

Abstract
The objective of the present study was to determifigne whether targeted-disruption of Npr1 gene (encoding for guanylyl cyclase/natriuretic peptide receptor-A; GC-A/NPRA) upregulates pro(renin) receptor (P)RR expression and leads to the activation of MAPKs in Npr1 gene-knockout mice. The Npr1 homozygous (Npr1−/−; 0-copy), heterozygous (Npr1+/−; 1-copy), wild-type (Npr1+/+; 2-copy), and gene-duplicated (Npr1++/++; 4-copy) mice were utilized. To identify the canonical pathway of (P)RR, we administered ACE-1 inhibitor (captopril), AT1R blocker (losartan), and MAPKs inhibitors (U0126 and SB203580) to all Npr1 mice genotypes. The renal expression of (P)RR mRNA was increased by 3-fold in 0-copy mice and 2-fold in 1-copy mice compared with 2-copy mice, which was also associated with significantly increased expression of ACE-1 and AT1R mRNA levels. Similarly, the phosphorylation of MAPKs (Erk1/2 and p-p38) was enhanced by 3.5-fold and 3.2-fold, respectively, in 0-copy mice with significant increases in 1-copy mice compared with 2-copy mice. The kidney and plasma levels of proinflammatory cytokines were significantly elevated in 0-copy and 1-copy mice. Treatment with captopril and losartan did not alter the expression of (P)RR in any of the Npr1 mice genotypes. Interestingly, losartan significantly reduced the phosphorylation of Erk1/2 and p38 in Npr1 mice. The present results suggest that the ablation of Npr1 upregulates (P)RR, MAPKs (Erk1/2 and p38), and proinflammatory cytokines in 0-copy and 1-copy mice. In contrast, the duplication of Npr1 exhibits the anti-inflammatory and antihypertensive effects by reducing the activation of MAPKs and inhibiting the expression levels of RAAS components and proinflammatory cytokines.
Keywords: Atrial natriuretic peptide receptor, (pro)renin receptor, mitogen-activated protein kinases, renin angiotensin system, proinflammatory cytokines
1. Introduction
The cardiac hormones, atrial and brain natriuretic peptides (ANP and BNP) elicit natriuretic, diuretic, vasorelaxant, and antimitogenic effects, all directed to the reduction of blood pressure (BP) and blood volume. [1–5]. Both ANP and BNP bind to guanylyl cyclase/natriuretic peptide receptor-A (GC-A/NPRA), which is considered a principal natriuretic peptide receptor that synthesizes the intracellular second messenger cGMP. [6–9]. Mice carrying targeted-disruption of Npr1 gene (encoding for the NPRA) exhibit hypertension, congestive heart failure, reduced kidney function, altered plasma renin and angiotensin II (Ang II) levels, and end-organ damage and dysfunction with sudden death occurring at the adult age. [5, 10–18]. We have previously reported that adult Npr1 gene-disrupted mice showed significant reduction in plasma renin concentration (PRC) and kidney renin content, whereas adrenal renin content was significantly increased in these animals [13]. Those previous findings implicated that the inhibitory responses on renal and systemic renin-angiotensin-aldosterone system (RAAS) might exert a compensatory effect that prevents excessive increases in BP in adult Npr1 gene-knockout mice [13, 14]. On the other hand, the greater renin and Ang II levels were observed in Npr1 gene-knockout newborn pups that provided the evidence that ANP/NPRA system exerts inhibitory effect on renin synthesis and release [13]. Previous studies showed that both glomerular filtration rate (GFR) and renal plasma flow are drastically reduced in Npr1 gene-disrupted mice (0-copy) during and after blood volume expansion, indicating that ANP/NPRA signaling responses to volume expansion that leads to a diminished excretion of sodium and water [14]. Collectively, ANP/NPRA system serves as an antagonist to RAAS [1, 10, 13, 16, 19–21].
Prorenin receptor (P)RR, a new member of RAAS is widely expressed in various tissues; including heart, kidney, brain, and adipose tissues [22–26]. The full (P)RR protein consists of a single transmembrane domain, an extracellular binding site, a signaling peptide, and a cytoplasmic region [23, 26–28]. It is increasingly evident that (P)RR not only mediates the formation of Ang II by enhancing the renin activity or activating prorenin, but also acts in a RAAS-independent manner [23, 29, 30]. Both the Ang II- dependent and -independent (P)RR signaling pathways have been linked to the pathogenesis of hypertension, diabetic nephropathy, and choroidal neovascularization [31–33]. Previous studies have reported that higher levels of (P)RR exhibited with worsening of chronic renal insufficiency, increased incidence of gestational diabetes, and proliferative retinopathy [34]. It has also been suggested that (P)RR is distributed in various renal structures and cell types such as podocytes, mesangial cells, proximal and distal tubules, collecting ducts, and renal vasculature [23, 29, 30, 35]. Additionally, both renin and prorenin bind specifically to (P)RR and the later induces nonproteolytic activation that leads to activation of mitogen-activated protein kinases (MAPKs) including extracellular signal-regulated kinases (Erk1/2), p38, and c-Jun N-terminal kinases (c-JNK), independently from the RAAS [23, 32, 36].
ANP/NPRA system has been shown to inhibit the Ang II-mediated induction of protein kinase C (PKC) and MAPKs in vascular smooth muscle and mesangial cells [24, 27, 28, 37–40]. Recent studies have demonstrated the involvement of (P)RR in the development of BP, kidney diseases, inflammation, and end organ damage [39, 41–43]. Our previous studies have elucidated the role of Npr1 in the regulation of RAAS in Npr1 gene-knockout mice [10, 13, 16, 20, 21]. However, the role of (P)RR in the absence of Npr1 in not known. In the present study, we report that the genetic disruption of Npr1 upregulates (P)RR and activates MAPKs involving the canonical pathways, independent of Ang II generation through the enhancement of renal production of pro-inflammatory cytokines.
2. Materials And Methods
2.1. Materials
Gene specific primers were purchased from Eurofins, MWG Operon (Huntsville, AL). Primary antibodies for angiotensin converting enzyme-1 (ACE-1), extracellular-regulated kinase-1/2 (Erk1/2), phosphorylated MAPKs (Erk1/2 and p38MAPK), and IgG-HRP conjugated secondary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Prorenin receptor (P)RR) antibody was purchased from Sigma-Aldrich (St. Louis MO). The RNeasy mini-kit for total RNA isolation was obtained from Qiagen, (Valencia, CA). SYBR Green RT-PCR kit was purchased from Roche Applied Science, (Indianapolis, IN). Captopril and losartan were purchased from Sigma Chemicals (St. Louis, MO). U0126 was purchased from Calbiochem (San Diego, CA) and SB203580 were purchased from Selleck Chemicals (Houston, TX). The direct cGMP correlate-EIA kit was purchased from Arbor Assay (Ann Arbor, MI). Multiplex kit for mouse cytokine assay was purchased from Millipore (Billerica, MA). All other chemicals were of molecular biology and analytical grade.
2.2. Animals
Npr1 gene-targeted mice were produced by homologous recombination in embryonic stem cells as previously described [11, 44]. These animals were bred and maintained at the animal facility of the Tulane University Health Sciences Center. Animals were handled under protocols approved by the Institutional Animal Care and Use Committee. The mouse colonies were maintained under 12h light/dark cycles at 25⁰C and fed regular chow (Purina laboratory) and tap water ad libitum. All experimental animals were littermate progenies of the C57/BL6 genetic background and were designated as Npr1 gene-disrupted homozygous null mutant (Npr1−/−, 0-copy), heterozygous (Npr1+/−, 1-copy), wild-type (Npr1+/+, 2-copy), and gene-duplicated (Npr1++/++, 4-copy) mice. The animals were genotyped by polymerase chain reaction (PCR) analysis of DNA isolated from tail biopsies using primer A (5’ GCTCTCTTGTCGCCGAATCT-3’), corresponding to 5’ sequences of the mouse Npr1 gene common to both alleles (Npr1+/+): primer B (5’ TGTCACCATGGTCTGATCGC-3’), corresponding to the exon 1 sequence present only in the intact allele (Npr1+/−), and primer C (5’ GCTTCCTCGTGCTTTACGGT-3’), corresponding to a sequence in the neomycin-resistance cassette present only in the null mutant allele (Npr1−/−) as previously described [13, 45]. The Npr1 gene duplicated mice were genotyped using upstream (5 ‘-CCT CTA GAT GCA TAC ATG TGCC-3’) and downstream (5 -’GGT CAA GTT AAG TGT ATT TTT TTC CC-3’) primers. PCR was carried out in 25 ¼l of reaction mixture containing 50 mM Tris-HCl (pH 8.3), 20 mM ammonium sulfate, 1.5 mM MgCl2, 10% DMSO, 100 µM dNTPs, 2 units of taq DNA polymerase, and 40 nM primers with a 60 sec denaturation step at 72⁰C for 35 cycles using the GeneAmp 9700 (Applied Biosystem) as previously described [45]. PCR product was resolved on 2% agarose gel with the endogenous band of 500 bp and the target band of 200 bp.
2.3. Drug treatment, BP analysis, and blood and tissue collection
Experiments were performed on 5 groups of 16–20 weeks old mice. All the 5 groups consisted of 4 genotypes; including homozygous null mutant (Npr1−/−, 0-copy), heterozygous (Npr1+/−, 1-copy), wild-type (Npr1+/+, 2-copy), and gene-duplicated (Npr1++/++, 4-copy) mice. Group I mice received saline and served as the positive control; group II mice received captopril (5 mg/kg/day) and group III mice received losartan (25 mg/kg/day) for 30 days by oral gavage, group 4 mice received Erk1/2 inhibitor, U0126 (10 mg/kg/day) administered intraperitoneal, and group 5 mice received p38 inhibitor, SB203580 (30 mg/kg/day) administered orally for 21 days [46, 47].
