact of rising levels of greenhouse gas emissions on climate change has resulted in increased interest in renewable forms of energy including bioenergy (Stocker et al., 2013). It has been demonstrated that a low carbon energy economy which meets future energy needs requires the development of a significant global bioenergy sector (Edenhofer et al., 2010; van Vuuren et al., 2010). Recently, it has been estimated that total global bioenergy demand could double by 2030 (IRENA, 2014). Within the European Union, demand for biomass is driven by the Renewable Energy Directive (2009/28/ec) which has set renewable energy targets for each member state. Member states were required to prepare national renewable energy action plans detailing how country-specific targets are going to be met. An analysis of these plans showed that 54.5 % of the EU renewable energy target will be met by biomass, with solid biomass providing 71 % of renewable heating and cooling by 2020 (Atanasiu, 2010).
An increasing demand for biomass has generated conflict between food and energy production and concern about the indirect effects of land use change (Wiegmann et al., 2008; Baffes and Haniotis, 2010). Using marginal land for biomass production could be a possible solution (Campbell et al., 2008; Dauber et al., 2012). Marginal land has been defined as land of poor quality for agriculture which yields poor economic returns for farmers (Wiegmann et al., 2008), and crops grown on marginal land can be subjected to a range of abiotic stresses (Jones et al., 2014).
Grass species exhibit a wide range of adaptability to a range of environments, but are also suitable as feedstock for combustion (Prochnow et al., 2009a) and for anaerobic digestion (Prochnow et al., 2009b). It has been demonstrated that the cultivation of grass on degraded or exhausted soils can restore organic carbon content and physical properties of the soil (Potter et al., 1999). In some instances, maximum biomass production and minimum environmental impact can be achieved by utilizing pre-existing grassland (Tilman et al., 2006). Perennial rhizomatous grasses (PRGs) have been suggested as good candidate energy crops (Clifton-Brown et al., 2007). Jones et al. (2014) provided evidence that some PRG species have good tolerance to abiotic stresses and identified the morphological and physiological traits which could be modified in order to improve stress tolerance and to maximize biomass production from marginal land. Growing perennial rhizomatous grasses on marginal land to produce bioenergy has the potential to mitigate the effects of climate change without conflicting with a growing global requirement for food, thus improving the environment and the ability to provide both food and fuel for the global population. However, optimizing biomass production on land subject to, often severe, abiotic stress represents a challenge and requires an understanding of how grass species respond to and minimize abiotic stress.
Plants can suffer stress either through the absence of water or because of waterlogging (hypoxia). On a global scale, drought-induced losses in yield exceed all other causes (Richards, 2004). Differences exist, however, in the extent to which different grass species can tolerate drought. For example, cocksfoot is well adapted to dry conditions and can survive soil water deficits more effectively than other grass species (Voltaire and Thomas, 1995). Flooding is a widely distributed problem and may possibly become more frequent as a result of climate change (Jones et al., 2014). Perennial rhizomatous grasses such as reed canary grass are considered to be well adapted to waterlogged conditions (McDonald et al., 2002). In contrast, cocksfoot is considered to be not very tolerant of waterlogged conditions, although Etherington (1984) found differences in flooding tolerance among genotypes of cocksfoot. Thus, different grass species can be chosen to suit the particular abiotic stress which may, in addition, be present on marginal land intended for bioenergy production. The sequencing of total mRNA samples via next-generation sequencing (RNA-seq) can provide a survey of transcriptome-wide changes in expression level. Studies in the model species arabidopsis and rice have investigated the response to abiotic stress, such as salinity and water stress (Rasmussen et al., 2013; Venu et al., 2013). About 61 % of the transcriptome changes in response to double stresses were not predicted from the responses to a single stress. Transcriptomic response to drought stress has been investigated in the model grass Brachypodium with an Affymetrix tiling array (Verelst et al., 2012) which led to the conclusion that transcriptome profiles of different developmental leaf zones respond differently to drought. RNA-seq is well suited to study differential expression in non-model organisms, and has recently been adapted to stress studies of grasses with few available genomic resources (heat stress in Festuca, Hu et al., 2014; and in Panicum, Li et al., 2013; salt stress in reed canary grass, Haiminen et al., 2015; and in Sporobolus, Yamamato et al., 2015), including first applications for water stress (Thinopyrum, Shu et al., 2015; Lolium arundinaceum, Talukder et al., 2015; Lolium multiflorum, Pan et al., 2018). Both cocksfoot and reed canary grass can be considered non-model organisms. They are allogamous grass species (Baumann et al., 2000) rendering each seedling within one cultivar different from each other. Cocksfoot (Stebbins, 1971) and reed canary grass (Anderson, 1961) are predominantly tetraploid. The factor ploidy has been shown to have an impact on gene expression comparing within-species relatives at different ploidy levels (Miller et al., 2012).
Thus we aimed to understand in this study intraspecific and interspecific differences in abiotic tolerance, and examined and compared the genetic regulation of drought and waterlogging stress tolerance in two grass species with differing adaptations to water stress. Reed canary grass is a rhizomatous species tolerant of waterlogged conditions, and cocksfoot is a non-rhizomatous species tolerant of drought but not of waterlogging. Both species lack a sequenced and annotated genome. Haiminen et al. (2015) have prepared a reed canary grass transcriptome of 18 682 transcripts for the read mapping of transcripts differentially expressed under salt stress conditions. For cocksfoot, approx. 65 000 expressed sequence tags (ESTs) from different cultivars and wild accessions are available in public databases (Bushman et al., 2011), but it was lacking an assembled reference transcriptome suitable for the study of differential gene expression.
Our overall objective in this study was to understand how grass species respond to abiotic stress in order to optimize biomass production on marginal land. The specific objectives of this study are to (1) construct more comprehensive reference transcriptomes for both species based on the combined next-generation sequencing reads from this experiment; (2) to identify differentially expressed genes associated with drought and waterlogging stress response for both species, including novel transcripts with unknown function; and (3) to compare the responses between the species, as reed canary grass is known to be more resilient when faced with waterlogged conditions.
MATERIALS AND METHODS
Plant materials
The experiments were carried out on two commercial varieties of cocksfoot (Dactylis glomerata ‘Sparta’) and reed canary grass (Phalaris arundinacea ‘Venture’). Both species are temperate C3 grass species, but are not closely related (Saarela et al., 2017). Cocksfoot is considered to be well adapted to dry conditions (Voltaire and Thomas, 1995), and reed canary to waterlogged conditions (McDonald et al., 2002).
Water stress experiment
Experiments were carried out in a glasshouse located at Teagasc Oak Park Crops Research Centre in Ireland. Prior to the treatments, grass seeds were germinated on moistened soil and transferred as single plants to 10 × 10 cm soil pots after 4 weeks in the glasshouse. The plants were then separated into single tillers and grown again for 4 weeks in 10 × 10 cm pots to approx. 30 cm height. Each of the treatments, drought, waterlogging, control 1 and control 2, was repeated twice in each of four blocks placed in a greenhouse in a randomized block design. During the experiment over 2 weeks, the plants were watered daily. Controls received 100 mL of water, with pots standing on capillary matting. Drought-treated plants received 40 mL of water, with pots standing on saucers to avoid uptake of water from the capillary matting. Pots with waterlogged plants were kept in trays with a continuous water level of between 5 and 8 cm by submerging the roots fully. The treatment was monitored by recording the soil moisture content four times throughout the treatment. The treatment effects on plants were measured via plant height, relative water content (RWC) of leaves, chlorophyll content of leaves and electrolyte leakage of leaves. At the end of the experiments, the plants were harvested above the soil, and weighed for the determination of fresh and dried biomass.
Soil moisture
Soil moisture was measured with a Theta Kit soil moisture instrument from Delta-T Devices Ltd, Cambridge, UK. Three measurements per pot were averaged to determine the soil moisture.
Chlorophyll content
The chlorophyll content of leaves was determined with a Minolta SPAD 502 SPAD chlorophyll meter (Minolta Camera Company, Azuchi-Machi, Osaka, Japan) with five measurements per plant averaged. A leaf is clamped into the sensor head and the optical density difference at two wavelengths, 650 nm and 940 nm, is measured by the device.
