

Abstract
The role of interferon and interferon stimulated genes (ISG) in limiting bacterial infection is controversial, and the role of individual ISGs in the control of the bacterial life-cycle is limited. Viperin, is a broad acting anti-viral ISGs, which restricts multiple viral pathogens with diverse mechanisms. Viperin is upregulated early in some bacterial infections, and using the intracellular bacterial pathogen, S. flexneri, we have shown for the first time that viperin inhibits the intracellular bacterial life cycle. S. flexneri replication in cultured cells induced a predominantly type I interferon response, with an early increase in viperin expression. Ectopic expression of viperin limited S. flexneri cellular numbers by as much as 80% at 5hrs post invasion, with similar results also obtained for the intracellular pathogen, Listeria monocytogenes. Analysis of viperins functional domains required for anti-bacterial activity revealed the importance of both viperin’s N-terminal, and its radical SAM enzymatic function. Live imaging of S. flexneri revealed impeded entry into viperin expressing cells, which corresponded to a loss of cellular cholesterol. This data further defines viperin’s multi-functional role, to include the ability to limit intracellular bacteria; and highlights the role of ISGs and the type I IFN response in the control of bacterial pathogens.
Subject terms: Antimicrobial responses, Innate immunity, Microbiology, Bacteriology
Introduction
The role of type II interferon (IFN-γ) in the control of bacterial infection is well established, however the role of both type I and III IFN, and their related interferon stimulated gene products, in a protective host anti-bacterial response remains controversial1,2. There are three distinct interferon families, each of which has varying roles in the restriction of viral and bacterial pathogens; Type I IFN which is produced by most cells in the body, type II IFN produced mainly by T cells and natural killer (NK) cells, and lastly type III IFN, which has restricted activity, due to the fact that its receptor is limited mainly to epithelial cells3,4. Both type I and III IFN’s have been shown to have a prominent role in the restriction of viral pathogens, inducing the upregulation of hundreds of interferon stimulated genes (ISGs), that create an anti-viral state in the infected cell as well as neighbouring uninfected cells; however, type I IFN (IFNα/β) expression has often been demonstrated to favour bacterial pathogenesis2, and very little is known about the role of type III IFN in control of an anti-bacterial response (reviewed in1). There has however been a small number of pathogenic bacteria, including S. pneumoniae, E. coli, H. pylori and S. pyogenes, for which type I IFN expression has been demonstrated to play a protective role in mouse models of infection5–8. Additionally, type I IFNs are also able to restrict the entry of the intracellular bacterial pathogens Shigella flexneri and Salmonella enterica, but the mechanisms involved remain unknown9,10.
Interferon stimulated genes (ISGs) are host effector molecules whose expression is induced via activation of the JAK-STAT pathway following expression of IFN. While the role of many ISGs are uncharacterised, they have been best described in reference to their ability to limit viral infection11. It has previously been demonstrated that bacterial pathogens are also able to induce the expression of multiple type I IFN induced genes12, and more recent work has shown that a small handful of these ISGs can have direct anti-bacterial effects. The anti-viral IFITM proteins (Interferon induced transmembrane proteins) have been shown to restrict Mycobacterium tuberculosis intracellular growth via endosomal maturation pathways, and the interferon inducible GTPases, IRG (immunity related GTPase) and GBP’s (guanylate binding proteins) can specifically target certain intracellular bacteria, destroying their vacuolar compartment required for replication13,14. Additionally, the chemokine CXCL10, also an ISG, has recently been demonstrated to induce bacterial killing of Bacillus anthracis via membrane disruption, and more recent work involving a cell-based screen of multiple ISGs against Listeria monocytogenes, demonstrated that Trim14 was able to restrict intracellular growth of L. monocytogenes via a transcriptionally independent manner, however the exact mechanisms are yet to be uncovered15,16.
Viperin is one of the broadest acting anti-viral ISGs and has been described to restrict the life cycles of multiple viral families (reviewed in17,18), as well as augment both the TLR7/TLR9 and dsDNA response to viral infection18,19. Viperin has previously been shown to also be upregulated early during bacterial infection19–22, however its role in the restriction of bacterial pathogens remains unknown. Viperin is a 42 kDa, interferon inducible protein that is highly evolutionarily conserved17,23, and is induced early in viral infection through both interferon dependent and independent mechanisms24–29. It is localised to both the endoplasmic reticulum, as well as the lipid droplet via its N-terminal amphipathic helix30–32, and is a member of the radical SAM enzyme family33. Viperin is known to directly restrict viral replication of the Flaviviridae family members, HCV, Dengue, Zika, West Nile virus and TBEV30,34–39 through interactions with both host and viral proteins, and has also been shown to restrict the egress of HIV, influenza and RSV40–42. Viperin’s capacity to inhibit the life cycles of multiple viruses with distinct mechanisms, and its induction upon bacterial infection, poses the question of whether it is able to also restrict the life cycle of intracellular bacteria. Using S. flexneri as a model of intracellular bacterial infection, we show that S. flexneri is able to induce expression of viperin. Furthermore, loss of viperin was shown to enhance intracellular bacterial levels, and its ectopic expression was demonstrated to restrict bacterial entry in vitro, which coincided with reduced cellular cholesterol; demonstrating a further protective role for type I IFN inducible genes in control of intracellular bacteria.
Results
S. flexneri infection of cells in vitro predominantly induces a type I IFN response
Previous reports have demonstrated that both type I and type II interferon can restrict S. flexneri growth in vitro and in vivo10,43, but there is limited work assessing the ability of these intracellular bacteria to induce interferon expression following cellular invasion. To confirm the ability of interferon to limit infection of S. flexneri into HeLa cells, we initially pre-treated cells with type I and II interferon for 24 hours prior to invasion assays. As can be seen in Fig. 1A, S. flexneri infection was significantly inhibited by up to 50% with IFN-β treatment (Type I IFN), and to a lesser extent with IFN-γ (Type II IFN) pre-treatment (24%).
Figure 1.

