

Abstract
Prolactin (PRL) plays an important role in trophoblast growth, placental angiogenesis and immunomodulation within the feto-maternal interface, where different cell types secrete PRL and express its receptor. During pregnancy, inflammatory signalling is a deleterious event that has been associated with poor fetal outcomes. The placenta is highly responsive to the inflammatory stimulus; however, the actions of PRL in placental immunity and inflammation remain largely unknown. The aim of this study was to evaluate PRL effects on the TLR4/NFkB signalling cascade and associated inflammatory targets in cultured explants from healthy term human placentas. An in utero inflammatory scenario was mimicked using lipopolysaccharides (LPS) from Escherichia coli. PRL significantly reduced LPS-dependent TNF-α, IL-1β and IL-6 secretion and intracellular levels. Mechanistically, PRL prevented LPS-mediated upregulation of TLR-4 expression and NFκB phosphorylation. In conclusion, PRL limited inflammatory responses to LPS in the human placenta, suggesting that this hormone could be critical in inhibiting exacerbated immune responses to infections that could threaten pregnancy outcome. This is the first evidence of a mechanism for anti-inflammatory activity of PRL in the human placenta, acting as a negative regulator of TLR-4/NFkB signaling.
Keywords: cytokines, inflammatory response, peptide hormones, placenta, Toll-like receptors
Introduction
Pregnancy favours a unique immunoendocrine milieu through the action of diverse cytokines and hormones, which coordinately participate in feto-maternal tolerance, pregnancy continuity and parturition (Napso et al., 2018). Endocrine, paracrine and autocrine placental mediators, including prolactin (PRL), are also involved in the immunomodulatory activity. PRL is a neuroendocrine hormone mainly synthesized by lactotrophs in the adenohypophysis, but other extra-pituitary tissues are also able to produce it (Featherstone et al., 2012). During pregnancy, the decidua is the main source of PRL, playing an important role for placental growth and angiogenesis (Stefanoska et al., 2013; Binart, 2016). In addition to the maternal decidua, fetal villous trees, columnar trophoblasts and cytotrophoblasts are placental sites for PRL and PRL-receptor (PRL-R) expression (Garzia et al., 2013; Stefanoska et al., 2013), which suggests that PRL could exert paracrine/autocrine roles in the placenta. The placenta is a complex organ with a known immune activity, of being able to produce diverse chemokines, cytokines and antimicrobial peptides in response to bacterial compounds, such as lipopolysaccharides (LPS), as a defensive mechanism (Olmos-Ortiz et al., 2018; Duval et al., 2019).
Normal pregnancy constitutes a physiologic hyperprolactinemic state, given that PRL levels fluctuate ~100 ng/mL to 500 ng/mL in the intervillous space and cord blood at term (Ferriani and Silva de Sa, 1988; Takser et al., 2004). Even so, fetal membranes have a central role in PRL accumulation into amniotic fluid, with levels as high as 3500 ng/mL (Cheng et al., 2011). These concentrations are substantially higher when compared to non-pregnant female serum (8–23 ng/mL) (Mayo-Clinic-Laboratories, 2018).
Regarding the effects of PRL in the placenta, it is known that this hormone stimulates trophoblast migration and invasion (Stefanoska et al., 2013). In addition, PRL may behave as a cytokine with tissue-specific immunomodulatory activities. For instance, PRL treatment was shown to induce interleukin (IL)-1β, gamma interferon and tumour necrosis factor (TNF)-α release by murine peritoneal macrophages in vitro (Sodhi and Tripathi, 2008). In contrast, our laboratory demonstrated that exogenous PRL downregulated IL-1β and TNF-α in cultured chorioamniotic membranes (Zaga-Clavellina et al., 2014; Flores-Espinosa et al., 2017). Taking into account the PRL-R and PRL expression in placenta, it is critical to understand the regulation of the placental immune profile by this hormone, especially under an inflammatory scenario. Inflammation could be related to infectious or non-infectious causes, and the resulting signaling is deleterious for pregnancy and is associated with poor fetal outcomes, such as premature rupture of membranes and preterm birth (Tchirikov et al., 2018). A great deal of evidence supports that Escherichia coli is one of the most common Gram-negative bacteria causing urinary tract infections during pregnancy, which in turn may result in preterm labor (Dautt-Leyva et al., 2018).
It has been clearly established that Toll-like receptor (TLR)-4 signaling can be triggered by LPS binding, which initiates a phosphorylation cascade of myeloid differentiation factor 88 (MyD88), the IL-1 receptor–associated kinases, the TNF receptor–associated factor and the IκB kinase (IKK) complex formed by IKKα, IKKβ and NEMO. IKK phosphorylates IκB proteins, which results in its ubiquitination and degradation. Consequently, IκB is dissociated from NF-κB, which can freely translocate to the nucleus and modulate genes containing NF-κB binding sites, such as the pro-inflammatory cytokine TNF-α (Mitchell et al., 2016). It is important to note that the placenta express TLR-4 throughout pregnancy (Beijar et al., 2006).
Given the pleiotropic and sometimes opposite effects that PRL exerts depending on the tissue, the main objective of the present work was to evaluate the immunomodulatory capacity of this hormone in the human placenta by studying the effect of PRL on levels of pro-inflammatory cytokines TNF-α, IL-1β and IL-6 in cultured placental explants exposed to LPS from E. coli. LPS was selected to emulate an inflammatory scenario for exploring the immunomodulatory ability of PRL. Our data show that PRL significantly inhibits the inflammatory status induced by LPS treatment and suggest that this effect may involve partial blockade of the TLR-4/NFκB pathway.
Methods
Ethics statement
This protocol was approved by the Biosafety, Ethical and Research Committee from the Instituto Nacional de Perinatología—Isidro Espinosa de los Reyes and from the Instituto Mexicano del Seguro Social and is registered under code numbers 212250-3210-21205-01-14 and R-2017-785-013, respectively. All methodological approaches were conducted according to the guidelines of the Declaration of Helsinki. A written informed consent was obtained voluntarily from each mother before caesarean section.
