

Abstract
A subset of patients with Ataxia-Telangiectasia (A-T) have dramatically reduced levels of IgG, IgA, and IgE with retained or elevated IgM levels. Several reports suggest that these A-T patients with a “hyper-IgM phenotype” (HIgM) suffer more clinical immunologic consequences than other A-T patients. The immunopathologic mechanism driving this phenomenon is unknown, making it difficult to predict response to immunomodulatory therapy. We describe an A-T patient with HIgM who underwent tumor necrosis factor (TNF) receptor blockade for cutaneous granuloma and after several months of successful therapy developed non-malignant lymphoproliferation, cytopenia, and increased serum immunoglobulin levels. This process was subsequently followed by an immune-complex-mediated intrarenal small vessel vasculitis that led to renal failure. The vasculitis was successfully treated with rituximab and corticosteroids. This case underscores the importance of HIgM as an unfavorable prognostic indicator in A-T and highlights the complexity of immunomodulatory treatment in this population, and the potential for a successful approach tailored to the immune defect.
Keywords: ataxia-telangiectasia, primary immunodeficiency, vasculitis, hyper IgM, granuloma, tumor necrosis factor, CD21lo
Introduction
The immunologic defects seen in Ataxia-Telangiectasia (A-T) are varied (1) and result in severe clinical morbidity in a subset of patients. One third of patients have no immunologic abnormality, whereas the remaining two-thirds have some combination of lymphopenia, immunoglobulin isotype or subclass deficiency, decreased naïve T cells, and abnormal vaccine responses (2). Despite laboratory evidence of immune abnormalities, most of these A-T patients suffer no more than repeated sinopulmonary infections without more serious infectious consequences (3). Approximately 10% of A-T patients exhibit a “hyper-IgM phenotype (HIgM),” with elevated IgM and severe deficiency of other immunoglobulin isotypes (4). Unlike the majority of A-T patients, HIgM A-T patients suffer potentially life threatening immune complications, such as autoimmunity and lymphoproliferative disease, and worse long term outcomes (5, 6).
Another minority of A-T patients develop severe cutaneous granulomas (7, 8). These lesions most frequently present as nodules on the face and extremities that can progress to large, ulcerating plaques (9). Their etiology is not well-defined, but reports have identified reduced naïve CD8 T cell and IgA levels within the blood of patients (8) and increased ratios of CD8 to CD4 T cells within the granulomas (10). More recently, many of these granulomas have been found to contain vaccine-strain rubella antigen (11). Treatment generally consists of corticosteroids, tumor necrosis factor (TNF) inhibition, or nitazoxanide with responses reported in the literature variable at best (8, 9, 12).
We report the clinical course of an A-T patient with HIgM in whom the TNF inhibitor, adalimumab, was used to treat chronic cutaneous granulomas. She experienced gradual improvement in the size and appearance of the granulomas; however, several months into her therapy, she developed severe immunologic dysregulation ultimately leading to immune-complex vasculitis and renal failure. Multiple immunomodulatory insults including anti-TNF therapy, chronic granuloma, and acute infection with norovirus may have contributed to the immune dysregulation seen in this patient. Nonetheless, the vasculitis was suppressed by a targeted approach, and the patient remains in remission.
Case Report
We report the extended follow up of a 6-year-old girl with HIgM A-T and a history of rubella-positive cutaneous granuloma whose initial evaluation was reported previously (13). Her initial immunological findings included elevated serum IgM levels with absent IgA and IgG, increased percentage of CD19+CD38loCD27−CD10−CD21−/low B cells, increased percentage of circulating T follicular helper cells (Tfh cells), and decreased percentage and function of regulatory T cells. Due to progression of her cutaneous granulomas while on topical and systemic corticosteroid therapy, she started adalimumab10 mg subcutaneously every 2 weeks. She experienced partial remission of the lesions, but 1 year into her treatment, she developed diffuse lymphadenopathy, splenomegaly, and cytopenia, and adalimumab was discontinued. An axillary lymph node biopsy showed predominant paracortical hyperplasia, sparse residual germinal centers with follicular dendritic cell networks, collections of Tfh cells, and abundant polytypic, plasmablasts (Figures 1A,B). There were no granulomas, and EBV was not detected. IgM and IgD were detected diffusely (Figures 1C,D), IgG staining was sparse, and IgA staining was absent. Serum IgM level at the time of biopsy had increased to 4,000 mg/dl, and her blood CD20+ B cells experienced a global decrease in surface IgD expression (Figures 2A,B). Oral corticosteroid treatment was initiated, and the lymphadenopathy and splenomegaly demonstrated mild improvement.