S. flexneri induces both a type I and type III IFN response upon invasion of epithelial cells. (A) HeLa cells were pre-treated with IFN 24 hours prior to bacterial invasion, and CFU counts performed at 5 hours post infection; p < 0.0001. (B) HeLa and 293T cells were transfected with an IFN-β luciferase promoter construct and associated controls 24 hrs prior to S. flexneri invasion; cells were harvested for luciferase quantitation 5 hours following invasion. S. flexneri invasion of Huh-7 and HeLa cells was performed, and either RNA extracted at both 3 and 5 hours following infection to quantitate (C) mRNA from IFN and selected ISGs or (D) the constitutively expressed ipaH gene from S. flexneri via real-time PCR. All graphs represent the mean and standard deviation of three independent experiments performed in triplicate, *p < 0.0001.
We next wanted to assess whether S. flexneri infection itself was able to induce activation of IRF3, to stimulate the IFN- β promoter in vitro. 5 hours post S. flexneri invasion of both HeLa and 293 T cell lines, a significant activation of the IFN-β promoter was observed (Fig. 1B); that was supported by increased expression of both interferon- β and -λ at the mRNA level (Fig. 1C). The regulation of IFN and the interferon stimulated genes (ISGs) IFIT1 and viperin, was assessed in the epithelial cell lines, HeLa and Huh-7 cells, with differences observed in gene induction between these two cell lines (Fig. 1C), despite there being no significant difference in the levels of bacteria present in each at both time points (Fig. 1D). Both Huh-7 and HeLa cells predominantly produced type I interferon in response to S. flexneri infection, with peak expression of both type I and III IFN seen at 5 hours post invasion in Huh-7 cells. Interestingly this was not mirrored in the HeLa cells, which showed a decrease in type I IFN between 3 and 5 hours post invasion, and an overall lack of type III induction. Huh-7 cells also demonstrated a greater increase in the ISG IFIT1, than HeLa cells, likely due to the enhanced IFN production overall in this cell line. Viperin expression was also assessed in the Huh-7 cells (HeLa cells do not regulate viperin41), and was shown to be markedly increased as early as 3 hours post bacterial invasion, with its earlier induction than IFIT1 potentially due to its ability to be directly regulated independently of IFN, via a direct IRF3 mediated mechanism26.
Viperin inhibits both S. flexneri and L. monocytogenes cellular infection in vitro
Viperin and other ISGs are known to be upregulated early in response to bacterial infection22, and as previously discussed we have shown that Huh-7 cells upregulate viperin as early as 3 hours post S. flexneri invasion (Fig. 1C). In order to assess the ability of viperin to limit bacterial infection we first expressed viperin transiently in both 293T and HeLa cells, 24 hours prior to invasion with S. flexneri. Viperin expression significantly decreased the levels of intracellular bacteria at both 3 hours and 5 hours post invasion by 82% and 87% respectively in 293T cells, and 57% and 33% respectively in HeLa cells (Fig. 2A); this was accompanied by a significant drop in intracellular colony forming units at both time points in 293T cells (80% and 67% respectively)(Fig. 2B). The differences in viperin’s ability to limit S. flexneri infection in the 2 different cell types is most likely due to the transfection efficiency and corresponding viperin expression, and as such 293T cells were used for further experimental analysis. Interestingly, the ability of viperin to inhibit bacterial infection was not limited to only S. flexneri, with inhibition rates of 84% and 85% also observed at 3 and 5 hours respectively post invasion of 293T cells with L. monocytogenes (Fig. 2C); indicating that viperin’s ability to limit the intracellular bacterial levels of S. flexneri may be a more general inhibition of intracellular bacteria, rather than a specific inhibition.
Figure 2.

Viperin expression inhibits intracellular bacteria infection of cells in vitro. (A) Viperin was transiently expressed 24 hours prior to invasion with S.flexneri in 293T and HeLa cells, and RNA harvested at the indicated times; graphs represent the mean and standard deviation of three independent experiments performed in triplicate. 293T cells were transiently transfected with viperin 24 hours prior to invasion with either (B) S. flexneri, or (C) L. monocytogenes, and CFU counts performed at the indicated times. Graphs represent the mean and standard deviation of three independent experiments performed in triplicate for S. flexneri and in quadruplicate for L. monocytogenes, *p < 0.0001.
To assess the impact of loss of viperin on S. flexneri infection in vitro, we utilised Huh-7 cells with impaired viperin expression as we have discussed previously30. Initial infection of the Huh-7 cells demonstrated that the control cells (shControl) were able to induce viperin mRNA expression at 5 hours post S. flexneri invasion, while no viperin expression was detected at any time point in the Huh-7 shViperin cell line (Fig. 3A). This induction of viperin coincided with a decreased ability of S. flexneri to infect shControl Huh-7 cells at 5 hours post invasion by approximately 45% (Fig. 3B), demonstrating that viperin expression impacts S. flexneri infection in vitro. To further confirm the role of viperin in the control of S. flexneri, we next assessed the ability of the bacterium to infect viperin knock-out murine embryonic fibroblasts (MEF’s) sourced from our previously described CRISPR/cas derived Vip -/- del32 strain in comparison to wild-type fibroblasts38. As can be seen in Fig. 3C, MEFs absent in viperin expression had a 3.4 fold higher rate of S. flexneri infection than wild-type MEFs, confirming the impact that viperin has on the outcome of successful S. flexneri infection of cells in vitro.
Figure 3.