Reagents
DMEM culture media and fetal bovine serum (FBS) were from Invitrogen (Carlsbad, CA, USA). LPS from E. coli 055:B5 was purchased from Sigma-Aldrich (St. Louis, MO, USA) and was diluted in PBS. Recombinant human PRL was from PeproTech (Rocky Hill, NJ, USA) and was reconstituted in H2O_0.1% bovine serum albumin (BSA). Alin is a commercial formulation of dexamethasone (DXM) that was purchased from Chinoin Laboratories (Mexico City, México); DXM is a classical anti-inflammatory drug used in the clinical practice and was used as an anti-inflammatory control. Trixilem is a commercial formulation of methotrexate (MTX) from Teva Laboratories (Toluca, Estado de Mexico, México) and was used as a pan-Jak inhibitor. Del-1-9-G129R-hPRL (herein abbreviated as Del-1-9-G129R) is an engineered variant of PRL that binds but does not activate the PRL-R; therefore, it acts as a competitive antagonist of endogenous PRL for PRL-R triggering; Del-1-9-G129R was used as a PRL antagonist and was produced as earlier described (Goffin et al., 1992). All other reagents were purchased from Sigma-Aldrich.
Cotyledon explant culture and experimental procedures
Exclusion criteria for this study comprised patients with endocrine, metabolic, infectious and other systemic diseases such as hypertension, diabetes mellitus or thyroid, liver or choric renal diseases. Also, patients allergic to penicillin or streptomycin as well as patients who suffered cervico-vaginal infections during the third trimester of pregnancy were excluded from this study. Placentas were obtained by caesarean sections performed at Instituto Nacional de Perinatología or Hospital Gineco-Obstetricia No. 4 ‘Luis Castelazo Ayala’ from term (37–40 weeks) uncomplicated pregnancies. All patients included in this study lived in Mexico City, they were Hispanic and of medium socioeconomic status. Specific information from the patients is described in Table I.
Table I.
Clinical data of mothers and newborns.
|
n = 8 (mean ± SD) |
Range (min − max) |
|
|---|---|---|
| Maternal age (years) | 28.7 ± 4.0 | 23–34 |
| Pre-gestational BMI (kg/m2) | 25.8 ± 2.6 | 22.3–28.9 |
| Gestational age (weeks) | 38.5 ± 0.7 | 37.3–39.5 |
| Number of pregnancies | 3 ± 0.9 | 2–5 |
| Newborn weight (grams) | 3091 ± 208 | 2820–3500 |
| Newborn length (cm) | 49.3 ± 0.8 | 48–51 |
| Newborn cephalic perimeter (cm) | 34.3 ± 0.5 | 33.5–35.0 |
| Newborn sex | ||
| Female (%) | 50% | |
| Male (%) | 50% |
For these experiments, we used a total of eight placentas; the cytokine concentrations by ELISA was measured in all them. However, only four placentas were processed and used for western blots.
Placental cotyledons were exhaustively washed with sterile 0.9% NaCl; blood clots, blood vessels, decidua and chorionic basal plate were removed. Cotyledons were cut into 3–5-mm fragments, placed into 24-well culture dishes and maintained for 24 h in DMEM supplemented culture media (DMEM + 10% FBS + 1% sodium pyruvate + 1% penicillin/streptomycin) in a humidified incubator at 37°C and 5% CO2–95% air. To mimic an infection, we used LPS (500 ng/mL), a major component of the outer membrane of Gram-negative bacteria that can induce a strong inflammatory response in animals primarily via TLR-4. PRL was used at 100 ng/mL, 300 ng/mL and 500 ng/mL, which corresponds to physiologic concentrations in the intervillous space and cord blood at term (Takser et al., 2004; Ferriani and Silva de Sa, 1988); DXM was used at 300 nm, MTX was used at 50 μm, and Del-1-9-G129R was used at 5-fold molar excess compared to PRL. Vehicle (Vh)-treated explants were incubated with PBS, H2O and 0.1% BSA. Additionally, untreated explants (U) were incubated without any experimental compound.
On the first day of incubation (0–24 h), we pretreated explants with PRL 0 ng/mL, 100 ng/mL, 300 ng/mL or 500 ng/mL, as indicated, to preserve a quiescent inflammatory profile. The next day (25–48 h), the media was changed, and cotyledon explants were treated with LPS 500 ng/mL and/or the indicated treatments. In parallel, some explants were co-incubated with PRL, DXM, MTX or Del-1-9-G129R. After 24 h of incubation, the culture media was frozen until cytokine quantification and cotyledon tissue was homogenized in protein lysis buffer for western blotting analysis.
Quantification of pro-inflammatory cytokines
After thawing, the amounts of TNF-α from culture media were quantified by an R&D system ELISA commercial kit (DY210, Minneapolis, MN, USA) with a 31 pg/mL detection limit. IL-1β and IL-6 were quantified by using Peprotech ELISA commercial kits (900-K95 and 900-K16), with detection limits of 8 pg/mL and 46 pg/mL, respectively. PRL quantification was determined with an R&D system ELISA commercial kit (DY682), with a detection limit of 16 pg/mL. Assays were performed according to the manufacturer’s instructions.
At the end of each experiment, the placental explant weight was measured, and the concentration of each analyte was normalized by 1 g of wet tissue.
Western blotting
After treatments, explants were washed and mechanically disrupted using a polytron homogenizer (OMNI International, Kennesaw, GA, USA) in the presence of cold lysis buffer (HEPES 10 mm, MgCl2 1.5 mm, KCl 10 mm, DTT 0.5 mm, Nonidet P-40 0.5%, Na3VO4 5 mM, NaF 20 mm and protease inhibitor cocktail Sigma-Aldrich P8340, 1:1000) and incubated for 30 min with shaking on ice. Then lysates were centrifuged at 7000 rpm for 1 min at 4°C. Supernatants and pellets were used as total protein lysates.
Total protein content was quantified by the Bradford method (Bradford, 1976). Equal amounts of protein from the homogenates were boiled for 10 min in Laemmli buffer and separated by electrophoresis. Samples to be assessed for TLR-4, phosphorylated-Jak-2 and phosphorylated-IΚB were separated using SDS-PAGE while those for IL-1β, TNF-α and IL-6 were separated using Tricine-SDS-PAGE, designed for separation of small molecular weight proteins (Schagger, 2006).
