

Abstract
Background –
Alzheimer’s disease (AD) is a multifactorial neurodegenerative disorder with important vascular and hemostatic alterations that should be taken into account during diagnosis and treatment.
Objectives –
This study evaluates whether anticoagulation with dabigatran, a clinically approved oral direct thrombin inhibitor with a low risk of intracerebral hemorrhage, ameliorates AD pathogenesis in a transgenic mouse model of AD.
Methods –
TgCRND8 AD mice and their wild type (WT) littermates were treated for one year with dabigatran etexilate or placebo. Cognition was evaluated using the Barnes maze, and cerebral perfusion was examined by arterial spin labeling (ASL). At the molecular level, western blot (WB) and histochemical analyses were performed to analyze fibrin content, amyloid burden, neuroinflammatory activity, and blood brain barrier (BBB) integrity.
Results –
Anticoagulation with dabigatran prevented memory decline, cerebral hypoperfusion, and toxic fibrin deposition in the AD mouse brain. In addition, long-term dabigatran treatment significantly reduced the extent of amyloid plaques, oligomers, phagocytic microglia, and infiltrated T cells by 23.7%, 51.8%, 31.3% and 32.2%, respectively. Dabigatran anticoagulation also prevented AD-related astrogliosis and pericyte alterations and maintained expression of the water channel aquaporin-4 (AQP4) at astrocytic perivascular endfeet of the BBB.
Conclusions –
Long-term anticoagulation with dabigatran inhibited thrombin and the formation of occlusive thrombi in AD, preserved cognition, cerebral perfusion, and BBB function and ameliorated neuroinflammation and amyloid deposition in AD mice. Our results open a field for future investigation on whether the use of direct oral anticoagulants might be of therapeutic value in AD.
Keywords: Cognitive impairment, oral anticoagulation, thrombin, thrombosis, neuroinflammation, animal models of human disease
Condensed Abstract
AD is a multifactorial disorder with a chronic pro-thrombotic component. This study combined physiological and molecular techniques to demonstrate that long-term anticoagulation with dabigatran ameliorated AD pathogenesis in an AD transgenic mouse model. Dabigatran treatment impeded fibrin deposition and was able to prevent cognitive decline, maintain optimal cerebral perfusion, curb neuroinflammation, decrease amyloid pathology, and maintain the integrity of the BBB in the AD mouse brain. The use of dabigatran might be a future candidate treatment to normalize the sustained pro-coagulant state present in AD patients. Further studies are needed to warrant its use in the clinic.
Introduction
AD is a progressive and multifactorial neurodegenerative disorder characterized by amyloid-β (Aβ) plaques, tau tangles, neuroinflammation, and brain atrophy (1). AD is strongly linked with cardiovascular risk factors, and is often accompanied by an important vascular component (2–4). The cerebrovascular pathology present in AD includes BBB disruption, neurovascular unit dysfunction, neurovascular uncoupling, and cerebral blood flow (CBF) alterations (5–7). Furthermore, chronic dysregulated hemostasis is present in AD, with increased thrombin generation, presence of activated platelets and leakage of plasma proteins into the brain parenchyma (8,9), favoring the formation and persistence of fibrin clots (10,11).
Fibrin(ogen) is upregulated early in AD (12), is found intra- and extra-vascularly in areas of synaptic dysfunction and amyloid pathology (10,11), and interacts with Aβ (13,14) inducing the formation of resistant clots (10,15). Since decreasing systemic fibrin(ogen) levels in AD mice ameliorates disease progression (10,11,16), therapeutics that normalize the pro-thrombotic environment present in AD might be useful in combination with other strategies (17). Indeed, traditional anticoagulants have been reported to be beneficial for dementia patients (18,19) and AD mouse models (20,21). However, to overcome their important limitations, such as the necessity for close monitoring and the high risk of bleeding, non-vitamin K oral anticoagulants (NOACs) have emerged as a useful alternative (22). Among these, dabigatran is a potent oral direct thrombin inhibitor already approved for several indications, such as the prevention of stroke in patients with non-valvular atrial fibrillation and the treatment of venous thromboembolism (23). Dabigatran has minimal drug-drug interactions (22), a low risk of intracranial bleeding (24,25), a potent anti-inflammatory effect (26), and an effective reversal agent available (27).
Here, we present evidence that long-term anticoagulation with dabigatran ameliorates multiple features of AD pathogenesis. Dabigatran treatment preserved memory and cerebral perfusion in transgenic AD mice, which was accompanied by improved BBB integrity, together with lower levels of fibrin, amyloid deposition and neuroinflammatory activity in the AD brain.
Methods
Mice
TgCRND8 AD mice and non-transgenic littermate controls were provided by The Tanz Centre for Research in Neurodegenerative Diseases, University of Toronto, Canada (28). Only female mice were used for this study due to aggressive behavior of TgCRND8 male littermates, and single-housing was not feasible for long-term and behavioral experiments. Seventy female mice were obtained through in vitro fertilization: 37 TgCRND8 and 33 WT mice. Five TgCRND8 mice died before weaning and 8 during placebo/dabigatran treatment, which means a 35% loss in TgCRND8 mice, in line with previous studies (28). Only 1 WT mouse died during placebo treatment. Placebo/dabigatran treatment was also conducted in smaller 30-week-old cohorts, and brains from these mice were extracted and included in the WB and immunohistochemical analyses. Group sizes for all experiments are indicated in each section. Animal protocols were approved by the corresponding IACUC committees.
Dabigatran Treatment.
Two-month-old female TgCRND8 mice and their non-transgenic WT littermates (n=13-18 mice/group) were randomized into groups receiving chow supplemented with 5 mg/g of the prodrug dabigatran etexilate (BIBR 1048) or placebo (both provided by Boehringer Ingelheim, Germany) and were treated until they were 30- (n=8-11 mice/group) or 60- (n=5-7 mice/group) week-old. Diluted thrombin time assays were performed to measure dabigatran plasma concentration (23).
Barnes Maze.
The Barnes maze was used to assess spatial memory in 25-week-old TgCRND8 mice and WT littermates treated with dabigatran/placebo for 17 weeks (n= 16 WT/placebo; n=14 AD/placebo; n=16 WT/dabigatran; n=12 AD/dabigatran mice/group) as described in Supplemental Methods.
Arterial Spin Labeling.
CBF was evaluated non-invasively by ASL-magnetic resonance imaging (MRI) on 40-week-old TgCRND8 mice and their WT littermates treated with dabigatran/placebo for 32 weeks (n= 7 WT/placebo; n=6 AD/placebo; n=6 WT/dabigatran; n=5 AD/dabigatran mice/group) as described in Supplemental Methods.
WB assays.
Soluble and insoluble (fibrin-containing) protein fractions were extracted from the cortex and hippocampus of mice in each experimental group (n=8 WT/placebo/30w; n=9 AD/placebo/30w; n=10 WT/dabigatran/30w; n=11 AD/dabigatran/30w; n=6 WT/placebo/60w; n=6 AD/placebo/60w; n=6 WT/dabigatran/60w; n=5 AD/dabigatran/60w mice/group) and subjected to WB analysis as described in Supplemental Methods.
Mouse Brain Staining and Quantification.
