

Abstract
Background:
KIWI ( NCT01705145) was a 24-week, single-arm, pharmacokinetics, safety, and efficacy study of ivacaftor in children aged 2 to 5 years with cystic fibrosis (CF) and a CFTR gating mutation. Here, we report the results of KLIMB ( NCT01946412), an 84-week, open-label extension of KIWI.
Methods:
Children received age- and weight-based ivacaftor dosages for 84 weeks. The primary outcome was safety. Other outcomes included sweat chloride, growth parameters, and measures of pancreatic function.
Results:
All 33 children who completed KIWI enrolled in KLIMB; 28 completed 84 weeks of treatment. Most adverse events were consistent with those reported during KIWI. Ten (30%) children had transaminase elevations >3 × upper limit of normal (ULN), leading to 1 discontinuation in a child with alanine aminotransferase >8 × ULN. Improvements in sweat chloride, weight and body mass index z scores and fecal elastase-1 observed during KIWI were maintained during KLIMB; there was no further improvement in these parameters.
Conclusions:
Ivacaftor was generally well tolerated for up to 108 weeks in children aged 2 to 5 years with CF and a gating mutation, with safety consistent with the KIWI study. Improvements in sweat chloride and growth parameters during the initial 24 weeks of treatment were maintained for up to an additional 84 weeks of treatment. Prevalence of raised transaminases remained stable and did not increase with duration of exposure during the open-label extension.
Keywords: cystic fibrosis, CFTR potentiator, ivacaftor, KLIMB, pediatrics, safety
1. INTRODUCTION
The pathophysiologic effects of cystic fibrosis (CF), including poor nutritional status and structural lung damage, typically begin in the first years of life (1). Early intervention is known to be clinically beneficial (2, 3), and thus treatment with cystic fibrosis transmembrane conductance regulator (CFTR) modulators early in life could potentially improve long-term outcomes. To date, no studies have been conducted on prolonged use of CFTR modulators in children with CF who are younger than 6 years of age.
Ivacaftor, a CFTR potentiator that enhances chloride transport by increasing the channel-open probability of CFTR at the cell surface (4, 5), has been shown to be safe and efficacious in patients aged 6 years and older with CF and specific CFTR mutations (6–9). The 24-week, open-label, 2-part, phase 3 KIWI study demonstrated that the pharmacokinetics, safety, and efficacy of ivacaftor in children aged 2 to 5 years with CF and a CFTR gating mutation are generally similar to those seen in older patients (10). Data from KIWI led to the approval of ivacaftor in the United States, European Union, Canada, and Australia for treatment of patients aged 2 years and older with CF and a CFTR gating/ivacaftor-responsive mutation.
Here, we report results from KLIMB, an 84-week extension study of ivacaftor in children aged 2 to 5 years with CF and a CFTR gating mutation who completed the 24-week KIWI study. The primary outcome was long-term safety. Other outcomes included changes in sweat chloride, growth parameters, and measures of pancreatic function.
2. METHODS AND MATERIALS
2.1. Study design and participants
KLIMB was an open-label extension study (ClinicalTrials.gov, number NCT01946412) in children who completed the 24-week, single-arm, open-label, phase 3 KIWI (part B) study of ivacaftor treatment (10). Children eligible for KIWI were aged 2 to 5 years (median, 3.0 years), weighed 8 kg or more, and had a confirmed diagnosis of CF (11) and a CFTR gating mutation (G551D, G178R, S549N, S549R, G551S, G970R, G1244E, S1251N, S1255P, G1349D) on at least 1 allele (10). After completion of the study’s design, the G970R mutation was discovered to be a class I splice mutation; none of the children in this study had this mutation. There was no interruption of ivacaftor treatment between KIWI and KLIMB. The primary outcome of KLIMB was long-term safety. Other outcomes included change from KIWI and KLIMB study baseline measurements in sweat chloride (assessed at clinic visits at day 1 and weeks 24, 48, 72, and 84) and weight, height, and body mass index (BMI; assessed at day 1 and weeks 12, 24, 36, 48, 60, 72, and 84). Exploratory endpoints included change from KIWI and KLIMB study baseline measurements in fecal elastase-1 (a measure of pancreatic exocrine function (13)), and percent predicted forced expiratory volume in 1 second (ppFEV1), all assessed at day 1 and weeks 12, 24, 36, 48, 60, 72, and 84. Immunoreactive trypsinogen (IRT; a serum-based marker of pancreatic insult) was assessed at day 1 and weeks 24, 48, 60, 72, and 84 (14). Neither the site staff nor enrolled children received study-specific training in preschool lung function testing, nor were certification or over-reading conducted for spirometric assessments. Because acceptable spirometric data were only obtained in a small number of children in KIWI, these results are not presented.