Proteins were transferred to nitrocellulose membranes, blocked with 5% non-fat dry milk (Bio-Rad, Hercules, CA, USA) and incubated overnight at 4°C with the following primary antibodies: phospho-Jak-2 (Cell Signaling, Boston, MA, USA. 3771S, 1:1500, rabbit), phospho-IκB (Cell Signaling 2859S, 1:1500, rabbit), IL-1β (Santa Cruz, Santa Cruz, CA, USA. sc-7884, 1:500, rabbit), IL-6 (Santa Cruz sc-7920,1:350, rabbit), TNF-α (Santa Cruz sc-1350, 1:350, goat), TLR-4 (Santa Cruz, sc-293072, 1:750, mouse), GAPDH (Abcam, Cambridge, MA, USA. ab8245, 1:8000, mouse) or β-actin (Abcam ab8226, 1:8000, mouse). The following day, membranes were incubated with their respective secondary antibodies: goat anti-rabbit IgG-HRP (Vector Laboratories, Burlingame, CA, USA. PI-1000, 1:6000), horse anti-goat IgG-HRP (Vector, PI-9500, 1:6000) or goat anti-mouse IgG-HRP (R&D Systems HAF007, 1:6000). Afterwards immune complexes were detected in membranes with the Immobilon Western chemiluminescent peroxidase/luminol 1:1 (Millipore, Burlington, MA, USA), and densitometric analysis was performed by using the Molecular Imager ChemiDoc XRS System and the Image Lab Software (Bio-Rad, Hercules, CA, USA).
NFκB activity ELISA assay
The commercial kit NFκB p65 InstantOne ELISA (eBioscience, 85-86083) was used for detection of total and phosphorylated NFκB in protein extracts. NFκB dimers containing p65 are activators of transcription. Total extracts were adjusted to 35 μg of protein, and NFκB activity was expressed as phosphorylated/total NFκB ratio. Plate readings were performed at 450 nm with correction to 650 nm.
PCR amplifications
The effect of LPS effect on PRL and PRL-R gene expression was studied by extracting total RNA from treated cotyledon explants using TRIzol reagent. In all cases, the amount and quality of RNA were estimated spectrophotometrically at 260/280 nm, and a constant amount of RNA (1 μg) was reverse transcribed using a RT assay, with previous DNAsa I treatment. Primers and probes for PCR amplifications were designed by TIB MOLBIOL, and the sequences are shown in Table II. Identical PCR conditions were performed for all genes, and in all cases, the results were normalized against GAPDH, used as housekeeping gene internal control.
TableII.
Primers and probes for PCR amplifications.
| Gene/accession number | Forward primer | Reverse primer | FL probe | LC probe | Amplicon (nt) |
|---|---|---|---|---|---|
| Prolactin/ NM_000948 |
gggAAACgAATg CCTgAT |
CAAACAggTCTCg AAgggT |
CCTgCTCCTgTgCC AgAgCgTg-FL |
LC-CCCCCTTgCCC ATCTgTCCCg—PH |
274 |
| Prolactin receptor/ NM_000949 |
TggTTCACgCTCCTgT ATgAA |
TggACTCCATgCA CTCCAgT |
TCAgCCTACATCCAgg ACAgAAATACCTTg—FL |
LC-CCAggTTCgCTg CAAACCAgAC--PH |
174 |
| GAPDH/NM_002046 | gAAggTgAAggTCgg AgTC |
gAAgATggTgATggg ATTTC |
AggggTCATTgATggC AACAATATCCA-FL |
LC-TTTACCAgAgTTAA AAgCAgCCCTggTg-p |
226 |
Amplifications were carried on the LightCycler 2.0 instrument (Roche), according to the following protocol: activation of Taq DNA polymerase and DNA denaturation at 95°C for 5 min, proceeded by 45 amplification cycles of 10 s at 95°C, 10 s at 60°C and 15 s at 72°C and a final cooling cycle for 2 min at 40°C.
Statistical analysis
Data were analysed by one-way ANOVA with Dunn, Dunnett or Holm–Sidak post hoc tests, depending on normality data distribution, as indicated in figure legends. Statistical differences were calculated using a specialized software package (SigmaPlot 11.0, Jandel Scientific). Differences were considered statistically significant at P < 0.05.