Relative water content of leaves
The top 5 cm of the topmost leaf were cut, and weighed in tinfoil for the fresh weight (f. wt). The leaf was submerged in 20 mL of distilled water and left in the refrigerator at 4 °C for 24 h. The turgid weight (t. wt) was determined by blotting the leaf dry and weighing. The leaf was dried for 48 h at 80 °C, and weighed again for the dry weight (d. wt). The RWC was calculated using the formula (f. wt – d. wt)/(t. wt – d. wt) × 100 = % RWC. RWC of leafs was measured four times during the experiment (the same schedule for both species as for soil water content laid out in Supplementary data File S1). Samples were taken in a 2 h interval between 11.00 and 13.00 h.
Electrolyte leakage
Two leaves were placed in a 50 mL polypropylene tube filled with distilled water. The tubes were closed, covered in tinfoil and left for 24 h at room temperature at approx. 20 °C. The conductivity in each tube was measured with a CDM80 conductivity meter (Radiometer A/S Copenhagen/Denmark). Afterwards, the tubes were capped and autoclaved (15 min, 121 °C, 15 psi). After cooling to room temperature, the conductivity of the solutions was measured again. The percentage electrolyte leakage was calculated as the ratio of conductivity before and after autoclaving; the value after represents 100 % leakage.
Biomass
One destructive biomass harvest was carried out at the end of the experiment. Fresh weight was determined for the total plant above the soil. Dry weight was determined after drying the fresh biomass for 48 h at 80 °C.
Statistical analysis
Data sets for reed canary grass and cocksfoot were combined, and plant species was used as a factor in analyses, allowing evaluation of the interaction between plant species and treatment. Block, treatment and plant were included in all analyses and, where multiple measurements of a response per subject were made over a number of time points, time was included in the analysis as a repeated measures factor and the correlations were modelled using a covariance structure in the Mixed procedure in SAS (2011). All factors in the analyses were fixed. Where appropriate, baseline measurements were also included as covariates. Tukey adjustments for multiplicity were used for means comparisons, and residuals were checked to ensure that the assumptions of the analyses were met.
RNA-seq
Leaf samples were taken during the experiment and flash-frozen in liquid nitrogen. The sample time points for both species are summarized in Supplementary data File S1. Samples were taken at the same time of the day to avoid any influence of the diurnal cycle on the samples. Samples were taken in a 2 h interval between 11.00 and 13.00 h. Total RNA was extracted using the RNeasy plant Mini kit from Qiagen according to the manufacturer’s instructions, including an on-column digest of residual genomic DNA. The total RNA was converted into an mRNA sequencing library using the Illumina TruSeq RNA Sample Preparation Kit (V2) according to the manufacturer’s instructions. The libraries were sequenced as 100 bp paired-end reads on an Illumina HiSeq 2000 sequencer. The libraries were multiplexed four times (cocksfoot) and six times (reed canary grass) in one sequencing flow cell lane each, using two lanes per species. All raw sequencing data were submitted to ENA, project number PRJEB16763.
Reference transcriptomes
We constructed reference transcriptomes by combining all available Illumina sequencing reads generated from treatment and control samples for cocksfoot (1 billion reads) and reed canary grass (800 million reads). The reads were quality trimmed, and cleaned of adaptor sequences by BBDuk from the BBMap package (Bushnell, 2015). For each sample, duplicate read fragments were identified and collapsed using BBDuk, and the remaining paired reads from all samples were combined. Reads were digitally normalized using bbnorm (BBMap package), with a k-mer coverage of 40 and a k-mer size of 31 bases. After this step, the data sets were reduced to approx. 15 % of their original size. The normalized read sets were assembled using the Trinity assembler with a k-mer of 25 bases (Grabherr et al., 2011). To reduce redundancy, highly similar transcripts (>95 % identity over their entire length) were collapsed into a single sequence using CD-HIT-EST. Transcripts were translated into open reading frames (ORFs). All transcripts where at least one of the ORFs had a significant BLAST hit against the Swissprot and/or Uniprot protein databases (UniProt Consortium, 2017) were retained. The most complete annotation per transcript was kept when more than one ORF from a transcript hit a known protein. The longest sequence was retained in the final assembled transcriptome, and sequences <100 amino acids were discarded. The final transcriptome comprised 22 634 transcripts for cocksfoot and 21 439 for reed canary grass.
Gene expression analysis
We mapped the treatment and control samples from each species against the respective reference using Burrows–Wheeler aligner (BWA) with the ‘mem’ option (Li and Durbin, 2009), using default parameters and only considering reads with unique concordantly paired hits against the reference. Hits were processed with samtools (Li et al., 2009), and reads with minimum mapping quality 30 were counted per transcript with BEDTools (Quinlan and Hall, 2010). As a result, 47–88 million reads per sample were mapped to cocksfoot and 32–41 million to reed canary grass. We used RoDEO (Haiminen et al., 2015) to quantify gene expression in each sample. RoDEO projects the read counts of transcripts to robust distributions of expression values in the range {1,2,…,P}, through a resampling and segmentation approach. These distributions per transcript are compared between stress and control samples to identify differentially expressed transcripts. We used the RoDEO parameters P ≤20 projection levels, I = 100 iterations and R = 106 reads per iteration. The overall bioinformatics workflow for identifying and visualizing differentially expressed transcripts is illustrated in Fig. 1.
Fig. 1.

Informatics analysis workflow. The diagram illustrates the bioinformatics steps starting from read mapping to the resulting figures and tables.
Parameter P selection for RoDEO as a function of sample size.
We developed novel methodology in order to compare accurately, with RoDEO, multiple samples with large variations in the numbers of total mapped reads. In this approach, we select the RoDEO parameter P separately for each sample, in a way that ensures all samples are directly comparable, regardless of their sequencing depth. The guiding principle in our methodology is: if max is the total sum of read counts in the largest sample, we assume that the smaller sample is a random sub-sample of its corresponding hypothetical sample of size max. The main stumbling block is the transcripts with lowest abundances rounding off to zero in the smaller sample. In our approach, this translates to using a reduced number of projected values for the smaller sample (decreasing the value for parameter P). The reduction in the number of segments is estimated by comparing the segment boundaries in the largest sample with the number of non-zero transcripts in the smaller sample. The method applied is illustrated in Fig. 2 and summarized as follows. (1) Segment the largest sample S with parameter P, where largest denotes the one with the most genes with non-zero counts. (2) Select parameter P′(i) for each of the smaller samples i as described below. Then carry out RoDEO projection using the respective parameters P for S and P′ for the smaller samples. Finally add [P – P′(i)] to all the projected values of the smaller sample i, ensuring that the largest projected value for each sample is P. Then perform the RoDEO DE (differential expression) score computation assuming P projected values. The parameter P′(i) for smaller sample i is selected as follows: find the segment boundary x = b in the largest sample S that is closest to the number of non-zero genes x = n in sample i. Let the boundary b separate the projected values v and v + 1. Then P′(i) = (P – v + 1).
Fig. 2.

Illustration of the method for selecting parameter P for RoDEO for samples with very different sequencing depths. The smaller sample A′ is made comparable to the larger sample A by using P′ = P – 5 and adding 5 to the segment labels in A′. If no scaling was applied, the rightmost segment of A′ (having label 6 in the figure) would have label 1 and would be compared with the segment indicated by the red arrow in A (having label 1 in A). Instead, the segments 6, 5, 4, 3 and 1 in A are higher resolution sub-segments in A that are all represented by the segment 6 in A′. The genes are ordered from highest to lowest expressed on the x-axis, and the horizontal segments represent genes with zero reads.
We have validated the methodology using samples from a data set with known answers from quantitaitve PCR (qPCR) which was also originally used for RoDEO validation (Haiminen et al., 2015); the details are in Supplementary data File S2.
Differential expression.