Figure 1.

Axillary lymph node biopsy from an HIgM A-T patient. (A) Lymph node biopsy with paracortical expansion (A, 20X) consisting of a mixture of small lymphocytes, plasma cells, and immunoblasts (B, 200X). Immunohistochemical staining for IgD (C) and IgM (D) demonstrate cytoplasmic staining of plasma cells and lymphocytes.
Figure 2.

Baseline B cell abnormalities worsened following TNF blockade. Two-dimensional flow cytometry plot of the patient's CD20+ B cells 1 year before (A) and 1 year after (B) initiating adalimumab therapy. The patient's plots are on top and underneath each plot is the same day normal control sample.
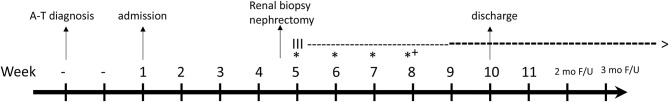
Two months after the onset of lymphoproliferation, the patient was admitted with fever and diarrhea and subsequently developed acute kidney failure. Infectious workup identified norovirus in the stool but was otherwise negative. Nitazoxanide was initiated with little impact on the fevers, progressive renal failure, or the cutaneous granulomas. The diarrhea gradually resolved. She became oliguric at week 4 of admission with a serum creatinine peak of 2.2 mg/dL (Table 1). Renal biopsy was complicated by uncontrollable bleeding necessitating nephrectomy. Histologic staining of the kidney demonstrated acute and chronic necrotic, inflamed arteries with thrombosis and various amounts of intimal thickening (Figures 3A,B,G,H), and strong IgG, IgM, C3, and C1q staining (Figures 3C–F). Serum C3 and C4 levels were decreased (Table 1). Serum immunoglobulin levels were remarkable for an IgM level of 326 mg/dl and normal levels of IgA (63 mg/dl), a change from prior examinations when her IgM was over 4,000 mg/dl and her IgA was below the limits of detection. Four weekly doses of rituximab were administered in conjunction with pulse and maintenance methylprednisone. Over the next 2 weeks, complement levels normalized, IgA levels dropped below the assay limit of detection, and renal function returned to normal. Just prior to hospital discharge, she was transitioned to oral prednisolone, started on mycophenolate, and continued monthly IVIG. One year later, the vasculitis remains in remission, but the cutaneous granulomas have worsened and are now detected in her bone marrow.
Table 1.
Results of immunologic studies and serum creatinine levels.
 | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Normal Ranges | ||||||||||||||||
| IgM (mg/dL) | 719 | >4,000 | 326 | 445 | 228 | 220 | 218 | 44–266 mg/dL | ||||||||
| IgA (mg/dL) | <10 | <40 | 63 | <10 | <10 | 43–253 mg/dL | ||||||||||
| C3 (mg/dL) | 49 | 77 | 93 | 110 | 154 | 170 | 166 | 138 | 70–206 mg/dL | |||||||
| C4 (mg/dL) | <8 | 21.2 | 30.6 | 31.9 | 37.7 | 33.8 | 11–61 mg/dL | |||||||||
| Cr (mg/dL) | 0.5 | 1.2 | 1.7 | 1.2 | 2.2 | 0.6 | 0.5 | 3.0 | 0.9 | 0.3–1 mg/dL | ||||||
| ESR | 99 | 28 | 0–20 mm/h | |||||||||||||
| PLT | 261 | 86 | 70 | 103 | 150–500 K/uL | |||||||||||
(*) rituximab 270 mg, (+) mycophenolate mofetil 300 mg twice daily, (I) high-dose methylprednisone, ( ) IV methylprednisone, (
) IV methylprednisone, ( ) oral prednisone.
