

Abstract
Ischemic AKI is predominantly mediated by the innate inflammatory response to DAMPS released during the reperfusion phase of the ischemic event. Here we show that pre-emptive IgM infusions to increase binding of natural IgM (nIgM) antibodies to leucocyte receptors (IgM-ALA), inhibit this inflammatory response and prevents AKI in mice. Similarly, AKI was prevented by pre-emptively infusing Bregs, induced ex-vivo by pre-treating pan-B cells with nIgM. Harnessing such a physiologic mechanism to inhibit inflammation and prevent ischemia induced AKI can have translational potential in humans. For example, one can pre-emptively infuse IgM or ex-vivo induced B regs in patients who have a high risk of developing ischemia induced AKI especially the subset of these patients with low levels of IgM-ALA or regulatory T cells (Tregs).
Introduction:
Experiments in rodent models have clearly demonstrated that ischemia induced acute kidney injury (AKI) result from two basic mechanisms. Firstly, acute cessation of blood flow for 25 to 35 minutes rapidly leads to hypoxia induced cell damage and death of renal endothelial and tubular epithelial cells. This hypoxia induced cell death mostly results from endoplasmic reticulum stress and mitochondrial damage and the extent of cell death is variable and depends on the level and duration of ischemia as well as innate cellular resistance to hypoxia. Secondly, during kidney reperfusion, damaged cells release intracellular substances, reactive oxygen species and glycolipids, commonly referred to as damage-associated molecular patterns (DAMPS), and DAMPS are presented by antigen presenting cells (APC) to NKT-1 cells to initiate an innate inflammatory response in the kidney (1, 3). NKT-1 cells comprise <2% of circulating T cells and unlike un-activated T cells, express high levels of receptors that recognize DAMPS and glycolipids. Hence, following ischemic injury, NKT-1 cells are the initial T cells which get activated and produce pro-inflammatory cytokines that recruit and activate other leucocytes (2). The resulting inflammatory response contributes significantly towards AKI as one observes minimal or no AKI, based on increase in plasma creatinine, if the inflammatory response is inhibited by pre-emptive anti-inflammatory therapies that are initiated before the ischemic event (3, 4, 5). However, initiation of anti-inflammatory therapy within a few hours post-ischemia, fails to rescue AKI as the rapid inflammatory response, occurring within 6 to 24 hours, causes extensive tubular and endothelial cell damage.
There is a good possibility that inflammation has a role in ischemia induced AKI in humans as high levels of circulating DAMPS have been observed in humans under conditions that are associated with ischemia induced AKI such as trauma and sepsis and it is very likely that DAMPS also activate human NKT-1 cells (6). However, humans, unlike inbred mice, manifest heterogeneity in their inflammatory response resulting in large part from heterogeneity in their regulatory mechanisms which include different leucocyte subsets e.g Tregs, cytokines and natural auto-antibodies such as IgM anti-leucocyte autoantibodies (IgM-ALA). For example, we have clearly shown that there is a striking heterogeneity in levels of natural IgM-ALA in ESRD patients with a third of patients lacking IgM-ALA (7). This heterogeneity in regulatory mechanisms may explain why <25% (and not more) high risk elderly patients with CKD¾, develop AKI following coronary bypass surgery.
Natural IgM:
B cells have recently been subdivided into B1 and B2 cells. B1 cells spontaneously produce natural IgM (nIgM) auto-antibodies from birth (reviewed in ref # 7) and B1 cells do not differentiate into plasma cells or switch their IgM isotype to IgG or IgA. These nIgM autoantibodies comprise >90% of circulating IgM, bind with low affinity to autoantigens and do not cause autoimmune disease. nIgM autoantibodies increase rapidly (< 3 days) following any infectious or inflammatory process and nIgM levels remain high over several weeks. B2 cells are activated by foreign antigens e.g an infectious pathogen and by day 10 to 14, start producing immune IgM that is specific for the foreign antigen and unlike nIgM does not bind to host antigens. Within 3–4 weeks, B2 cells differentiate into plasma cells and switch to producing IgG and this explains the low level (<10%) of circulating immune IgM in normal individuals. B2 cells can also produce pathogenic IgG autoantibodies as for example in SLE (7).