Diaminobenzidine immunohistochemical and double immunofluorescence analyses were performed in free-floating sections as previously described (29). All analyses were performed blindly. Experimental group sizes are the same as described for WB. Details can be found in Supplemental Methods.
Statistics.
All numerical values are shown as mean±standard error of the mean (SEM). Graphs, outlier calculations and statistics were generated using GraphPad Prism 8.0.1 software. Comparison of two groups (indicated by a horizontal line in the corresponding graph) was performed using the Student’s t-test. To determine whether the effect of genotype, age, or treatment was significant, two-way analyses of variance (ANOVA) and Tukey’s post-test for multiple comparisons was performed. Significance is reported as follows: *p≤0.05, **p≤0.01 and ***p≤0.001.
Results
TgCRND8 female AD mice and their non-transgenic littermates were fed chow supplemented with dabigatran etexilate or placebo at 8 weeks-of-age, immediately prior to amyloid deposition (Figure 1). Based on food ingestion per mouse (~5 g), animal weights throughout the study (26.5 g ±4.0), and the low bioavailability of dabigatran etexilate (6.5%) (30), we estimated that mice received an average dose of ~60 mg/kg of dabigatran etexilate over 24 hours. Diluted thrombin time assays showed delayed clot formation in plasma from dabigatran-treated mice (graph in Figure 1), and the average active plasma dabigatran concentration throughout treatment was 141.2±72.5 ng/ml in the WT/dabigatran group and 125.7±64.5 ng/ml in the AD/dabigatran group. During dabigatran/placebo treatment spatial memory and CBF were evaluated by Barnes maze and ASL-MRI, respectively (Figure 1). Mice were sacrificed at 30- or 60-week-old, and tissue was collected for histology and WB (Figure 1).
Figure 1. Outline of the experimental design.
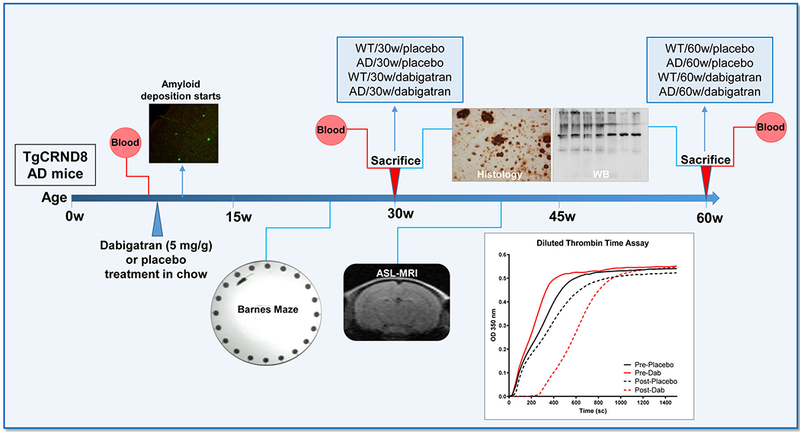
TgCRND8 AD mice and WT littermates (n=13-18 female mice/group) started dabigatran/placebo treatment at 8 weeks-of-age, and were sacrificed after 22 weeks (at 30-week-old, n=8-11 mice/group) or 52 weeks of treatment (at 60-week-old, n=5-7 mice/group). Histology and WB assays were performed in the 8 experimental groups (WT/30w/placebo; AD/30w/placebo; WT/30w/dabigatran; AD/30w/dabigatran; WT/60w/placebo; AD/60w/placebo; WT/60w/dabigatran and AD/60w/dabigatran). Blood was extracted before and after treatment to measure dabigatran concentration by diluted thrombin time assays (representative graph is shown). Barnes maze to assess spatial memory performance was performed 17 weeks after the treatment started (at 25-week-old, n=12-16 mice/group). Cerebral perfusion was measured non-invasively by ASL-MRI 32 weeks after treatment started (at 40-week-old, n=5-7 mice/group). AD=Alzheimer’s disease; ASL=Arterial Spin Labeling, MRI=Magnetic Resonance Imaging, WT=wild type, WB=western blot.
Dabigatran treatment prevents spatial memory decline in TgCRND8 AD mice.
Twenty-five-week-old TgCRND8 AD mice and their WT littermates treated with dabigatran/placebo were tested in the Barnes maze task to evaluate spatial memory. During training, all mice showed comparable decreases in the latency to explore the escape hole, indicating a similar acquisition of spatial learning (Figure 2A). However, during the probe trial, when the escape hole was blocked and spatial memory retention was challenged, TgCRND8 AD mice treated with placebo did not remember where the escape box was and spent similar time near target and non-target holes (Figure 2B). Dabigatran-treated AD mice performed comparably to WT mice, spending significantly more time in target than in non-target areas (Figure 2B), demonstrating that dabigatran treatment in AD mice had a beneficial effect on spatial memory retention.
Figure 2. AD mice treated with dabigatran showed no cognitive decline.
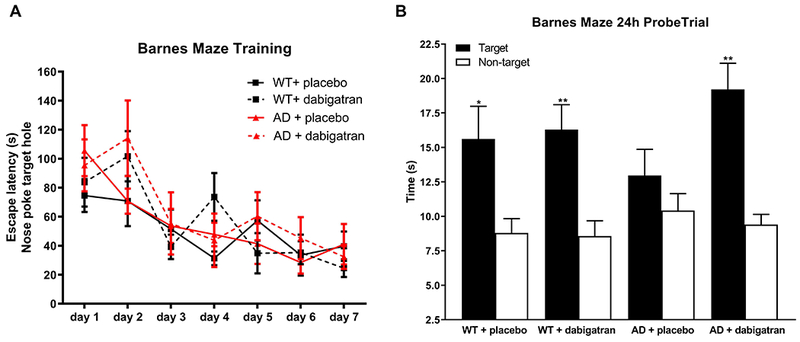
TgCRND8 AD mice and WT controls treated with dabigatran/placebo were subjected to the Barnes maze task to assess spatial memory at 25-week-old. (A) Mice were trained for 7 days, and latency to explore the target hole (nose poke) was measured and plotted. All groups learned the position of the escape box during training. (B) Time spent near the escape and adjoining holes (target quadrant) and average time spent near other holes (non-target quadrants) during the probe trial was measured and plotted. Placebo-treated AD mice did not show a preference for the target area, while dabigatran-treated AD mice explored the target region significantly more than the non-target locations. Graphs show mean±SEM. *p≤0.05, **p≤0.01 target vs. non-target quadrants in each experimental group. n=12-16 mice/group. Abbreviations as in Figure 1.
CBF is preserved in AD mice treated with dabigatran.
ASL-MRI experiments were performed on 40-week-old TgCRND8 and WT mice treated with dabigatran/placebo to quantify CBF (Figure 3A). We detected significant cortical hypoperfusion in TgCRND8 AD mice, with a ~15% decrease in CBF compared to WT littermates (Figure 3B; 100%±4.2 WT/placebo vs. 85.8%±3.4 AD/placebo). This hypoperfusion was prevented by dabigatran treatment (Figure 3B; 85.8%±3.4 AD/placebo vs. 112.7%±9.6 AD/dabigatran).
Figure. 3. Cerebral perfusion was maintained in AD mice treated with dabigatran.