BP was measured by a noninvasive computerized tail-cuff method using Visitech 2000 and was calculated as the average of three to five sessions per day for 7 consecutive days as previously described [13, 15]. Initially, all the mice were trained for 7 days. Later, actual systolic BP (SBP) was measured on 24th day until 30th day of the treatment and data were collected. Blood was collected by cardiac puncture under CO2 anesthesia in cold tubes containing 10 ¼l of heparin (1,000 USP units/ml). Plasma was centrifuged at 3,000 x g for 20 min at 4⁰C and stored at −80⁰C until use. Animals were euthanized by high concentration of CO2 gas and the kidney was harvested. The half kidney was collected for the use of protein and total RNA extraction and stored at −80⁰C and the other half kidney was fixed in 10% buffered formaldehyde solution for immunohistochemical staining. Renal protein and mRNA levels were quantified with Western blot and semi-quantitative real-time PCR (qPCR), respectively.
2.4. Preparation of cytoplasmic extracts
Cytosolic proteins were extracted from the frozen kidney tissues as previously described with some modifications [15, 48]. Tissues were homogenized in ice-cold 10 mM Tris-HCl buffer (pH 8.0) containing 0.32 M sucrose, 3 mM calcium chloride (CaCl2), 2 mM magnesium acetate (Mg0Ac), 0.1 mM ethylenediaminetetraacetic acid (EDTA), 0.5% Nonidet P-40, 1 mM dithiothreitol (DTT), 0.5 mM phenylmethylsulfonyl fluoride (PMSF), and 4 μg/ml each of leupeptin, aprotinin, and pepstatin. The homogenate was centrifuged at 800 × g and the supernatant was separated and saved as a cytosolic fraction.
2.5. Western blot analyses
Whole cell lysate was mixed with an equal volume of 2X sodium dodecyl sulfate (SDS) sample loading buffer containing 125 mM Tris-HCl, 4% SDS, 20% glycerol, 100 mM DTT, and 0.2% bromophenol blue, and then separated in a 10% polyacrylamide gel. Proteins were then electrotransferred onto a polyvinyldifluoride (PVDF) membrane. The membrane was blocked with 1 X Tris-buffered saline-Tween 20 (TBST; 25 mM Tris, 500 mM NaCl, and 0.05% Tween 20, pH 7.5) containing 5% fat-free milk and incubated overnight in TBST (Tris, borate, saline, and Tween-20) containing 3% fat-free milk at 4⁰C with primary antibodies at 1:200 dilution (Table 1). The membrane was finally treated with corresponding secondary horseradish peroxidase-conjugated antibodies at 1:5000 dilution. Protein bands were visualized by enhanced chemiluminescence (ECL) plus detection system with Alpha Innotech phosphoimager.
TABLE 1:
The list of the antibodies with specific details of reference numbers and suppliers utilized in the Western blot analyses in the current work.
| Protein | Description | Source |
|---|---|---|
| ACE | Cat# SC-23908 | Santa Cruz, Biotechnology (Santa Cruz, CA) |
| Beta-actin | Cat # SC-47778HRP | Santa Cruz, Biotechnology (Santa Cruz, CA) |
| Erk1/2 | Cat # SC-514302 | Santa Cruz, Biotechnology (Santa Cruz, CA) |
| p-Erk1/2 | Cat # SC-514302 | Santa Cruz, Biotechnology (Santa Cruz, CA) |
| p38 | Cat # SC-7973 | Santa Cruz, Biotechnology (Santa Cruz, CA) |
| p-p38 | Cat # SC-166182 | Santa Cruz, Biotechnology (Santa Cruz, CA) |
| (P)RR | Cat # SAB4100475 | Sigma-Aldrich (St. Louis, MO) |
2.6. Semi-quantitative real-time PCR
Total RNA was extracted from mouse kidney tissues using RNeasy mini kit. First-strand cDNA was synthesized from 1¼g of total RNA in a total volume of 20 ¼l using the SYBR Green RT-PCR kit. All the primers were designed using Primer 3.0 software. The primers used for mouse (P)RR were 5’-TCGAATCCCAGACGTAGCTG-3’ (sense) and 5’-GGTGGAATAGGTTACCCACGG-3’, (antisense) for mouse ACE-1 – 5-’AGGTTGGGCTACTCCAGGAC-3’, (sense) and 5-’ GGTGAGTTGTTGTCTGGCTTC-3’, (antisense) for mouse AT1R 5’-TTGTCCACCCGATGAAGTCTC-3’ (sense) and 5’-AAAAGCGCAAACAGTGATATTGG-3’ (antisense). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control with 5’-TCCCTCAAGATTGTCAGCAA-3’ (sense) and 5’- AGATCCACAAACGGATACATT-3’ (antisense) primers. Real-time reverse transcription was done with the Mx3000P system (Stratagene, La Jolla, CA) using the SYBR green qPCR master mix (Roche) according to the manufacturer’s instructions. Cycling conditions were 95⁰C for 10 min and then 40 cycles consisting of 15 sec at 95⁰C, 60 sec at 60⁰C and 30⁰C at 72⁰C. Relative expression was calculated using MxPro software according to the 2-ΔΔCT method. After PCR amplification, a melting curve of each amplicon was determined to verify its accuracy.
2.7. Assay of plasma and renal levels of proinflammatory cytokines
Commercially available kit was used to measure the concentrations of proinflammatory cytokines. The concentrations of TNF-α and IL1-β in plasma and kidney tissue homogenate were measured using the Bioplex system, which are multiplex bead-based assays used with the Luminex Xmap technology according to the manufacturer’s instructions (Bio-Rad Laboratories, Hercules, CA). Eight-point standard curves were performed for each cytokine using the same Luminex bead technology as previously described [49, 50].
2.8. Immunohistochemistry
Paraffin-embedded 5 μm kidney sections were washed in xylene twice for 5 min each and rehydrated by successive rinsing in 100%, 95%, 70%, 50%, and 10% ethanol followed by phosphate-buffered saline (PBS), and antigen retrieval was performed using 10 mmol/l sodium citrate (pH 6.0) with 0.1% triton X −100 for 20 min at 95⁰C. Slides were incubated at room temperature with normal blocking serum for 30 min, primary antibody (rabbit polyclonal (P)RR antibody) diluted in PBS containing 1% bovine serum albumin and incubated overnight at 4⁰C. The sections were washed and incubated with secondary biotin- conjugated rabbit anti-mouse IgG for 30 min, after which peroxidase activity was visualized using the ABC Vectastain kit (Vector laboratories, Burlingame, CA). The slides were then counterstained with hematoxylin and mounted using aqueous mounting medium and cover slipped. Immunohistochemical stained slides were visualized using an Olympus BX51 camera and photographed with integrated Magnafire SP Digital Firewire camera software.
2.9. Assay of plasma and renal cGMP levels
Blood samples were collected in tubes containing EDTA and immediately centrifuged at 3,000 x g for 10 min at 4ºC. Plasma were separated and stored at −70ºC until used. Frozen kidney tissue samples were homogenized in 10 volumes of 0.1 M HCl containing 1% Triton x-100. The homogenate was heated at 95⁰C for 5 min and centrifuged at 600 × g at 22⁰C, after which the supernatant was collected. The levels of cGMP in the kidney tissues and plasma samples were analyzed using a direct cGMP immunoassay kit (Arbor Assay, Ann Arbor, MI) according to the manufacturer’s protocols as previously described [5, 51]. The results were expressed as picomoles of cGMP/mg of protein.
2.10. Statistical Analysis
Statistical analysis of differences between groups was performed by ANOVA and subsequent Dunnett test for multiple comparisons using GrahPad Prism software (La Jolla, CA). All data are expressed as mean ± SD. Comparisons with a P < 0.05 were considered as statistically significant.
3. Results
The results showed that SBP was 36 mmHg higher (138.2 ± 4.8, P < 0.001) in 0-copy (Npr1−/−) mice and 15.0 ± mmHg higher (117 ± 4.6, P < 0.001) in 1-copy (Npr1+/−) mice compared with 2-copy (Npr1+/+; 102 ± 3.3) mice (Table 2). The SBP was decreased by 23 mmHg (79.0 ± 3.8) in gene-duplicated 4-copy (Npr1++/++; P < 0.001) mice compared with 2-copy mice. Treatment with captopril and losartan significantly decreased SBP in 0-copy mice (110.8 ± 3.0 and 106.5 ± 3.2, respectively) compared with vehicle-treated 0-copy (138.2 ± 4.8) mice (P<0.001). Similarly, SBP was also significantly reduced in 1-copy (92.3 ± 5.2; 91.8 ± 4.8 vs. 117 ± 4.6), 2-copy (83.1 ±4.3; 82.0 ± 4.0 vs. 102 ± 3.3), and 4-copy mice (66.0 ± 3.8; 63.2 ± 2.9 vs. 79.0 + 3.2) compared with their respective control groups (Table 2). However, SBP was not significantly changed after treatments with Erk1/2 inhibitor, U0126 and p38MAPK inhibitor, SB 203580 in either 0-copy, 1-copy, 2-copy, or 4-copy mice (Table 2).
TABLE 2:
Comparison of systolic blood pressure (SBP) and cGMP concentrations in plasma and kidney tissues in Npr1 gene-targeted mice.
| Parameters | Npr1 Mice Genotypes | |||
|---|---|---|---|---|
| 0-Copy | 1-copy | 2-copy | 4-copy | |
| Control SBP (mmHg) | 138.2±4.8** | 122.0±4.6** | 102.0±3.3 79.0±3.8** | 79.0±3.8** |
| Captopril SBP (mmHg) | 110.8±3.0** | 104.3±5.2** | 83.1±4.3* | 72.0±3.8** |
| Losartan SBP (mmHg) | 106.5±3.2** | 97.8±4.8** | 82.4±4.0* | 71.2±2.9** |
| U0126 SBP (mmHg) | 138±4.6** | 121±4.1 ** | 101.0±4.5 ** | 78.6±3.5 ** |
| SB203580 SBP (mmHg) | 137.2±5.0 ** | 120.8±4.9 ** | 99.8±4.7 ** | 78.1±4.0 ** |
| Plasma cGMP (pmol/ml) | 4.9±1.9** | 11.0±2.2* | 17.5±2.8 | 24.9±3.2* |
| Kidney cGMP (pmol/mg) | 5.1±2.9** | 17.7±5.5** | 38.5±6.1 | 54.7±4.6** |
Systolic blood pressure was measured by non-invasive computerized tail-cuff method as described under Materials and Methods section. The concentrations of cGMP in the plasma and kidney tissues were estimated by our established procedures as stated in the Materials and Methods section. Values are expressed as mean ± SEM. n = 8 mice per group.