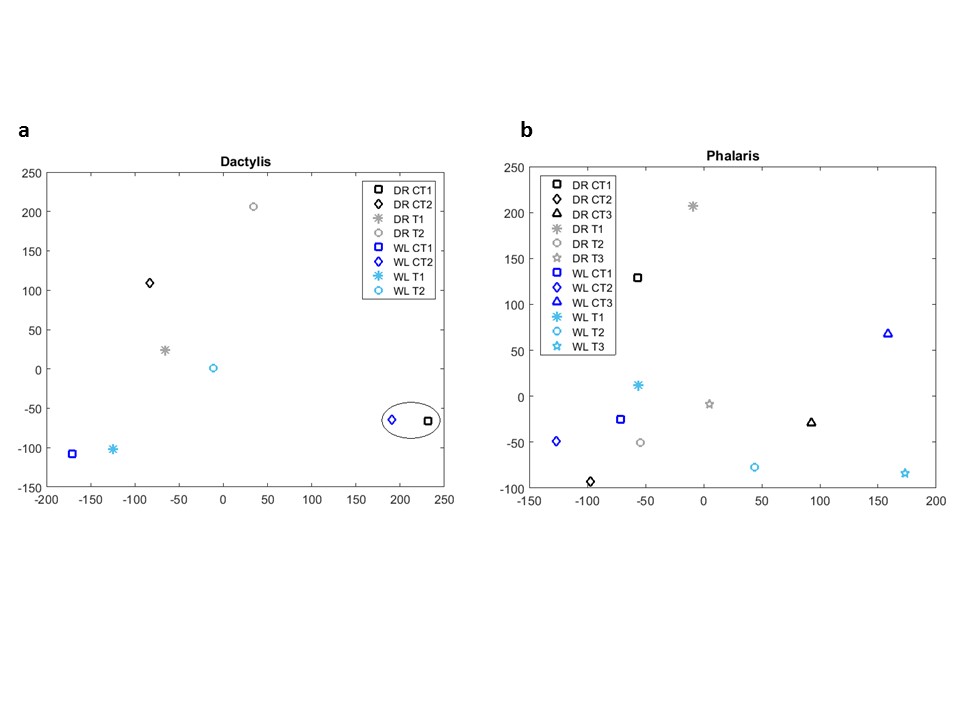
We identified differentially expressed genes between each stress condition and the control samples. The control samples were derived from four cocksfoot and six reed canary grass, and they include one duplicated genotype (biological replicate) for cocksfoot. Each stress sample represents a different genotype. The genotypes and the sampling scheme for RNA collection are included in Supplementary data File S1. We first studied the overall expression landscape of all the samples. For this, we used multidimensional scaling (MDS) of the Euclidean distance of the average RoDEO projections of transcript abundances. The corresponding two-dimensional MDS plots are shown in Supplementary data Fig. S1.
Note that the duplicated genotype control samples in cocksfoot are closely clustered in the MDS plot, indicating good biological reproducibility as well as technical reproducibility in the RNA collection and sequencing process. The control samples exhibit varying genotype-specific expression, as they are not all clustering together in the MDS plots. We modelled this variation by using all the controls together as replicates, representing baseline transcript expression capturing the naturally occurring genotype-specific variation. Each stress sample was then compared with this baseline of controls. The DE score for transcript g in a stress sample was computed by comparing the stress sample’s RoDEO projection of g [stress distribution S(g)]with the RoDEO projections of g in the control samples [baseline distribution B(g)]: DE(g) = max norm distance × mean distance of these distributions S(g) and B(g). The baseline distribution S(g) was constructed by summing all the control samples’ RoDEO projected distributions for g. The duplicated cocksfoot genotype control samples’ distributions were multiplied by 1/2 when adding to the other control distributions.
Significance of differential expression scores.
In order to set a meaningful threshold for the DE scores to convey significant differential expression, we defined an empirical P-value for DE score y as the fraction of genes that have DE scores ≥y in the comparisons of a control sample with the other control samples. P(y) = [c(y) + 1]/(sN), where s is the number of control samples, N is the number of genes and c(y) is the total count of scores ≥y in the s comparisons. For cocksfoot, where one control genotype was sequenced twice, each duplicated genotype control sample was compared with only the two other controls. Avoiding comparison between controls of the same genotype is a conservative approach as it reduces any tendency towards smaller DE scores due to genotype similarities. When comparing a non-duplicated genotype control with the other controls, each duplicated genotype’s RoDEO projection distribution was multiplied by 1/2 so that together they have the same impact on the baseline distribution as each of the other controls. For the differential expression results, a threshold P ≤ 0.005 was set for a transcript to be called differentially expressed.
Transcript clustering.
Transcripts were clustered with k-means clustering and Euclidean distance using ValWorkBench (Giancarlo et al., 2015), and the number of clusters was estimated by the Gap Statistic (Tibshirani et al., 2001). Cocksfoot transcripts were transformed to four-dimensional vectors for clustering, where each entry corresponds to the DE score in one stress sample compared with all the control samples. The elements of the vector for each gene are (1) DE score of drought at T1; (2) DE score of drought at T2; (3) DE score of waterlogging at T1; and (4) DE score of waterlogging at T2. Reed canary grass transcripts were respectively transformed to six-dimensional vectors as RNA for the species was sequenced at three time points for each stress condition (see Supplementary data File S1).
Identification of frequent protein domains in differentially expressed transcript sets.
The differentially expressed transcripts (Supplementary Data File 3a, cocksfoot; Supplementary Data File 3b, reed canary grass) for both species were used to search for coding domains for drought and waterlogging conditions (Supplementary data File 4). The frequency of domains at each time point was calculated manually and visualized using Venny (Oliveros, 2007).
RESULTS
The plants were kept for 2 weeks under a differential watering regime. Figure 3 illustrates how these conditions affected the soil moisture content of control, waterlogged and drought-treated plants. The watering regime consistently kept the available water in separate ranges for the treatments, resulting in distinct growth conditions without creating very severe stress conditions which would cause irreversible damage.
Fig. 3.

Soil water content for cocksfoot (Dactylis glomerata) and reed canary grass (Phalaris arundinacea); day 1 is the beginning of differential watering between treatments and controls.
Biomass
Reed canary grass showed increased accumulation of dry and fresh biomass under waterlogging conditions, followed by control and drought (Table 1). In contrast, for cocksfoot, both waterlogging and drought conditions stressed the plant, with a follow on effect on reduced biomass accumulation (Table 1).
Table 1.
Physiological reaction of reed canary grass and cocksfoot subjected to water stress measured by the factors biomass fresh and dry weights (one measurement per pot at the end of the experiment), leaf relative water content (three measurements per pot) and chlorophyll content (five measurements per pot) at the end of the experiment (n = 4 except for control n = 8)
| Treatment | Least squares means | ||||
|---|---|---|---|---|---|
| Reed canary grass | Cocksfoot | ||||
| Mean* | s.e. | Mean* | s.e. | ||
| Biomass fresh (g) | Control | 18.8125b | 1.2493 | 6.5613d | 1.0095 |
| Drought | 13.0500b | 1.7668 | 4.7950d | 1.4277 | |
| Waterlogging | 32.9583a | 2.1235 | 5.0775d | 1.4277 | |
| Biomass dry (g) | Control | 3.4000 b | 0.3693 | 0.4850d | 0.1338 |
| Drought | 3.1000b | 0.5223 | 0.3350d | 0.1892 | |
| Waterlogging | 5.7778a | 0.6277 | 0.3250d | 0.1892 | |
| Chlorophyll† | Control | 33.7313ab | 0.7448 | 32.7000d | 1.1126 |
| Drought | 31.1000a | 1.0533 | 34.3625d | 1.5734 | |
| Waterlogging | 35.1625b | 1.0533 | 32.9750d | 1.5734 | |
| Leaf RWC (%)‡ | Control | 96.9138a | 0.8440 | 95.9500d | 0.7802 |
| Drought | 91.9350b | 1.1935 | 95.7275d | 1.1034 | |
| Waterlogging | 96.5075ab | 1.1935 | 94.5300d | 1.1034 | |
*Means having letters in common are not statistically different.
†Chlorophyll content in relative units.
‡Relative water content (RWC) of leaf.
Chlorophyll content
For reed canary grass, the better growing condition under waterlogging treatment was also reflected in the chlorophyll measurements taken using the SPAD meter: waterlogged plants had significantly higher levels of chlorophyll than drought or control plants. No significant differences were detected in cocksfoot plants.
Leaf relative water content
For cocksfoot, leaf RWC agrees well with treatment at later time points 2 and 3. At time point 1, differences are not significant, presumably because treatment effects have not yet manifested themselves in the plants (data not shown).