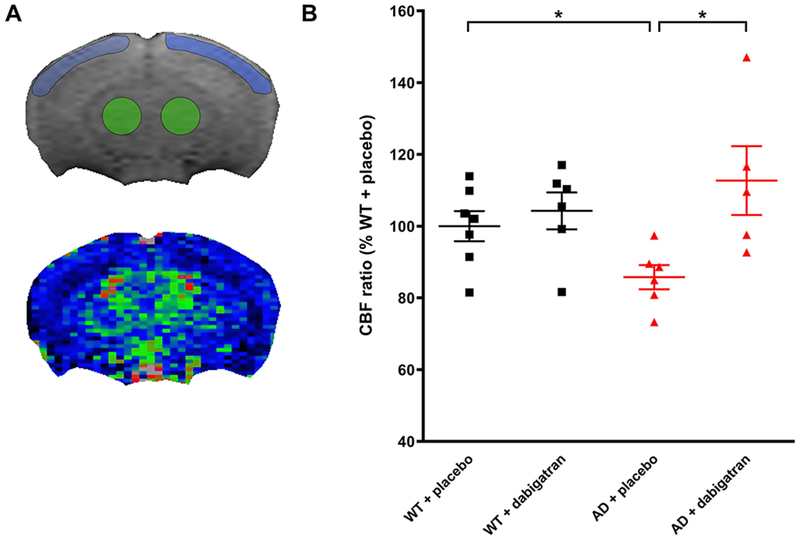
CBF was measured by ASL-MRI in 40-week-old TgCRND8 and WT mice treated with dabigatran/placebo. Anatomical MRI was acquired and used to delineate the cortical (blue) and thalamic (green) ROIs (A, top panel) that were used to measure CBF in the perfusion map (A, bottom panel). (B) AD mice showed a significant decrease in the CBF ratio (cortical CBF/thalamic CBF) compared to WT mice, while dabigatran-treated AD mice presented similar levels of cerebral perfusion as WT mice. CBF ratio of the WT/placebo group average was considered to be 100% and mice from the other groups were represented as relative percentages. Each dot represents an individual mouse. Graph shows mean±SEM. *p≤0.05, n=5-7 mice/group. CBF=cerebral blood flow, ROI=region of interest. Other abbreviations as in Figure 1.
Dabigatran treatment prevents fibrin deposition in the AD brain.
We next compared the amount of insoluble fibrin present in the cortex and hippocampus of 30- and 60-week-old TgCRND8 and WT mice treated with dabigatran/placebo. WB assays showed that brain fibrin deposition increased as mice aged and as AD pathology progressed (Figure 4) (11). However, after long-term anticoagulation with dabigatran there was less fibrin in the AD brain (Figure 4), indicating that inhibition of thrombin prevented pathological fibrin clot formation and accumulation in the mouse brain during the course of the disease.
Figure 4. Dabigatran treatment prevented fibrin deposition in the AD brain.
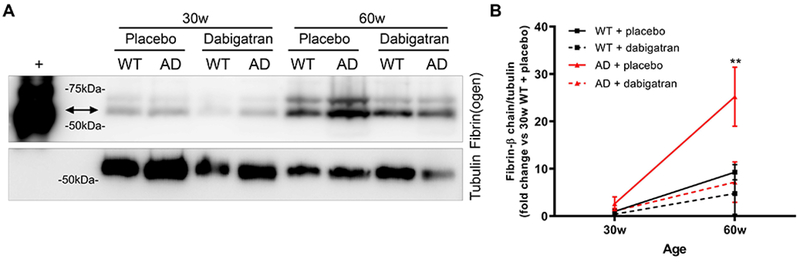
Fibrin was extracted from the cortex/hippocampus of 30- and 60-week-old TgCRND8 AD and WT mice treated with dabigatran/placebo. (A) Equal amounts of the fibrin-containing fraction were pooled and subjected to WB using a specific fibrin(ogen) antibody (top panel), and then re-probed with a tubulin antibody as loading control (bottom panel). An in vitro fibrin clot was used as positive control (+). (B) The amount of the fibrin β-chain (double arrow) and tubulin were quantified in 8 different WBs, averaged, and plotted. Fibrin content in the brain increased with age and AD pathology, but dabigatran treatment prevented brain fibrin deposition in 60-week-old AD mice. Graph shows mean±SEM. Effect of treatment (p<0.0001) and age (p<0.0001) are significant. **p≤0.01 AD/60w/placebo vs. AD/60w/dabigatran. n=5-11 mice/group. Abbreviations as in Figure 1.
Dabigatran treatment ameliorates amyloid burden in AD mice.
The extent of amyloid pathology was measured by immunohistochemistry using the 6E10 Aβ antibody. 6E10 staining showed widespread brain amyloid pathology in 30-week-old TgCRND8 mice that increased at 60 weeks-of-age (Figure 5A). Dabigatran-treated TgCRND8 AD mice also presented Aβ pathology at both ages (Figure 5A), but quantification showed that the cortical amyloid burden was significantly ameliorated by ~24% in the 60-week-old dabigatran group compared to placebo-treated mice (Figure 5B; 3.2%±0.2 AD/60w/placebo vs. 2.5%±0.2 AD/60w/dabigatran).
Figure 5. Long-term anticoagulation with dabigatran ameliorated amyloid burden in AD mice.
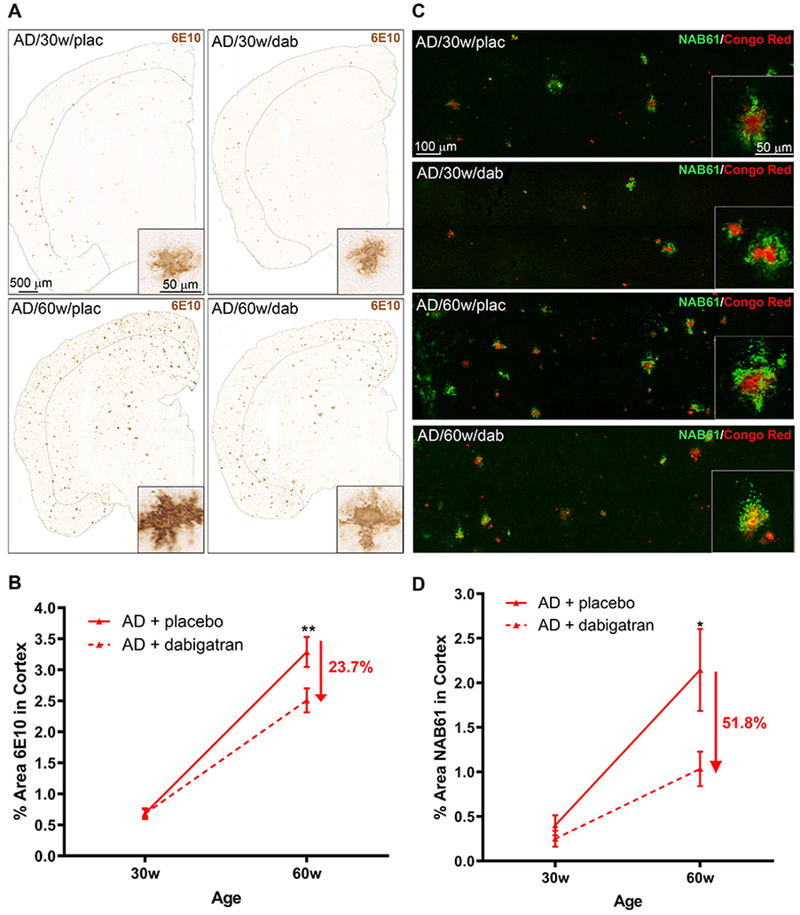
Aβ brain pathology was analyzed in TgCRND8 and WT mice treated with dabigatran/placebo. (A) Aβ immunohistochemistry (6E10) showed widespread amyloid pathology throughout the brains of 30- and 60-week-old TgCRND8 AD mice (insets show high magnification of cortical amyloid plaques). Sections and quantified cortical areas are outlined for clarity. (B) 6E10-positive cortical area was decreased by 23.7% in 60-week-old dabigatran-treated TgCRND8 mice compared to placebo. (C) Aβ oligomer immunofluorescence analysis (NAB61, green) showed staining around fibrillar amyloid plaques (Congo Red, red) in the brains of TgCRND8 mice (see insets for high magnification). Confocal tile-scan Z-stacks were acquired over the parietal cortex and quantification of the NAB61-positive area showed dabigatran treatment decreased by 51.8% the amount of oligomeric Aβ in the cortex of 60-week-old AD mice (D). Graphs show mean±SEM. Effect of treatment (p=0.0089 for 6E10 & p=0.0083 for NAB61) and age (p˂0.0001 for both) are significant. *p≤0.05, **p≤0.01 AD/60w/placebo vs. AD/60w/dabigatran. n=5-11 mice/group. Abbreviations as in Figure 1.