) oral prednisone.
Figure 3.
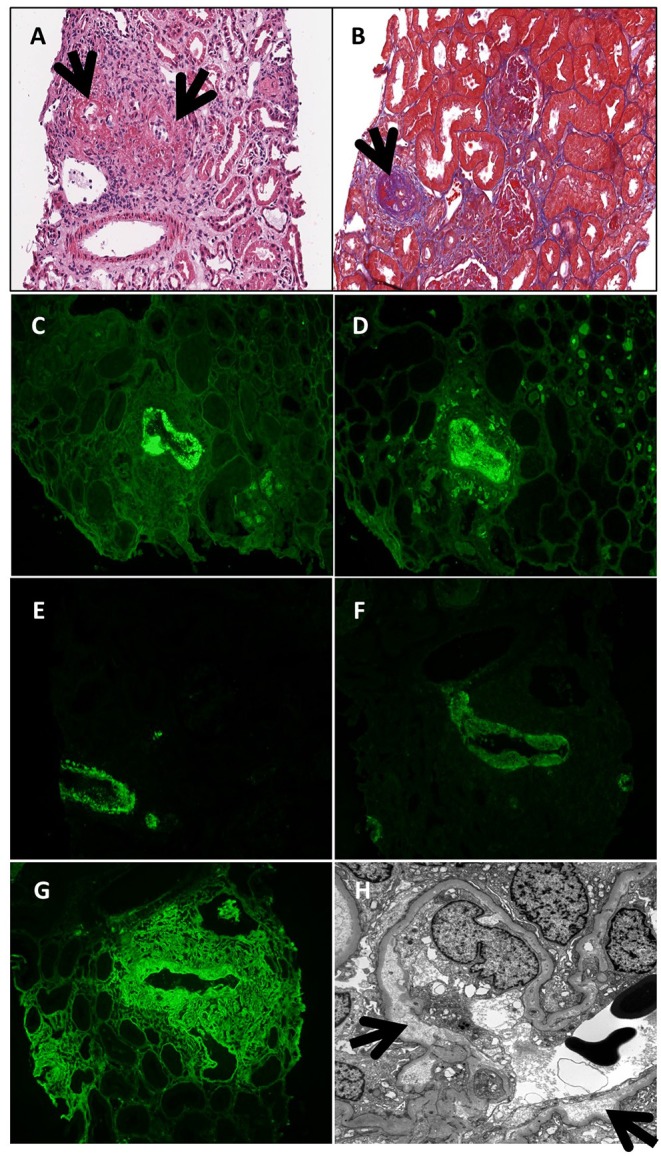
Renal biopsy at age 6 years. Necrotic and inflamed arteries (black arrows; A—hematoxylin eosin stain; B—trichrome stain). IgG (C), IgM (D), C3 (E), C1q (F), and fibrinogen (G) deposition seen in a small necrotic artery by immunofluorescent staining. (H) Electron micrograph demonstrating subendothelial widening along a glomerular capillary loop (black arrows).
Discussion
The HIgM phenotype of A-T is associated with more severe immunologic disease, including autoimmunity and lymphoproliferative disease, and worse outcomes than the remainder of A-T patients (4, 5, 14–21). As illustrated in the presented case, the poorer clinical outcomes are often preceded by immune dysregulation and progress to autoimmune or inflammatory organ damage that can be fatal. While the reason for such severe clinical deterioration is poorly understood, it appears from this case that multiple factors contribute to clinical worsening. At baseline, the patient had lymphopenia and abnormal class switching. Superimposed on that background, she had vaccine-strain-rubella-containing cutaneous granulomas, a source of chronic immunologic stimulation. Her immune system was then subject to TNF blockade, a state that could have influenced both her baseline immunologic abnormalities and her response to chronic rubella antigen stimulation. Finally, she was infected with norovirus, an additional external stimulus too her immune system. It is likely that combination of these events set the stage for the immune-mediated vasculitis.