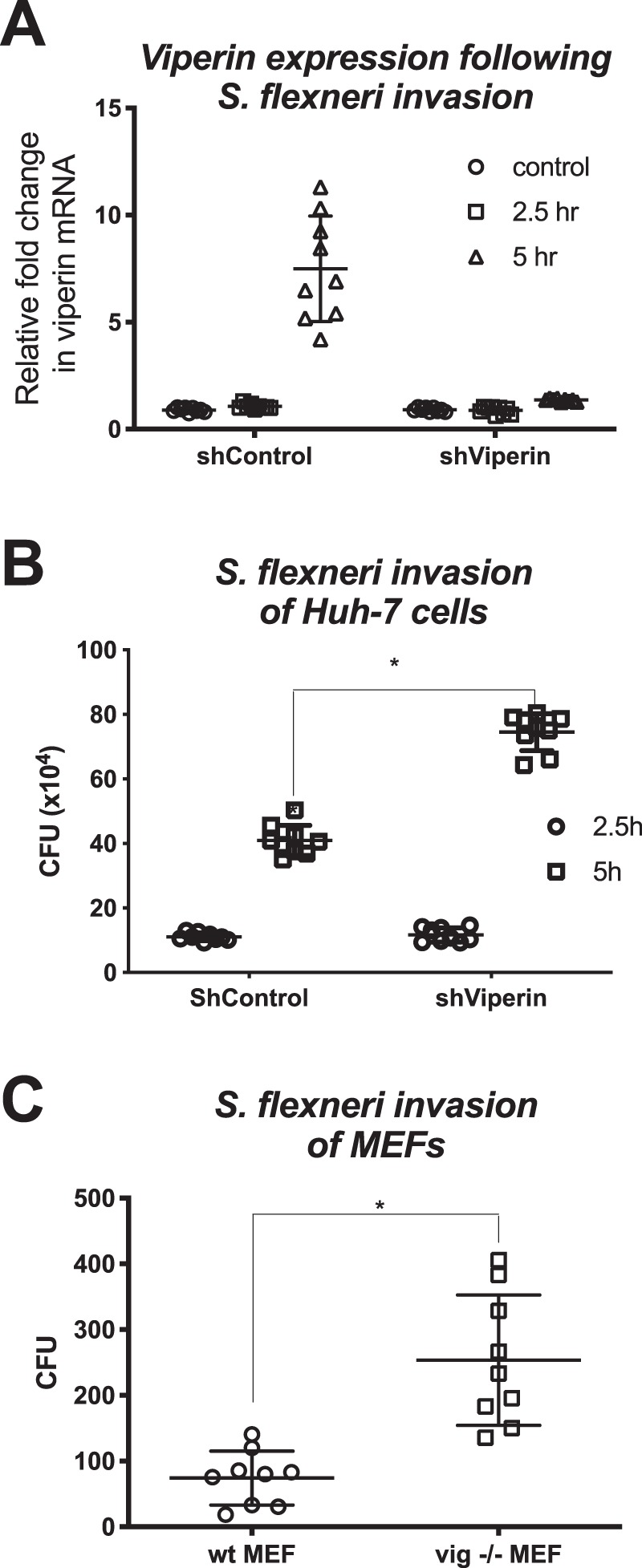
Loss of viperin enhances S. flexneri in vitro. (A) Viperin shRNA or control shRNA expressing Huh7 cells were invaded with S. flexneri and either RNA collected for real-time analysis or (B) cell lysate collected for CFU counts, at the indicated time points. (C) Primary MEFs harvested from either wild-type or viperin KO mice were invaded with S. flexneri and cell lysates collected for CFU counts at 5 hours post invasion. Graphs represent the mean and standard deviation of three independent experiments performed in triplicate, *p < 0.0001.
Viperin’s enzymatic activity impacts its ability to limit S. flexneri infection in vitro
Viperin is a radical SAM enzyme, that has recently been demonstrated to catalyse the conversion of CTP to the novel molecule, ddhCTP (3′-deoxy-3′,4′-didehydro-CTP), which acts a chain terminator of RNA dependent RNA polymerases of selected members of the Flaviviridae viral family39. However, viperin has antiviral activity beyond its enzymatic activity17, and this prompted us to determine whether viperins’ enzymatic activity was required for its ability to limit intracellular bacteria. Viperin mutants harbouring point mutations of conserved cysteine residues in the S1 domain (SAM M130) were expressed transiently in 293T cells in parallel with wild-type viperin, and S. flexneri and L. monocytogenes invasion assessed. The ability of SAM M1 viperin to limit both S. flexneri and L. monocytogenes was completely abrogated in cells expressing the SAM M1 mutant (Fig. 4A). To further analyse the ability of the radical SAM enzymatic domain of viperin to play a role in limiting intracellular bacterial infection, we performed invasion assays in the presence of cycloleucine, a known inhibitor of cellular SAM synthesis, and an inhibitor in general of radical SAM enzymatic activity44. Cycloleucine did not impact cell viability at concentrations at or lower than 20 mM when added to the culture media of 293T cells (Fig. 4B), neither did it have a significant impact on S. flexneri infection of 293T cells 3 hours post invasion (Fig. 4C). However, 293T cells pre-treated with cycloleucine prior to S. flexneri invasion, showed a significant difference in intracellular bacterial numbers 3 hours post invasion, with viperin expression decreasing bacterial counts by approximately 70%, but only 13% in the presence of cycloleucine; reinforcing the fact that viperin’s radical SAM enzymatic activity is important for its ability to limit S. flexneri infection of cells in vitro.
Figure 4.

Viperin requires its radical SAM domain to limit intracellular bacterial numbers. (A) 293T cells were transiently transfected with either a viperin wt expressing plasmid, or a SAM domain viperin mutant 24 hours prior to invasion with either S. flexneri or L. monocytogenes; cell lysates were harvested for CFU counts at 3 hours post invasion. (B) 293T cells were treated with the indicated concentrations of cycloleucine, and a cell viability assay performed at the indicated time points. (C) Transfection of 293T cells with either a control or a viperin expressing plasmid was performed 24 hours prior to cycloleucine or control treatment for 24 hours. Cells were then invaded with S. flexneri for 3 hours prior to cell lysate harvesting for CFU counts. (D) 293T cells were transfected with either a control, viperin wt, or viperin truncation mutants 24 hours prior to invasion with S. flexneri for 3 hours. Graphs represent the mean and standard deviation of three independent experiments performed in triplicate, *p < 0.0001.
A number of domains/regions of importance have been classified within viperin, including its N-terminal amphipathic helix, which we and others have previously shown to localise viperin to lipid droplets and the endoplasmic reticulum30,32, as well as its C-terminal region which has been shown to be vital in its ability to restrict some viral infections (reviewed in17). To assess the importance of these two domains, truncated viperin mutants were expressed transiently prior to invasion of 293T cells with S. flexneri. Both the amphipathic helix and the C-terminal region of the viperin protein were found to be vital in the ability of viperin to limit S. flexneri bacterial counts at 3 hours post bacterial invasion, with the truncated viperin mutants showing no significant difference in bacterial counts from the controls (Fig. 4D).