We also analyzed whether oligomers, one of the most toxic species of Aβ (31), were also reduced by dabigatran treatment. Immunostaining using the Aβ-oligomer antibody NAB61 (32) revealed a characteristic halo of oligomeric staining surrounding amyloid plaques in AD mice (Figure 5C) (33). As expected, the amount of Aβ oligomers increased with age in TgCRND8 placebo-treated mice, but this increase was attenuated by 51.8% in dabigatran-treated AD mice (Figure 5D; 2.1%±0.5 AD/60w/placebo vs. 1.0%±0.2 AD/60w/dabigatran). These results provide evidence that long-term treatment with dabigatran significantly decreased amyloid burden and Aβ oligomer pathology in AD mice.
Long-term anticoagulation with dabigatran decreases neuroinflammation.
AD has a robust inflammatory component, mainly orchestrated by microglia and astrocytes (34). CD68 immunohistochemistry showed clusters of phagocytic microglia in the brains of TgCRND8 mice that increased with age and pathological progression (Figure 6A&B; 0.5%±0.1 AD/30w/placebo vs. 1.3%±0.2 AD/60w/placebo). Anticoagulation reduced neuroinflammatory activity as there was a 31% decrease in cortical CD68 levels in 60-week-old dabigatran-treated AD mice compared to placebo (Figure 6A&B; 1.3%±0.2 AD/60w/placebo vs. 0.9%±0.1 AD/60w/dabigatran). We next analyzed whether AD-related astrogliosis was also ameliorated by long-term anticoagulation. WB assays showed that the overall levels of two markers elevated in reactive astrocytes, vimentin and glial fibrillary acidic protein (GFAP), were significantly decreased by 50% and 32.3%, respectively, in TgCRND8 60-week-old dabigatran-treated mice compared to placebo (Figure 6C&D).
Figure 6. Dabigatran treatment ameliorated neuroinflammation in AD mice.
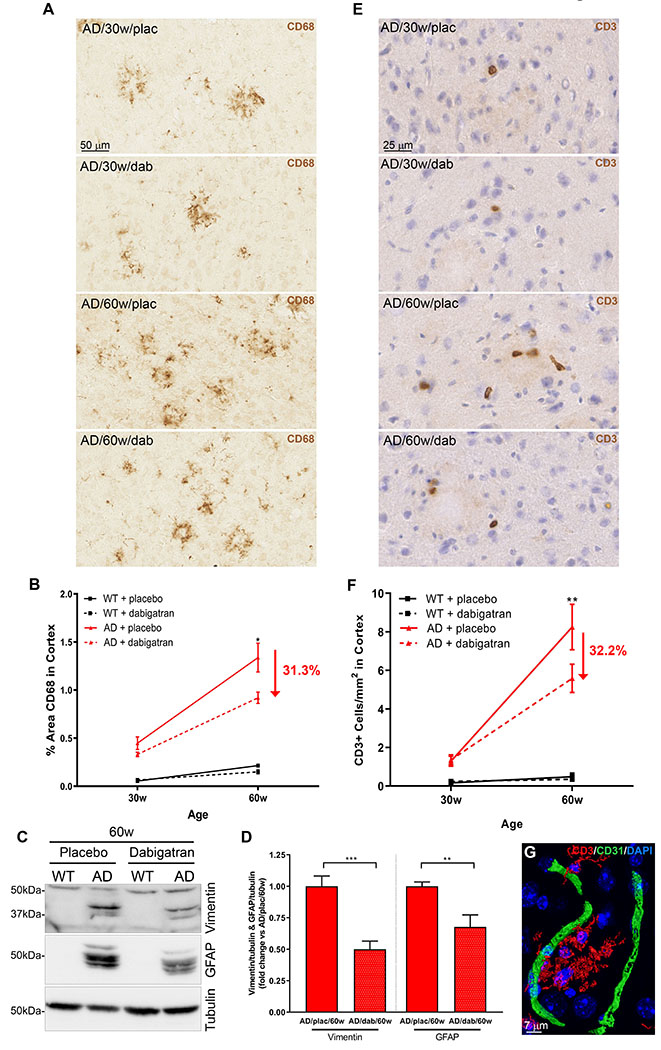
(A) Immunohistochemical analysis showed CD68-positive microglial clusters in 30- and more numerously in 60-week-old AD mice. (B) Quantification of the CD68-positive cortical area showed a 31.3% decrease in the amount of phagocytic microglia in 60-week-old dabigatran-treated AD mice compared to placebo. (C) Soluble proteins from 60-week-old mice were extracted, pooled, and subjected to WB using a vimentin antibody (top panel), and then re-probed with a GFAP antibody (middle panel) and a tubulin antibody (loading control, bottom panel). Six different WBs were quantified, averaged and plotted (D). Degradation products of vimentin and GFAP astrocytic markers drastically increased in AD mice compared to WT mice (53) but their levels were ameliorated in dabigatran-treated AD mice by 50% and 32.3%, respectively. (E) Immunohistochemical staining showed scattered CD3-positive cells in 30-week-old AD brains that increased with age and pathology. (F) Quantification of CD3-positive cells in the cortex showed that infiltrated T cells significantly decreased by 32.2% in dabigatran-treated AD mice compared to placebo. (G) Double immunofluorescence showed that CD3-positive cells (red) were not inside blood vessels (CD31, green) but rather were extravasated into the brain parenchyma. (B&F) The cortical area quantified was similar to the one outlined in the low power images in Figure 5A. Graphs show mean±SEM. The effect of treatment and age were significant (p˂0.0001 for both). *p≤0.05; **p≤0.01; ***p≤0.001 AD/60w/placebo vs. AD/60w/dabigatran. n=5-11 mice/group (A-F) and n=5-6 mice/group (C&D). GFAP=glial fibrillary acidic protein. Other abbreviations as in Figure 1.