As noted previously (13), the patient had elevated numbers of Tfh, reduced Tregs, and an abundance of CD21lo B cells—all of which showed evidence of chronic IFNγ stimulation. This immunophenotypic pattern is frequently seen in rheumatic disease and complicated common variable immune deficiency (CVID) (22, 23); however, this pattern has not yet been studied in a large group of HIgM A-T patients. As a result, it is unknown whether this subgroup of A-T is inherently predisposed to this pattern of immune dysregulation, or if additional immunologic perturbations are required. Beyond the HIgM A-T phenotype, the discussed patient had a source of chronic antigen stimulation, rubella-associated cutaneous granulomatous disease. Tfh have been identified at the site of granuloma formation within mycobacteria-infected lung tissue (24), suggesting their role in controlling chronic intracellular infection. It is therefore worth asking if the expanded Tfh pattern as seen in this patient resulted from cutaneous granulomatous disease, HIgM phenotype, or the combination of the two? Additional research will be required to answer this question.
We also question whether the clinical developments were related to the institution of the anti-TNFα therapy used to control her progressive granulomatous disease? There are several reports of similar outcomes in patients treated with an anti-TNFα including a case of a patient with psoriasis who developed IgA glomerulonephritis after 18 months of TNF blockade with adalimumab (25, 26). Additionally, a small retrospective study of 8 patients reported development of cutaneous and systemic small-vessel vasculitis associated with the use of TNFα inhibitors (27). Unlike the previously reported anti-TNF-associated vasculitis patients who experienced a remission of vasculitis following anti-TNF discontinuation, this patient was off adalimumab for 2 months before the vasculitis developed. If TNF blockade played such a role in this patient, it would imply an irreversible change in the immune system, an adverse outcome that has not previously been described.
The mechanism that could explain such a relationship between TNF blockade and vasculitis is unclear. TNF-α is produced by activated B cells and supports their expansion in an autocrine manner (28), and there is evidence that T-cell-independent immunoglobulin production may be supported by TNF blockade (29–31). In addition, TNF inhibitors augment B-cell expression of TACI and TLR9, receptors capable of transmitting potent activation signals (32). Given these findings, it is conceivable that abnormal and possibly autoreactive B cell populations were somehow rescued by TNF inhibition in a T-cell-independent manner, particularly since the predominant B cell population was CD19+CD38loCD27−CD10−CD21−/low, a subset typically enriched for autoreactive B cell receptors (33). However, in the context of autoimmune disease, rather than augmenting germinal center activity, blockade of TNF disrupts germinal center reactions and impairs T-cell-dependent vaccine responses (29, 34). The patient had a dramatic increase in Tfh cells, suggesting that T-cell help was an important component of her progressive autoimmunity. How TNF inhibition influences Tfh inside granulomas to our knowledge has not been explored. It seems unlikely that TNF blockade indirectly augments Tfh activity by generating more antigen to which T cells can react. In this case, adalimumab did not result in rubella dissemination, and in fact, the clinical appearance of the skin granulomas improved while on adalimumab. Therefore, while Tfh are implicated in granuloma formation and control of chronic infection, we do not conclude that TNF inhibition indirectly augmented Tfh activity by increasing availability of rubella antigens. It is also unlikely that TNF blockade directly affected the Tfh compartment, as it does not appear to be important for Tfh development (35). TNF inhibition is therefore difficult to implicate as the sole cause of Tfh expansion or Tfh-mediated autoimmunity in this case.
The immune dysregulation seen in the HIgM subset of A-T frequently leads to fatal outcomes. In this case, immune-mediated vasculitis was successfully treated with a combination of B-cell and T-cell targeted therapy. Rituximab was chosen because of the implication of autoantibodies in the disease process. The resulting elimination of an almost completely phenotypically abnormal B-cell compartment provided reassurance that ongoing autoantibody production was halted; however, the persistence of an expanded Tfh compartment required additional immunosuppression. Maintenance treatment with mycophenolate was prescribed at the time of discharge. She has not experienced relapse in the year following her discharge.
This case highlights the complexity of immune dysregulation and difficulty in utilizing immunomodulatory therapy in the HIgM subset of A-T. The complexity may be enhanced in the presence of chronic rubella-containing chronic granulomas in the face of dysregulation of both B and T cell compartments. The impact of immunomodulatory therapy and infection on these dysregulated immune cells can be difficult to predict, and patients should be monitored closely. Treatment of severe immune complications should be tailored to the clinical circumstance, where detailed immune system characterization can support designing specific treatment regimens.