Viperin limits initial entry of S. flexneri in vitro
In order to gain insight into the life cycle stage at which viperin inhibits S. flexneri, we used microscopy to gain a broader view of the potential interactions between viperin and bacterially infected and uninfected cells in vitro. Experiments investigating infection of an mCherry expressing S. flexneri (MLRM107,45) into viperin pre-transfected Huh-7 cells, displayed an almost complete exclusion of bacteria 3 hours post invasion, in many cells expressing viperin (Fig. 5A). In order to quantitate exclusion at an earlier time point, Huh-7 cells were transfected with either a viperin-GFP expression plasmid, or GFP only, 24 hours prior to invasion of the cells with mCherry labelled S. flexneri. Cells were fixed 15 minutes post infection, and then visualised using microscopy. Whole fields were imaged, and cells counted at random until a minimum of 300 viperin-GFP or GFP positive and negative cells were assessed for the number of intracellular S. flexneri per cell. In cells expressing GFP only, we were able to visualise on average, 3.66 bacteria per Huh-7 cell, in comparison to 3.68 bacteria per cell in GFP negative cells (Fig. 5B), demonstrating that there was no significant difference between the ability of S. flexneri to invade Huh-7 cells whether or not they were expressing the GFP protein. Interestingly, in the presence of viperin-GFP expression, the average number of S. flexneri present per cell was significantly reduced to 0.89 bacteria, in comparison to 3.41 bacteria per cell in the viperin-GFP negative cells (Fig. 5C), equating to an approximate 3.8 fold reduction in bacterial invasion per cell. When taking into account the actual number of cells that contained at least one bacterium, irrespective of the total amount of bacteria per cell, we can see that in cells expressing viperin, approximately 54% of cells were positive for early bacterial invasion, in comparison to approximately 86–88% of cells in the presence or absence of GFP (Fig. 5D), indicating that the Huh-7 cells expressing viperin where overall less permissive for S. flexneri invasion at early time points following infection.
Figure 5.

Viperin expression inhibits the entry of S. flexneri in vitro. (A) Huh-7 cells transiently expressing viperin-GFP where invaded with an S. flexneri strain expressing mCherry and imaged 5 hours post invasion. (B,C) Huh-7 cells transiently expressing either viperin-GFP or GFP alone, where invaded with an S. flexneri strain expressing mCherry for 15 minutes prior to fixation and imaging on a Nikon TiE inverted microscope; Whole fields were counted at random and assessed for the number of intracellular S. flexneri per cell (n = 300 cells per group, counted over 3 biological replicates), as well as (D) categorised as either S. flexneri infected or not, to assess percentage of cells invaded by the bacteria. *p < 0.0001.
Viperin expression impacts cholesterol levels in vitro
S. flexneri intracellular invasion is known to be supported by an interaction between host cell CD44 and the bacterial invasin IpaB, that occurs on lipid rafts enriched in cholesterol and sphingolipids46. Viperin expression has previously been identified to alter the fluidity of lipid rafts in vitro, and to reduce cholesterol levels in BHK cell lines41,47. We next assessed the ability of transient viperin expression to lower the levels of cholesterol in both HeLa and Huh-7 cells. As can be seen in Fig. 6, the expression of viperin was able to significantly lower the levels of cholesterol in both HeLa and Huh-7 cells by approximately 55% and 33% respectively, with the difference likely reflecting the transfection efficiency of these two cell types.
Figure 6.

Viperin expression lowers cellular cholesterol levels in vitro. Huh-7 and HeLa cells transiently expressing either viperin or transfected with a control plasmid were assessed 24 hours post transfection for their cellular levels of cholesterol. Graphs represent the mean and standard deviation of three independent experiments performed in triplicate, *p < 0.05, **p < 0.001.
Discussion
The role of the classical anti-viral cytokines, type I (IFN-α,β) and type III IFN (IFN-λ), and the interferon stimulated genes that they upregulate, in the control of bacterial infection remains controversial1,2. Multiple extracellular and intracellular bacterial species are known to upregulate these interferons following infection of host cells, with the activation of type I IFN being mainly via STING dependent mechanisms, and through bacterial LPS activation of TLR4, with a small number of bacterial species being shown to also induce type I IFN via engagement of NOD1/NOD248. The induction of type III IFN by bacteria is less well studied, but is also induced by a range of both intracellular and extracellular pathogens. Both TLR7 and MAVS activation have been proven to be crucial for type III IFN induction by Borrelia burgdorfei and Listeria monocytogenes infection respectively, with the mechanisms of induction by other bacterial species not being well characterised to date1. Despite the knowledge that bacterial infection of a host induces these cytokines, their respective ability to restrict bacterial pathogenesis and directly inhibit the bacterial life-cycle remains contentious.
Hundreds of interferon stimulated genes are induced following activation of the JAK-STAT pathway by type I and III IFNs, with only a small handful of these to date being demonstrated to directly inhibit the life-cycle of some bacterial species. Viperin is an interferon stimulated gene that is upregulated via multiple pathways, including via IFN-independent means17. It belongs to the radical SAM family, which are enzymes that bind iron-sulphur clusters to mediate their reactions; and interestingly is one of a handful of mammalian radical SAM enzymes, with the majority of known radical SAM members belonging to bacterial family members49. Viperin is known to be induced following cellular invasion of the intracellular bacterial pathogens, Mycobacterium tuberculosis, Listeria monocytogenes and Salmonella typhimurium19,21. Additionally, in a fish muscle model of bacterial infection, Vibrio vulnificus colony forming units were significantly reduced when a plasmid expressing viperin was electroporated into fish muscle prior to bacterial infection, although the exact mechanisms behind viperin’s potential anti-bacterial activity were not elucidated50. Here we show that S. flexneri infection of cultured cells in vitro is significantly hampered by the presence of IFN, and that cellular infection by the bacteria is able to upregulate viperin expression in epithelial cell lines in vitro (Fig. 1C). Interestingly, S. flexneri appeared to illicit a more dominant type I IFN response in the two epithelial cell lines tested, which coincides with previous work demonstrating that S. flexneri is a weak inducer of Type III IFN species in epithelial cells51.