Neuroinflammation in AD is also characterized by infiltration of peripheral T cells into the brain (35). We detected an increase in CD3-positive cells inside the brains of TgCRND8 mice compared to non-transgenic controls, and this number was augmented during pathological progression with age (Figure 6E&F; 1.3±0.3 CD3+ cells/mm2 AD/30w/placebo vs. 8.2±1.2 CD3+ cells/mm2 AD/60w/placebo). The majority of CD3-positive cells were extravasated into the brain parenchyma (Figure 6G), and this recruitment to the brain was decreased by 30% in aged AD mice treated with dabigatran (Figure 6E&F; 8.2±1.2 CD3+ cells/mm2 AD/60w/placebo vs. 5.6±0.7 CD3+ cells/mm2 AD/60w/dabigatran). All these results demonstrate that long-term anticoagulation with dabigatran ameliorated AD-related neuroinflammation.
Dabigatran treatment preserved BBB integrity in AD mice.
We next analyzed the expression and localization of AQP4, a water channel selectively present in astrocytic perivascular endfeet and essential for Aβ clearance and neurovascular coupling (36). AQP4 was localized around brain blood vessels in 60-week-old WT mice (Figure 7A&B). However, in age-matched AD mice, astrocyte depolarization was found with AQP4 expression shifting from the astrocytic endfeet surrounding vessels to areas of neuropil near the amyloid plaques (Figure 7A). Quantification showed that perivascular AQP4 decreased by 30% in AD mice compared to age-matched WT controls (Figure 7C; 100%±5.6 WT/60w/placebo vs. 67.4%±2.5 AD/60w/placebo). Dabigatran treatment partially prevented the redistribution of AQP4 expression from endfeet to non-endfeet areas in AD mice (Figure 7C; 67.4%±2.5 AD/60w/placebo vs. 88.0%±3.3 AD/60w/dabigatran).
Figure 7. Dabigatran treatment preserved AQP4 expression and pericyte morphology at the BBB.
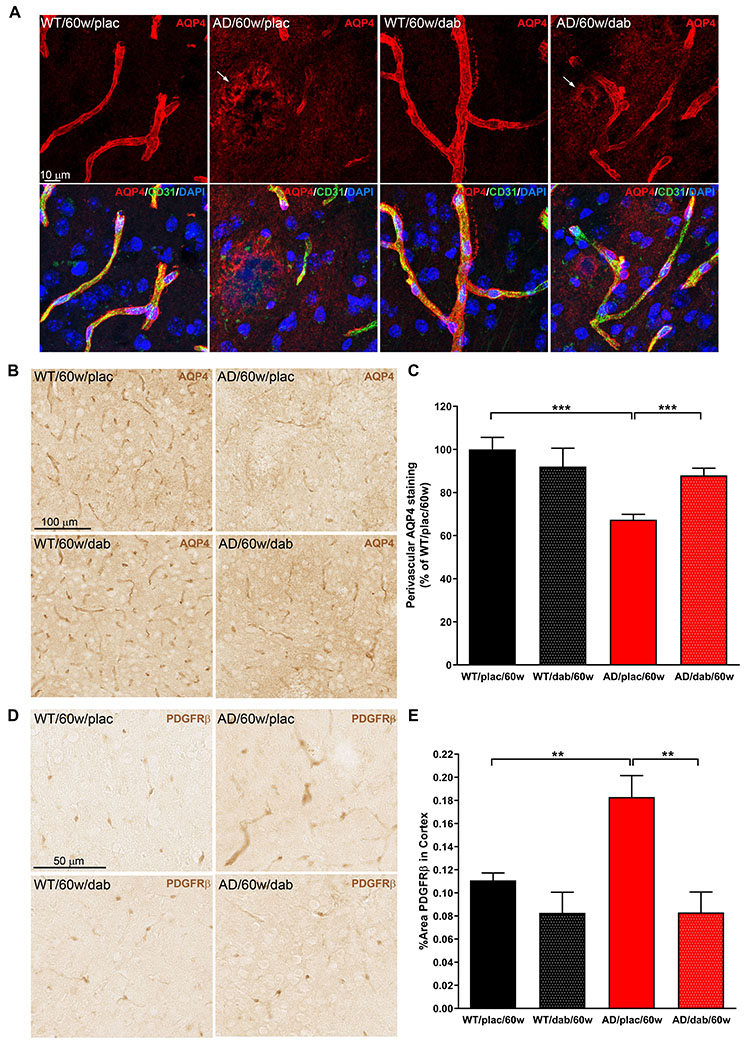
Immunostaining was performed on brain slices from 60-week-old TgCRND8 AD and WT mice treated with dabigatran/placebo to detect expression and localization of AQP4 and PDGFRβ. (A) Double immunofluorescence showed that AQP4 expression (red) was found around blood vessels (CD31, green) in the astrocytic endfeet of WT mice. However, AQP4 staining shifted from its perivascular location to neuropil areas surrounding amyloid plaques in AD mice (arrows). (B) Immunohistochemical analysis was performed to segment and quantify the amount of perivascular AQP4 in all experimental groups (C). AD mice treated long-term with dabigatran presented significantly more perivascular AQP4 staining than placebo-treated AD mice. (D) PDGFRβ immunohistochemistry demonstrated hypertrophic pericytic processes outlining the capillaries of TgCRND8 AD mice. (E) Quantification showed a robust increase of PDGFRβ-positive area in the cortex of 60-week-old AD mice compared to WT mice. This abnormal pericyte staining was normalized by dabigatran treatment. Graphs show mean±SEM. **p≤0.01; ***p≤0.001. n=5-6 mice/group. AQP4=Aquaporin 4; PDGFRβ=Platelet-Derived Growth Factor Receptor-β. Other abbreviations as in Figure 1.
Since pericytes are also an integral part of the BBB, we performed immunohistochemical analysis using the pericyte marker platelet-derived growth factor receptor-β (PDGFRβ). We observed that pericytes in the TgCRND8 AD mice presented abnormal hypertrophic processes surrounding capillaries (Figure 7D), similar to what has been described in other transgenic AD mouse lines (37). These elongated processes significantly increased the overall PDGFRβ-positive area in the cortex of 60-week-old AD mice compared to WT littermates (Figure 7E; 0.11%±0.006 WT/60w/placebo vs. 0.18%±0.02 AD/60w/placebo). Dabigatran treatment ameliorated these alterations in pericyte morphology and normalized the PDGFRβ staining in TgCRND8 AD mice (Figure 7D&E; 0.18%±0.02 AD/60w/placebo vs. 0.08%±0.02 AD/60w/dabigatran).
Taken together, our results indicate that the beneficial effects observed after long-term anticoagulation with dabigatran could be partially due to improved preservation of BBB structure and integrity.
Discussion
Increasing evidence supports a chronic pro-coagulant state in AD with important implications for disease onset and progression. Here, we present behavioral, physiological, and molecular evidence that targeting the thrombotic component of this disorder with a NOAC ameliorates different AD pathological hallmarks in an AD mouse model (Central Illustration). Treatment with dabigatran preserved cerebral perfusion and prevented memory decline in TgCRND8 AD mice. These functional improvements were accompanied in the long-term by inhibition of fibrin deposition, amelioration of amyloid burden and neuroinflammatory activity, and enhanced preservation of the BBB in the AD mouse brain.