Results
PRL and LPS biological activity in placental explants
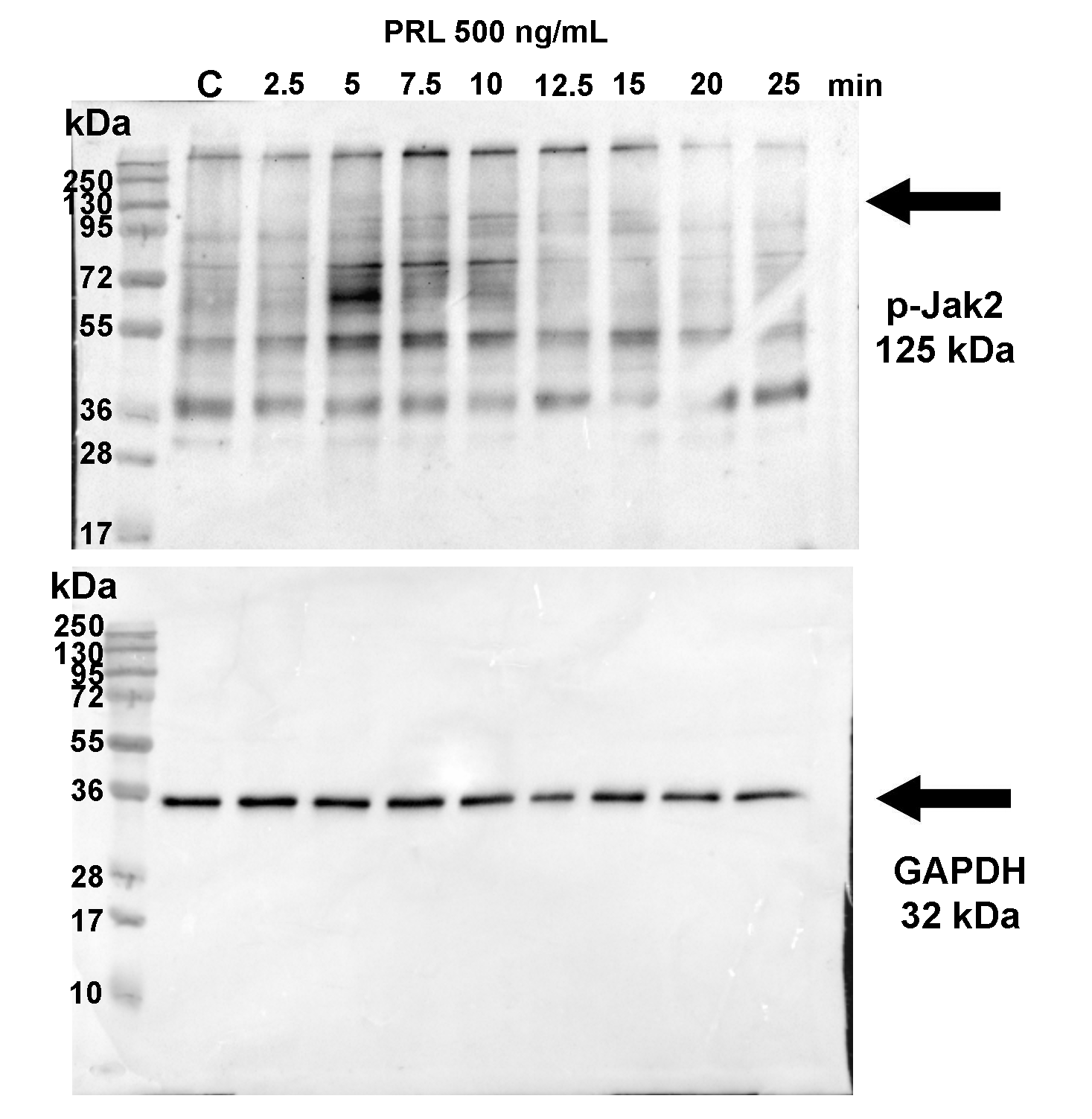
First, we aimed to assess the biological activity of PRL and LPS used in this study. To that end, we evaluated the level of phosphorylation of Jak-2 and IκB, two hallmarks of intracellular signalling triggered by PRL and LPS treatments, respectively. Significant accumulation of intracellular phospho-Jak-2 was observed in cotyledon explants after 5-(P = 0.035) to 10-minute (P = 0.003) treatment with PRL 500 ng/mL, reflecting PRL-R activation (Supplementary Figs S1A and S3). Similarly, a significant accumulation of phospho-IκB was observed after 10 min (P = 0.043) to 15 min (P = 0.031) of LPS treatment, reflecting activation of the pro-inflammatory TLR-4 pathway (Supplementary Figs S1B and S4). These data indicate that the signaling pathways activated by PRL and LPS are functional in our biological model of placental cotyledon explants.
PRL reduced the LPS-dependent pro-inflammatory response in placental explants
After LPS stimulation, an expected, positive and significant induction of TNF-α (Fig. 1A), IL-1β (Fig. 1B) and IL-6 (Fig. 1C) secretion into culture media was observed in comparison to U and Vh controls (P < 0.001). Additionally, we observed that without the LPS-stimulus, PRL at the highest concentration tested (500 ng/mL) had no effect on the modulation of these pro-inflammatory cytokines. However, when LPS-treated explants were co-incubated with PRL, the latter significantly inhibited TNF-α, IL-1β and IL-6 secretion compared to explants treated with LPS alone (P < 0.035, P = 0.031 and P < 0.045, respectively). This profile of the PRL-mediated anti-inflammatory effect was cytokine selective, since IL-6 secretion was already affected by the lowest PRL concentration (100 ng/mL), while downregulation of TNF-α and IL-1β required higher PRL concentrations (300–500 ng/mL). Of interest, the latter effects were comparable to the anti-inflammatory activity of DXM in comparison to LPS (P = 0.038 for TNF-α, P = 0.002 for IL-1β and P < 0.001 for IL-6). On the other hand, when co-incubating PRL with MTX or Del-1-9-G129R in LPS-exposed explants, the significant anti-inflammatory effect of PRL (500 ng/mL) on IL-1β and TNF-α secretion was lost; however, no effect was observed by these antagonists on IL-6 secretion (Fig. 1).
Figure 1.
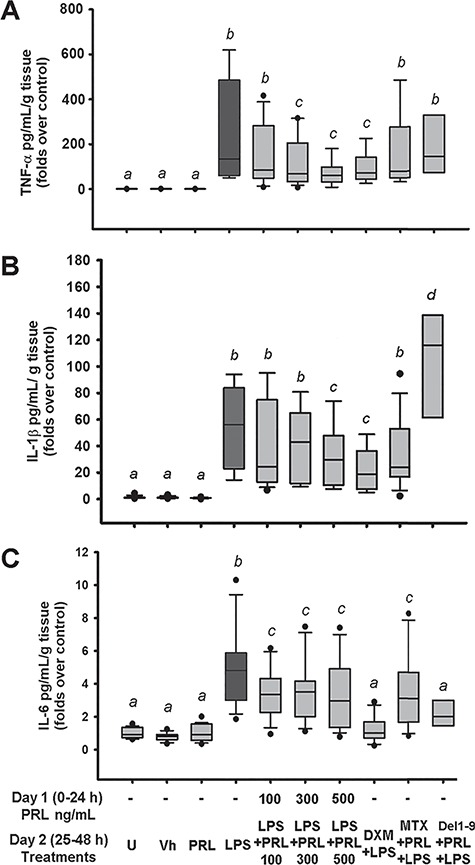
PRL attenuates pro-inflammatory cytokine secretion induced by LPS in human placenta. PRL co-treatment reduces (A) TNF-α, (B) IL-1β and (C) IL-6 secretion into culture media. Since the data showed no normal distribution, they are presented as boxes and whiskers: box lines indicate 25, 50 and 75 percentiles, and whiskers indicate 5 and 95 percentiles. Outliers are indicated in closed circles. U = untreated, Vh = vehicle, PRL = prolactin 500 ng/mL unless another concentration is indicated, LPS = lipopolysaccharide 500 ng/mL. DXM = dexamethasone 300 nm, MTX = methotrexate 50 μm, Del1–9 = Del-1-9-G129R 2500 ng/mL. One-way ANOVA and Dunn post hoc test. Different letters indicate a significant difference (P < 0.05) between them. n = 8 independent experiments in triplicate.