This study was conducted at 15 sites in the United States, United Kingdom, and Canada from December 2013 to December 2015. Children received weight-based ivacaftor as granules (Vertex Pharmaceuticals Incorporated, Boston, MA) every 12 hours (q12h) at a dose of 50 mg q12h (weight <14 kg) and 75 mg q12h (weight ≥14 kg). Children who turned 6 years of age during KLIMB received ivacaftor 150 mg q12h as tablets (Vertex Pharmaceuticals Incorporated, Boston, MA). Dose was adjusted as necessary based on weight at each study visit. Safety assessments consisted of adverse events primarily defined using Common Terminology Criteria for Adverse Events, version 4.0 (15), clinical laboratory values, vital signs, 12-lead electrocardiogram readings, and physical and ophthalmological examinations. Standardized eye examinations were conducted by a licensed ophthalmologist at baseline and at specified intervals after dosing. Serious adverse events were defined per the International Conference on Harmonization guidelines (16).
An independent ethics committee or institutional review board for each site approved the study protocol, and an independent data monitoring committee monitored study safety data. Written informed consent was obtained from each child’s parent or legal guardian.
2.2. Statistical analyses
Safety and efficacy were assessed among all children who received at least 1 dose of ivacaftor in KLIMB. A mixed-effects model for repeated measures (MMRM) was used to analyze the absolute change from baseline in sweat chloride; height, weight, and BMI z scores; and fecal elastase-1 at each study visit. For IRT, as it was not normally distributed, the Wilcoxon Signed Rank Test was used. There was no adjustment for multiple comparisons in the analyses of absolute change from baseline in study endpoints due to the small sample size. All P values are nominal. Additional analyses performed using descriptive statistics and 95% CI for group comparisons are provided. Analyses were performed using SAS®, version 9.2 (Cary, NC).
3. RESULTS
3.1. Study population
Thirty-four children were enrolled in KIWI part B, and 33 (97%) completed the 24 weeks of treatment (Supplementary Figure 1). All 33 who completed KIWI enrolled in KLIMB, with 28 (84.8%) completing the 84-week open-label treatment period. Of the 5 children who discontinued study drug before week 84, 2 switched to commercial ivacaftor, 1 had an adverse event (elevated alanine aminotransferase [ALT] >8 × upper limit of normal [ULN] and aspartate aminotransferase [AST] >3 × ULN), 1 had difficulty swallowing the ivacaftor 150-mg tablet, and 1 withdrew because of inability to tolerate further blood tests.
Baseline characteristics at the start of KLIMB are presented in Supplementary Table 1. Mean (SD) age was 3.7 (1.0) years. At the beginning of KLIMB, 5 children (mean [SD] age, 2.4 [0.6] years; mean [SD] weight, 12.8 [0.8] kg) received ivacaftor 50 mg q12h; 27 children (mean [SD] age, 3.9 [0.9] years; mean [SD] weight, 17.5 [2.0] kg) received ivacaftor 75 mg q12h, and one 6-year-old child (weight, 23.7 kg) received ivacaftor 150 mg q12h.
3.2. Primary outcome: safety
Adverse events are summarized in Table 1. All children reported at least 1 adverse event during the 84 weeks. The most common adverse events occurring in ≥30% of children were cough (72.7%), pyrexia (39.4%), vomiting (39.4%), and pulmonary exacerbation (30.3%), events that commonly occur in the pediatric CF population. Twenty-one serious adverse events occurred in 11 children. Serious adverse events considered related to ivacaftor were elevated ALT and AST levels that occurred in 2 children. One child had elevated ALT levels >8 × ULN and elevated AST levels >5 × ULN on the same day. The second had elevated ALT/AST levels >8 × ULN on the same day.