Electrolyte leakage
For both species, electrolyte leakage values were not significantly different between treatments (data not shown). The experimental conditions were not harsh enough to elicit an increased electrolyte leakage in the leaves of stressed plants.
Characterizing the reference transcriptomes
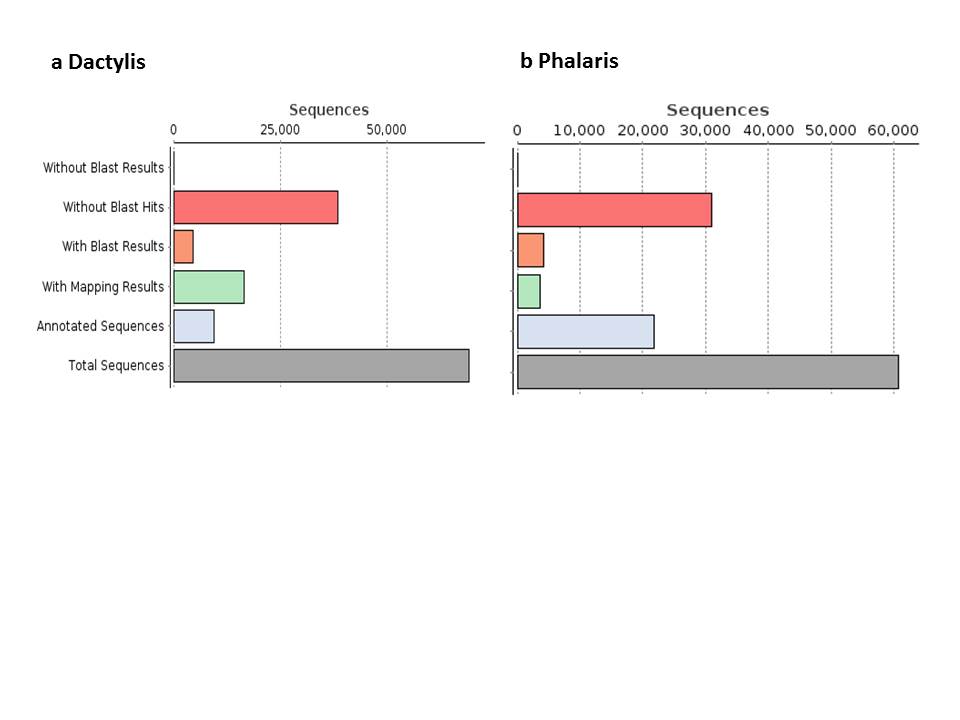
Reference transcriptomes were constructed from a total of 800 million (reed canary grass) and 1 billion (cocksfoot) Illumina sequencing reads generated for this experiment, resulting, respectively, in 21 439 and 22 634 non-redundant transcripts. For cocksfoot, approx. 65 000 ESTs were available at the start of this experiment; however, we decided to restrict the new assembly to the newly generated sequence data: they were all based on ‘Sparta’, and inclusion of sequences from different accessions would have complicated the assembly from sequence polymorphisms likely to be found between cultivars. For reed canary grass, we had constructed earlier a reference transcriptome from approx. 500 000 reads of a normalized library sequenced on the Roche 454 platform which had resulted in approx. 18 000 transcripts (Haiminen et al., 2015). The present transcriptome based on 800 million Illumina reads represents an improvement over the earlier transcriptome and achieved a higher representation of sequences in an individual sample aligned to the assembly: approx. 80 % of the samples’ reads were represented in the Illumina read-based transcriptome, compared with approx. 50 % representation in the assembly constructed from the Roche 454 reads (Haiminen et al., 2015). The transcriptomes were annotated by a BLASTx search of individual translated transcripts against the non-redundant NCBI protein database and adopting existing annotations (Supplementary data Fig. S2).
In particular, many shorter sequences did not have a match with the database after translation. Most top hits were concentrated among the well-annotated species Hordeum vulgare, Aegilops tauschii and Brachypodium distachyon (Fig. 4) which are taxonomically related to the C3 grasses of this study. The overall number of BLAST hits was highest in the more distant grasses Oryza sativa and Zea mays, reflecting the high level of database entries in these species.
Fig. 4.

Species distribution of BLAST top hits for the individual transcripts of the two transcriptomes.
Differential gene expression
The DE scores for each individual sample compared with all the controls are presented in Supplementary data Files 3a and b (for 1000 transcripts with the highest DE scores), and the overall score distribution is visualized in Supplementary data Fig. S3. In the files, the P-value for each transcript is also reported and the sub-set of differentially expressed transcripts with P ≤ 0.005 can be extracted according to the column ‘P-value’. From Supplementary data Fig. S3, it is evident that stress samples (DR and WL) compared with all the controls yield similar DE scores to control samples (DR CT and WL CT) compared with all the other controls. Thus the global distribution of DE scores does not show enrichment for high DE scores in stress, as might have been expected. Possible explanations for this are the different genotypes of the sequenced samples, as is evident from the MDS plots (Supplementary data Fig. S1); sometimes the control samples are more distant from each other than from some stress samples. The only notable exception is the drought stress first time point (T1) in reed canary grass, which shows many high DE scores.
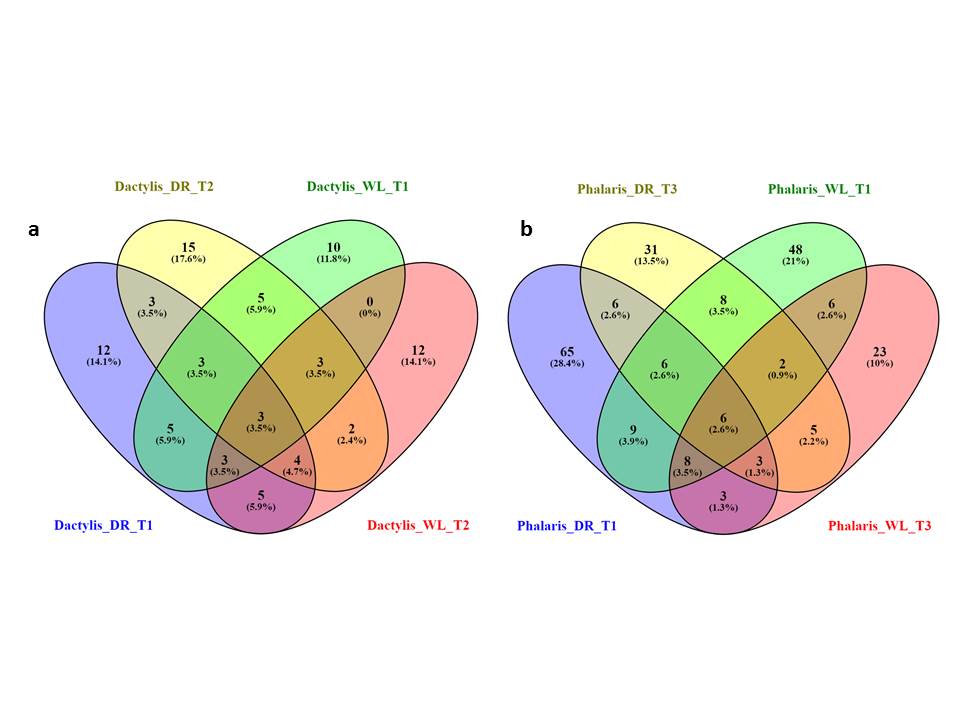
Figure 5 shows the numbers of and the overlaps between top differentially expressed transcripts for each species and each stress condition, given a threshold P-value ≤ 0.005. The total number of differentially expressed transcripts is 55–85 for Dactylis and 124–296 for Phalaris, and they can be found in Supplementary data Files S3a and b (by considering the transcripts highlighted in green in the P-value column). Note that although we were unable to easily assess the top differentially expressed transcript overlaps between the species, as there is no clear homology mapping between their transcripts, we observed similar annotations between the species via PFAM domains (see next sub-sections and Supplementary data File 4).
Fig. 5.