P<0.01,
P<0.001. (0-copy, 1-copy, and 4-copy mice were compared with 2-copy control mice).
Plasma cGMP levels were significantly reduced by 80% and 50% in Npr1 0-copy and 1- copy mice, respectively, compared with Npr1 2-copy mice (P<0.001). In the 4-copy mice, there was 42% increase in cGMP levels compared with 2-copy control mice. Similarly, renal cGMP level was drastically reduced by more than 90% and 50% in 0-copy and 1-copy mice, respectively, compared with Npr1 2-copy control mice. There was 40–45 % increase in cGMP level in gene-duplicated Npr1 4-copy mice compared with 2-copy control mice (Table 2).
The protein expression of (P)RR and ACE-1 is shown in Fig 1, A–C. The expression level of (P)RR was significantly increased in Npr1 0-copy (3-fold, P < 0.001) and 1-copy mice (2-fold, P < 0.01) compared with 2-copy mice (Fig. 1 A). The expression level of (P)RR was decreased by 2.5-fold in Npr1 4-copy mice compared with 2-copy mice (Fig. 1 B). Similarly, the expression of ACE-1 was increased 3-fold (P < 0.001) in Npr1 0-copy and 2-fold (P<0.01) in 1-copy mice compared with those of Npr1 2-copy control mice. Although significant changes were not observed in ACE-1 protein levels in 4-copy mice compared with 2-copy mice (Fig. 1 C).
Figure 1: Western blot analyses of (P)RR and ACE-1 proteins in the kidney tissues of Npr1 gene-targeted mice:
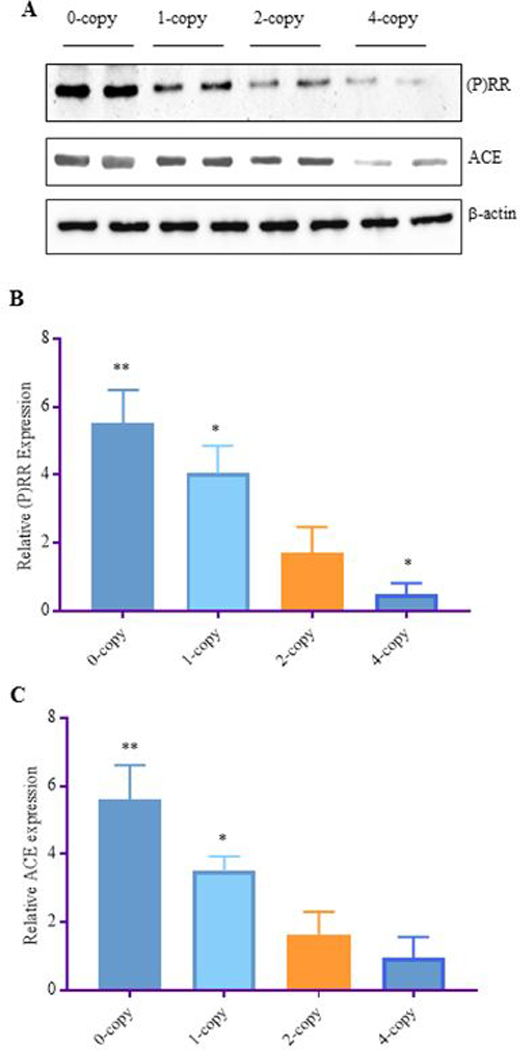
Renal expression levels of (P)RR and ACE-1 proteins in Npr1 homozygous mutant (0-copy), heterozygous (1-copy), wild-type (2-copy) and gene duplicated (4-copy) mice as determined by Western blot analysis. β-actin was used as a loading control (A). Densiometric analyses of the respective bands were done by Alpha Innotech phosphoimager are shown in panels B-C for each protein: panel (B) (P)RR and panel (C) ACE-1. The data for 0-copy, 1-copy, and 4-copy mice were compared with 2-copy control mice. Values are expressed as mean ± SEM. *P<0.01, **P<0.001; n = 8 mice per group.
The representative real-time qPCR analysis of renal mRNA expression for (P)RR, ACE-1, and AT1R are shown in Fig 2, A-C. The expression of (P)RR (3-fold, P < 0.001), ACE-1 (3.5-fold, P < 0.01), and AT1R (2.8-fold, P < 0.001) mRNA levels, were significantly increased in Npr1 0-copy mice compared with 2-copy counterparts. Similar changes were also observed in the Npr1 1-copy mice. There was no significant change in the expression of ACE-1, while the expression of (P)RR, and AT1R was significantly reduced by more than 2-fold in the Npr1 4-copy mice compared with 2-copy control mice (Fig. 2 A-C). After staining kidney sections from each group of mice with anti-(P)RR antibody, we observed an increased levels of (P)RR immunolocalization in 0-copy (4-fold) mice and 1-copy (2.5-fold) mice as compared with Npr1 2-copy mice (Fig. 3 A-C, E). Immunohistochemical staining of the kidney tissues indicated that the immunoreactive (P)RR was localized in the glomerulus apparatus, mesangial cells, and collecting duct of 0-copy (Fig. 3 A ). Similarly, in the kidney of 1-copy mice, the immunoreactive (P)RR was localized largely in the glomerulus apparatus (Fig. 3 A, B). The immunoreactivity of (P)RR in 4-copy mice was decreased by almost 50% compared with 2-copy mice (Fig. 3 C-E).
Figure 2.
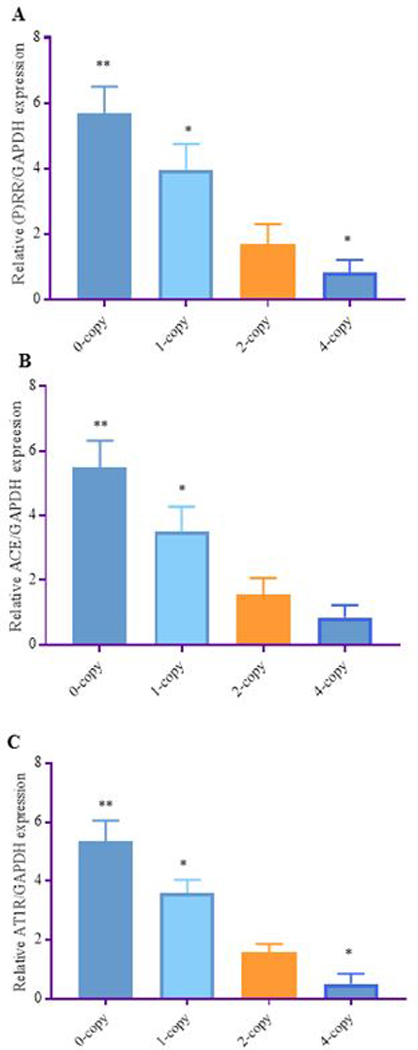
A: Analyses of mRNA expression levels of (P)RR, ACE-1, and AT1R in the kidney tissues of Npr1 gene-targeted mice: Relative expression of renal mRNA are shown in (A) (P)RR, (B) ACE-1, and (C) AT1R in Npr1 mice genotypes as determined by real time RT-PCR method. The expression levels in 0-copy, 1-copy and 4-copy mice were compared with 2-copy control mice. Values are expressed as mean ± SEM. *P<0.01, **P<0.001; n = 6 mice per group.
Figure 3: Immunohistochemistry of (pro)renin receptor expression in kidney sections of Npr1 mice genotypes:
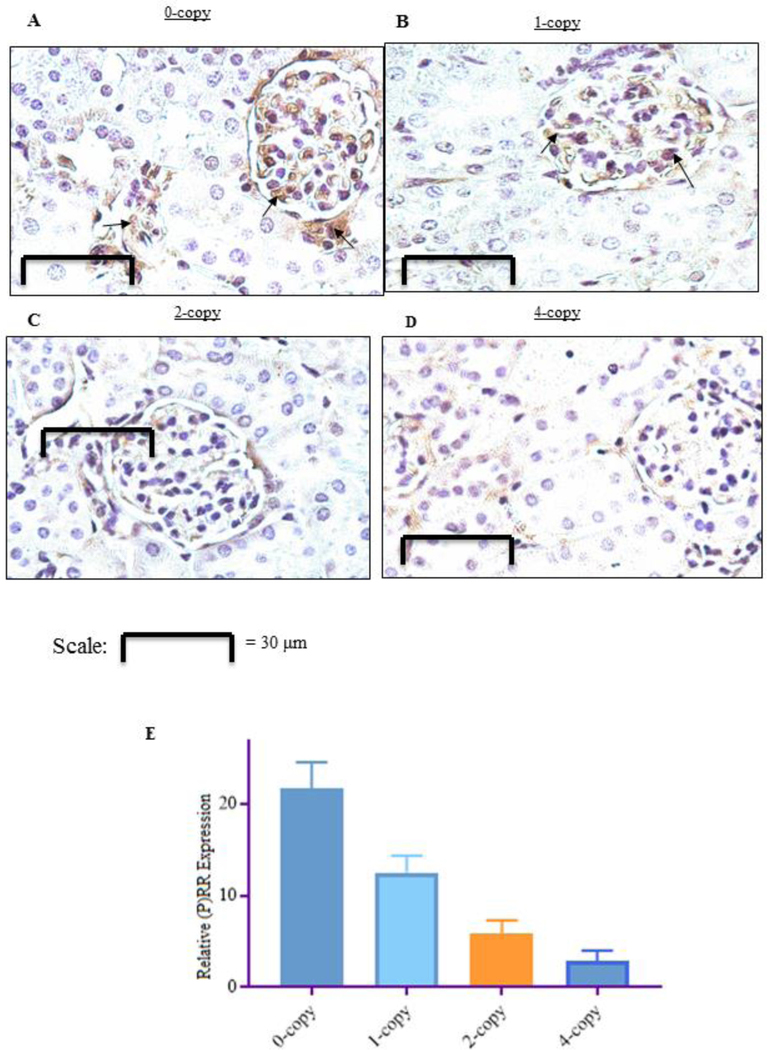
Kidney sections were stained with rabbit polyclonal primary antibody against (P)RR from Npr1 gene-targeted mice. Panel (A) homozygous mutant (0-copy), (B) heterozygous mutant (1-copy), panel (C) wild-type (2-copy), and panel (D) gene-duplicated (4-copy) mice. Panels A and B show a marked (P)RR staining (denoted by arrows) in Npr1 0-copy and 1-copy mice compared with 2-copy and 4-copy mice (C and D). Immunoreactive (P)RR was localized in the glomerulus apparatus, mesangial cells, and tubular collecting duct of kidney sections of 0-copy mice. Similarly, the immunoreactive (P)RR staining was also observed largely in the glomerulus apparatus of 1-copy mice (A and B). Panel E shows the semi-quantitative measurements of (P)RR protein expression in Npr1 0-copy, 1-copy, and 4-copy mice compared with 2-copy control mice. Values are expressed as mean ± SEM. *P<0.01; **P<0.001; All panels are representative photographs of 6 experiments.