To strengthen these results, we then evaluated the intracellular levels of the three cytokines by western blot. The intracellular levels of TNF-α, IL-1β and IL-6 were very similar to those reported for cytokines secreted into culture media (Fig. 2; Supplementary Fig. S5). As expected, LPS treatment strongly induced the protein expression of TNF-α and IL-1β precursors as well as IL-6 (P = 0.048, P = 0.002 and P = 0.016, respectively), and this effect was prevented by PRL (P < 0.001, P = 0.007 and P < 0.029, respectively). Again, a significant inhibitory effect on IL-6 expression compared to LPS alone was observed from the lowest PRL concentration, while pro-TNF-α and pro-IL-1β were significantly inhibited only at 500 ng/mL PRL. As expected, DXM treatment significantly diminished LPS-dependent pro-inflammatory cytokine synthesis (P = 0.008 for TNF-α, P = 0.003 for IL-1β and P = 0.016 for IL-6). And, MTX blocked the PRL-dependent inhibition of TNF-α, IL-1β and IL-6 synthesis.
Figure 2.
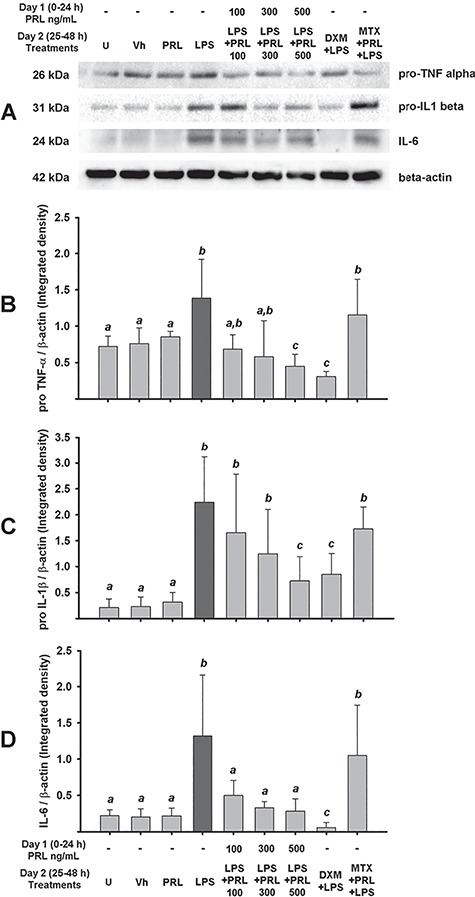
PRL diminishes pro-inflammatory cytokines expression induced by LPS in human placenta. (A) Representative western blotting for the immunodetection of pro-TNF-α, pro-IL-1β, IL-6 and β-actin in cotyledon explants. Integrated data for (B) pro-TNF-α, (C) pro-IL-1β and (D) IL-6. In all cases, the data were normalized against β-actin. Data are presented as mean ± SD from four independent experiments. U = untreated, Vh = vehicle, PRL = prolactin 500 ng/mL unless another concentration is indicated, LPS = lipopolysaccharide 500 ng/mL. DXM = dexamethasone 300 nm, MTX = methotrexate 50 μm. One-way ANOVA and Dunnett post hoc test. Different letters indicate a significant difference (P < 0.05) between them.
PRL reduced inflammatory signaling in human placenta through a lower expression of TLR-4 and the NFκB signaling
In order to obtain insights into the mechanistic regulation of the anti-inflammatory activity exerted by PRL under an inflammatory scenario in the human placenta, we evaluated TLR-4 protein expression and phospho-NFκB activity. As shown in Fig. 3A, PRL treatment inhibited TLR-4 levels, with a significant effect achieved at PRL 500 ng/mL (P = 0.005) (Fig. 3A; Supplementary Fig. S6). Therefore, a possible impact on LPS-dependent NFκB phosphorylation was expected. Indeed, Fig. 3B shows that NFκB phosphorylation was significantly higher in LPS-treated explants (P = 0.002). As estimated, a significant inhibitory effect of NFκB was observed at 500 ng/mL PRL (P = 0.034), which was similar to that observed in DXM-treated placental explants exposed to LPS (P = 0.006). However, MTX blockade of PRL-R signaling did not prevent the PRL-dependent inhibition of phospho-NFκB (P = 0.015).
Figure 3.