Overlaps of up- (+) and down- (–) regulated differentially expressed transcripts in each species and stress condition for those transcripts with a P-value ≤0.005, for cocksfoot (Dactylis glomerata) (A) and reed canary grass (Phalaris arundinacea) (B). Note that there are no transcripts that are up (+) in one stress and down (–) in the other for cocksfoot. For reed canary grass, there are few such transcripts up in drought and down in waterlogging. For cocksfoot, there are two time points measured (T1 and T2) while for reed canary grass there are three time points (T1, T2 and T3). For example, there are 12 transcripts upregulated in drought T1 and upregulated in waterlogging T2 in cocksfoot (dark pink square in the figure).
In addition to looking individually at the stress samples, we also combined the stress samples in an all vs. all differential expression comparison of the stress and control samples which were aimed at comparing the specific stress responses within species, as reed canary grass is known to be more resilient when faced with waterlogged conditions (transcript sequences are not comparable across species). Figure 6 visualizes the global distribution of all vs. all DE scores in cocksfoot and reed canary grass as a heatmap of transcript density per DE score combination in drought and waterlogging. Both species show an overall trend towards having a similar direction of differential expression in both stress conditions, compared with the controls, observed as higher values near the diagonal.
Fig. 6.

Global view of the DE scores in cocksfoot (Dactylis glomerata) (A) and reed canary grass (Phalaris arundinacea) (B) for drought and waterlogging. The colour indicates the number of transcripts per DE score combination (yellow being high and blue low) number of transcripts.
Some examples of the differentially expressed transcripts are visualized in Fig. 7, showing transcripts that are differentially expressed in both drought and waterlogging at some time point. Annotations are included for those transcripts for which they exist.
Fig. 7.

Transcripts appearing differentially expressed during both drought and waterlogging stress. Red asterisks mark the (transcript, sample) pairs that are differentially expressed (P ≤ 0.005). Colour corresponds to RoDEO-processed expression value averages, yellow = high and blue = low. The control samples for drought and waterlogging are on the left. Annotations were added from BLAST matches to known proteins.
In order to obtain a global view of the behaviour of the differentially expressed transcripts across stress conditions and time points, their DE scores were clustered and visualized. MDS of the differentially expressed transcripts coloured by cluster identity is shown in Supplementary data Fig. S4, and the cluster centroids are visualized in Supplementary data Fig. S5.
For the cocksfoot experiment, the genes that are differentially expressed at both time points can be grouped into two well-separated clusters (Supplementary data Fig. S6a). The 78 genes in cluster 1 are upregulated in waterlogging and downregulated in drought, while the 150 genes in cluster 2 have the opposite behaviour (Supplementary data Fig. S5a). A total of 54 out of 78 (approximately two-thirds) genes in cluster 1 were denoted as uncharacterized proteins. Four genes are related to disease resistance genes (RPM1 and RPP13). In cluster 2, two-thirds of the genes were also denoted as uncharacterized proteins. The remaining genes have a variety of functions and no clear pattern can be deduced from the annotations. For reed canary grass, the genes that are differentially expressed in at least two out of three time points can be grouped into six clusters (Supplementary data Fig. S5b). The six clusters varied in size (cluster 1 = 114 genes. cluster 2 = 281 genes, cluster 3 = 65 genes, cluster 4 = 157 genes, cluster 5 = 73 genes and cluster 6 = 130 genes). Overall, again two-thirds of the genes are uncharacterized proteins. In this case, the behaviour of the clusters is partially overlapping, e.g. several clusters have near zero DE scores for one or the other stress condition. Examples of interesting cluster patterns include monotonic trends across time points: increasing expression for drought in cluster 1 and decreasing expression in cluster 2, compared with the control plants. An interesting candidate in cluster 1 is the two-component response regulator ARR11 which is a transcriptional activator that binds specifically to the DNA sequence 5′-[AG]GATT-3′. The phosphorylation of the aspartate residue in the receiver domain activates the ability of the protein to promote the transcription of target genes, e.g. genes involved in stress response cascades.
Most frequently found PFAM domains in the cocksfoot data set.
For the drought samples at time point 1, a total of 57 PFAM domains were identified (Supplementary data File 4), while 62 were identified for the second drought time point, 45 for waterlogging time point 1 and 39 for waterlogging time point 2. Across all drought- and waterlogging-stressed cocksfoot plants, differentially expressed genes with NB-ARC domains were found in high frequencies (e.g. 11 times at drought time point 1 and nine times in drought time point 2).
Leucine-rich repeat domains and protein kinase domains were found in both types of stress condition. Protein tyrosine kinases were found more frequently in drought samples. Besides known protein domains, several proteins of unknown function also featured frequently, DUF707, DUF1618, DUF1668, DUF1785 and DUF3615. DUF3615 featured especially in waterlogged samples. Many of the domains had only one hit.
Most frequently found PFAM domains in the reed canary grass data set
In reed canary grass under drought and waterlogging, in general more domains were identified per sampling time point (161 at drought time point 1; 103 in drought time point 2; 91 in drought time point 3; 142 in waterlogging time point 1; 84 at waterlogging time point 2; and 87 at waterlogging time point 3; Supplementary data File 4; Supplementary data Fig. S6). The frequent domains found per time point in reed canary grass were the same as in cocksfoot, e.g. NB-ARC domains, leucine-rich repeats and protein kinases.
DISCUSSION
Role of species and genotypes within species for biomass potential
Grassland species growing in river floodplains and other areas are regularly subjected to floods which reduce their growth rate and biomass production (van Eck et al., 2004). However, some grassland species are able to cope with such conditions by physiological and morphological adaptations such as shoot elongation, development of adventitious roots above soil, continuation of photosynthesis under water and the maintenance of oxygen concentrations within tissues (Blom and Voesenek, 1996; Voesenek et al., 2002). Reed canary grass was found to be the most flooding tolerant of 19 species studied by McManmon and Crawford (1971). Flooding tolerance among these 19 species was related to changes in the activity of enzymes associated with glycolytic and respiratory metabolism. Similarly, higher yields were obtained from reed canary grass, a flood-tolerant species, compared with a flood-intolerant grass species (cocksfoot) when the flooding tolerance of different grass cultivars were compared in an experiment in Australia (Norton et al., 2004). Reed canary grass is generally found in floodplain communities as a result of its flooding tolerance, whereas less flood-tolerant species such as cocksfoot are generally not found in floodplain communities but in communities unaffected by flooding (Jung et al., 2009). Greet et al. (2015) found that the prevalence of cocksfoot is reduced following flooding. Kercher and Zedler (2004) suggested that rapid early growth and the maintenance of high levels of root airspace contribute to the flooding tolerance of reed canary grass. Coops et al. (1996) also found that airspace in the rhizome of reed canary grass remained constant with depth of immersion in a study in which reed canary grass survived for 2 years after immersion in depths of water up to 80 cm. Flooding was followed by an increase in mean stem length and the formation of adventitious roots from lower nodes. In our study, reed canary grass responded very well to flooding treatment and nearly doubled its fresh biomass compared with control conditions (Table 1).
Drought-induced losses of yield exceed losses from all other causes (Jones, 1988; Richards, 2004). Cocksfoot is well adapted to dry conditions and can survive soil water deficits more effectively than most forage grasses (Thomas, 1986). The ability of cocksfoot to survive drought has been related to tiller survival, slower shoot growth, greater root density, high leaf RWC, osmotic adjustment in leaf bases, higher concentration of water-soluble carbohydrates (WSCs), greater ability to export WSCs from dying leaves, maintenance of phosphorus status and a lower proline:amino acid ratio (Volaire and Thomas, 1995). Additionally, Volaire (2002) reported that drought survival in cocksfoot was related to the accumulation of specific dehydrins. In our study, cocksfoot adapted relatively well to drought, with only a minor drop in fresh biomass production compared with the control (Table 1). Reed canary grass has been reported to have good tolerance to drought in New Zealand (Kemp and Culvenor, 1994) although its roots still require contact with water in order for the plant to survive. Jung et al. (2009) found no significant difference in biomass production when reed canary grass communities were subjected to drought. Also in our study, reed canary grass under drought conditions had a similar biomass production to that in control conditions.
Given the fact that global bioenergy demand could double by 2030 (IRENA, 2014), it is evident that the development of previously unused or underutilized biomass potential is essential not only to meet demand but also to avoid potential conflict with food production. The use of marginal land for biomass production represents a realistic strategy for increasing bioenergy production while avoiding conflicts with food production (Dauber et al., 2012). The results of this study have shown that biomass production from marginal land can be optimized by the selection of species with specific features to optimize growth and yield under abiotic stress. Improving potential for bioenergy production from marginal land, hitherto not used or little used for energy or food production, can help in mitigating the effects of climate change while improving the ability of the planet to produce both energy and food for a growing global population.