Furthermore, to identify the Ang II-dependent and -independent mechanisms of (P)RR, we utilized ACE-1 inhibitor (captopril) and AT1R antagonist (losartan), which were administered to all Npr1 mice genotype groups. Both captopril and losartan did not significantly alter the expression of (P)RR protein levels (Fig. 4 A, B, D, E) but decreased the protein levels of ACE-1 (2.5-fold, p<0.001) in Npr1 0-copy mice compared with Npr1 2-copy mice (Fig. 4 A, C, D). Similarly, treatment with captopril and losartan also attenuated the expression of ACE-1 (2-fold, P < 0.01) in 1-copy mice compared with the vehicle-treated control mice (Fig. 4 A, C, D). The protein levels of ACE-1 in Npr1 2-copy and 4-copy mice was significantly reduced after treatment with captopril compared with the control groups (Fig. 4 A, C, D). However, losartan treatment did not exhibit any significant changes in (P)RR levels in either 0-copy, 1-copy 2-copy, or 4-copy mice (Fig. 4 E).
Figure 4: The effect of Captopril and Losartan on the protein levels of (P)RR and ACE-1 in the kidney tissues of Npr1 gene-targeted mice:
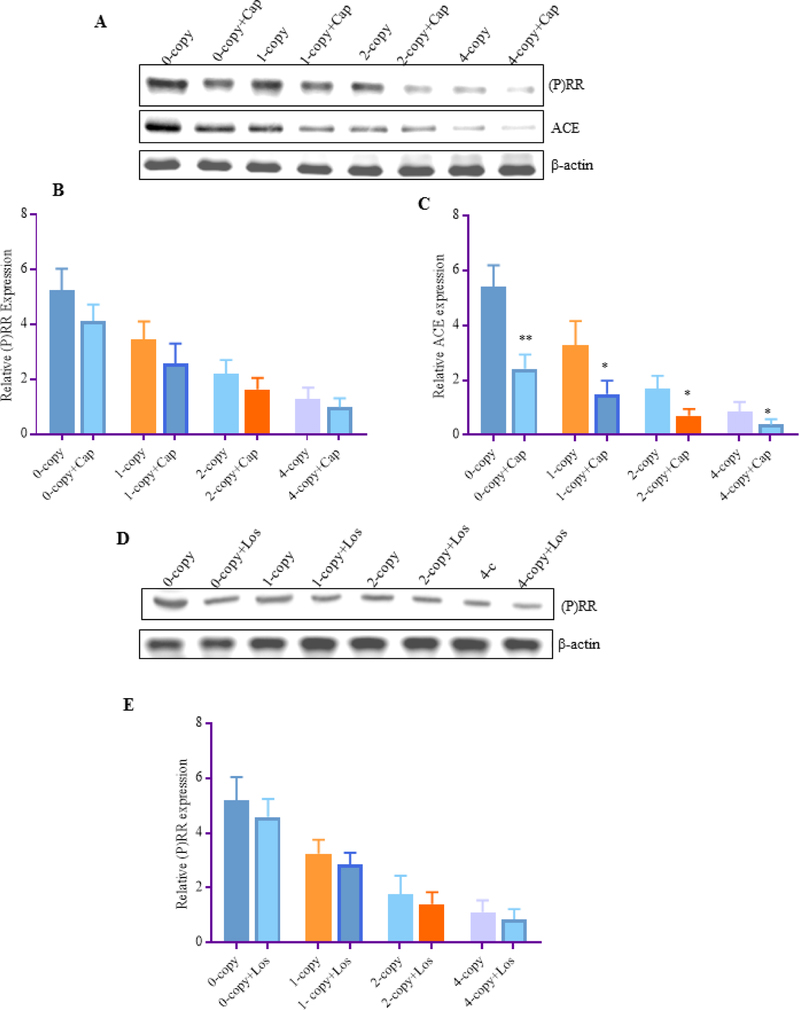
Expression of renal (P)RR and ACE-1 in Npr1 genotypes mice after treatments with Captopril and Losartan. (A) Renal protein expression of (P)RR and ACE-1 were determined by Western blot after captopril treatment, (B) represents densitometric analysis of (P)RR, and (C) represents the densitometric analysis of ACE-1. (D) represents protein levels of (P)RR and (E) represents densitometric analysis of (P)RR. β-actin was used as a loading control. Representative densitometry analysis of the respective bands were done by Alpha Innotech phosphoimager for each protein. The treated 0-copy, 1-copy, 2-copy, and 4-copy mice were compared with their respective untreated control groups. Values are expressed as mean ± SEM. *P<0.05, **P<0.01; n = 6 mice per group.
In Npr1 0-copy mice, the phosphorylation of extracellular-regulated kinases (Erk1/2) was increased by 3.5-fold (P < 0.001) and p38 by 3-fold (P < 0.001) and in 1-copy mice, phosphorylation of both Erk1/2 and p38 was increased by 2-fold (p<0.01) compared with 2-copy control mice (Fig. 5 A-C). A significant decrease in the phosphorylation of both the Erk1/2 (60% P < 0.01) and p38 (40%; P<0.05) was observed in Npr1 4-copy mice compared with Npr1 2-copy control mice (Fig. 5 A-C).
Figure 5: Western blot analyses of phosphorylated Erk1/2 and p38 MAPKs in the kidney tissues of Npr1 gene-targeted mice:
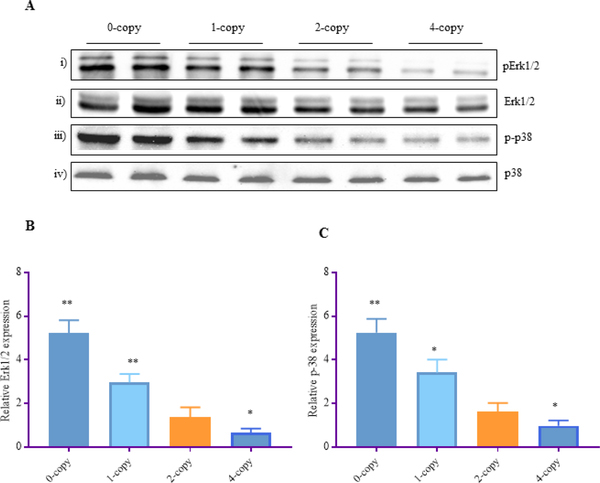
(A) Renal expression levels of phosphorylated-Erk1/2 and phosphorylated-p38 protein bands. Total Erk1/2 and total p38 was used as a loading control. Densitometric analysis of the respective bands were done by Alpha Innotech phosphoimager and are shown in (B, C) for each protein. The protein levels in 0-copy, 1-copy, and 4-copy mice were compared with 2-copy control mice. Values are expressed as mean ± SEM. *P<0.05, **P<0.0; n = 8 mice per group.
Treatment with Erk1/2 inhibitor, U0126 and p38 inhibitor, SB203580 significantly attenuated the phosphorylated levels of Erk1/2 and p38 by almost 60–65% in all the Npr1 mice genotypes as compared to their vehicle treated control groups (Fig. 6 A-D). In contrast, captopril did not alter the phosphorylation of Erk1/2 and p38 in any of the treated mice groups (Fig. 7 A-C). Interestingly, losartan significantly reduced the phosphorylation of Erk1/2 (48%, P < 0.01) and p38 (52%, P < 0.05) in Npr1 1-copy mice and as well as in 2-copy and 4-copy mice compared to their vehicle treated control groups (Fig. 7 D-F).