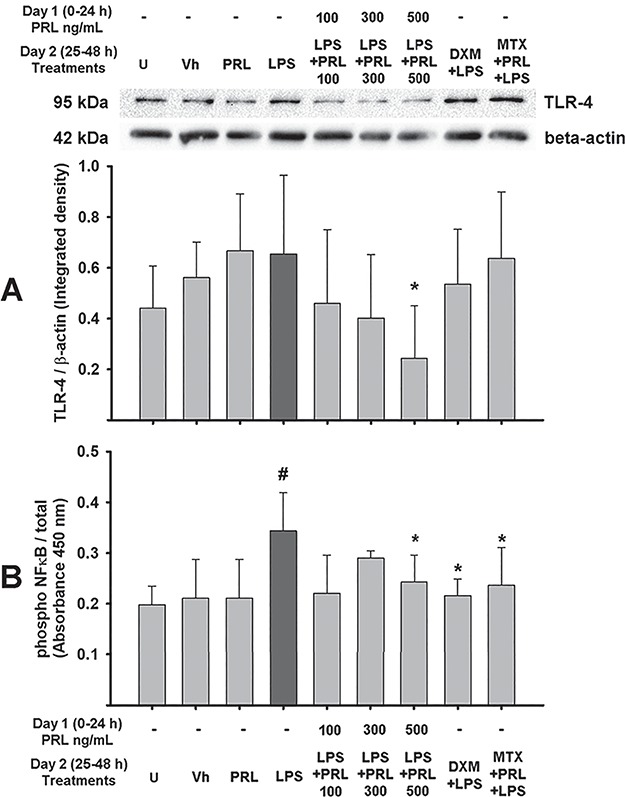
PRL downregulates TLR-4 expression and blocks LPS-dependent NFκB phosphorylation in human placenta. (A) Upper panel: representative western blotting for the immunodetection of TLR-4 and β-actin in cotyledon explants. Lower panel: integrated data for TLR-4 expression normalized against β-actin. (B) Assay for NFκB activity expressed as a ratio of phosphorylated/total NFκB. Data are presented as mean ± SD from four to six independent experiments. U = untreated, Vh = vehicle, PRL = prolactin 500 ng/mL unless another concentration is indicated, LPS = Lipopolysaccharide 500 ng/mL, DXM = dexamethasone 300 nm, MTX = methotrexate 50 μm. One-way ANOVA and Holm–Sidak post hoc test. *P < 0.05 vs. LPS; #P < 0.05 vs. control.
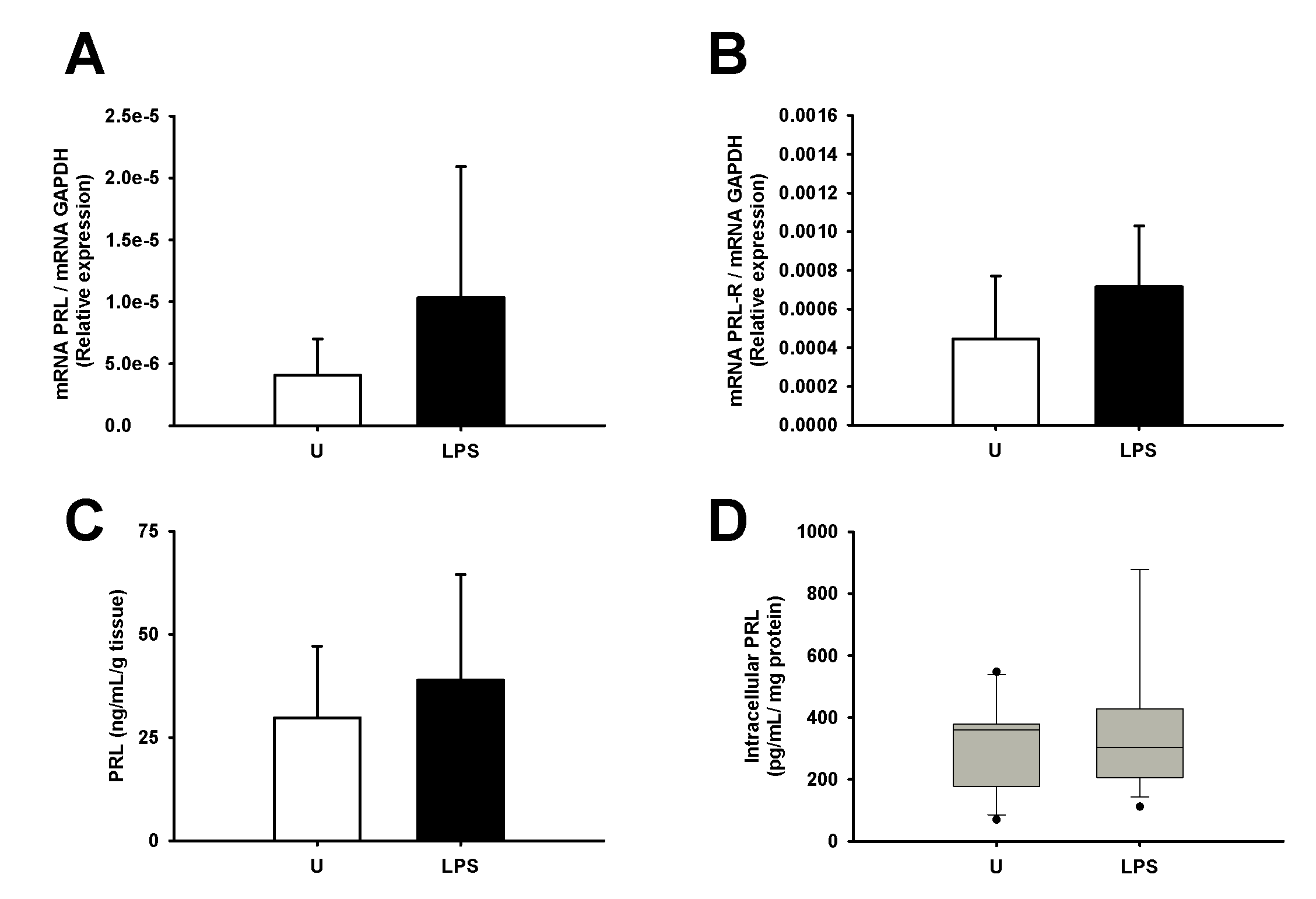
Furthermore, we investigated whether LPS may modulate the PRL gene or protein synthesis, as another modulatory loop between LPS and PRL. There was no regulation of PRL synthesis by LPS in the explant cotyledon culture at neither the mRNA expression (Supplementary Fig. S2A) nor protein level (Supplementary Fig. S2C and D). Additionally, we also analysed the expression of PRL receptor, and the results show that in comparison with basal conditions, the incubation with LPS did not modify the PRL-R mRNA level in the explants (Supplementary Fig. S2B).
Discussion
PRL is a pleiotropic hormone exerting multiple biological functions related to growth and development, endocrinology and metabolism, brain and behaviour, as well as reproduction and lactation (Costanza et al., 2015). Importantly, it also acts in a cytokine-like manner regulating the immune response (Díaz et al., 2013). In the professional immune system, PRL is synthesized by macrophages, lymphocytes and natural killer cells, where it exerts autocrine and paracrine biological effects such as monocytes differentiation, T-cell activation and production of pro-inflammatory cytokines (Díaz et al., 2013).