Concluding Remarks
This case highlights the importance of hyper IgM as an unfavorable prognostic indicator in A-T, explores the factors that preceded almost fatal severe immune disease, and describes the treatment approach that induced clinical remission.
Data Availability Statement
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Ethics Statement
The subject of this case report was enrolled in a research study approved by the National Jewish Health Institutional Review Board. Written informed consent was obtained from the parents for the publication of any potentially identifiable images or data included in this article.
Author Contributions
AM and JA co-wrote the manuscript. TN and RZ contributed pathological slides and edited the manuscript. JC, MB, and EG cared for the patient, discussed the manuscript, and edited the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. Funding for publication was provided by National Jewish Health, Department of Pediatrics.
References
- 1.Jason JM, Gelfand EW. Diagnostic considerations in ataxia-telangiectasia. Arch Dis Child. (1979) 54:682–86. 10.1136/adc.54.9.682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nowak-Wegrzyn A, Crawford TO, Winkelstein JA, Carson KA, Lederman HM. Immunodeficiency and infections in ataxia-telangiectasia. J Pediatr. (2004) 144:505–11. 10.1016/j.jpeds.2003.12.046 [DOI] [PubMed] [Google Scholar]
- 3.Micol R, Ben Slama L, Suarez F, Le Mignot L, Beauté J, Mahlaoui N, et al. Morbidity and mortality from ataxia-telangiectasia are associated with ATM genotype. J Allergy Clin Immunol. (2011) 128:382–9.e1. 10.1016/j.jaci.2011.03.052 [DOI] [PubMed] [Google Scholar]
- 4.Ghiasy S, Parvaneh L, Azizi G, Sadri G, Zaki Dizaji M, Abolhassani H, et al. The clinical significance of complete class switching defect in Ataxia telangiectasia patients. Expert Rev Clin Immunol. (2017) 13:499–505. 10.1080/1744666X.2017.1292131 [DOI] [PubMed] [Google Scholar]
- 5.Noordzij JG, Wulffraat NM, Haraldsson A, Meyts I, van't Veer LJ, Hogervorst FB, et al. Ataxia-telangiectasia patients presenting with hyper-IgM syndrome. Arch Dis Child. (2009) 94:448–9. 10.1136/adc.2008.149351 [DOI] [PubMed] [Google Scholar]
- 6.van Os NJH, Jansen AFM, van Deuren M, Haraldsson A, van Driel NTM, Etzioni A, et al. Ataxia-telangiectasia: immunodeficiency and survival. Clin Immunol. (2017) 178:45–55. 10.1016/j.clim.2017.01.009 [DOI] [PubMed] [Google Scholar]
- 7.Rothblum-Oviatt C, Wright J, Lefton-Greif MA, McGrath-Morrow SA, Crawford TO, Lederman HM. Ataxia telangiectasia: a review. Orphanet J Rare Dis. (2016) 11:159. 10.1186/s13023-016-0543-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woelke S, Valesky E, Bakhtiar S, Pommerening H, Pfeffermann LM, Schubert R, et al. Treatment of granulomas in patients with Ataxia telangiectasia. Front Immunol. (2018) 9:2000. 10.3389/fimmu.2018.02000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leclerc-Mercier S, Moshous D, Neven B, Mahlaoui N, Martin L, Pellier I, et al. Cutaneous granulomas with primary immunodeficiency in children: a report of 17 new patients and a review of the literature. J Eur Acad Dermatol Venereol. (2019) 33:1412–20 10.1111/jdv.15568 [DOI] [PubMed] [Google Scholar]
- 10.Harp J, Coggshall K, Ruben BS, Ramírez-Valle F, He SY, Berger TG. Cutaneous granulomas in the setting of primary immunodeficiency: a report of four cases and review of the literature. Int J Dermatol. (2015) 54:617–25. 10.1111/ijd.12765 [DOI] [PubMed] [Google Scholar]