Central Illustration. Dabigatran treatment ameliorates AD pathogenesis in the TgCRND8 mouse model.

Long-term treatment with dabigatran prevents fibrin deposition in the AD brain, ameliorating amyloid pathology and neuroinflammation and preserving BBB integrity and CBF. Aβ=amyloid-β; AD=Alzheimer’s disease; BBB=Blood-Brain Barrier.
Pathological accumulation of cerebral fibrin in the brain parenchyma and inside cerebral blood vessels plays a role on AD pathogenesis (10,11,38). Dabigatran treatment prevented the abnormal deposition of fibrin (Figure 4), which may have facilitated the cerebral circulation in the AD mouse brain (Figure 3). This result is remarkable since CBF alterations precede the onset of dementia (39). Additionally, long-term anticoagulation decreased amyloid plaques, and, more importantly, halved levels of Aβ oligomers (Figure 5), the toxic Aβ species recognized as a main contributor to AD synaptic dysfunction (31). Since Aβ strongly interacts with fibrin (15) and fibrinogen (13), and this interaction induces formation of blood clots resistant to degradation (10,15), decreasing the amount of fibrin by long-term anticoagulation may have prevented Aβ binding and subsequent entrapment into degradation-resistant clots.
Hemostatic and non-hemostatic mechanisms were likely involved on dabigatran’s amelioration of AD neuroinflammation (Figure 6). Reduction in fibrin clot formation, and the subsequent improvement in CBF, may have allowed proper nutrient and oxygen delivery to the brain, supporting neural health and function. In addition, dabigatran also inhibits thrombin’s effect on platelet aggregation (23), hence contributing to decrease primary hemostasis and recruitment of white blood cells into the AD brain. Furthermore, thrombin, fibrin, and Aβ act as pro-inflammatory mediators in AD (9,34,40,41). Therefore, the inhibition of thrombin and the resulting decrease in fibrin and Aβ accumulation in the dabigatran-treated AD mice possibly contributed to the amelioration of neuroinflammation. Studies using other AD mouse models have shown that thrombin inhibition diminishes the neuroinflammatory response (40,42), further supporting the role of thrombin in AD-related inflammation.
The BBB is compromised in AD, which translates into neurovascular uncoupling (6), together with leakage of plasma components (e.g. thrombin and fibrin) into the cerebral parenchyma, failure of proper elimination of waste products (e.g Aβ), and infiltration of peripheral immune cells into the brain (5). Long-term anticoagulation with dabigatran ameliorated all of these pathological features, possibly through preservation of pericytes and astrocytic endfeet, integral parts of the BBB (Figure 7). Dabigatran specifically prevented the redistribution of perivascular AQP4, one of the key players in the uptake, clearance, and active transport of Aβ through the BBB (36), which may have facilitated Aβ drainage and contributed to the decrease in amyloid burden (Figure 5). Dabigatran treatment also prevented pericyte alterations in the AD mouse brain. Considering that pericyte degeneration in AD contributes to BBB disruption (43), fibrin extravasation (44) and depolarization of astrocyte endfeet (45), preservation of pericyte structure and/or function by dabigatran may have contributed to the observed reduction in fibrin content in the AD brain (Figure 4) and the maintenance of AQP4 at astrocytic endfeet (Figure 7). The imbalance between Aβ production and clearance through an impaired BBB is considered one of the main pathological processes underlying AD (1). Our results further support the hypothesis that fibrin and altered hemostasis may be one of the missing links between AD and vascular pathology (8,17).
The amelioration of AD-related neuroinflammation, amyloid burden, and BBB dysfunction was observed to be significant only after long-term anticoagulation. Since the pro-coagulant state in AD is a chronic, sustained process, and fibrin deposition accumulates in the brain as pathology advances with age (Figure 4 and (11)), the effect of dabigatran treatment was more robust after longstanding treatment. Indeed, recent epidemiological studies have shown reduced incidence of dementia in atrial fibrillation patients that undergo long-term anticoagulation (46). Interestingly, we also observed that treatment with dabigatran was accompanied by preservation of cognition (Figure 2), which is essential for any molecule to be considered a promising therapeutic approach in AD.
Our results indicate that the normalization of the AD pro-thrombotic state with dabigatran ameliorated AD pathogenesis. We expect that other clinically approved NOACs, such as Factor Xa inhibitors, may have a similar beneficial effect on AD. Repurposing an already approved drug, such as a NOAC, to a new disease indication, such as AD, translates to a quicker transition from bench to bedside and a greater clinical impact.
AD is a multifactorial disease, and efforts should be focused towards the development of multidrug personalized therapies targeting the different mechanisms contributing to an individual’s pathology instead of the “one target, one treatment” approach that has not been successful thus far. Our studies indicate that a pathological mechanism worth targeting in AD is the pro-coagulant state, although other contributing factors should be also treated accordingly.
Study Limitations
One of the limitations of anticoagulation in AD is the increased incidence of intracerebral hemorrhage in AD patients due to the presence of cerebral microbleeds (47) and Aβ deposition in vessels as cerebral amyloid angiopathy (CAA) (48). The incidence of both of these vasculopathies is also linked with worse cognitive performance and the appropriate use of anti-thrombotics in this population is under debate (49). Dabigatran presents a low risk of intracranial bleeding (24,25) and does not promote the formation of cerebral microbleeds in AD mice (50). Furthermore, we observed no hemorrhages or incidents of intracerebral bleeding in any of our animals (data not shown). Nevertheless, use of dabigatran or other anti-thrombotics in AD patients with widespread CAA and cerebral microbleeds will need to be carefully evaluated by a heart-brain team of experts to ensure its use outweighs any bleeding risk (49). Yet, if dabigatran is used early in the course of the disease, Aβ may be cleared more efficiently through the BBB, hence decreasing its deposition in the cortical and leptomeningeal vessels forming CAA. Also, since increased clot formation in AD is a chronic process, long-term treatment with low doses of dabigatran may be sufficient to have a therapeutic effect in AD. The dose used in the present study is lower than what has been used in other animal studies (26,42,51). Since the half-life of dabigatran etexilate in mice is short (~15 min) and dabigatran is two-fold less effective inhibiting mouse than human thrombin (23), a supra-therapeutic dose in mice is needed to achieve similar plasma concentrations of the drug as in humans. Although further studies are required to carefully extrapolate the long-term anticoagulation achieved in our AD mice to humans, the dose we used rendered plasma dabigatran concentrations similar to what has been reported in patients undergoing long-term anticoagulation (52). Additionally, even though dabigatran presents a predictable pharmacokinetic profile (22), long-term anticoagulation in the frail, aging and comorbid AD population may benefit from a drug-tailored program.
Conclusions
Long-term anticoagulation with dabigatran inhibits thrombin and the abnormal deposition of fibrin in the AD brain, hence preserving blood flow and facilitating oxygen and nutrient delivery to the brain. This drug, in turn, has a beneficial effect on controlling neuroinflammation, maintaining the integrity and functionality of the BBB, and facilitating Aβ clearance (Central Illustration). Therefore, therapeutics aimed at normalizing the pro-thrombotic environment present in AD, in combination with other disease-modifying compounds, might be instrumental in improving AD pathogenesis.