Table 1.
Adverse Events
| Adverse events (AE), n (%) | Overall (N=33) |
||
|---|---|---|---|
| Children with ≥1 adverse event | 33 (100) | ||
|
Treatment-emergent adverse events in ≥10% of children | |||
| Cough | 24 (72.7) | ||
| Pyrexia | 13 (39.4) | ||
| Vomiting | 13 (39.4) | ||
| Pulmonary exacerbation | 10 (30.3) | ||
| Nasal congestion | 7 (21.2) | ||
| Increased ALT | 7 (21.2) | ||
| Increased AST | 6 (18.2) | ||
| Otitis media | 6 (18.2) | ||
| Rhinorrhea | 6 (18.2) | ||
| Abdominal pain | 5 (15.2) | ||
| Sinusitis | 5 (15.2) | ||
| Viral upper respiratory tract infection | 5 (15.2) | ||
| Viral gastroenteritis | 4 (12.1) | ||
| Streptococcal pharyngitis | 4 (12.1) | ||
| Rash | 4 (12.1) | ||
|
Serious adverse events (SAEs) | |||
| Pulmonary exacerbationb | 6 (18.2) | Patient # 1, 2, 5, 6, 7, 9 | |
| ALT >8 × ULNa | 2 (6.1) | Patient # 10, 11 | |
| AST >5 × ULNa | 2 (6.1) | Patient # 10, 11 | |
| Pyrexiab | 2 (6.1) | Patient # 3, 7 | |
| Enterovirus infectionb | 1 (3.0) | Patient # 4 | |
| Respiratory syncytial virus infectionb | 1 (3.0) | Patient # 1 | |
| Staphylococcal infectionb | 1 (3.0) | Patient # 2 | |
| Adenovirus test positiveb | 1 (3.0) | Patient # 3 | |
| Dehydrationb | 1 (3.0) | Patient # 3 | |
| Anoxic Seizureb | 1 (3.0) | Patient # 8 | |
ALT, alanine transaminase; AST, aspartate transaminase; ULN, upper limit of normal.
These were the same 2 children. An episode of transaminase >8 × ULN was observed in a total of 5 children (see Table 2); in only 2 cases did the investigator report the event as an SAE.
Resulting in hospitalization.
Elevated ALT and/or AST levels >3 × ULN were documented in 10 children (30%) during 84 weeks in KLIMB (Table 2). 4 of these 10 children also had transaminase elevations during KIWI, and a history of liver function test (LFT) elevations before enrollment in KIWI. In KLIMB, LFT elevations were reported as adverse events in 7 of the 10 children. Per protocol, treatment was interrupted in all 5 children who reported ALT and/or AST elevations >8 × ULN. Ivacaftor dosing was successfully resumed in 4 of the 5 children. One child permanently discontinued ivacaftor because of LFT elevations after ivacaftor was reinitiated.
Table 2.
Summary of Liver Function Test Elevations During the 84-Week KLIMB Study
| Maximum on-treatment ALT or AST (U/L), n (%)a | N=33 |
|---|---|
| >3 to ≤5 × ULN | 1 (3.0) |
| >5 to ≤8 × ULN | 4 (12.1) |
| >8 × ULN | 5 (15.2) |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; ULN, upper limit of normal.
7 of the 10 total events were reported as AEs; 2 of the 5 events with ALT or AST >8 x ULN were reported as SAEs
Figure 1 illustrates the prevalence of transaminase elevations over time during KLIMB. There was no evidence of increased prevalence of elevated transaminases with prolonged exposure to ivacaftor.
Figure 1.