Additionally, PRL is secreted by decidual cells and to a lesser extent by trophoblasts. PRL accumulates in amniotic fluid or intervillous blood in a compartmentalized way, suggesting a key role in the maintenance of immune privilege. Since the effects of PRL on placental cells are not completely understood, in this study, we explored its effects in cultured, LPS-stimulated explants from human term placenta. Our results provide strong evidence to support an immunomodulatory role of this hormone in the fetal compartment.
Indeed, physiologic PRL concentrations during pregnancy significantly prevented the LPS-dependent induction of IL-1β, TNF-α and IL-6, in a similar manner as that observed using the well-known anti-inflammatory drug DXM. These results were further supported by western blots showing that PRL was able to hinder the rise in IL-1β and TNF-α precursor, as well as IL-6 expression, in LPS-challenged explants. These findings were not unexpected, given a previous study from our group showing similar results in cultured human fetal membranes (Flores-Espinosa et al., 2017). They are consistent with a reciprocal negative feedback regulatory loop at the fetoplacental unit, since it is known that inflammatory cytokines such as TNF-α, IL-1β and IL-2, among others, inhibit decidual PRL expression (Jikihara and Handwerger, 1994). A tissue-specific regulation was also suggested by our results, since in immune cells PRL preferentially stimulates, rather than inhibits, expression of proinflammatory cytokines (Sodhi and Tripathi, 2008). Therefore, in the placental interface, a complex interplay seems to take place between decidual PRL, fetal trophoblasts, fetal membranes and cytokines produced by the immune cells that populate/infiltrate the placental bed. Of note, our findings are similar to those in a previous study showing that PRL significantly repressed IL-6 expression in mouse decidua (Bao et al., 2007). Interestingly, in the same work, PRL was also shown to inhibit the expression of 20-hydroxysteroid dehydrogenase. Given that this enzyme catabolizes progesterone, a potent immunosuppressive hormone, the fact that PRL prevents progesterone inactivation supports an additional anti-inflammatory role of placental PRL.
Mechanistically, the PRL-dependent anti-inflammatory effect may involve inhibition of the TLR-4/NFκB signaling cascade, which is well known to promote inflammation. In support of this hypothesis, we showed that PRL prevented LPS-induced upregulation of both TLR-4 protein expression and NFκB phosphorylation. Until now, there have been no studies that report on TLR regulation or activation by PRL in the human. This is the first evidence of a mechanism for anti-inflammatory activity of PRL in the human placenta, acting as a negative regulator of TLR-4/NFkB signaling.
In this study, LPS endotoxin was used to mimic an infectious scenario, as corroborated by increased cytokine secretion in LPS-treated explants. Considering that PRL prevented this effect, it is reasonable to speculate that this hormone plays an important role in preventing an exacerbated immune reaction to infection and/or inflammation in the fetal part of the placenta. Further studies aimed at exploring PRL effects on different feto-maternal tissues and immune cells should help in our understanding of the complex immune–endocrine interactions required to privilege the continuity of gestation.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors thank the participating women for donating their placentas for research. We thank Mayra Hernández-Pérez and Estefanía-Núñez Sánchez for helping in sample collection. We thank Irma Sosa González and Graciela Villeda Gabriel from the Infectology and Immunology Branch for providing all microbiology analyses. We acknowledge, with thanks, the Instituto Nacional de Perinatología (212250-3210-21205-01-14) and the Hospital de Gineco-Obstetricia ‘Luis Castelazo Ayala’ (R-2017-785-013) for assistance in placenta sampling and collaborative work. Additionally, we thank Dr. Adalberto Parra-Covarrubias for his contribution in formulating the original hypothesis of this project and its possible clinical implications. We also thank Dr Sebastian Carranza-Lira from Hospital de Ginecolgia-Obstetricia No. 4, Luis Castelazo Ayala (HGOLCA) for his constant support during the project registration and the collection of biological samples.
Finally, we thank David Wheaton and Daudi Langat for editorial assitence and specially to Patricia Arce for her invaluable support in each stage of this work.
Authors’ roles
A.O.-O. and M. D.G. developed ELISAS and western blotting assays and contributed to the methodological design and acquisition, analysis and interpretation of data. E.P.-M. and L.B.-M. contributed to the analysis and interpretation of cytokine secretion data. P.F.-E. and I.M.-H. contributed to the analysis and interpretation of cytokine western blotting data. C.I. and A.C.H.-R. contributed to the analysis and interpretation of phosphorylation/activation data. B.Q.-R. supervised placental sampling in Hospital Gineco-Obstetricia 4. V. Goffin advised on the Del-1-9-G129R-hPRL data analysis. L.D. provided critical analysis and supervised the draft design. V.Z.-C. contributed to conception of the scientific question, the methodological design, the acquisition, analysis and interpretation of data, supervision of the article draft and critical revision of the content of the manuscript. All authors read and approved the final version of the manuscript.
Funding
This study was supported by the Instituto Nacional de Perinatologia—Isidro Espinosa de los Reyes (grant no. 212250-3210-21205-01-14 to V.Z.C.) and by the National Council of Science and Technology of Mexico (CONACyT) (grant no. CB2014–24162 to V.Z.-C.). M.D.G. received fellowship 27158 from CONACyT. The funder had no role in the study design, data collection and analysis, the decision to publish or the preparation of the manuscript.
Conflict of interest
The authors declare no conflict of interests.