Both ectopic viperin expression and loss of viperin expression significantly impacted the growth of the gram-negative pathogen, S. flexneri in multiple cell lines in vitro, and this observation was also extended to the gram-positive intracellular pathogen, L. monocytogenes. The ability of viperin to impact the growth of these intracellular bacteria relied on its radical SAM enzymatic activity, as well as the presence of its N-terminal amphipathic helical domain, and its extreme C-terminal domain (Fig. 4). Viperin’s N-terminus contains both an amphipathic helix which is responsible for both its localisation to the lipid droplet, and the endoplasmic reticulum30, as well as a binding region for one of its iron sulphur targeting factors, CIA2A, with the others (CIA1/2B) binding to its C-terminal region52. Viperin’s enzymatic activity has recently been elucidated to involve the formation of the novel ribonucleotide, 3’-deoxy-3’, 4’-didehydro-CTP (ddhCTP) via catalysing the conversion of cytidine triphosphate (CTP)39. This novel compound was shown to act in an anti-viral manner by restricting polymerase activity of a small handful of members from the flavivirus genus, however its enzymatic activity has been shown to be redundant for its ability to limit other viral family members30,39. It is clear that viperin is a highly multifunctional protein, and recent work has demonstrated that viperin can also augment the STING dependent dsDNA signalling pathway, with this function being dependent on its co-factor binding sites, and the presence of its radical SAM enzymatic domains, perhaps indicating that viperin’s radical SAM enzymatic activity has multiple host cell purposes53.
Prior ectopic expression of viperin in epithelial cells in vitro appeared to exclude bacterial replication in these cells (Fig. 5A), and subsequent infection assays revealed that the presence of intracellular levels of viperin reduced the numbers of S. flexneri in cells by almost 4 fold (Fig. 5C). Viperin has been shown previously to inhibit the egress of influenza, HIV and respiratory syncytial virus virions, with the common mechanism being changes in cholesterol levels, or lipid raft membrane fluidity40–42, and here we were able to demonstrate that viperin expression significantly reduces the levels of cholesterol in both Huh-7 and HeLa epithelial cell lines (Fig. 6). Lipid raft cholesterol is essential for entry of Shigella spp. into epithelial cells, where its invasin, IpaB, is known to interact with hyaluronan receptor, CD44, and ultimately cause redistribution of plasma membrane cholesterol to the sites of bacterial invasion46,54. Given’s viperin’s previously known ability to limit viral egress via interruption of cholesterol in membranes, in particularly altering plasma membrane fluidity41, it is likely that viperin expression might also be disrupting S. flexneri entry through the diminished presence of plasma membrane cholesterol, however further studies need to be performed to elucidate the exact mechanism that viperin utilises to impede entry of S. flexneri, and whether it alters the ability of S. flexneri to utilise cellular entry receptors. Another limitation of this study, is the inability to compare bacterial growth and spread kinetics between a viperin competent and a deficient model system, due to the very low baseline of the protein, and the time it takes to be induced following bacterial infection (Fig. 3A). However, we would hypothesise that in a spreading infection a bystander effect would likely occur, where interferons produced by an S. flexneri infected cell, would upregulate viperin in surrounding as yet uninfected cells, and potentially impact bacterial entry.
Although the potential mechanisms at play in the control of bacterial pathogenesis by type I and III IFN remain in their infancy, recently it has been shown that type III IFNs can reinforce epithelial barriers during bacterial infection, and prevent invasion from intracellular bacteria such as Shigella spp.55, although the ISGs responsible for this effect remain unknown. Despite similar signalling pathways, type I and III IFNs are known to selectively induce ISGs, with differential kinetics of expression, as well as an altered ISG expression profile depending on cell type56. Viperin is known to be expressed in intestinal epithelial cells, and is a dominantly expressed protein in multiple cell types, regardless of its induction by type I or III IFN17,57,58. Here we present data for the first time that the interferon stimulated gene, viperin, not only exhibits broadly acting anti-viral capabilities, but is also able to restrict the infection of the intracellular pathogen, S. flexneri. This work paves the ways for future studies investigating the role of other multifunctional ISGs and their ability to restrict bacterial pathogens, and possibly help unravel the role of both type I and III IFNs in direct control of bacterial infection.
Methods
Cells and bacterial stocks
Huh-7 human hepatoma cells, which are a well differentiated hepatocyte-derived carcinoma cell line (a kind gift from Stanley Lemon, The University of Texas Medical Branch); HeLa human epithelial cells which a cervical carcinoma cell line (laboratory stock gifted from the Institute of Medical and Veterinary Science, Adelaide), and 293T cells, which are a derivative of the 293 human embryonic kidney cell line (a kind gift from Murray Whitelaw, University of Adelaide), were maintained as previously described59, and at 37 °C with 5% CO2 in DMEM containing 10% FCS, penicillin and streptomycin. Huh-7 shControl cells, and the shViperin cells were maintained as above, and are described in30. Murine embryonic fibroblasts (MEF’s) were isolated from both wild-type and viperin knock-out mice and maintained as previously38. The isolation of all MEFs was carried out in accordance with the relevant guidelines and regulations of the National Health and Medical Research Council Australian Code of Practice for the Care and Use of Animals for Scientific Purposes, and approved by the University of Adelaide Animal Ethics Committee. The S. flexneri strains 2457 T and MLRM10745, were grown from a Congo Red positive colony as described previously60, and cultured in Luria Bertani (LB) broth and on LB agar. L. monocytogenes was cultured in Luria Bertani (LB) broth and on LB agar. Bacteria were grown in LB media for 16 hours, prior to being subcultured 1:20, and grown to mid-exponential growth phase for 2 hours at 37 °C with aeration. MLRM107 cultures were supplemented with tetracycline (10 μg/l). Cell viability assays were performed using a Cell Titre Blue assay kit (Promega) as previously61.
S. flexneri invasion and detection
Cell lines were seeded at 8 × 104 in 12 well plates, 24 hours prior to invasion with S. flexneri 2457 T. Cells were washed twice in PBS, followed by one wash in complete DMEM lacking antibiotics. Mid-exponential phase S. flexneri (4.8 × 107 bacteria per well) were added to the cells in a 200 μl volume of complete DMEM without antibiotics, and the plates spun at 2,000 g for 7 minutes. After a one hour incubation at 37 °C with 5% CO2, the cells were washed 3 times with PBS, and the media replaced with DMEM containing 40 μg/ml gentamicin. At the indicated time points cells were washed three times with PBS and were either harvested for RNA extraction, or lysed with 0.1% (v/v) Triton X-100 in PBS for 5 minutes and the bacteria enumerated on LB plates. All experiments were repeated in at least triplicate.
RNA extraction and RT-PCR
Total cellular RNA was isolated from cells using Trizol (Invitrogen, Calsbad, USA), DNase treated and quantitated by spectrophotometry. cDNA synthesis and subsequent real-time PCR was performed on an ABI 7000 as previously described for the detection of IFN’s, ISGs and the house keeping gene, RPLPO mRNA62. Primer sequences used to detect S. flexneri iPAH mRNA were 5′-TTGGTCGCCCTACCTTTTCA and 5′-CTCGATAAGCGCAGAGAAATGA.