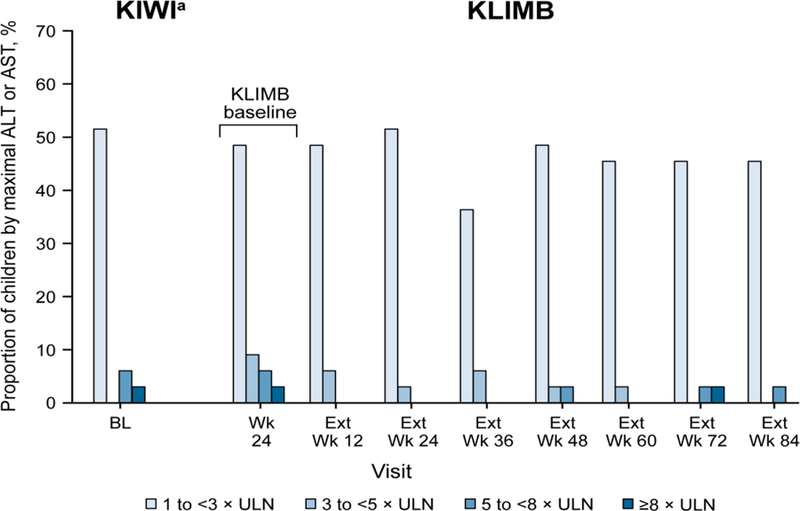
Prevalence of elevated transaminase measurements over time in KLIMB, including proportion of children with ALT or AST elevations 3 × ULN and greater during the 84-week study. ALT, alanine aminotransferase; AST, aspartate aminotransferase; BL, baseline; Ext, extension; ULN, upper limit of normal. aData shown for children who enrolled in KLIMB.
No abnormalities were detected on serial electrocardiograms. No meaningful changes in visual acuity from baseline occurred throughout the study. One child with a history of astigmatism developed a lens opacity at week 84 that was considered not visually significant and possibly related to ivacaftor. Drug withdrawal was not recommended.
3.3. Secondary outcomes
3.3.1. Sweat chloride
Ivacaftor treatment during KIWI had led to a significant reduction in sweat chloride that was maintained over 24 weeks of treatment (10)]. Continued treatment of these children with ivacaftor in KLIMB maintained this reduction in sweat chloride until the end of the study at week 84 (Figure 2 and Table 3).
Figure 2.

Mean absolute change from KIWI baseline in (A) sweat chloride and (B) BMI z score and mean values for (C) fecal elastase-1 levels by visit. Means were calculated for each visit from the number of children contributing data at that time point. BL, baseline; Ext, extension; SE, standard error. aData shown for all children who enrolled in KLIMB. Data for intermediate visits from KIWI are not shown for fecal elastase-1. *P<0.05. †P<0.01. ‡P<0.0001. All P values are for absolute change from KIWI baseline.
Table 3.
Absolute Change in Secondary and Tertiary Endpoints at Extension Week 84
| Endpoints | Mean absolute change at extension week 84 (95% CI) | P value for mean absolute change from KIWI baselinea | |
|---|---|---|---|
| Secondary endpoints | From KIWI baseline | From KLIMB baseline | |
| Sweat chloride, mmol/L | −54.7 (−65.4, −43.9) | −8.5 (−18.9, 1.8) | <0.0001 |
| Tertiary endpoints | |||
| BMI z score | 0.27 (0.04, 0.50) | −0.08 (−0.31, 0.15) | 0.0229 |
| Weight z score | 0.20 (−0.05, 0.44) | 0.00 (−0.20, 0.20) | 0.1119 |
| Height z score | 0.12 (−0.06, 0.29) | 0.14 (0.00, 0.29) | 0.1800 |
| Fecal elastase-1, µg/g | 128.8 (45.7, 211.9) | 56.8 (−22.2, 135.8) | 0.0050 |
|
Median absolute change (min,max) at extension week 84 | |||
| IRT, ng/mL | −8.1 (−71.1, 21.9) | 1.0 (−16.4, 35.6) | 0.0103b |
BMI, body mass index; IRT, immunoreactive trypsinogen.
All P values are nominal.
From a Wilcoxon Signed Rank Test.
3.3.2. Nutrition
Improvements in weight z score (+0.2 [SD, 0.3]; P<0.0001) and BMI z score (+0.4 [SD, 0.4]; P<0.001) were observed during the initial 24-week KIWI study (10)]. During the 84-week KLIMB extension, the weight z score from KIWI baseline was unchanged but the BMI z score continued to be significantly better than at KIWI baseline (0.27 [95% CI: 0.04, 0.50]; Table 3).