Supplementary Material
Perspectives.
Competency In Medical Knowledge And Patient Care
AD is a multifactorial disease, and efforts should be focused toward development of combinational and individualized therapies targeting the different mechanisms contributing to pathology. Our studies indicate that one of the mechanisms worth targeting in AD is the pro-coagulant state.
Translational Outlook
This is a study performed in a transgenic AD mouse line. Additional studies in other rodents and in larger animal models will support the beneficial role of anti-thrombotics as treatment for AD patients.
Acknowledgements:
The authors thank the Centre for Research in Neurodegenerative Diseases at University of Toronto (Toronto, Canada) and the Center for Neurodegenerative Disease Research at University of Pennsylvania (Philadelphia, PA, USA) for kindly providing the TgCRND8 mice and the NAB61 antibody, respectively. The 7-Tesla MRI used is part of the ReDiB from Infraestructuras Científico-Técnico Singulares (ICTS). Authors also thank Dr. Jay L. Degen for supplying the fibrin(ogen) antibody, Drs. Ashley Goss and Joanne Van Ryn from Boehringer Ingelheim Pharma for providing the chow and information regarding dabigatran’s mechanism of action, Marina Saiz for assisting during her CICERONE summer project, Carlos Galan-Arriola for his invaluable help with the Central Illustration and CNIC Histopathology and Microscopy Units for their support with brain staining.
Funding:
Proof-of-Concept Award from the Robertson Therapeutic Development Fund, The Rockefeller University, New York, US.
NINDS/NIH (NIS106668), The Rockefeller University, New York, US.
European Union’s Seventh Framework Programme (FP7-PEOPLE-2013-IIF). Grant agreement n° PIIF-GA-2013-624811, CNIC, Madrid, Spain.
The CNIC is supported by the Instituto de Salud Carlos III (ISCIII), the Spanish Ministerio de Ciencia, Innovación y Universidades (MCNU) and the Pro CNIC Foundation, and is a Severo Ochoa Center of Excellence (SEV-2015-0505). CIC biomaGUNE is a Maria de Maeztu Unit of Excellence (MDM-2017-0720).
Miguel Servet type I research contract (CP16/00174 & MS16/00174), ISCIII, CNIC, Madrid, Spain.
Iniciativa de Empleo Juvenil (PEJ16/MED/TL-1231 & PEJ-2018-AI/BMD-11477) from Consejería de Educación, Juventud y Deporte de la Comunidad de Madrid. CNIC, Madrid, Spain.
European Regional Development Funds (FEDER “Una manera de hacer Europa”) & European Social Funds (FSE “El FSE invierte en tu futuro”). CNIC, Madrid, Spain.
Abbreviations and Acronyms
- Aβ
Amyloid-β
- AD
Alzheimer’s Disease
- AQP4
Aquaporin 4
- ASL
Arterial Spin Labeling
- BBB
Blood-Brain Barrier
- CAA
Cerebral Amyloid Angiopathy
- CBF
Cerebral Blood Flow
- GFAP
Glial Fibrillary Acidic Protein
- MRI
Magnetic Resonance Imaging
- NOAC
Non-vitamin K Oral Anticoagulant
- PDGFRβ
Platelet-Derived Growth Factor Receptor-β
- ROI
Regions of Interest
- WB
Western Blot
- WT
Wild Type
Footnotes
Disclosures: Javier Sanchez-Gonzalez is a full-time Philips employee. The remaining authors have nothing to disclose.
References
- 1.Lane CA, Hardy J, Schott JM. Alzheimer’s disease. Eur J Neurol 2018;25:59–70. [DOI] [PubMed] [Google Scholar]
- 2.Iadecola C, Gottesman R. Cerebrovascular Alterations in Alzheimer Disease: Incidental or Pathogenic? Circ Res 2018;123:406–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Picano E, Bruno RM, Ferrari GF, Bonuccelli U. Cognitive impairment and cardiovascular disease: so near, so far. Int J Cardiol 2014;175:21–9. [DOI] [PubMed] [Google Scholar]
- 4.de la Torre JC. Alzheimer’s Turning Point A Vascular Approach to Clinical Prevention. 1 ed: Springer International Publishing, 2016. [Google Scholar]
- 5.Sweeney MD, Sagare AP, Zlokovic BV. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol 2018;14:133–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kisler K, Nelson AR, Montagne A, Zlokovic BV. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat Rev Neurosci 2017;18:419–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iadecola C The pathobiology of vascular dementia. Neuron 2013;80:844–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cortes-Canteli M, Zamolodchikov D, Ahn HJ, Strickland S, Norris EH. Fibrinogen and altered hemostasis in Alzheimer’s disease. J Alzheimers Dis 2012;32:599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zamolodchikov D, Strickland S. A possible new role for Abeta in vascular and inflammatory dysfunction in Alzheimer’s disease. Thromb Res 2016;141 Suppl 2:S59–61. [DOI] [PubMed] [Google Scholar]
- 10.Cortes-Canteli M, Paul J, Norris EH et al. Fibrinogen and beta-amyloid association alters thrombosis and fibrinolysis: a possible contributing factor to Alzheimer’s disease. Neuron 2010;66:695–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cortes-Canteli M, Mattei L, Richards AT, Norris EH, Strickland S. Fibrin deposited in the Alzheimer’s disease brain promotes neuronal degeneration. Neurobiol Aging 2015;36:608–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kitamura Y, Usami R, Ichihara S et al. Plasma protein profiling for potential biomarkers in the early diagnosis of Alzheimer’s disease. Neurol Res 2017;39:231–8. [DOI] [PubMed] [Google Scholar]
- 13.Ahn HJ, Zamolodchikov D, Cortes-Canteli M, Norris EH, Glickman JF, Strickland S. Alzheimer’s disease peptide {beta}-amyloid interacts with fibrinogen and induces its oligomerization. Proc Natl Acad Sci U S A 2010;107:21812–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zamolodchikov D, Berk-Rauch HE, Oren DA et al. Biochemical and structural analysis of the interaction between β-amyloid and fibrinogen. Blood 2016;128:1144–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zamolodchikov D, Strickland S. Abeta delays fibrin clot lysis by altering fibrin structure and attenuating plasminogen binding to fibrin. Blood 2012;119:3342–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paul J, Strickland S, Melchor JP. Fibrin deposition accelerates neurovascular damage and neuroinflammation in mouse models of Alzheimer’s disease. J Exp Med 2007;204:1999–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Strickland S Blood will out: vascular contributions to Alzheimer’s disease. The Journal of Clinical Investigation 2018;128:556–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walsh AC, Walsh BH, Melaney C. Senile-presenile dementia: follow-up data on an effective psychotherapy-anticoagulant regimen. J Am Geriatr Soc 1978;26:467–70. [DOI] [PubMed] [Google Scholar]
- 19.Ratner J, Rosenberg G, Kral VA, Engelsmann F. Anticoagulant therapy for senile dementia. J Am Geriatr Soc 1972;20:556–9. [DOI] [PubMed] [Google Scholar]