Plasmids, transfections and cell stimulants
Viperin wt and mutant plasmids were as described previously30. Cell lines were seeded at 7 × 104 into 12 well plates, 24 hours prior to transfection with DNA vectors using Fugene6 (Roche, Penzburg, DEU) as per manufacturer’s instructions, or stimulation with IFN-γ, β and IFN-α (Peprotech, Israel) for 24 hours. All experiments were repeated in at least triplicate.
Luciferase experiments
Cells were seeded at 7 × 104 in 12 well plates 24 hours prior to transfection with an IFN-β promoter driven luciferase plasmid (pIF(−125/ + 75)lucter (a kind gift from Dr Liu, Royal Children’s Hospital, QLD) and the pRL-TK constitutively expressing renilla luciferase plasmid to control for transfection efficiency. Twenty four hours following transfection, cells were invaded with 4.8 × 104 bacteria per well of S. flexneri 2457 T as above, for 5 hours prior to luciferase detection using the Dual-Luciferase Reporter Assay System (Promega, Fitchberg, USA) as per manufacturer’s instructions. Luciferase measurements were taken on a Promega GloMax 96 luminometre. Experiments were performed in triplicate.
Inhibition of radical SAM enzyme function
To inhibit viperin’s radical SAM enzyme function, cells were treated with cycloleucine (Sigma, St. Louis USA) for 24 hours as previously described37, 24 hours post DNA plasmid transfection. Cells were washed twice in PBS, prior to invasion with S. flexneri 2457 T for 3 hours, as described above. Cell viability assays were performed using a Cell Titre Blue Assay kit (Promega, Fitchberg, USA) as previously described61.
S. flexneri early entry assay
Huh-7 cells were seeded at 2 × 104 cells per well of a 24 well plate, containing a sterile gelatin coated glass coverslip in the bottom. Cells were transfected with either viperin-GFP or GFP alone as described previously30, and 24 hours later invaded with 2.4 × 107 bacteria per well of the mCherry fluorescing S. flexneri MLRM107 strain. Cells were spun at 2,000 g for 7 minutes and incubated for a further 15 minutes at 37 °C with 5% CO2. Cells were then washed vigorously 3 times with PBS, prior to fixation with 4% paraformaldehyde for 20 minutes. Coverslips were mounted, and florescence was visualised using a Nikon TiE inverted microscope (Nikon, Japan). Cells were visualised using a 40x objective, and whole fields counted at random until a minimum of 300 GFP positive and negative cells were assessed for the number of intracellular S. flexneri per cell. Experiments were performed in triplicate.
Assessment of cellular cholesterol levels
Cells were seeded at 6 × 104 cells per well of a 12 well plate 24 hours prior to transient transfection using Lipofectamine 3000, with either a viperin expression plasmid or an empty vector. 24 hours following transfection, cells were harvested and lysed with 0.1% triton X-100. The cholesterol content of the cell lysates was quantified with the cholesterol quantification kit (40006; AAT Bioquest, USA) according to the manufacturer’s specifications. Each condition was performed at least 8 times and on four independent occasions. Fluorescent signal was monitored at Ex/Em = 540/590 nm (cholesterol) using a microplate reader (CLARIOstar® BMG LABTECH).
Immunostaining and co-localisation studies
Cells were cultured on gelatin coated glass coverslips, fixed in acetone:methanol (1:1) and stored at −20 °C. Slides were washed in PBS, before blocking in 5% (w/v) bovine serum albumin (BSA) in Hanks buffered salts solution (Gibco BRL, NY). Cells were stained using a rabbit anti-mCherry antibody (BioVision, CA) together with a goat anti-rabbit IgG-Alexa 555, secondary antibody (Molecular probes, CA). Bodipy 493/503 (Invitrogen) was prepared as a stock solution of 1 mg/ml in ethanol and used at a 1:1000 concentration PBS. Florescence was visualised using a Nikon TiE inverted microscope (Nikon, Tokyo, Japan).
Statistical analysis
Mann-Whittney U tests (2 tailed) were utilised to analyse the distributions of two data sets and all experiments were performed a minimum of three times and in at least triplicate. Statistical analysis was performed using PRISM8 (GraphPad software). The data points for all figures can be found in Supplementary Table 1.
Supplementary information
Author contributions
K.H., M.R.B. and R.M. conceived the experiments; K.H., M.Y.T., E.A.M., K.M.C., E.N.T. and A.J.S. performed the experiments; K.H. analysed the data and wrote the manuscript; all authors participated in reviewing and editing the final manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-019-52130-8.