3.4. Exploratory outcomes
3.4.1. Pancreatic exocrine function
In the KIWI study, mean fecal elastase-1 increased by 99.8 µg/g from baseline (SD, 138.4; P<0.001). Additionally, 23% (P=0.0504) had a fecal elastase-1 that improved to greater than 200 µg/g, the cutoff value for pancreatic exocrine insufficiency. Improvements in fecal elastase-1 levels observed during KIWI were maintained during KLIMB (Figure 2). At week 84, the mean (95% CI) absolute increase in fecal elastase-1 was 128.8 (45.7, 211.9) µg/g from KIWI baseline. During the 84-week open-label extension, fecal elastase-1 continued to increase, but the change from KLIMB baseline was not statistically significant (56.8 µg/g; 95% CI: −22.2, 135.8; Table 3). Seventeen children had paired fecal elastase-1 data at KIWI baseline and week 84 of KLIMB. At the start of the KIWI study, 1 of 17 (6%) children had fecal elastase-1 levels ≥200 µg/g. By week 84 of KLIMB, 6 of 17 (35%) children had fecal elastase-1 ≥200 µg/g.
In KIWI, IRT levels decreased from baseline to week 24 by a mean of 20.7 ng/mL (SD, 24), suggesting reduced pancreatic inflammation/stress from KIWI study entry (10). This decrease was maintained in KLIMB: at week 84, the mean (SD) absolute decrease from KIWI baseline was −15.9 (25.2) ng/mL. The median (range) absolute decrease from KIWI baseline was −8.1 ng/mL (−71.1, 21.9), with no significant change during the KLIMB extension (1.0 ng/mL; −16.4, 35.6).
4. DISCUSSION
The results of this open-label extension study suggest that ivacaftor is generally well tolerated in children aged 2 to 5 years with CF and a gating mutation for up to 108 weeks. The safety profile was consistent with previous clinical trials of ivacaftor in children and adults. There were no clinical safety concerns identified in assessments of hematology laboratory parameters, vital signs, or electrocardiograms in this small sample..
While there are limited data on the prevalence of LFT elevations in children with CF, studies indicate a natural propensity for transaminase elevations in children with CF in the first 2 to 3 years of life (18, 19). Our findings on transaminase elevations seem to be consistent with the published data. Thirty percent of children aged 2 to 5 years in KLIMB had transaminase elevations >3 × ULN on at least 1 occasion across 84 weeks. Transaminase elevations were generally asymptomatic, did not require permanent treatment discontinuation except in 1 child, and occurred more often in children with a history of transaminase elevations prior to ivacaftor exposure and in KIWI. The lack of a placebo arm in this study makes interpretation of our results difficult, with uncertainty as to whether the prevalence of elevated transaminases reflects an effect of ivacaftor, ascertainment bias due to increased monitoring during a clinical trial, or the natural history of LFT elevations in this age group. This long-term extension study is important in demonstrating that the prevalence of LFT elevations did not appear to increase with the length of exposure to ivacaftor. However, it is recommended that LFTs be assessed before initiation of ivacaftor and monitored during treatment, particularly in children with a history of elevated transaminases.
It is difficult to interpret the potential risk of the non-visually significant lens opacity seen in one patient at week 84 in association with astigmatism. Baseline assessment for lens opacities and periodic monitoring while on therapy is recommended.
The improvements observed in sweat chloride concentrations during the 24-week KIWI study were maintained during this 84-week extension trial, demonstrating maintenance of improved CFTR function. Similarly, improvements in BMI z scores observed in KIWI were generally maintained in KLIMB, although without further improvement.