- 11.Bodemer C, Sauvage V, Mahlaoui N, Cheval J, Couderc T, Leclerc-Mercier S, et al. Live rubella virus vaccine long-term persistence as an antigenic trigger of cutaneous granulomas in patients with primary immunodeficiency. Clin Microbiol Infect. (2014) 20:O656–63. 10.1111/1469-0691.12573 [DOI] [PubMed] [Google Scholar]
- 12.Perelygina L, Buchbinder D, Dorsey MJ, Eloit M, Hauck F, Hautala T, et al. Outcomes for nitazoxanide treatment in a case series of patients with primary immunodeficiencies and rubella virus-associated granuloma. J Clin Immunol. (2019) 39:112–7. 10.1007/s10875-019-0589-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Minto H, Mensah KA, Reynolds PR, Meffre E, Rubtsova K, Gelfand EW. A novel ATM mutation associated with elevated atypical lymphocyte populations, hyper-IgM, and cutaneous granulomas. Clin Immunol. (2019) 200:55–63. 10.1016/j.clim.2019.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rawat A, Imai K, Suri D, Gupta A, Bhisikar S, Saikia B, et al. Ataxia telangiectasia masquerading as hyper IgM syndrome. Indian J Pediatr. (2016) 83:270–1. 10.1007/s12098-015-1852-x [DOI] [PubMed] [Google Scholar]
- 15.Ghosh S, Schuster FR, Binder V, Niehues T, Baldus SE, Seiffert P, et al. Fatal outcome despite full lympho-hematopoietic reconstitution after allogeneic stem cell transplantation in atypical ataxia telangiectasia. J Clin Immunol. (2012) 32:438–40. 10.1007/s10875-012-9654-7 [DOI] [PubMed] [Google Scholar]
- 16.Pietrucha BM, Heropolitanska-Pliszka E, Wakulinska A, Skopczynska H, Gatti RA, Bernatowska E. Ataxia-telangiectasia with hyper-IgM and Wilms tumor: fatal reaction to irradiation. J Pediatr Hematol Oncol. (2010) 32:e28–30. 10.1097/MPH.0b013e3181bfd3d9 [DOI] [PubMed] [Google Scholar]
- 17.Aghamohammadi A, Imai K, Moazzami K, Abolhassani H, Tabatabaeiyan M, Parvaneh N, et al. Ataxia-telangiectasia in a patient presenting with hyper-immunoglobulin M syndrome. J Investig Allergol Clin Immunol. (2010) 20:4. [PubMed] [Google Scholar]
- 18.Soresina A, Meini A, Lougaris V, Cattaneo G, Pellegrino S, Piane M, et al. Different clinical and immunological presentation of ataxia-telangiectasia within the same family. Neuropediatrics. (2008) 39:43–45. 10.1055/s-2008-1076736 [DOI] [PubMed] [Google Scholar]
- 19.Etzioni A, Ben-Barak A, Peron S, Durandy A. Ataxia-telangiectasia in twins presenting as autosomal recessive hyper-immunoglobulin M syndrome. Isr Med Assoc J. (2007) 9:406–7. [PubMed] [Google Scholar]
- 20.Meyts I, Weemaes C, De Wolf-Peeters C, Proesmans M, Renard M, Uyttebroeck A, et al. Unusual and severe disease course in a child with Ataxia-telangiectasia. Pediatr Allergy Immunol. (2003) 14:330–3. 10.1034/j.1399-3038.2003.00037.x [DOI] [PubMed] [Google Scholar]
- 21.Tangsinmankong N, Wayne AS, Howenstine MS, Washington KR, Langston C, Gatti RA, et al. Lymphocytic interstitial pneumonitis, elevated IgM concentration, and hepatosplenomegaly in ataxia-telangiectasia. J Pediatr. (2001) 138:939–41. 10.1067/mpd.2001.113356 [DOI] [PubMed] [Google Scholar]
- 22.Deng J, Wei Y, Fonseca VR, Graca L, Yu D. T follicular helper cells and T follicular regulatory cells in rheumatic diseases. Nat Rev Rheumatol. (2019) 15:475–90. 10.1038/s41584-019-0254-2 [DOI] [PubMed] [Google Scholar]
- 23.Unger S, Seidl M, van Schouwenburg P, Rakhmanov M, Bulashevska A, Frede N, et al. The TH1 phenotype of follicular helper T cells indicates an IFN-γ-associated immune dysregulation in patients with CD21low common variable immunodeficiency. J Allergy Clin Immunol. (2018) 141:730–40. 10.1016/j.jaci.2017.04.041 [DOI] [PubMed] [Google Scholar]