Molecular mechanism of differentially regulated genes and detoxification of xenobiotics
The transcriptomics literature in monocots on flooding tolerance is still scarce (reviewed by Mustroph, 2018). This study on the transcriptional regulation of reed canary grass and cocksfoot is one of the first to report on genes up- and downregulated under flooding conditions in perennial C3 grasses. Rice is the best studied model of the grasses using a quiescence strategy to survive flooding conditions. Transcription factors belonging to group VII ethylene response factors (VII-ERFs) have been related either to restriction of growth under water (SUB1A-1; Xu et al., 2006) or to enhanced growth (SNORKEL1/2; Hattori et al., 2009). Several of these ethylene-responsive transcription factors have been found for the waterlogged treatment for both species in our study. The transcriptomics literature on drought tolerance is still scarce for cocksfoot and reed canary grass, but a good number of reports have been reported for another C3 forage grass species, Lolium multiflorum (Pan et al., 2018), and a lot of headway has been made in arable crop grasses such as maize, rice and wheat (reviewed by Pegler et al., 2018). In L. multiflorum, several differentially expressed proteins and transcripts related to core metabolism have been identified under drought stress, which links in with previous drought physiological studies in reflecting the impact on core metabolism with, for example, higher concentration of WSCs and a lower proline:amino acid ratio (Volaire and Thomas, 1995). We have identified in our study for both species under drought an overlap with the core metabolism differentially related transcripts as reported by Pan et al. (2018), e.g. branched-chain amino acid aminotransferases and lipoxygenases.
The three stage step-wise scheme of detoxification and degradation of xenobiotics proposed by Richard Williams (Neuberger and Smith, 1983) can be applied in interpreting the top differentially expressed genes in flooding and drought response. Supplementary data Files 3a and b contain details for the top 1000 differentially expressed genes per species and treatment. The metabolism of xenobiotics is often divided into three phases: modification, conjugation and excretion. The three phases aim to detoxify xenobiotics and remove them from cells. Several gene families involved in those three steps have been identified in our top differentially expressed gene lists. These involve members of glucosyltransferases, glutathione S-transferases (GSTs) and P450 mono-oxygenases.
In the first step (Phase I) of this pathway, a variety of enzymes act to introduce reactive and polar groups into their substrates. Xenobiotics are altered so that certain functional groups are exposed, enabling Phase II enzymes to bind to them. Oxidation is a common Phase I process, which may be carried out by cytochrome P450 mono-oxygenases (Schuler and Werck-Reichhart, 2003). Several P450 PFAM domain-containing differentially expressed genes were found in our top differentially expressed genes list.
Phase II detoxification involves the action of glycosyl transferases and GSTs to conjugate hydrophilic members to the previously activated molecules. This results in products that can be recognized by Phase III (Bowles et al., 2005). Several glycosyl transferases and GSTs have been identified in our data sets.
In Phase III, the conjugated molecule is moved into the vacuole or extracellular space. This is carried out by ATP-binding cassette (ABC) family transporters (Klein et al., 2006). In data sets for both species, cocksfoot and reed canary grass, ABC transporters have been identified amongst the top differentially expressed genes. Detoxification involves further degradation of the conjugation product in the vacuolar or extracellular space. In our reed canary grass data set, several genes involved extracellularly according to Gene Ontology terms have been identified (Supplementary data File 3b).
Genes involved in the top 20 PFAM domains
The most frequent gene family across both species’ top differentially expressed genes is the NB-ARC family. The NB-ARC domain is a novel signalling motif found in bacteria and eukaryotes, shared by plant resistance gene products and regulators of cell death in animals (Van der Biezen and Jones, 1998). While this domain is thought to bind and hydrolyse ATP, only ADP binding has been experimentally verified (https://www.ebi.ac.uk/interpro/entry/IPR002182). Four of these proteins have been identified in human and mouse, but in plants they are much more frequent, e.g. 1339 identified proteins in rice and 653 in arabidopsis. Most of the plant NB-ARC genes are involved in dealing with the responses to pathogen attacks. Resistance (R) proteins in plants are involved in pathogen recognition and subsequent activation of innate immune responses. Most resistance proteins contain a central nucleotide-binding domain, NB-ARC. The NB-ARC domain consists of three sub-domains: NB, ARC1 and ARC2. The NB-ARC domain is a functional ATPase domain, and its nucleotide binding state is proposed to regulate activity of the R protein (Van Ooijen et al., 2008). NB-ARC domains have also been reported to be involved in the response to abiotic stress resistance such as drought. As such it is not surprising that a large number of differentially expressed genes with those domains were identified under drought and waterlogging experiments for cocksfoot and reed canary grass.
Protein kinases have also been previously reported in conjunction with abiotic stress such as drought (Wei et al., 2014). Protein kinases were amongst the most frequently classes of domains in the drought and waterlogging experiments carried out in this study.
Several proteins with domains of unknown function (DUF) (Bateman et al., 2010) were identified in this study, such as DUF1618, DUF1668, DUF3615, DUF4220 and DUF4413. Several of those have been reported previously in other plant genome projects (e.g. DUF4220 in a genomic region related to yield and drought resistance in wheat: Hen-Avivi et al., 2016), but their function still requires to be elucidated. DUFs are still a large set of uncharacterized protein families (Bateman et al., 2010). Some of those have been shown to be specific to monocots, e.g. DUF1618 which has been shown to be expressed in rice under certain stress and hormone conditions (Wang et al., 2014). For DUF3616, a higher expression in rice roots has been shown when subjected to salt and drought stress (Campo et al., 2014). These DUF domains could be components of the interesting genes to answer how plants cope with anoxia as in waterlogging and drought. However, functional genomics approaches would be required to verify the function of those DUFs under drought and waterlogging/anoxia.
Cocksfoot (Stebbins, 1971) and reed canary grass (Anderson, 1961) are predominantly tetraploid inherited. The factor ploidy has been shown to have an impact on gene expression comparing relatives in arabidopsis of different ploidy levels (Miller et al., 2012); however, in our study, we compared gene expression differences amongst genotypes of the same cultivars and species without an interference of different ploidy levels amongst genotypes of the same varieties within species. We therefore did not consider ploidy as a valid treatment level in the analysis of our gene expression data.
Pathway towards future implementation of findings in molecular crop improvement programmes
Modern plant breeding allows us to improve our crops at accelerated rates by selecting varieties with resistance to biotic stress, higher yields and tolerance to abiotic stresses (Małyska and Jacobi, 2018). The most prominent example of successful agronomical application of knowledge on a flooding tolerance trait comes from rice. Quantitative trait locus (QTL) analyses and subsequent molecular investigations, including transcriptomic studies, have revealed the underlying ethylene-responsive factor (VII-ERFs) genes (Xu et al., 2006; Hattori et al., 2009). The genetic trait, the ability to induce quiescence, has been successfully introduced into a molecular breeding programme for crop improvement (Ismail et al., 2013; Singh et al., 2017). It is not inconceivable to make such progress in crop improvement in the two grass species reported in this study. If genome editing techniques were to become acceptable as non-genetically modified techniques in the future, abiotic stress could also be tackled by following the genome editing route, picking the most important differentially regulated genes under stress for trait improvement.
SUPPLEMENTARY DATA
Supplementary data are available online at https://academic.oup.com/aob and consist of the following. Figure S1: multidimensional scaling for RoDEO-processed cocksfoot and reed canary grass samples. Figure S2: annotation of the transcriptomes of cocksfoot and reed canary grass. Figure S3: distribution of DE scores for cocksfoot and reed canary grass drought and waterlogging experiments. Figure S4: multidimensional scaling of differentially expressed transcripts coloured by cluster membership, where the clusters are defined by the genes’ DE scores for each time point and stress condition. Figure S5: cluster centroids of stress patterns by species. Figure S6: visualization of overlapping PFAM domains in the cocksfoot and reed canary grass data sets using VENNY. File S1: time line of the experiments and list of genotypes used. File S2: description of RoDEO scaling evaluation. File S3: cocksfoot and reed canary grass top differential gene expression gene list. File S4: cocksfoot and reed canary grass PFAM domain frequencies.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
FUNDING
This project has been funded through the FP7 grant GrassMargins (FP7-KBBE-2011-5-289461).