The changes in the exploratory endpoints of fecal elastase-1 and serum IRT observed during KIWI were maintained during KLIMB, suggesting that early, effective CFTR modulation has the potential to delay deterioration in pancreatic function. Further support is lent to this hypothesis by the improvements in markers of pancreatic exocrine function (fecal elastase-1) and pancreatic insult (IRT, amylase, and lipase) reported recently in children 12 to <24 months treated with ivacaftor (20). The mechanism by which ivacaftor might improve exocrine pancreatic function is unclear. In a recent study employing mouse models of Sjogren’s syndrome and auoimmune pancreatitis, both of which involve decreased expression and mislocalization of CFTR in the pancreatic ducts, treatment with ivacaftor and a CFTR corrector rescued CFTR expression and localization, resulting in decreased acinar inflammation, fibrosis, and tissue damage (21). These results (albeit not from a CF model) suggest that the effect of ivacaftor on the pancreas in infants and toddlers with CF may be mediated through restoration of ductal function, in turn improving acinar cell function and allowing some normalization of pancreatic secretions (enzymes, bicarbonate, fluid). In older children and adults, in whom ivacaftor does not improve fecal elastase, the improved nutritional status associated with ivacaftor appears to be the result of normalization of intestinal pH and CFTR-mediated bicarbonate secretion (22) as well as decreased intestinal inflammation, resulting in improved absorption of fat (23).
The current study had several limitations. The age of the population and relative rarity of the CFTR gating mutations led to the decision to perform the original KIWI study as an open-label rather than a placebo-controlled trial. Because KLIMB was designed as a single-arm extension of this safety and pharmacokinetic trial, there was no placebo group. The lack of a control group limits our interpretation of both safety and therapeutic benefit. In addition, most participants had at least 1 G551D mutation, limiting our ability to assess the effects of ivacaftor treatment in persons with rarer gating mutations. The small sample size also limited our ability to detect rare adverse events. Finally, because of the challenge of obtaining accurate spirometry data from children aged 2 to 5 years (24), very little acceptable spirometry data were collected, precluding our ability to analyze those data. Alternative lung function measures such as the lung clearance index from multiple-breath washout may also be considered in the future (25).
5. CONCLUSIONS
This is the first study reporting long-term safety and efficacy of ivacaftor in children aged 2 to 5 years with a CFTR gating mutation. Ivacaftor was generally safe and well tolerated for up to 108 weeks. Increases in transaminases did not become more frequent with prolonged exposure to the drug. The reduction in sweat chloride concentration, growth benefits, and improvements in markers of pancreatic exocrine function observed during 24 weeks in KIWI were maintained for an additional 84 weeks in KLIMB.
Supplementary Material
HIGHLIGHTS.
Ivacaftor was generally well tolerated in 2–5 year-olds up to 84 weeks in KLIMB
AST and/or ALT >3 × ULN occurred in 30% of children on ≥1 occasion
Sweat chloride improvements seen in KIWI were maintained through 84 weeks of KLIMB
Weight and BMI z score gains seen in KIWI were sustained but not further elevated
Improvements in pancreatic function seen in KIWI were maintained during KLIMB
ACKNOWLEDGMENTS
The authors thank all the patients and their families for participating in the study. The authors also thank Linda T. Wang, MD, and Daniel Campbell, PhD, for their critical review of the manuscript. LTW and DC are employees of Vertex Pharmaceuticals Incorporated and may own stock or stock options in that company. The authors acknowledge the contributions of all KLIMB (VX11–770-109) study site investigators and coordinators: Frank Accurso, MD (Children’s Hospital Colorado, Aurora, CO), Philip Black, MD (Children’s Mercy Hospital, Kansas City, MO), Barbara Chatfield, MD (University of Utah Primary Children’s Medical Center, Salt Lake City, UT), Theresa Laguna, MD (University of Minnesota, Minneapolis, MN), Gregory Montgomery, MD (Riley Hospital for Children at Indiana University Health, Indianapolis, IN), Howard Schmidt, MD (Virginia Commonwealth University Health Systems, Nelson Clinic, Richmond, VA), and Seth Walker, MD (The Emory Clinic Children’s Healthcare of Atlanta at Egleston, Atlanta, GA) and acknowledge Andrew Fall, MBChB, and Debbie Miller from the NHS Lothian Children’s Clinical Research Facility at the Royal Hospital for Sick Children, Edinburgh, Scotland; Katie Brand from the Child Health Research Unit at the University of Alabama at Birmingham; Melissa Richmond from the BC Children’s Hospital, Vancouver, BC, Canada; and Rebecca Dobra, MBChB, and Sandra Scott, PhD, Royal Brompton and Harefield NHS Foundation Trust, London, UK. This project was supported by the NIHR Respiratory Disease Biomedical Research Unit at the Royal Brompton and Harefield NHS Foundation Trust and Imperial College London. The views expressed in this publication are those of the authors and not necessarily those of the NHS, The National Institute for Health Research, or the Department of Health.