Natural IgM autoantibodies, induced by an infection, are polyreactive and bind not only to conserved antigens on the infecting agent but also to several host autoantigens including (i) neo-antigens released by damaged cells (e.g DNA, cytoplasmic enzymes, actin/myosin) and (ii) specific receptors on all live leucocytes and endothelial cells. Several studies clearly demonstrated that such a rapid response by B1 cells provides the first line of defense against the infecting agent and also provides a homeostatic mechanism to prevent autoimmunity as nIgM masks exposed host neo-antigens and forms immune complexes that are rapidly phagocytosed (7). However, the functional role for the increase in nIgM that bind to receptors on live leucocytes and endothelial cells has, until recently, eluded investigators. Since these low affinity binding nIgM anti-leucocyte autoantibodies (IgM-ALA) are not cytolytic at body temperature (37°C), despite the presence of complement, we hypothesized that IgM-ALA, by binding to specific leucocyte receptors, regulates leucocyte function. Supporting such a hypothesis were observations we made in ESRD patients receiving kidney allografts where recipients with no or low levels of IgM-ALA, at time of transplant, had significantly more cell-mediated allograft rejections (i.e. an inflammatory response to allo-antigens) and graft loss when compared to recipients with high levels of IgM-ALA (7,8).
Anti-inflammatory role of IgM-ALA in ischemic AKI:
We initially demonstrated that IgM-ALA bind to (i) certain co-stimulatory receptors i.e. CD4, CD86, CD40 and PD1 and (ii) chemokine receptors. However, these polyreactive IgM-ALA autoantibodies manifest some form of specificity as they do not randomly bind to glycoproteins on other cell receptors i.e.CD8, CD80, CD40L, PDL1, CD28, CD1d and HLA receptors (5, 8, 9). We have not examined the binding of IgM-ALA to other leucocyte receptors but other investigators have shown that IgM-ALA also bind to TcR, CD3 and CD45 (10). Secondly, with in-vitro studies on murine and human leucocytes, we demonstrated that IgM-ALA, by binding to TcR, CD3, co-stimulatory receptors and chemokine receptors (i) inhibit glycolipid (i.e. α-gal-ceramide) induced activation and IFNγ production of NKT-1 cells (ii) inhibit effector T cell proliferation and production of pro-inflammatory cytokines in response to alloantigens (MLR) or anti-CD3/28 and (iii) inhibit leucocyte chemotaxis. IgM-ALA do not inhibit Treg function (5, 8).
We next determined if IgM-ALA could inhibit the in-vivo inflammatory response and prevent C57BL/6 mice (WT-B6) from developing ischemia induced AKI (5). Male mice received one intravenous dose (150 to 200 μg) of purified IgM 24 hours before the 32 minute cross-clamping of both renal pedicles and AKI was quantitated at 24 hours. IgM infusion increased baseline plasma levels of IgM to 400–500μg/ml, which is at the upper limit of normal (200 to 420μg/ml) for WT-B6 mice. Figure 1 clearly demonstrates that high physiologic levels of nIgM prevents ischemia induced AKI by inhibiting inflammation. Protection is mediated by IgM-ALA as removing IgM-ALA from nIgM with leucocyte adsorption abolishes the nIgM mediated protection. Importantly, AKI in this model is primarily mediated by the inflammatory response as in the IgM treated mice, with IgM-ALA mediated inhibition of the inflammatory response, one observes no significant increase in plasma creatinine over sham treated mice even though the IgM treated mice were subjected to 32 minutes of kidney ischemia.
Figure 1.
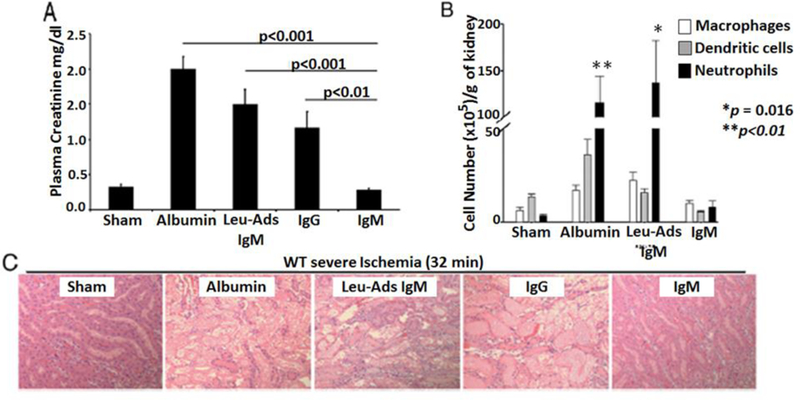
Pre-emptive nIgM infusion Prevents Ischemia-Induced Inflammation and AKI.