Figure 6: The effect of MAPKs inhibitors on the phosphorylated state of Erk1/2 and p38 MAPKs in the kidney tissues of Npr1 gene-targeted mice:
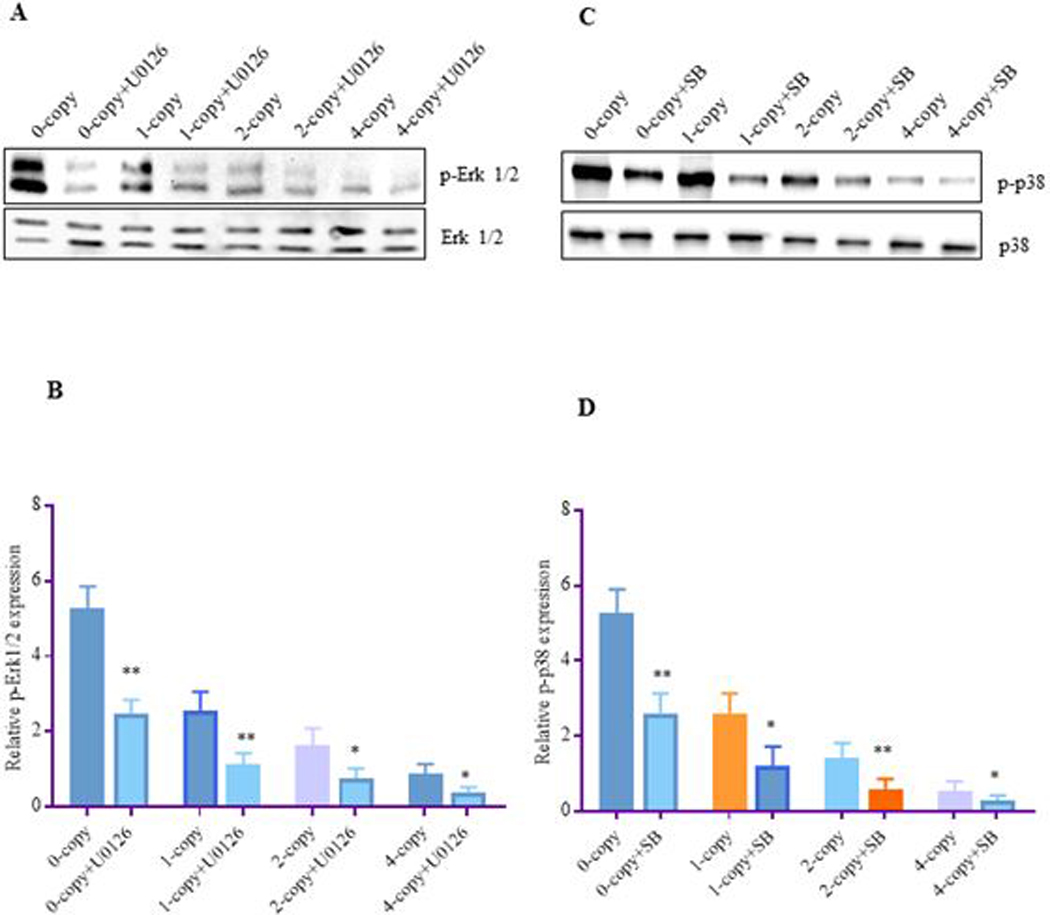
Renal expression levels of phosphorylated-Erk1/2, and phosphorylated-p38 proteins in Npr1 mice genotypes were determined by Western blot analysis after treatment with Erk1/2 inhibitor U0126 and p38 inhibitor SB 203580. Total Erk1/2 and p38 was used as a loading control. Densitometric analysis of the respective bands was done by Alpha Innotech phosphoimager for each protein. (A) Represents the renal protein bands of Erk1/2 and p-Erk1/2 after U0126 treatment, (B) represents densitometric analysis of Erk1/2, (C) represents the renal protein bands of p38 and p-p38, after treatment with SB203580, and (D) represents densitometric analysis of p38. The protein levels of MAPKs in the inhibitor-treated 0-copy, 1-copy, 2-copy, and 4-copy mice were compared with their respective vehicle-treated control groups. Values are expressed as mean ± SEM. *P<0.05, **P<0.01; n = 6 mice per group.
Figure 7: The effect of captopril and losartan on the phosphorylated Erk1/2 and p38 MAPKs in the kidney tissues of Npr1 gene-targeted mice:
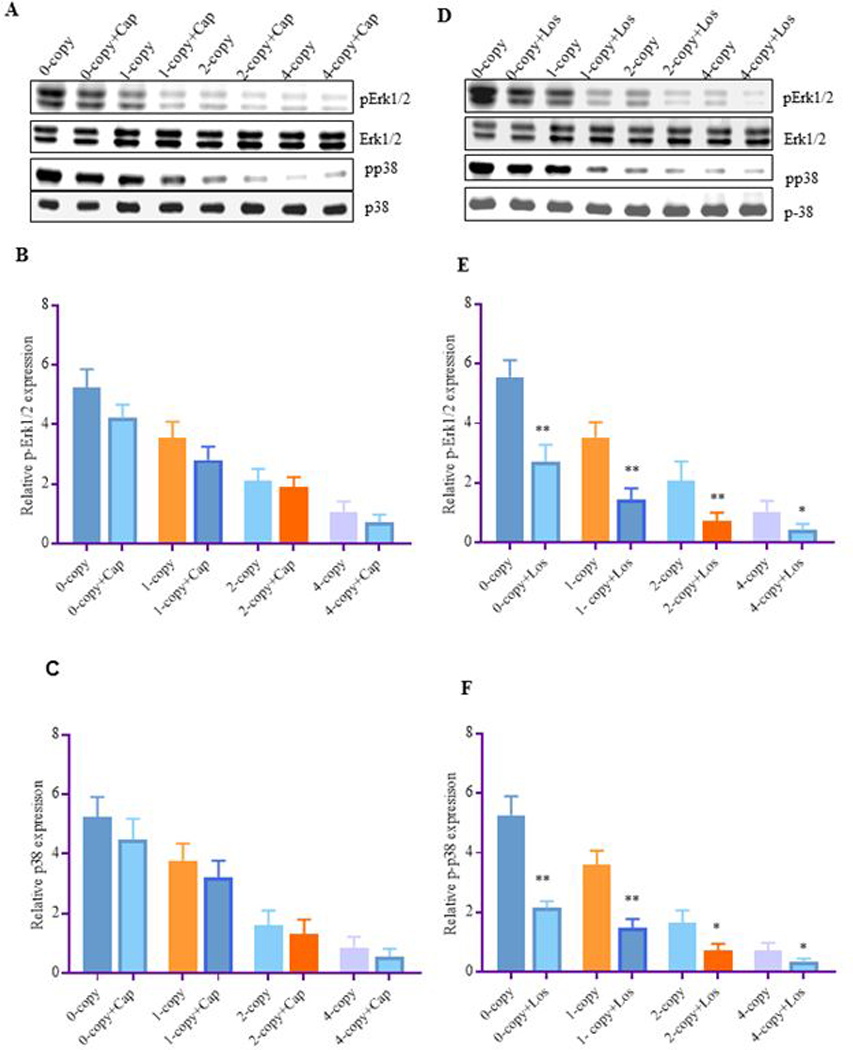
Expression levels of renal p-Erk1/2 and p-p38 proteins in Npr1 mice genotypes after treatment with captopril and losartan. Total Erk1/2 and total p38 was used as a loading control. Representative densitometric analysis of the respective bands were done by Alpha Innotech phosphoimager for each protein. (A) Represents the renal protein bands of Erk1/2 and p38 after captopril treatment, (B) represents the densitometric analysis of Erk1/2, (C) represents the densitometric analysis of p38, (D) represents the renal protein bands of Erk1/2 and p38 after losartan treatment, (E) represents the densitometric analysis of Erk1/2, and (F) represents the densitometric analysis of p38. The treated 0-copy, 1-copy, 2-copy and 4-copy mice were compared with their respective vehicle-treated groups. Values are expressed as mean ± SEM. *P<0.05, **P<0.01; n = 6 mice per group.
To determine whether Erk1/2 and p38 activation could also triggered the expression of critical proinflammatory pathways, we analyzed key cytokines, including tumor necrosis factor-alpha (TNF-α) and interleukin 1-beta (IL1-β) in the plasma and kidney tissues of Npr1 mice. Plasma TNF-α level was increased 4-fold in Npr1 0-copy and 2-fold in 1-copy mice compared with Npr1 2-copy mice. Treatment with U0126 and SB203580 significantly reduced plasma TNF-α level in Npr1 0-copy mice and Npr1 1-copy mice by 2-fold compared with their respective vehicle-treated control mice (Fig. 8 A). Similar to plasma, renal TNF-α concentrations were significantly increased by 5- fold and 2-fold in Npr1 0-copy and Npr1 1-copy mice, respectively, compared with their vehicle-treated control groups. The TNF-α level in the kidney was also significantly decreased in Npr1 0-copy and 1-copy mice after treatment with U0126 and SB203580 compared with the untreated Npr1 0-copy and 1-copy mice (Fig. 8 B). In gene-duplicated Npr1 4-copy mice, plasma and renal TNF-α levels were significantly lower and even undetectable after treatment with U0126 and SB203580. In addition, plasma IL1-β levels were increased to 7-fold in Npr1 0-copy and 4-fold in 1-copy mice compared with 2-copy control mice. Treatment with small molecule inhibitors such as Erk1/2 inhibitor (U0126) and p-38 inhibitor (SB203580) significantly reversed the increase in IL1-β level in both Npr1 0-copy and Npr1 1-copy mice compared with their respective control groups.
Figure 8: Effect of mitogen-activated protein kinase inhibitors on the proinflammatory cytokines levels in the kidney tissues of Npr1 gene-targeted mice:
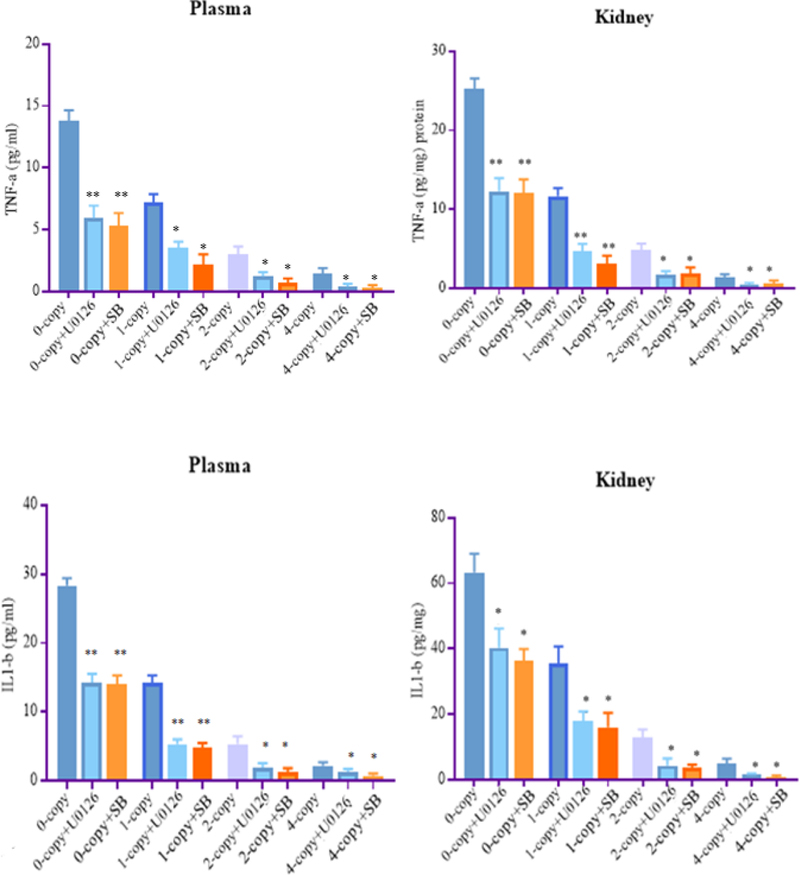
Plasma and renal proinflammatory cytokine levels in Npr1 gene-targeted mice were analyzed after treatment with Erk1/2 inhibitor U0126 and p38 inhibitor SB203580. (A) and (B) panels show plasma and kidney tissue level of TNF-α and (C) and (D) panels show plasma and kidney tissue level of IL1-β of 0-copy, 1-copy, 2-copy, and 4-copy mice. Values are expressed as mean ± SEM. *P<0.05, **P<0.01; n = 8 mice per group.