Medical writing and editorial support were provided by Tejendra Patel, PharmD an employee of Vertex Pharmaceuticals Incorporated and Stephanie Vadasz, PhD, of Ashfield Healthcare Communications, which received funding from Vertex Pharmaceuticals Incorporated.
Funding:This study was sponsored by Vertex Pharmaceuticals Incorporated. Medical writing and editorial support and coordination were funded by Vertex Pharmaceuticals Incorporated.
ABBREVIATIONS
- ALT
alanine transaminase
- AST
aspartate transaminase
- BMI
body mass index
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- IRT
immunoreactive trypsinogen
- LFT
liver function test
- MMRM
mixed-effect model for repeated measures
- q12h
every 12 hours
- SD
standard deviation
- SE
standard error
- ULN
upper limit of normal
Footnotes
Data from this study were previously presented in part at the 30th Annual North American Cystic Fibrosis Conference; October 27–29, 2016; Orlando, FL, and at the British Thoracic Society Winter Meeting 2016; December 7–9, 2019; London, UK.
Declaration of interest: MR has received research grants and served as a consultant for Vertex Pharmaceuticals Incorporated, for which her institution received payment. SC has received fees paid by the UK Cystic Fibrosis Trust made to his institution for his contribution to the UKCFT Pharmacovigilance Programme. WTH reports grants from Vertex Pharmaceuticals, during the conduct of the study; grants from National Institutes of Health (NIH), grants from Cystic Fibrosis Foundation, and grants from Gilead Sciences outside the submitted work. WER and MC have served as site principal investigators for trials, institutions received fees from Vertex Pharmaceuticals Incorporated. GSS has received consulting fees from Vertex Pharmaceuticals Incorporated and his institution has received financial support from Vertex Pharmaceuticals Incorporated, Gilead Sciences, and Genentech. MH and ST are employees of Vertex Pharmaceuticals Incorporated and may own stock or stock options in that company. JC is a former employee of Vertex Pharmaceuticals Incorporated and may own stock or stock options in that company. JCD has served on advisory boards, undertaken educational activities, and served as a national/site principal investigator for trials, for which her institution received fees from Vertex Pharmaceuticals Incorporated. AL and KWS have no declarations of interest.
REFERENCES
- 1.Ranganathan SC, Hall GL, Sly PD, Stick SM, Douglas TA, Australian Respiratory Early Surveillance Team for Cystic Fibrosis. Early lung disease in infants and pre-school children with cystic fibrosis: what have we learned and what should we do about it? Am J Respir Crit Care Med 2017;195:1567–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yen EH, Quinton H, Borowitz D. Better nutritional status in early childhood is associated with improved clinical outcomes and survival in patients with cystic fibrosis. J Pediatr 2013;162(3):530–5 e1. [DOI] [PubMed] [Google Scholar]
- 3.VanDevanter DR, Kahle JS, O’Sullivan AK, Sikirica S, Hodgkins PS. Cystic fibrosis in young children: A review of disease manifestation, progression, and response to early treatment. Journal of Cystic Fibrosis 15(2):147–57. [DOI] [PubMed] [Google Scholar]
- 4.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A 2009;106(44):18825–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van Goor F, Yu H, Burton B, Hoffman BJ. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J Cyst Fibros 2014;13(1):29–36. [DOI] [PubMed] [Google Scholar]
- 6.Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Drevinek P, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 2011;365(18):1663–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davies JC, Wainwright CE, Canny GJ, Chilvers MA, Howenstine MS, Munck A, et al. Efficacy and safety of ivacaftor in patients aged 6 to 11 years with cystic fibrosis with a G551D mutation. Am J Respir Crit Care Med 2013;187(11):1219–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Boeck K, Munck A, Walker S, Faro A, Hiatt P, Gilmartin G, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J Cyst Fibros 2014;13(6):674–80. [DOI] [PubMed] [Google Scholar]