- 24.Slight SR, Rangel-Moreno J, Gopal R, Lin Y, Fallert Junecko BA, Mehra S, et al. CXCR5+ T helper cells mediate protective immunity against tuberculosis. J Clin Invest. (2013) 123:712–26. 10.1172/JCI65728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wei SS, Sinniah R. Adalimumab (TNF α Inhibitor) therapy exacerbates IgA glomerulonephritis acute renal injury and induces lupus autoantibodies in a psoriasis patient. Case Rep Nephrol. (2013) 2013:812781. 10.1155/2013/812781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Piga M, Chessa E, Ibba V, Mura V, Floris A, Cauli A, et al. Biologics-induced autoimmune renal disorders in chronic inflammatory rheumatic diseases: systematic literature review and analysis of a monocentric cohort. Autoimmun Rev. (2014) 13:873–9. 10.1016/j.autrev.2014.05.005 [DOI] [PubMed] [Google Scholar]
- 27.Sokumbi O, Wetter DA, Makol A, Warrington KJ. Vasculitis associated with tumor necrosis factor-α inhibitors. Mayo Clin Proc. (2012) 87:739–45. 10.1016/j.mayocp.2012.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boussiotis VA, Nadler LM, Strominger JL, Goldfeld AE. Tumor necrosis factor alpha is an autocrine growth factor for normal human B cells. Proc Natl Acad Sci USA. (1994) 91:7007–11. 10.1073/pnas.91.15.7007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salinas GF, De Rycke L, Barendregt B, Paramarta JE, Hreggvidsdottir H, Cantaert T, et al. Anti-TNF treatment blocks the induction of T cell-dependent humoral responses. Ann Rheum Dis. (2013) 72:1037–43. 10.1136/annrheumdis-2011-201270 [DOI] [PubMed] [Google Scholar]
- 30.Di Sabatino A, Rosado MM, Cazzola P, Biancheri P, Tinozzi FP, Laera MR, et al. Splenic function and IgM-memory B cells in Crohn's disease patients treated with infliximab. Inflamm Bowel Dis. (2008) 14:591–6. 10.1002/ibd.20374 [DOI] [PubMed] [Google Scholar]
- 31.Souto-Carneiro MM, Mahadevan V, Takada K, Fritsch-Stork R, Nanki T, Brown M, et al. Alterations in peripheral blood memory B cells in patients with active rheumatoid arthritis are dependent on the action of tumour necrosis factor. Arthritis Res Ther. (2009) 11:R84. 10.1186/ar2718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moura RA, Quaresma C, Vieira AR, Gonçalves MJ, Polido-Pereira J, Romão VC, et al. B-cell phenotype and IgD-CD27- memory B cells are affected by TNF-inhibitors and tocilizumab treatment in rheumatoid arthritis. PLoS ONE. (2017) 12:e0182927. 10.1371/journal.pone.0182927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Isnardi I, Ng YS, Menard L, Meyers G, Saadoun D, Srdanovic I, et al. Complement receptor 2/CD21- human naive B cells contain mostly autoreactive unresponsive clones. Blood. (2010) 115:5026–36. 10.1182/blood-2009-09-243071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anolik JH, Ravikumar R, Barnard J, Owen T, Almudevar A, Milner EC, et al. Cutting edge: anti-tumor necrosis factor therapy in rheumatoid arthritis inhibits memory B lymphocytes via effects on lymphoid germinal centers and follicular dendritic cell networks. J Immunol. (2008) 180:688–92. 10.4049/jimmunol.180.2.688 [DOI] [PubMed] [Google Scholar]
- 35.Crotty S. T follicular helper cell biology: a decade of discovery and diseases. Immunity. (2019) 50:1132–48. 10.1016/j.immuni.2019.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.