Similarly, renal IL1-β level was increased to 5.5-fold and 2.5-fold in Npr1 0-copy and Npr1 1-copy mice, respectively, compared with their vehicle-treated control mice group. A significant attenuation in proinflammatory cytokines levels was observed in both Npr1 0-copy and 1-copy mice after treatment with the MAPKs inhibitors. However, the levels were considerably reduced in Npr1 4-copy mice compared with 2-copy mice and were even undetectable after treatment with inhibitors.
4. Discussion
The results of the present study, for the first time, showed an association between (P)RR and Npr1 gene copy number in the context of renal expression of proinflammatory signal activation and regulation of BP. Although, our previous studies elucidated the role of RAAS in Npr1 gene-knockout mice, the importance of (P)RR remained largely unknown [10, 12–16, 52]. The results of the present study demonstrate that the disruption of Npr1 activates the expression of RAAS components including; (P)RR, ACE-1, and AT1R in Npr1 gene-knockout mice. The mRNA expression and protein levels of (P)RR were significantly increased in Npr1 gene-knockout mice, which in turn seem to be associated with increased expression levels of both mRNA and protein levels of ACE-1 and mRNA expression of AT1R, accompanied with elevated BP in these animals. Previous studies from our laboratory showed that the absence of Npr1 gene allows higher renin and Ang II levels in Npr1 null mutant newborn pups as compared to their age-matched wild-type control pups [13]. In contrast, the circulating renin and Ang II levels were considerably decreased in adult Npr1 homozygous null mutant mice as compared to the wild-type control mice [13]. The decrease in renin activity in adult Npr1 0-copy mice is probably due to the progressive elevation in arterial pressure, leading to inhibition of renin synthesis and release from the juxtaglomerular cells [13]. Interestingly, those previous studies have also shown that the adrenal renin content, mRNA levels, Ang II, and aldosterone concentrations were elevated in adult Npr1 null mutant 0-copy mice as compared with wild-type 2-copy mice. Evidence suggests that the circulating levels of prorenin were almost 10-fold higher than those of circulating levels of renin and may reach up to 100-fold higher during the pathophysiological conditions [53].
The results of the present study showed that decreased plasma and renal cGMP levels correlated with increased BP and increased expression of RAAS components in an Npr1 gene-dose- dependent manner. On the other hand, increased cGMP levels correlated with decreased BP and reduced expression of mRNA and protein levels of (P)RR and ACE-1, and reduced mRNA expression of AT1R in Npr1 gene-duplicated 4-copy mice. Furthermore, renal RAAS components were significantly activated in the kidneys of Npr1 0-copy mice similar to those seen in in the heart of Npr1 null mutant mice; however, in those previous studies (P)RR was not determined [15, 16].
It has been shown that binding of prorenin to (P)RR contributes to the generation of Ang II, resulting in the activation of RAAS [23, 54]. On the other hand, it also stimulates (P)RR-mediated signal transduction in an Ang II-independent manner, which in turn activates Erk1/2 and p38MAPK and fibrotic pathways [22, 23, 30, 54–56]. To elucidate Ang II-dependent and -independent mechanism of (P)RR, all the Npr1 mice genotypes were treated with captopril and losartan, both drugs did not have any significant effect in down-regulation of (P)RR in Npr1 null mutant mice. The present findings suggest that increased expression of (P)RR has contributing role in the development of BP and proinflammatory pathway in the kidneys through the activation of (P)RR, and important component of RAAS in the absence of functional Npr1 gene. Increased expression of (P)RR in Npr1 gene-knockout mice might therefore be related to kidney damage through the activation of tissue RAAS and by direct stimulation of MAPKs signaling pathways. ANP/NPRA system has been shown to inhibit the Ang II-mediated induction of protein kinase C (PKC) and MAPKs in vascular smooth muscle and mesangial cells [38, 40, 57–59]. It has also been shown that ANP/NPRA negatively regulates MAPKs (Erk1/2 and p38MAPK) activity in a cGMP-dependent manner in mesangial cells [5, 12, 38, 60]. The present results are consistent with those previous in vitro findings that a decreased phosphorylation of Erk1/2 and p38 exhibited a progressive decrease in MAPKs signaling with increased Npr1 gene-copy number. Small molecule inhibitors such as Erk1/2 inhibitor (U0126) and p38 inhibitor (SB203580) significantly blocked the activation of Erk1/2 and p38 in Npr1 null mutant mice. Previous findings have indicated that activation of (P)RR and AT1R by Ang II, transactivates MAPKs (Erk1/2, p38, and JNK) in different cell types [38, 42, 61–63]. The evidence suggests that the phosphorylated MAPKs (Erk1/2, p38, and JNK) were significantly activated in the kidneys of wild-type diabetic and AT1R gene-knockout mice [28]. Interestingly, ACE-1 inhibitor, imidapril or AT1R gene-knockout mice did not completely abolish the activation of MAPKs whereas, handle region peptide infusion reversed the increased levels of Erks, p38, and JNK in both diabetic wild-type and AT1R gene-knockout mice [28, 64]. The present data suggest that (P)RR expression follows Npr1 gene-copy number and is not changed by RAAS blockade and the resulting BP status. Moreover, ACE expression seem to follow the changes in Erks and p38 phosphorylation without changing (P)RR expression. Thus, (P)RR expression seems to be independent of the RAAS and BP. Since, the treatment with captopril and losartan did not alter the expression of (P)RR, suggesting that the increased expression is largely regulated in Ang II-independent manner. Further, the discrepancy between captopril and losartan effect on MAPKs remains unexplained but could be caused by an ACE-independent sources of Ang II in these animals.
In the present study, captopril did not show any significant effect in reducing the expression of Erk1/2 and p38 in Npr1 null mutant mice. Surprisingly, losartan decreased the expression of both MAPKs (Erk1/2 and p38) in Npr1 0-copy and 1-copy mutant mice. It is evident that the observed inhibition of Erk1/2 and p38 could be due to the losartan-dependent inhibitory efforts on Ang II effects. The results of our study demonstrate that MAPKs activation in Npr1 gene-knockout mice is not completely related to Ang II-dependent pathway; however, it might also be due to at least, in part, to the direct activation of (P)RR, which is independent of Ang II mechanism. In addition to phosphorylation of Erk1/2 and p38, we also observed increased up-regulation of both plasma and tissue levels of pro-inflammatory cytokines (TNF-α and IL1-β). It has been demonstrated that systemic disruption of Npr1 gene activates cardiac and renal NF-kB pathways in Npr1 null mutant mice and transactivates the expression of pro-inflammatory cytokines. [10, 15, 16, 49, 50, 65]. In the present study, we observed a sizeable reduction of proinflammatory cytokines levels, including TNF-α and IL1-β in Npr1 gene-duplicated mice, suggesting that increased production of cGMP exhibits anti-inflammatory effects. The inhibitors of MAPKs (U0126 and SB203580) reduced the levels of proinflammatory cytokines in Npr1 null mutant and heterozygous mice; however, undetectable in gene-duplicated mice treated with U0126 or SB203580. Interestingly, both MAPKs inhibitors, U0126 or SB203580 did not exert any significant effect on lowering the BP; however, both inhibitors significantly reduced the proinflammatory cytokines levels. Previous studies demonstrated that binding of prorenin or renin to (P)RR might directly activate Erk1/2 and p38, thereby, leading to the activation of TNF-α and IL1-β, involving the Ang II-independent pathway [43, 66]. It is also possible that the influence of (P)RR in the up-regulation of pro-inflammatory cytokines could in part be mediated via Ang II-independent mechanisms. However, inhibition of (P)RR with appropriate blocker or with small interference RNA could explain its cascade of intracellular signal transduction mechanisms, including activation of MAPKs and subsequent production of proinflammatory cytokines.
In conclusion, (P)RR is up-regulated with subsequent increased expression of other RAAS components, such as ACE-1 and AT1R in the kidneys of Npr1 gene-knockout mice. Treatments with captopril and losartan did not alter the expression of (P)RR, implicating that its enhanced expression in Npr1 0-copy and 1-copy mice is mediated by Ang II-independent mechanisms. Furthermore, captopril failed to attenuate the increased phosphorylation of Erk1/2 and p38 in Npr1 null mutant mice. On the other hand, it is noteworthy that losartan decreased the expression of MAPKs (Erk1/2 and p-38) in Npr1 mice genotypes. Since, the treatments with captopril or losartan did not change the expression of (P)RR, suggesting that the Ang II-independent signaling pathway mainly triggers its enhanced expression; however, the (P)RR is proportionately increased with decreasing Npr1 gene copy number.
Highlights.
-
■
(P)RR expression is increased with decreasing Npr1 gene copy number.