References
- Bao L, Tessier C, Prigent-Tessier A, Li F, Buzzio OL, Callegari EA, Horseman ND, Gibori G. Decidual prolactin silences the expression of genes detrimental to pregnancy. Endocrinology 2007;148:2326–2334. [DOI] [PubMed] [Google Scholar]
- Beijar EC, Mallard C, Powell TL. Expression and subcellular localization of TLR-4 in term and first trimester human placenta. Placenta 2006;27:322–326. [DOI] [PubMed] [Google Scholar]
- Binart N. Prolactin and pregnancy in mice and humans. Ann Endocrinol (Paris) 2016;77:126–127. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–254. [DOI] [PubMed] [Google Scholar]
- Cheng PJ, Wang TH, Huang SY, Kao CC, Lu JH, Hsiao CH, Shaw SW. Differential proteomics analysis of amniotic fluid in pregnancies of increased nuchal translucency with normal karyotype. Prenat Diagn 2011;31:274–281. [DOI] [PubMed] [Google Scholar]
- Costanza M, Binart N, Steinman L, Pedotti R. Prolactin: a versatile regulator of inflammation and autoimmune pathology. Autoimmun Rev 2015;14:223–230. [DOI] [PubMed] [Google Scholar]
- Dautt-Leyva JG, Canizalez-Roman A, Acosta Alfaro LF, Gonzalez-Ibarra F, Murillo-Llanes J. Maternal and perinatal complications in pregnant women with urinary tract infection caused by Escherichia coli. J Obstet Gynaecol Res 2018;44:1384–1390. [DOI] [PubMed] [Google Scholar]
- Díaz L, Díaz-Muñoz M, González L, Lira-Albarrán S, Larrea F, Méndez I. Prolactin in the immune system In: Nagy GM. (ed). Prolactin. InTech/Janeza, Rijeka, Croatia, 2013 [Google Scholar]
- Duval C, Brien ME, Gaudreault V, Boufaied I, Baker B, Jones RL, Girard S. Differential effect of LPS and IL-1beta in term placental explants. Placenta 2019;75:9–15. [DOI] [PubMed] [Google Scholar]
- Featherstone K, White MR, Davis JR. The prolactin gene: a paradigm of tissue-specific gene regulation with complex temporal transcription dynamics. J Neuroendocrinol 2012;24:977–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferriani RA, Silva de Sa MF. Prolactin levels in blood from the intervillous space of the human placenta. Gynecol Obstet Invest 1988;26:73–76. [DOI] [PubMed] [Google Scholar]
- Flores-Espinosa P, Preciado-Martinez E, Mejia-Salvador A, Sedano-Gonzalez G, Bermejo-Martinez L, Parra-Covarruvias A, Estrada-Gutierrez G, Vega-Sanchez R, Mendez I, Quesada-Reyna B et al. . Selective immuno-modulatory effect of prolactin upon pro-inflammatory response in human fetal membranes. J Reprod Immunol 2017;123:58–64. [DOI] [PubMed] [Google Scholar]
- Garzia E, Clauser R, Persani L, Borgato S, Bulfamante G, Avagliano L, Quadrelli F, Marconi AM. Prolactin and proinflammatory cytokine expression at the fetomaternal interface in first trimester miscarriage. Fertil Steril 2013;100:108–115e1–2. [DOI] [PubMed] [Google Scholar]
- Gibori G, Richards JS. Dissociation of two distinct luteotropic effects of prolactin: regulation of luteinizing hormone-receptor content and progesterone secretion during pregnancy. Endocrinology 1978;102:767–774. [DOI] [PubMed] [Google Scholar]
- Goffin V, Norman M, Martial JA. Alanine-scanning mutagenesis of human prolactin: importance of the 58-74 region for bioactivity. Mol Endocrinol 1992;6:1381–1392. [DOI] [PubMed] [Google Scholar]
- Jikihara H, Handwerger S. Tumor necrosis factor-alpha inhibits the synthesis and release of human decidual prolactin. Endocrinology 1994;134:353–357. [DOI] [PubMed] [Google Scholar]
- Mayo-Clinic-Laboratories Prolactin Serum Reference Values. 2018. https://endocrinology.testcatalog.org/show/PRL.
- Mitchell S, Vargas J, Hoffmann A. Signaling via the NFkappaB system. Wiley Interdiscip Rev Syst Biol Med 2016;8:227–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napso T, Yong HEJ, Lopez-Tello J, Sferruzzi-Perri AN. The role of placental hormones in mediating maternal adaptations to support pregnancy and lactation. Front Physiol 2018;9:1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olmos-Ortiz A, Garcia-Quiroz J, Avila E, Caldino-Soto F, Halhali A, Larrea F, Diaz L. Lipopolysaccharide and cAMP modify placental calcitriol biosynthesis reducing antimicrobial peptides gene expression. Am J Reprod Immunol 2018;79:e12841. [DOI] [PubMed] [Google Scholar]
- Schagger H. Tricine-SDS-PAGE. Nat Protoc 2006;1:16–22. [DOI] [PubMed] [Google Scholar]
- Sodhi A, Tripathi A. Prolactin and growth hormone induce differential cytokine and chemokine profile in murine peritoneal macrophages in vitro: involvement of p-38 MAP kinase, STAT3 and NF-kappaB. Cytokine 2008;41:162–173. [DOI] [PubMed] [Google Scholar]
- Stefanoska I, Jovanovic KM, Vasilijic S, Cujic D, Vicovac L. Prolactin stimulates cell migration and invasion by human trophoblast in vitro. Placenta 2013;34:775–783. [DOI] [PubMed] [Google Scholar]
- Takser L, Mergler D, de Grosbois S, Smargiassi A, Lafond J. Blood manganese content at birth and cord serum prolactin levels. Neurotoxicol Teratol 2004;26:811–815. [DOI] [PubMed] [Google Scholar]
- Tchirikov M, Schlabritz-Loutsevitch N, Maher J, Buchmann J,Naberezhnev Y, Winarno AS, Seliger G. Mid- trimester preterm premature rupture of membranes (PPROM): etiology, diagnosis, classification, international recommendations of treatment options and outcome. J Perinat Med 2018;46:465–488. [DOI] [PubMed] [Google Scholar]
- Zaga-Clavellina V, Parra-Covarrubias A, Ramirez-Peredo J, Vega-Sanchez R, Vadillo-Ortega F. The potential role of prolactin as a modulator of the secretion of proinflammatory mediators in chorioamniotic membranes in term human gestation. Am J Obstet Gynecol 2014;211:48 e1–48 e6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.