Role of nIgM induced pan-Bregs in inhibiting ischemia induced innate inflammation and AKI:
Since we observed 8 to 10 fold more binding of IgM-ALA to splenic pan-B cells (B1 and B2) when compared to T cells, we investigated whether IgM-ALA, by binding and cross-linking co-stimulatory molecules
(CD86 and CD40) on pan-B cells, regulated the APC function of these cells (11). Pan-B cells utilize their CD1d receptor to present glycolipids to NKT-1 cells. This hypothesis was tested by pre-treating isolated splenic pan-B cells with or without nIgM for 48 hours at 37°C and then intravenously infusing these ex-vivo pretreated B cells (0.5 × 106 B cells/mouse) 24 hours before inducing 26 minute bilateral renal ischemia. At 24 hours post-ischemia, mice infused with nIgM pretreated B cells were protected from AKI when compared to mice infused with non-pretreated B cells (plasma creatinine 0.4 vs 2.4, p<0.01). Infusing nIgM pretreated CD1d ko B cells or CD86 ko B cells failed to protect mice indicating that protection requires interaction of nIgM induced B regs with in-vivo NKT-1 cells. Infused Bregs act as APC and render in-vivo NKT-1 anergic and non-responsive to autologous APC presenting DAMPS 24 hours later (Fig 2) (11).
Figure 2.
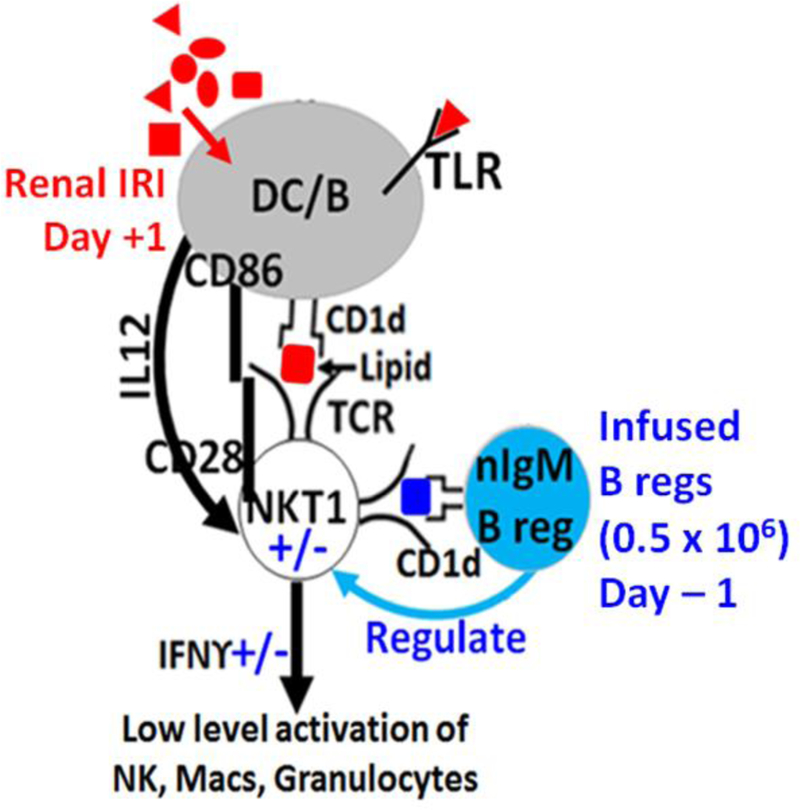
Infusion of B Cells pretreated Ex-vivo with nIgM regulate in vivo NKT1 and prevent renal IRI.