-
■
(P)RR expression seems to be independent of RAAS and blood pressure.
-
■
MAPKs and proinflammatory cytokines are upregulated in Npr1−/− mice.
-
■
Captopril and losartan did not alter expression of (P)RR.
Acknowledgements
The authors wish to thank Vickie Nguyen, Whitney Nolan, and Megan Bloodworth for excellent technical assistance and Mrs. Kamala Pandey for the help in the preparation of this manuscript. The authors are indebted to late Professor Oliver Smithies and Nobuyo Maeda (University of North Carolina, Chapel Hill, NC) for providing with the initial breeding pairs of Npr1 gene-targeted mice. This work was supported by the National Institutes of Health grants (HL057531 and HL062147).
Footnotes
Conflict of Interest
The authors declare that there is no conflict of interest.
References
- [1].Brenner BM, Ballermann BJ, Gunning ME, Zeidel ML, Diverse biological actions of atrial natriuretic peptide, Physiol Rev 70(3) (1990) 665–99. [DOI] [PubMed] [Google Scholar]
- [2].Garbers DL, Guanylyl cyclase receptors and their endocrine, paracrine, and autocrine ligands, Cell 71(1) (1992) 1–4. [DOI] [PubMed] [Google Scholar]
- [3].de Bold AJ, Atrial natriuretic factor: a hormone produced by the heart, Science 230(4727) (1985) 767–70. [DOI] [PubMed] [Google Scholar]
- [4].Levin ER, Gardner DG, Samson WK, Natriuretic peptides N Engl J Med 339(5) (1998) 321–8. [DOI] [PubMed] [Google Scholar]
- [5].Pandey KN, Biology of natriuretic peptides and their receptors, Peptides 26(6) (2005) 901–32. [DOI] [PubMed] [Google Scholar]
- [6].Drewett JG, Garbers DL, The family of guanylyl cyclase receptors and their ligands, Endocr Rev 15(2) (1994) 135–62. [DOI] [PubMed] [Google Scholar]
- [7].Pandey KN, Inagami T, Misono KS, Atrial natriuretic factor receptor on cultured Leydig tumor cells: ligand binding and photoaffinity labeling, Biochemistry 25(26) (1986) 8467–72. [DOI] [PubMed] [Google Scholar]
- [8].Schulz S, Singh S, Bellet RA, Singh G, Tubb DJ, Chin H, Garbers DL, The primary structure of a plasma membrane guanylate cyclase demonstrates diversity within this new receptor family., Cell 58 (1989) 1155–1162. [DOI] [PubMed] [Google Scholar]
- [9].Pandey KN, Singh S, Molecular cloning and expression of murine guanylate cyclase/atrial natriuretic factor receptor cDNA, The Journal of biological chemistry 265(21) (1990) 12342–8. [PubMed] [Google Scholar]
- [10].Das S, Au E, Krazit ST, Pandey KN, Targeted disruption of guanylyl cyclase-A/natriuretic peptide receptor-A gene provokes renal fibrosis and remodeling in null mutant mice: role of proinflammatory cytokines, Endocrinology 151(12) (2010) 5841–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Oliver PM, Fox JE, Kim R, Rockman HA, Kim HS, Reddick RL, Pandey KN, Milgram SL, Smithies O, Maeda N, Hypertension, cardiac hypertrophy, and sudden death in mice lacking natriuretic peptide receptor A, Proceedings of the National Academy of Sciences of the United States of America 94(26) (1997) 14730–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Pandey KN, Emerging roles of antriuretic peptides and their receptors in pathophysiology of hypertension and cardiovascular regulation, J. Am. Soc. Hypert 2(4) (2008) 210–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Shi SJ, Nguyen HT, Sharma GD, Navar LG, Pandey KN, Genetic disruption of atrial natriuretic peptide receptor-A alters renin and angiotensin II levels, American journal of physiology. Renal physiology 281(4) (2001) F665–73. [DOI] [PubMed] [Google Scholar]
- [14].Shi SJ, Vellaichamy E, Chin SY, Smithies O, Navar LG, Pandey KN, Natriuretic peptide receptor A mediates renal sodium excretory responses to blood volume expansion, American journal of physiology. Renal physiology 285(4) (2003) F694–702. [DOI] [PubMed] [Google Scholar]
- [15].Vellaichamy E, Khurana ML, Fink J, Pandey KN, Involvement of the NF-kappa B/matrix metalloproteinase pathway in cardiac fibrosis of mice lacking guanylyl cyclase/natriuretic peptide receptor A, The Journal of biological chemistry 280(19) (2005) 19230–42. [DOI] [PubMed] [Google Scholar]
- [16].Vellaichamy E, Zhao D, Somanna N, Pandey KN, Genetic disruption of guanylyl cyclase/natriuretic peptide receptor-A upregulates ACE and AT1 receptor gene expression and signaling: role in cardiac hypertrophy, Physiological genomics 31(2) (2007) 193–202. [DOI] [PubMed] [Google Scholar]
- [17].Kishimoto I, Tokudome T, Nakao K, Kangawa K, Natriuretic peptide system: an overview of studies using genetically engineered animal models, FEBS J 278(11) (2011) 1830–41. [DOI] [PubMed] [Google Scholar]
- [18].Pandey KN, Molecular and genetic aspects of guanylyl cyclase natriuretic peptide receptor-A in regulation of blood pressure and renal function, Physiological genomics 50(11) (2018) 913–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Pandey KN, The functional genomics of guanylyl cyclase/natriuretic peptide receptor-A: perspectives and paradigms, FEBS J 278(11) (2011) 1792–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zhao D, Das S, Pandey KN, Interactive roles of NPR1 gene-dosage and salt diets on cardiac angiotensin II, aldosterone and pro-inflammatory cytokines levels in mutant mice, Journal of hypertension 31(1) (2013) 134–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhao D, Vellaichamy E, Somanna NK, Pandey KN, Guanylyl cyclase/natriuretic peptide receptor-A gene disruption causes increased adrenal angiotensin II and aldosterone levels, American journal of physiology. Renal physiology 293(1) (2007) F121–7. [DOI] [PubMed] [Google Scholar]
- [22].Nguyen G, Delarue F, Berrou J, Rondeau E, Sraer JD, Specific receptor binding of renin on human mesangial cells in culture increases plasminogen activator inhibitor-1 antigen, Kidney international 50(6) (1996) 1897–903. [DOI] [PubMed] [Google Scholar]
- [23].Nguyen G, Delarue F, Burckle C, Bouzhir L, Giller T, Sraer JD, Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin, J Clin Invest 109(11) (2002) 1417–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Muller DN, Binger KJ, Riediger F, Prorenin receptor regulates more than the renin-angiotensin system, Ann Med 44 Suppl 1 (2012) S43–8. [DOI] [PubMed] [Google Scholar]
- [25].Quadri S, Siragy HM, (Pro)renin receptor contributes to regulation of renal epithelial sodium channel, Journal of hypertension 34(3) (2016) 486–94; discussion 494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sihn G, Rousselle A, Vilianovitch L, Burckle C, Bader M, Physiology of the (pro)renin receptor: Wnt of change?, Kidney international 78(3) (2010) 246–56. [DOI] [PubMed] [Google Scholar]
- [27].Nguyen G, Contrepas A, Physiology and pharmacology of the (pro)renin receptor, Curr Opin Pharmacol 8(2) (2008) 127–32. [DOI] [PubMed] [Google Scholar]
- [28].Ichihara A, Suzuki F, Nakagawa T, Kaneshiro Y, Takemitsu T, Sakoda M, Nabi AH, Nishiyama A, Sugaya T, Hayashi M, Inagami T, Prorenin receptor blockade inhibits development of glomerulosclerosis in diabetic angiotensin II type 1a receptor-deficient mice, Journal of the American Society of Nephrology : JASN 17(7) (2006) 1950–61. [DOI] [PubMed] [Google Scholar]
- [29].Huang Y, Wongamorntham S, Kasting J, McQuillan D, Owens RT, Yu L, Noble NA, Border W, Renin increases mesangial cell transforming growth factor-beta1 and matrix proteins through receptor-mediated, angiotensin II-independent mechanisms, Kidney international 69(1) (2006) 105–13. [DOI] [PubMed] [Google Scholar]
- [30].Batenburg WW, Krop M, Garrelds IM, de Vries R, de Bruin RJ, Burckle CA, Muller DN, Bader M, Nguyen G, Danser AH, Prorenin is the endogenous agonist of the (pro)renin receptor. Binding kinetics of renin and prorenin in rat vascular smooth muscle cells overexpressing the human (pro)renin receptor, Journal of hypertension 25(12) (2007) 2441–53. [DOI] [PubMed] [Google Scholar]
- [31].Matavelli LC, Huang J, Siragy HM, In vivo regulation of renal expression of (pro)renin receptor by a low-sodium diet, American journal of physiology. Renal physiology 303(12) (2012) F1652–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Feng Y, ANG II-independent prorenin/(pro)renin receptor signaling pathways in the central nervous system, Am J Physiol Heart Circ Physiol 309(5) (2015) H731–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yang T, Xu C, Physiology and Pathophysiology of the Intrarenal Renin-Angiotensin System: An Update, Journal of the American Society of Nephrology : JASN 28(4) (2017) 1040–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kanda A, Noda K, Saito W, Ishida S, (Pro)renin receptor is associated with angiogenic activity in proliferative diabetic retinopathy, Diabetologia 55(11) (2012) 3104–13. [DOI] [PubMed] [Google Scholar]