References
- 1.Cohen TS, Parker D. Microbial pathogenesis and type III interferons. Cytokine Growth Factor Rev. 2016;29:45–51. doi: 10.1016/j.cytogfr.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I interferons in infectious disease. Nat Rev Immunol. 2015;15:87–103. doi: 10.1038/nri3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lazear HM, Nice TJ, Diamond MS. Interferon-lambda: Immune Functions at Barrier Surfaces and Beyond. Immunity. 2015;43:15–28. doi: 10.1016/j.immuni.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee AJ, Ashkar AA. The Dual Nature of Type I and Type II Interferons. Front Immunol. 2018;9:2061. doi: 10.3389/fimmu.2018.02061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Watanabe T, et al. Activation of type I IFN signaling by NOD1 mediates mucosal host defense against Helicobacter pylori infection. Gut Microbes. 2011;2:61–65. doi: 10.4161/gmic.2.1.15162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gratz N, et al. Type I interferon production induced by Streptococcus pyogenes-derived nucleic acids is required for host protection. PLoS Pathog. 2011;7:e1001345. doi: 10.1371/journal.ppat.1001345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.LeMessurier KS, Hacker H, Chi L, Tuomanen E, Redecke V. Type I interferon protects against pneumococcal invasive disease by inhibiting bacterial transmigration across the lung. PLoS Pathog. 2013;9:e1003727. doi: 10.1371/journal.ppat.1003727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mancuso G, et al. Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J Immunol. 2007;178:3126–3133. doi: 10.4049/jimmunol.178.5.3126. [DOI] [PubMed] [Google Scholar]
- 9.Bukholm G, Berdal BP, Haug C, Degre M. Mouse fibroblast interferon modifies Salmonella typhimurium infection in infant mice. Infect Immun. 1984;45:62–66. doi: 10.1128/iai.45.1.62-66.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Niesel DW, Hess CB, Cho YJ, Klimpel KD, Klimpel GR. Natural and recombinant interferons inhibit epithelial cell invasion by Shigella spp. Infect Immun. 1986;52:828–833. doi: 10.1128/iai.52.3.828-833.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boxx GM, Cheng G. The Roles of Type I Interferon in Bacterial Infection. Cell Host Microbe. 2016;19:760–769. doi: 10.1016/j.chom.2016.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meunier E, Broz P. Interferon-inducible GTPases in cell autonomous and innate immunity. Cell Microbiol. 2016;18:168–180. doi: 10.1111/cmi.12546. [DOI] [PubMed] [Google Scholar]
- 14.Ranjbar S, Haridas V, Jasenosky LD, Falvo JV, Goldfeld AE. A Role for IFITM Proteins in Restriction of Mycobacterium tuberculosis Infection. Cell Rep. 2015;13:874–883. doi: 10.1016/j.celrep.2015.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Margulieux, K. R., Fox, J. W., Nakamoto, R. K. & Hughes, M. A. CXCL10 Acts as a Bifunctional Antimicrobial Molecule against Bacillus anthracis. MBio7, 10.1128/mBio.00334-16 (2016). [DOI] [PMC free article] [PubMed]
- 16.Perelman SS, et al. Cell-Based Screen Identifies Human Interferon-Stimulated Regulators of Listeria monocytogenes Infection. PLoS Pathog. 2016;12:e1006102. doi: 10.1371/journal.ppat.1006102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Helbig KJ, Beard MR. The role of viperin in the innate antiviral response. J Mol Biol. 2014;426:1210–1219. doi: 10.1016/j.jmb.2013.10.019. [DOI] [PubMed] [Google Scholar]
- 18.Crosse KM, Monson EA, Beard MR, Helbig KJ. Interferon-Stimulated Genes as Enhancers of Antiviral Innate Immune Signaling. J Innate Immun. 2018;10:85–93. doi: 10.1159/000484258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saitoh T, et al. Antiviral protein Viperin promotes Toll-like receptor 7- and Toll-like receptor 9-mediated type I interferon production in plasmacytoid dendritic cells. Immunity. 2011;34:352–363. doi: 10.1016/j.immuni.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 20.Charrel-Dennis M, et al. TLR-independent type I interferon induction in response to an extracellular bacterial pathogen via intracellular recognition of its DNA. Cell Host Microbe. 2008;4:543–554. doi: 10.1016/j.chom.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manzanillo PS, Shiloh MU, Portnoy DA, Cox JS. Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell Host Microbe. 2012;11:469–480. doi: 10.1016/j.chom.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weiss G, et al. Lactobacillus acidophilus induces virus immune defence genes in murine dendritic cells by a Toll-like receptor-2-dependent mechanism. Immunology. 2010;131:268–281. doi: 10.1111/j.1365-2567.2010.03301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Milic NL, et al. Sequence analysis and characterisation of virally induced viperin in the saltwater crocodile (Crocodylus porosus) Dev Comp Immunol. 2015;51:108–115. doi: 10.1016/j.dci.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 24.Boudinot P, et al. Vesicular stomatitis virus and pseudorabies virus induce a vig1/cig5 homologue in mouse dendritic cells via different pathways. J Gen Virol. 2000;81:2675–2682. doi: 10.1099/0022-1317-81-11-2675. [DOI] [PubMed] [Google Scholar]
- 25.Chin KC, Cresswell P. Viperin (cig5), an IFN-inducible antiviral protein directly induced by human cytomegalovirus. Proc Natl Acad Sci USA. 2001;98:15125–15130. doi: 10.1073/pnas.011593298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grandvaux N, et al. Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. J Virol. 2002;76:5532–5539. doi: 10.1128/JVI.76.11.5532-5539.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hinson ER, et al. Viperin is highly induced in neutrophils and macrophages during acute and chronic lymphocytic choriomeningitis virus infection. J Immunol. 2010;184:5723–5731. doi: 10.4049/jimmunol.0903752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stirnweiss A, et al. IFN regulatory factor-1 bypasses IFN-mediated antiviral effects through viperin gene induction. J Immunol. 2010;184:5179–5185. doi: 10.4049/jimmunol.0902264. [DOI] [PubMed] [Google Scholar]
- 29.White LK, et al. Chikungunya virus induces IPS-1-dependent innate immune activation and protein kinase R-independent translational shutoff. J Virol. 2011;85:606–620. doi: 10.1128/JVI.00767-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Helbig KJ, et al. The antiviral protein viperin inhibits hepatitis C virus replication via interaction with nonstructural protein 5A. Hepatology. 2011;54:1506–1517. doi: 10.1002/hep.24542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hinson ER, Cresswell P. The N-terminal amphipathic alpha-helix of viperin mediates localization to the cytosolic face of the endoplasmic reticulum and inhibits protein secretion. J Biol Chem. 2009;284:4705–4712. doi: 10.1074/jbc.M807261200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hinson ER, Cresswell P. The antiviral protein, viperin, localizes to lipid droplets via its N-terminal amphipathic alpha-helix. Proc Natl Acad Sci USA. 2009;106:20452–20457. doi: 10.1073/pnas.0911679106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duschene KS, Broderick JB. The antiviral protein viperin is a radical SAM enzyme. FEBS Lett. 2010;584:1263–1267. doi: 10.1016/j.febslet.2010.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Helbig KJ, et al. Viperin is induced following dengue virus type-2 (DENV-2) infection and has anti-viral actions requiring the C-terminal end of viperin. PLoS Negl Trop Dis. 2013;7:e2178. doi: 10.1371/journal.pntd.0002178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Helbig KJ, Lau DT, Semendric L, Harley HA, Beard MR. Analysis of ISG expression in chronic hepatitis C identifies viperin as a potential antiviral effector. Hepatology. 2005;42:702–710. doi: 10.1002/hep.20844. [DOI] [PubMed] [Google Scholar]