Conclusion and potential therapeutic use:
Infusing natural IgM or pan-Bregs, induced ex-vivo with nIgM, have an inhibitory effect on leucocyte function and chemotaxis and can prevent the ischemia induced inflammation that has a major role in contributing towards AKI in rodent models. Currently available immunoglobulin preparations e.g IVIG are devoid of IgM or IgA (<1%) and in our rodent AKI model, IgG, unlike IgM, was ineffective in inhibiting the ischemia induced inflammatory response or ameliorating AKI (5). Hence IgM isolated from pooled serum and ex-vivo induced pan-Bregs has translational potential in humans. One can use pooled IgM or ex-vivo induced pan-Bregs to (a) prevent AKI in high risk patients with CKD-3 undergoing elective coronary bypass surgery or (b) prevent ischemia induced delayed graft function (DGF) of deceased donor allografts when transplanted. Harnessing such a physiological mechanism to inhibit the ischemia induced inflammatory response would be particularly important in the subset of coronary bypass patients or transplant recipients with low levels of IgM-ALA or Tregs. The latter studies are relevant as we and others clearly demonstrated significantly higher allograft rejection rates in kidney and heart transplant recipients with low levels of IgM-ALA (7, 8 ). Additionally, both IgM and Bregs have a short half-life (< 1 week in mice) making excess immunosuppression less of a problem.
References
- 1.Zuk A, Bonventre JV. Acute Kidney Injury. Annu Rev Med 2016. January 14;67:293–307. doi: 10.1146/annurev-med-050214-013407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li L, Huang L, Sung SS, Lobo PI, Brown MG, Gregg RK, et al. NKT cell activation mediates neutrophil IFN-gamma production and renal ischemia-reperfusion injury. J Immunol 2007. May 1;178(9):5899–5911. [DOI] [PubMed] [Google Scholar]
- 3.Zuk A, Gershenovich M, Ivanova Y, MacFarland RT, Fricker SP, Ledbetter S. CXCR4 antagonism as a therapeutic approach to prevent acute kidney injury. Am J Physiol Renal Physiol 2014. October 1;307(7):F783–97. 10.1152/ajprenal.00685.2013 [DOI] [PubMed] [Google Scholar]
- 4.Gigliotti JC, Huang L, Ye H, Bajwa A, Chattrabhuti K, Lee S, Klibanov AL, Kalantari K, Rosin DL, and Okusa MD. Ultrasound Prevents Renal Ischemia-Reperfusion Injury by Stimulating the Splenic Cholinergic Anti-Inflammatory Pathway. J Am Soc Nephrol 2013;24(9):1451–1460; DOI: 10.1681/ASN.2013010084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lobo PI, Bajwa A, Schlegel KH, Vengal J, Lee SJ, Huang L, et al. Natural IgM anti-leukocyte autoantibodies attenuate excess inflammation mediated by innate and adaptive immune mechanisms involving Th-17. J Immunol 2012. February 15; 188(4):1675–1685. http://www.jimmunol.org/content/188/4/1675. DOI: 10.4049/JIMMUNOL.1101762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharma SK, Naidu G. The role of danger-associated molecular patterns (DAMPs) in trauma and infections. J Thorac Dis 2016. July; 8(7): 1406–1409. doi: 10.21037/jtd.2016.05.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lobo PI. Role of Natural Autoantibodies and Natural IgM Anti-Leucocyte Autoantibodies in Health and Disease. Front. Immunol, 06 June 2016. | 10.3389/fimmu.2016.00198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lobo PI, Schlegel KH, Spencer CE, Okusa MD, Chisholm C, McHedlishvili N, et al. Naturally occurring IgM anti-leukocyte autoantibodies (IgM-ALA) inhibit T cell activation and chemotaxis. J Immunol 2008. February 1;180(3):1780–1791. http://www.jimmunol.org/content/180/3/1780. [DOI] [PubMed] [Google Scholar]
- 9.Lobo PI, Schlegel KH, Bajwa A, Huang L, Kurmaeva E, Wang B, et al. Natural IgM Switches the Function of Lipopolysaccharide-Activated Murine Bone Marrow-Derived Dendritic Cells to a Regulatory Dendritic Cell That Suppresses Innate Inflammation. J Immunol 2015. October 30;195 (11):5215 http://www.jimmunol.org/content/195/11/5215. DOI: 10.4049/jimmunol.1500052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mimura T, Fernsten P, Jarjour W, Winfield JB. 1990. Autoantibodies specific for different isoforms of CD45 in systemic lupus erythematosus. J Exp Med 1990;172(2):653–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lobo PI, Schlegel KH, Bajwa A, Huang L, Okusa MD. Natural IgM and TLR Agonists Switch Murine Splenic Pan-B to “Regulatory” Cells That Suppress Ischemia-Induced Innate Inflammation via Regulating NKT-1 Cells. Front. Immunol, 2017. August 23;8:974 10.3389/fimmu.2017.00974. [DOI] [PMC free article] [PubMed] [Google Scholar]