LITERATURE CITED
- Anderson D. 1961. Taxonomy and distribution of the genus Phalaris. Iowa State Journal of Science 36: 1–96. [Google Scholar]
- Atanasiu B. 2010. The role of bioenergy in the National Renewable Energy Action Plans: a first identification of issues and uncertainties. Institute for European Environmental Policy; https://ieep.eu/uploads/articles/attachments/208dab71-7833-4016-87b1-8cb15c3f41dc/bioenergy_in_NREAPs.pdf?v=63664509743. Accessed 14 September 2018. [Google Scholar]
- Baffes J, Haniotis T. 2010. Placing the 2006/08 commodity price boom into perspective. Policy Research Working Paper. The World Bank. [Google Scholar]
- Bateman A, Coggill P, Finn RD. 2010. DUFs: families in search of function. Acta Crystallographica Section F: Structural Biology and Crystallization Communications 66: 1148–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann U, Juttner J, Bian X, Langridge P. 2000. Self-incompatibility in the grasses. Annals of Botany 85 (Suppl A): 203–209. [Google Scholar]
- Blom CW, Voesenek LA. 1996. Flooding: the survival strategies of plants. Trends in Ecology & Evolution 11: 290–295. [DOI] [PubMed] [Google Scholar]
- Bowles D, Isayenkova J, Lim EK, Poppenberger B. 2005. Glycosyltransferases: managers of small molecules. Current Opinion in Plant Biology 8: 254–263. [DOI] [PubMed] [Google Scholar]
- Bushman BS, Larson SR, Tuna M, et al. . 2011. Orchardgrass (Dactylis glomerata L.) EST and SSR marker development, annotation, and transferability. Theoretical and Applied Genetics 123: 119–129. [DOI] [PubMed] [Google Scholar]
- Bushnell B. 2015. BBMap short read aligner, and other bioinformatic tools. http://sourceforge.net/projects/bbmap. Accessed 14 September 2018. [Google Scholar]
- Campbell JE, Lobell DB, Genova RC, Field CB. 2008. The global potential of bioenergy on abandoned agriculture lands. Environmental Science & Technology 42: 5791–5794. [DOI] [PubMed] [Google Scholar]
- Campo S, Baldrich P, Messeguer J, Lalanne E, Coca M, Segundo BS. 2014. Overexpression of a calcium-dependent protein kinase confers salt and drought tolerance in rice by preventing membrane lipid peroxidation. Plant Physiology 165: 688–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifton-Brown JC, Breuer J, Jones MB. 2007. Carbon mitigation by the energy crop, Miscanthus. Global Change Biology 13: 2296–2307. [Google Scholar]
- Coops H, van den Brink FWB, van der Velde G. 1996. Growth and morphological responses of four helophyte species in an experimental water-depth gradient. Aquatic Botany 54: 11–24. [Google Scholar]
- Dauber J, Brown C, Fernando A, et al. . 2012. Bioenergy from ‘surplus’ land: environmental and socio-economic implications. BioRisk 7: 5–50. [Google Scholar]
- Edenhofer O, Knopf B, Barker T, et al. . 2010. The economics of low stabilization: model comparison of mitigation strategies and costs. The Energy Journal 31: 11–48. [Google Scholar]
- Etherington JR. 1984. Relationship between morphological adaptation to grazing, carbon balance and waterlogging tolerance in clones of Dactylis glomerata L. New Phytologist 98: 647–658. [Google Scholar]
- Giancarlo R, Scaturro D, Utro F. 2015. ValWorkBench: an open source Java library for cluster validation with applications to microarray data analysis. Computer Methods and Programs in Biomedicine 118: 207–217. [DOI] [PubMed] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, et al. . 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nature Biotechnology 29: 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greet J, Webb JA, Cousens RD. 2015. Floods reduce the prevalence of exotic plant species within the riparian zone: evidence from natural floods. Applied Vegetation Science 18: 503–512. [Google Scholar]
- Haiminen N, Klaas M, Zhou Z, et al. . 2015. Comparative exomics of Phalaris cultivars under salt stress. BMC Genomics 15 (Supplement 6): S18. doi: 10.1186/1471-2164-15-S6-S18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori Y, Nagai K, Furukawa S, et al. . 2009. The ethylene response factors SNORKEL1 and SNORKEL2 allow rice to adapt to deep water. Nature 460: 1026–1030. [DOI] [PubMed] [Google Scholar]
- Hen-Avivi S, Savin O, Racovita RC, et al. . 2016. A metabolic gene cluster in the wheat W1 and the barley Cer-cqu loci determines β-diketone biosynthesis and glaucousness. The Plant Cell 28: 1440–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu T, Sun X, Zhang X, Nevo E, Fu J. 2014. An RNA sequencing transcriptome analysis of the high-temperature stressed tall fescue reveals novel insights into plant thermotolerance. BMC Genomics 15: 1147. doi: 10.1186/1471-2164-15-1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IRENA. 2014. Global bioenergy supply and demand projections. A working paper for RE map 2030. http://www.irena.org/remap/IRENA_REmap_2030_Biomass_paper_2014.pdf. Accessed 14 September 2018. [Google Scholar]
- Ismail AM, Singh US, Singh S, Dar MH, Mackill DJ. 2013. The contribution of submergence-tolerant (Sub1) rice varieties to food security in flood-prone rainfed lowland areas in Asia. Field Crops Research 152: 83–93. [Google Scholar]
- Jones MB. 1988. Water stress. In: Jones MB, Lazenby A, edrs. The grass crop—the physiological basis of production. London: Chapman and Hall, 205–236. [Google Scholar]
- Jones MB, Finnan J, Hodkinson T. 2014. Morphological and physiological traits for higher biomass production in perennial rhizomatous grasses grown on marginal land. GCB Bioenergy 7: 375–385. [Google Scholar]
- Jung V, Hoffmann L, Muller S. 2009. Ecophysiological responses of nine floodplain meadow species to changing hydrological conditions. Plant Ecology 201: 589–598. [Google Scholar]
- Kemp DR, Culvenor RA. 1994. Improving the grazing and drought tolerance of temperate perennial grasses. New Zealand Journal of Agricultural Research 37: 365–378. [Google Scholar]
- Kercher SM, Zedler JB. 2004. Flood tolerance in wetland angiosperms: a comparison of invasive and noninvasive species. Aquatic Botany 80: 89–102. [Google Scholar]
- Klein M, Burla B, Martinoia E. 2006. The multidrug resistance-associated protein (MRP/ABCC) subfamily of ATB-binding cassette transporters in plants. FEBS Letters 580: 1112–1122. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler Transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, et al. . 2009. The sequence alignment/map (SAM) format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YF, Wang Y, Tang Y, Kakani VG, Mahalingam R. 2013. Transcriptome analysis of heat stress response in switchgrass (Panicum virgatum L.). BMC Plant Biology 13: 153. doi: 10.1186/1471-2229-13-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Małyska A, Jacobi J. 2018. Plant breeding as the cornerstone of a sustainable bioeconomy. New Biotechnology 40: 129–132. [DOI] [PubMed] [Google Scholar]
- McDonald MP, Galway NW, Colmer TD. 2002. Similarity and diversity in adventitious root anatomy as related to aeration among a range of wetland and dryland grass species. Plant, Cell & Environment 25: 441–451. [Google Scholar]
- McManmon M, Crawford RMM. 1971. Metabolic theory of flooding tolerance: the significance of enzyme distribution and behaviour. New Phytologist 70: 299–306. [Google Scholar]
- Małyska A, Jacobi J. 2018. Plant breeding as the cornerstone of a sustainable bioeconomy. New Biotechnology 40: 129–132. [DOI] [PubMed] [Google Scholar]