- [35].Gonzalez AA, Liu L, Lara LS, Seth DM, Navar LG, Prieto MC, Angiotensin II stimulates renin in inner medullary collecting duct cells via protein kinase C and independent of epithelial sodium channel and mineralocorticoid receptor activity, Hypertension 57(3) (2011) 594–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Oshima Y, Morimoto S, Ichihara A, Roles of the (pro)renin receptor in the kidney, World J Nephrol 3(4) (2014) 302–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kiemer AK, Weber NC, Furst R, Bildner N, Kulhanek-Heinze S, Vollmar AM, Inhibition of p38 MAPK activation via induction of MKP-1: atrial natriuretic peptide reduces TNF-alpha-induced actin polymerization and endothelial permeability, Circulation research 90(8) (2002) 874–81. [DOI] [PubMed] [Google Scholar]
- [38].Pandey KN, Nguyen HT, Li M, Boyle JW, Natriuretic peptide receptor-A negatively regulates mitogen-activated protein kinase and proliferation of mesangial cells: role of cGMP-dependent protein kinase, Biochem Biophys Res Commun 271(2) (2000) 374–9. [DOI] [PubMed] [Google Scholar]
- [39].Burckle CA, Jan Danser AH, Muller DN, Garrelds IM, Gasc JM, Popova E, Plehm R, Peters J, Bader M, Nguyen G, Elevated blood pressure and heart rate in human renin receptor transgenic rats, Hypertension 47(3) (2006) 552–6. [DOI] [PubMed] [Google Scholar]
- [40].Kumar R, Cartledge WA, Lincoln TM, Pandey KN, Expression of guanylyl cyclase-A/atrial natriuretic peptide receptor blocks the activation of protein kinase C in vascular smooth muscle cells. Role of cGMP and cGMP-dependent protein kinase, Hypertension 29(1 Pt 2) (1997) 414–21. [DOI] [PubMed] [Google Scholar]
- [41].Hirose T, Hashimoto M, Totsune K, Metoki H, Asayama K, Kikuya M, Sugimoto K, Katsuya T, Ohkubo T, Hashimoto J, Rakugi H, Takahashi K, Imai Y, Association of (pro)renin receptor gene polymorphism with blood pressure in Japanese men: the Ohasama study, American journal of hypertension 22(3) (2009) 294–9. [DOI] [PubMed] [Google Scholar]
- [42].Kaneshiro Y, Ichihara A, Sakoda M, Takemitsu T, Nabi AH, Uddin MN, Nakagawa T, Nishiyama A, Suzuki F, Inagami T, Itoh H, Slowly progressive, angiotensin II-independent glomerulosclerosis in human (pro)renin receptor-transgenic rats, Journal of the American Society of Nephrology : JASN 18(6) (2007) 1789–95. [DOI] [PubMed] [Google Scholar]
- [43].Matavelli LC, Huang J, Siragy HM, (Pro)renin receptor contributes to diabetic nephropathy by enhancing renal inflammation, Clinical and experimental pharmacology & physiology 37(3) (2010) 277–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Oliver PM, John SW, Purdy KE, Kim R, Maeda N, Goy MF, Smithies O, Natriuretic peptide receptor 1 expression influences blood pressures of mice in a dose-dependent manner, Proc Natl Acad Sci U S A 95(5) (1998) 2547–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Pandey KN, Oliver PM, Maeda N, Smithies O, Hypertension associated with decreased testosterone levels in natriuretic peptide receptor-A gene-knockout and gene-duplicated mutant mouse models, Endocrinology 140(11) (1999) 5112–9. [DOI] [PubMed] [Google Scholar]
- [46].Zhou WD, Yang HM, Wang Q, Su DY, Liu FA, Zhao M, Chen QH, Chen QX, SB203580, a p38 mitogen-activated protein kinase inhibitor, suppresses the development of endometriosis by down-regulating proinflammatory cytokines and proteolytic factors in a mouse model, Hum Reprod 25(12) (2010) 3110–6. [DOI] [PubMed] [Google Scholar]
- [47].Ma X, Wang Q, Liu Y, Chen Y, Zhang L, Jiang M, Li X, Xiang R, Miao RQ, Duan Y, Han J, Inhibition of tumor growth by U0126 is associated with induction of interferon-gamma production, Int J Cancer 136(4) (2015) 771–83. [DOI] [PubMed] [Google Scholar]
- [48].Dignam JD, Martin PL, Shastry BS, Roeder RG, Eukaryotic gene transcription with purified components, Methods in enzymology 101 (1983) 582–98. [DOI] [PubMed] [Google Scholar]
- [49].Das S, Periyasamy R, Pandey KN, Activation of IKK/NF-kappaB provokes renal inflammatory responses in guanylyl cyclase/natriuretic peptide receptor-A gene-knockout mice, Physiological genomics 44(7) (2012) 430–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kumar P, Gogulamudi VR, Periasamy R, Raghavaraju G, Subramanian U, Pandey KN, Inhibition of HDAC enhances STAT acetylation, blocks NF-kappaB, and suppresses the renal inflammation and fibrosis in Npr1 haplotype male mice, American journal of physiology. Renal physiology 313(3) (2017) F781–F795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Pandey KN, Nguyen HT, Sharma GD, Shi SJ, Kriegel AM, Ligand-regulated internalization, trafficking, and down-regulation of guanylyl cyclase/atrial natriuretic peptide receptor-A in human embryonic kidney 293 cells, The Journal of biological chemistry 277(7) (2002) 4618–27. [DOI] [PubMed] [Google Scholar]
- [52].Vellaichamy E, Das S, Subramanian U, Maeda N, Pandey KN, Genetically altered mutant mouse models of guanylyl cyclase/natriuretic peptide receptor-A exhibit the cardiac expression of proinflammatory mediators in a gene-dose-dependent manner, Endocrinology 155(3) (2014) 1045–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Danser AH, Deinum J, Renin, prorenin and the putative (pro)renin receptor, Hypertension 46(5) (2005) 1069–76. [DOI] [PubMed] [Google Scholar]
- [54].Huang J, Siragy HM, Regulation of (pro)renin receptor expression by glucose-induced mitogen-activated protein kinase, nuclear factor-kappaB, and activator protein-1 signaling pathways, Endocrinology 151(7) (2010) 3317–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Batenburg WW, Lu X, Leijten F, Maschke U, Muller DN, Danser AH, Renin- and prorenin-induced effects in rat vascular smooth muscle cells overexpressing the human (pro)renin receptor: does (pro)renin-(pro)renin receptor interaction actually occur?, Hypertension 58(6) (2011) 1111–9. [DOI] [PubMed] [Google Scholar]
- [56].Schefe JH, Neumann C, Goebel M, Danser J, Kirsch S, Gust R, Kintscher U, Unger T, Funke-Kaiser H, Prorenin engages the (pro)renin receptor like renin and both ligand activities are unopposed by aliskiren, Journal of hypertension 26(9) (2008) 1787–94. [DOI] [PubMed] [Google Scholar]
- [57].Tripathi S, Pandey KN, Guanylyl cyclase/natriuretic peptide receptor-A signaling antagonizes the vascular endothelial growth factor-stimulated MAPKs and downstream effectors AP-1 and CREB in mouse mesangial cells, Mol Cell Biochem 368(1–2) (2012) 47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kumar R, von Geldern TW, Calle RA, Pandey KN, Stimulation of atrial natriuretic peptide receptor/guanylyl cyclase- A signaling pathway antagonizes the activation of protein kinase C-alpha in murine Leydig cells, Biochim Biophys Acta 1356(2) (1997) 221–8. [DOI] [PubMed] [Google Scholar]
- [59].Sharma GD, Nguyen HT, Antonov AS, Gerrity RG, von Geldern T, Pandey KN, Expression of atrial natriuretic peptide receptor-A antagonizes the mitogen-activated protein kinases (Erk2 and P38MAPK) in cultured human vascular smooth muscle cells, Molecular and cellular biochemistry 233(1–2) (2002) 165–73. [DOI] [PubMed] [Google Scholar]
- [60].Sugimoto T, Haneda M, Togawa M, Isono M, Shikano T, Araki S, Nakagawa T, Kashiwagi A, Guan KL, Kikkawa R, Atrial natriuretic peptide induces the expression of MKP-1, a mitogen-activated protein kinase phosphatase, in glomerular mesangial cells, J Biol Chem 271(1) (1996) 544–7. [DOI] [PubMed] [Google Scholar]
- [61].Eguchi S, Inagami T, Signal transduction of angiotensin II type 1 receptor through receptor tyrosine kinase, Regulatory peptides 91(1–3) (2000) 13–20. [DOI] [PubMed] [Google Scholar]
- [62].Sakoda M, Ichihara A, Kaneshiro Y, Takemitsu T, Nakazato Y, Nabi AH, Nakagawa T, Suzuki F, Inagami T, Itoh H, (Pro)renin receptor-mediated activation of mitogen-activated protein kinases in human vascular smooth muscle cells, Hypertens Res 30(11) (2007) 1139–46. [DOI] [PubMed] [Google Scholar]
- [63].Uraoka M, Ikeda K, Nakagawa Y, Koide M, Akakabe Y, Nakano-Kurimoto R, Takahashi T, Matoba S, Yamada H, Okigaki M, Matsubara H, Prorenin induces ERK activation in endothelial cells to enhance neovascularization independently of the renin-angiotensin system, Biochemical and biophysical research communications 390(4) (2009) 1202–7. [DOI] [PubMed] [Google Scholar]
- [64].Feldt S, Batenburg WW, Mazak I, Maschke U, Wellner M, Kvakan H, Dechend R, Fiebeler A, Burckle C, Contrepas A, Jan Danser AH, Bader M, Nguyen G, Luft FC, Muller DN, Prorenin and renin-induced extracellular signal-regulated kinase 1/2 activation in monocytes is not blocked by aliskiren or the handle-region peptide, Hypertension 51(3) (2008) 682–8. [DOI] [PubMed] [Google Scholar]
- [65].Subramanian U, Kumar P, Mani I, Chen D, Kessler I, Periyasamy R, Raghavaraju G, Pandey KN, Retinoic acid and sodium butyrate suppress the cardiac expression of hypertrophic markers and proinflammatory mediators in Npr1 gene-disrupted haplotype mice, Physiological genomics 48(7) (2016) 477–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Huang J, Siragy HM, Glucose promotes the production of interleukine-1beta and cyclooxygenase-2 in mesangial cells via enhanced (Pro)renin receptor expression, Endocrinology 150(12) (2009) 5557–65. [DOI] [PMC free article] [PubMed] [Google Scholar]