- 9.Rowe SM, Daines C, Ringshausen FC, Kerem E, Wilson J, Tullis E, et al. Tezacaftor–Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. New England Journal of Medicine 2017;377(21):2024–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davies JC, Cunningham S, Harris WT, Lapey A, Regelmann WE, Sawicki GS, et al. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2–5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study. Lancet Respir Med 2016;4(2):107–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farrell PM, White TB, Ren CL, Hempstead SE, Accurso F, Derichs N, et al. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J Pediatr 2017;181s:S4–S15.e1. [DOI] [PubMed] [Google Scholar]
- 12.Seibert FS, Linsdell P, Loo TW, Hanrahan JW, Riordan JR, Clarke DM. Cytoplasmic loop three of cystic fibrosis transmembrane conductance regulator contributes to regulation of chloride channel activity. The Journal of biological chemistry 1996;271(44):27493–9. [DOI] [PubMed] [Google Scholar]
- 13.Borowitz D, Baker SS, Duffy L, Baker RD, Fitzpatrick L, Gyamfi J, et al. Use of fecal elastase-1 to classify pancreatic status in patients with cystic fibrosis. J Pediatr 2004;145(3):322–6. [DOI] [PubMed] [Google Scholar]
- 14.Weintraub A, Blau H, Mussaffi H, Picard E, Bentur L, Kerem E, et al. Exocrine pancreatic function testing in patients with cystic fibrosis and pancreatic sufficiency: a correlation study. Journal of pediatric gastroenterology and nutrition 2009;48(3):306–10. [DOI] [PubMed] [Google Scholar]
- 15.Common Terminology Criteria for Adverse Events (CTCAE) version 4.0: National Cancer Institute, Cancer Therapy Evaluation Program; [Available from: http://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm.
- 16.Post-approval safety data management: definitions and standards for expedited reporting: International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use; 2003. [updated November 23, 2003 E2D:[Available from:http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E2D/Step4/E2D_Guideline.pdf. [Google Scholar]
- 17.Farrell PM, Rosenstein BJ, White TB, Accurso FJ, Castellani C, Cutting GR, et al. Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report. J Pediatr 2008;153(2):S4–S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lindblad A, Glaumann H, Strandvik B. Natural history of liver disease in cystic fibrosis. Hepatology (Baltimore, Md) 1999;30(5):1151–8. [DOI] [PubMed] [Google Scholar]
- 19.Woodruff SA, Sontag MK, Accurso FJ, Sokol RJ, Narkewicz MR. Prevalence of elevated liver enzymes in children with cystic fibrosis diagnosed by newborn screen. J Cyst Fibros 2017;16(1):139–45. [DOI] [PubMed] [Google Scholar]
- 20.Rosenfeld M, Wainwright CE, Higgins M, Wang LT, McKee C, Campbell D, et al. Ivacaftor treatment of cystic fibrosis in children aged 12 to <24 months and with a CFTR gating mutation (ARRIVAL): a phase 3 single-arm study. Lancet Respir Med 2018;6(7):545–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zeng M, Szymczak M, Ahuja M, et al. Correction of Ductal CFTR Activity Rescues Acinar Cell and Pancreatic and Salivary Gland Functions in Mouse Models of Autoimmune Disease. Gastroenterology 2017;153:1148–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gelfond D, Heltshe S, Ma C, et al. Impact of CFTR Modulation on Intestinal pH, Motility, and Clinical Outcomes in Patients With Cystic Fibrosis and the G551D Mutation Clinical and Translational Gastroenterology (2017) 8, e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stallings V, Sainath N, Oberle M, et al. Energy Balance and Mechanisms of Weight Gain with Ivacaftor Treatment of Cystic Fibrosis Gating Mutations. J Pediatr 2018;201:229–37. [DOI] [PubMed] [Google Scholar]
- 24.Gaffin JM, Shotola NL, Martin TR, Phipatanakul W. Clinically Useful Spirometry in Preschool-Aged Children: Evaluation of the 2007 American Thoracic Society Guidelines. The Journal of asthma : official journal of the Association for the Care of Asthma 2010;47(7):762–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horsley A Lung clearance index in the assessment of airways disease. Respir Med 2009;103(6):793–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.