- Miller M, Zhang C, Chen ZJ. 2012. Ploidy and hybridity effects on growth vigor and gene expression in Arabidopsis thaliana hybrids and their parents. G3 (Bethesda) 2: 505–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustroph A. 2018. Improving flooding tolerance of crop plants. Agronomy 8: 160. doi: 10.3390/agronomy8090160. [DOI] [Google Scholar]
- Neuberger A, Smith RL. 1983. Richard Tecwyn Williams: the man, his work, his impact. Drug Metabolism Reviews 14: 559–607. [DOI] [PubMed] [Google Scholar]
- Norton M, Koetz E, Stewart G. 2004. Perennial grasses for waterlogging prone, summer dry environments. Cahiers Options Mediterraneennes 62: 121–124. [Google Scholar]
- Oliveros JC. 2007. Venny. An interactive tool for comparing lists with Venn’s diagrams. http://bioinfogp.cnb.csic.es/tools/venny/index.html. Accessed 14 September 2018. [Google Scholar]
- Pan L, Meng C, Wang J, et al. . 2018. Integrated omics data of two annual ryegrass (Lolium multiflorum L.) genotypes reveals core metabolic processes under drought stress. BMC Plant Biology 18: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegler JL, Grof CPL, Eamens AL. 2018. Profiling of the differential abundance of drought and salt stress-responsive microRNAs across grass crop and genetic model plant species. Agronomy 8: 118. doi: 10.3390/agronomy8070118. [DOI] [Google Scholar]
- Potter KN, Torbert HA, Johnson HB, Tischler CR. 1999. Carbon storage after long term grass establishment on degraded soils. Soil Science 164: 718–725. [Google Scholar]
- Prochnow A, Heiermann M, Plochl M, et al. . 2009. a Bioenergy from permanent grassland – a review: 1. Biogas. Bioresource Technology 100: 4931–4944. [DOI] [PubMed] [Google Scholar]
- Prochnow A, Heiermann M, Plochl M, Amon T, Hobbs PJ. 2009b Bioenergy from permanent grassland – a review: 2. Combustion. Bioresource Technology 100: 4945–5954. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen S, Barah P, Suarez-Rodriguez MC, et al. . 2013. Transcriptome responses to combinations of stresses in Arabidopsis. Plant Physiology 161: 1783–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards RA. 2004. Physiological traits used in the breeding of new cultivars for water-scarce environments. In: Fischer T, Turner N, Angus J, et al. , eds. New directions for a diverse planet. Proceedings of the 4th International Crop Science Congress. The Regional Institute Ltd, Gosford, Australia: 1–12. [Google Scholar]
- Saarela JM, Bull RD, Paradis MJ, et al. . 2017. Molecular phylogenetics of cool-season grasses in the subtribes Agrostidinae, Anthoxanthinae, Aveninae, Brizinae, Calothecinae, Koeleriinae and Phalaridinae (Poaceae, Pooideae, Poeae, Poeae chloroplast group 1). PhytoKeys 87: 1–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAS Institute Inc. 2011. SAS/STAT® 9.3 User’s Guide. Cary, NC: SAS Institute Inc. [Google Scholar]
- Schuler MA, Werck-Reichhart D. 2003. Functional genomics of P450s. Annual Review of Plant Biology 54: 629–667. [DOI] [PubMed] [Google Scholar]
- Shu Y, Zhang J, Ao Y, Song L, Guo C. 2015. Analysis of the Thinopyrum elongatum transcriptome under water deficit stress. International Journal of Genomics 2015, Article ID 265791. doi: 10.1155/2015/265791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Septiningsi EM, Balyan HS, Singh NK, Rai V. 2017. Genetics, physiological mechanism and breeding of flood-tolerant rice (Oryza sativa L.). Plant &Cell Physiology 58: 185–197. [DOI] [PubMed] [Google Scholar]
- Stebbins GL. 1971. Chromosomal evolution in higher plants. London: Edward Arnold. [Google Scholar]
- Talukder SK, Azhaguvel P, Mukherjee S, et al. . 2015. De novo assembly and characterization of tall fescue transcriptome under water stress. The Plant Genome 8: doi: 10.3835/plantgenome2014.09.0050. [DOI] [PubMed] [Google Scholar]
- Tibshirani R, Walther G, Hastie T. 2001. Estimating the number of clusters in a dataset via the Gap Statistics. Journal of the Royal Statistical Society B 2: 411–423. [Google Scholar]
- Thomas H. 1986. Water use characteristics of Dactylis glomerata L., Lolium perenne L. and L. multiflorum Lam. plants. Annals of Botany 57: 211–223. [Google Scholar]
- Tilman D, Hill J, Lehman C. 2006. Carbon-negative biofuels from low-input high-diversity grassland biomass. Science 314: 1598–1600. [DOI] [PubMed] [Google Scholar]
- UniProt Consortium. 2017. UniProt: the universal protein knowledgebase. Nucleic Acids Research 45: D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Biezen EA, Jones JD. 1998. Plant disease-resistance proteins and the gene-for-gene concept. Trends in Biochemical Sciences 23: 454–456. [DOI] [PubMed] [Google Scholar]
- Van Eck WHJM, Van De Steeg M, Blom CWPM, De Kroon H. 2004. Is tolerance to summer flooding correlated with distribution patterns in river floodplains? A comparative study of 20 terrestrial grassland species. Oikos 107: 393–405. [Google Scholar]
- Van Ooijen G, Mayr G, Kasiem MMA, Albrecht M, Cornelissen BJC, Takken FLW. 2008. Structure–function analysis of the NB-ARC domain of plant disease resistance proteins. Journal of Experimental Botany 59: 1383–1397. [DOI] [PubMed] [Google Scholar]
- Venu RC, Sreerekha MV, Madhav MS, et al. . 2013. Deep transcriptome sequencing reveals the expression of key functional and regulatory genes involved in the abiotic stress signaling pathways in rice. Journal of Plant Biology 56: 216–231. [Google Scholar]
- Verelst W, Bertolini E, De Bodt S, et al. . 2012. Molecular and physiological analysis of growth-limiting drought stress in Brachypodium distachyon leaves. Molecular Plant 6: 311–322. [DOI] [PubMed] [Google Scholar]
- Voesenek LACJ, Rijnders JHGM, Peeters AJM, van de Steeg HM, de Kroon H. 2002. Plant hormones regulate fast shoot elongation under water: from genes to communities. Ecology 85: 16–27. [Google Scholar]
- Volaire F. 2002. Drought survival, summer dormancy and dehydrin accumulation in contrasting cultivars of Dactylis glomerata. Physiologia Plantarum 116: 42–51. [DOI] [PubMed] [Google Scholar]
- Volaire F, Thomas H. 1995. Effects of drought on water relations, mineral-uptake, water soluble carbohydrate accumulation and survival of two contrasting populations of cocksfoot (Dactylis glomerata L.). Annals of Botany 75: 513–524. [Google Scholar]
- van Vuuren DP, Bellevrat E, Kitous A, Isaac M. 2010. Bio-energy use and low stabilization scenarios. The Energy Journal 31 (special issue 1): 193–222. [Google Scholar]
- Wang L, Shen R, Chen LT, Liu YG. 2014. Characterization of a novel DUF1618 gene family in rice. Journal of Integrative Plant Biology 56: 151–158. [DOI] [PubMed] [Google Scholar]
- Wei K, Wang Y, Zhong X, Pan S. 2014. Protein kinase structure, expression and regulation in maize drought signalling. Molecular Breeding 34: 583–602. [Google Scholar]
- Wiegmann K, Hennenberg KJ, Fritsche UR. 2008. Degraded land and sustainable bioenergy feedstock production. In: Joint international workshop on high nature value criteria and potential for sustainable use of degraded lands. http://np-net.pbworks.com/f/OEKO,+RSB,+UNEP+et+al+(2008)+Degraded+land+and+sustainable+bioenergy+feedstock+production.pdf. Accessed 14 September 2018. [Google Scholar]
- Xu K, Xu X, Fukao T, et al. . 2006. Sub1A is an ethylene-response-factor-like gene that confers submergence tolerance to rice. Nature 442: 705–708. [DOI] [PubMed] [Google Scholar]
- Yamamoto N, Takano T, Tanaka K, et al. . 2015. Comprehensive analysis of transcriptome response to salinity stress in the halophytic turfgrass Sporobolus virginicus. Frontiers in Plant Sciences 6: 241. doi: 10.3389/fpls.2015.00241. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.