- 36.Szretter KJ, et al. The interferon-inducible gene viperin restricts West Nile virus pathogenesis. J Virol. 2011;85:11557–11566. doi: 10.1128/JVI.05519-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Upadhyay AS, et al. Viperin is an iron-sulfur protein that inhibits genome synthesis of tick-borne encephalitis virus via radical SAM domain activity. Cell Microbiol. 2014;16:834–848. doi: 10.1111/cmi.12241. [DOI] [PubMed] [Google Scholar]
- 38.Van der Hoek KH, et al. Viperin is an important host restriction factor in control of Zika virus infection. Sci Rep. 2017;7:4475. doi: 10.1038/s41598-017-04138-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gizzi AS, et al. A naturally occurring antiviral ribonucleotide encoded by the human genome. Nature. 2018;558:610–614. doi: 10.1038/s41586-018-0238-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nasr N, et al. HIV-1 infection of human macrophages directly induces viperin which inhibits viral production. Blood. 2012;120:778–788. doi: 10.1182/blood-2012-01-407395. [DOI] [PubMed] [Google Scholar]
- 41.Wang X, Hinson ER, Cresswell P. The interferon-inducible protein viperin inhibits influenza virus release by perturbing lipid rafts. Cell Host Microbe. 2007;2:96–105. doi: 10.1016/j.chom.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 42.Jumat MR, et al. Viperin protein expression inhibits the late stage of respiratory syncytial virus morphogenesis. Antiviral Res. 2015;114:11–20. doi: 10.1016/j.antiviral.2014.11.007. [DOI] [PubMed] [Google Scholar]
- 43.Way SS, Borczuk AC, Dominitz R, Goldberg MB. An essential role for gamma interferon in innate resistance to Shigella flexneri infection. Infect Immun. 1998;66:1342–1348. doi: 10.1128/iai.66.4.1342-1348.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caboche M, Hatzfeld J. Methionine metabolism in BHK cells: preliminary characterization of the physiological effects of cycloleucine, an inhibitor of s-adenosylmethionine biosynthesis. J Cell Physiol. 1978;97:361–370. doi: 10.1002/jcp.1040970311. [DOI] [PubMed] [Google Scholar]
- 45.Lum M, Attridge SR, Morona R. Impact of dynasore an inhibitor of dynamin II on Shigella flexneri infection. PLoS One. 2013;8:e84975. doi: 10.1371/journal.pone.0084975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lafont F, Tran Van Nhieu G, Hanada K, Sansonetti P, van der Goot FG. Initial steps of Shigella infection depend on the cholesterol/sphingolipid raft-mediated CD44-IpaB interaction. EMBO J. 2002;21:4449–4457. doi: 10.1093/emboj/cdf457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tang HB, et al. Viperin inhibits rabies virus replication via reduced cholesterol and sphingomyelin and is regulated upstream by TLR4. Sci Rep. 2016;6:30529. doi: 10.1038/srep30529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kovarik P, Castiglia V, Ivin M, Ebner F, Type I. Interferons in Bacterial Infections: A Balancing Act. Front Immunol. 2016;7:652. doi: 10.3389/fimmu.2016.00652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Broderick JB, Duffus BR, Duschene KS, Shepard EM. Radical S-adenosylmethionine enzymes. Chem Rev. 2014;114:4229–4317. doi: 10.1021/cr4004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee SH, et al. Characterization of tilapia (Oreochromis niloticus) viperin expression, and inhibition of bacterial growth and modulation of immune-related gene expression by electrotransfer of viperin DNA into zebrafish muscle. Vet Immunol Immunopathol. 2013;151:217–228. doi: 10.1016/j.vetimm.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 51.Bierne H, et al. Activation of type III interferon genes by pathogenic bacteria in infected epithelial cells and mouse placenta. PLoS One. 2012;7:e39080. doi: 10.1371/journal.pone.0039080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Upadhyay AS, et al. Cellular requirements for iron-sulfur cluster insertion into the antiviral radical SAM protein viperin. J Biol Chem. 2017;292:13879–13889. doi: 10.1074/jbc.M117.780122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Crosse, K. M. et al. The Antiviral Protein Viperin Enhances The Host Interferon Response Following STING Activation. BbioRxiv, 10.1101/493098 (2018).
- 54.Mounier J, et al. Shigella effector IpaB-induced cholesterol relocation disrupts the Golgi complex and recycling network to inhibit host cell secretion. Cell Host Microbe. 2012;12:381–389. doi: 10.1016/j.chom.2012.07.010. [DOI] [PubMed] [Google Scholar]
- 55.Odendall C, Voak AA, Kagan JC. Type III IFNs Are Commonly Induced by Bacteria-Sensing TLRs and Reinforce Epithelial Barriers during Infection. J Immunol. 2017;199:3270–3279. doi: 10.4049/jimmunol.1700250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ingle, H., Peterson, S. T. & Baldridge, M. T. Distinct Effects of Type I and III Interferons on Enteric Viruses. Viruses10, 10.3390/v10010046 (2018). [DOI] [PMC free article] [PubMed]
- 57.Pervolaraki K, et al. Differential induction of interferon stimulated genes between type I and type III interferons is independent of interferon receptor abundance. PLoS Pathog. 2018;14:e1007420. doi: 10.1371/journal.ppat.1007420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guo L, et al. Human Intestinal Epithelial Cells Release Antiviral Factors That Inhibit HIV Infection of Macrophages. Front Immunol. 2018;9:247. doi: 10.3389/fimmu.2018.00247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Helbig KJ, George J, Beard MR. A novel I-TAC promoter polymorphic variant is functional in the presence of replicating HCV in vitro. J Clin Virol. 2005;32:137–143. doi: 10.1016/j.jcv.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 60.Van den Bosch L, Manning PA, Morona R. Regulation of O-antigen chain length is required for Shigella flexneri virulence. Mol Microbiol. 1997;23:765–775. doi: 10.1046/j.1365-2958.1997.2541625.x. [DOI] [PubMed] [Google Scholar]
- 61.McCartney EM, et al. Alcohol metabolism increases the replication of hepatitis C virus and attenuates the antiviral action of interferon. J Infect Dis. 2008;198:1766–1775. doi: 10.1086/593216. [DOI] [PubMed] [Google Scholar]
- 62.Monson EA, Crosse KM, Das M, Helbig KJ. Lipid droplet density alters the early innate immune response to viral infection. PLoS One. 2018;13:e0190597. doi: 10.1371/journal.pone.0190597. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.