- 20.Bergamaschini L, Rossi E, Storini C et al. Peripheral treatment with enoxaparin, a low molecular weight heparin, reduces plaques and beta-amyloid accumulation in a mouse model of Alzheimer’s disease. J Neurosci 2004;24:4181–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Timmer NM, van Dijk L, van der Zee CE, Kiliaan A, de Waal RM, Verbeek MM. Enoxaparin treatment administered at both early and late stages of amyloid beta deposition improves cognition of APPswe/PS1dE9 mice with differential effects on brain Abeta levels. Neurobiol Dis 2010;40:340–7. [DOI] [PubMed] [Google Scholar]
- 22.Shameem R, Ansell J. Disadvantages of VKA and requirements for novel anticoagulants. Best Practice & Research Clinical Haematology 2013;26:103–14. [DOI] [PubMed] [Google Scholar]
- 23.van Ryn J, Goss AM, Hauel N et al. The Discovery of Dabigatran Etexilate. Front Pharmacol 2013;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garnock-Jones KP. Dabigatran etexilate: a review of its use in the prevention of stroke and systemic embolism in patients with atrial fibrillation. Am J Cardiovasc Drugs 2011;11:57–72. [DOI] [PubMed] [Google Scholar]
- 25.Hart RG, Diener HC, Yang S et al. Intracranial hemorrhage in atrial fibrillation patients during anticoagulation with warfarin or dabigatran: the RE-LY trial. Stroke 2012;43:1511–7. [DOI] [PubMed] [Google Scholar]
- 26.Bogatkevich GS, Ludwicka-Bradley A, Nietert PJ, Akter T, van Ryn J, Silver RM. Antiinflammatory and antifibrotic effects of the oral direct thrombin inhibitor dabigatran etexilate in a murine model of interstitial lung disease. Arthritis Rheum 2011;63:1416–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pollack CV, Jr., Reilly PA, van Ryn J et al. Idarucizumab for Dabigatran Reversal - Full Cohort Analysis. N Engl J Med 2017;377:431–41. [DOI] [PubMed] [Google Scholar]
- 28.Chishti MA, Yang DS, Janus C et al. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem 2001;276:21562–70. [DOI] [PubMed] [Google Scholar]
- 29.Cortes-Canteli M, Luna-Medina R, Sanz-Sancristobal M, Alvarez-Barrientos A, Santos A, Perez-Castillo A. CCAAT/enhancer binding protein beta deficiency provides cerebral protection following excitotoxic injury. J Cell Sci 2008;121:1224–34. [DOI] [PubMed] [Google Scholar]
- 30.Stangier J, Clemens A. Pharmacology, Pharmacokinetics, and Pharmacodynamics of Dabigatran Etexilate, an Oral Direct Thrombin Inhibitor. Clinical and Applied Thrombosis/Hemostasis 2009;15:9S–16S. [DOI] [PubMed] [Google Scholar]
- 31.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol 2007;8:101–12. [DOI] [PubMed] [Google Scholar]
- 32.Lee EB, Leng LZ, Zhang B et al. Targeting amyloid-beta peptide (Abeta) oligomers by passive immunization with a conformation-selective monoclonal antibody improves learning and memory in Abeta precursor protein (APP) transgenic mice. J Biol Chem 2006;281:4292–9. [DOI] [PubMed] [Google Scholar]
- 33.Koffie RM, Meyer-Luehmann M, Hashimoto T et al. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A 2009;106:4012–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heneka MT, Carson MJ, Khoury JE et al. Neuroinflammation in Alzheimer’s disease. The Lancet Neurology 2015;14:388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jimenez S, Baglietto-Vargas D, Caballero C et al. Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer’s disease: age-dependent switch in the microglial phenotype from alternative to classic. J Neurosci 2008;28:11650–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang C, Huang X, Huang X et al. Aquaporin-4 and Alzheimer’s Disease. J Alzheimers Dis 2016;52:391–402. [DOI] [PubMed] [Google Scholar]
- 37.Park L, Zhou J, Zhou P et al. Innate immunity receptor CD36 promotes cerebral amyloid angiopathy. Proc Natl Acad Sci U S A 2013;110:3089–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Canobbio I, Visconte C, Oliviero B et al. Increased platelet adhesion and thrombus formation in a mouse model of Alzheimer’s disease. Cell Signal 2016;28:1863–71. [DOI] [PubMed] [Google Scholar]
- 39.Wierenga CE, Hays CC, Zlatar ZZ. Cerebral blood flow measured by arterial spin labeling MRI as a preclinical marker of Alzheimer’s disease. J Alzheimers Dis 2014;42 Suppl 4:S411–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tripathy D, Sanchez A, Yin X, Luo J, Martinez J, Grammas P. Thrombin, a mediator of cerebrovascular inflammation in AD and hypoxia. Front Aging Neurosci 2013;5:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Petersen MA, Ryu JK, Akassoglou K. Fibrinogen in neurological diseases: mechanisms, imaging and therapeutics. Nat Rev Neurosci 2018;19:283–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Marangoni MN, Braun D, Situ A et al. Differential effects on glial activation by a direct versus an indirect thrombin inhibitor. J Neuroimmunol 2016;297:159–68. [DOI] [PubMed] [Google Scholar]
- 43.Winkler EA, Sagare AP, Zlokovic BV. The pericyte: a forgotten cell type with important implications for Alzheimer’s disease? Brain Pathol 2014;24:371–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sengillo JD, Winkler EA, Walker CT, Sullivan JS, Johnson M, Zlokovic BV. Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer’s disease. Brain Pathol 2013;23:303–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Armulik A, Genové G, Mäe M et al. Pericytes regulate the blood-brain barrier. Nature 2010;468:557. [DOI] [PubMed] [Google Scholar]
- 46.Friberg L, Rosenqvist M. Less dementia with oral anticoagulation in atrial fibrillation. Eur Heart J 2018;39:453–60. [DOI] [PubMed] [Google Scholar]
- 47.Martinez-Ramirez S, Greenberg SM, Viswanathan A. Cerebral microbleeds: overview and implications in cognitive impairment. Alzheimers Res Ther 2014;6:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Banerjee G, Carare R, Cordonnier C et al. The increasing impact of cerebral amyloid angiopathy: essential new insights for clinical practice. J Neurol Neurosurg Psychiatry 2017;88:982–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DeSimone CV, Graff-Radford J, El-Harasis MA, Rabinstein AA, Asirvatham SJ, Holmes DR, Jr. Cerebral Amyloid Angiopathy: Diagnosis, Clinical Implications, and Management Strategies in Atrial Fibrillation. J Am Coll Cardiol 2017;70:1173–82. [DOI] [PubMed] [Google Scholar]
- 50.Marinescu M, Sun L, Fatar M et al. Cerebral Microbleeds in Murine Amyloid Angiopathy: Natural Course and Anticoagulant Effects. Stroke 2017;48:2248–54. [DOI] [PubMed] [Google Scholar]
- 51.Kadoglou NP, Moustardas P, Katsimpoulas M et al. The beneficial effects of a direct thrombin inhibitor, dabigatran etexilate, on the development and stability of atherosclerotic lesions in apolipoprotein E-deficient mice : dabigatran etexilate and atherosclerosis. Cardiovasc Drugs Ther 2012;26:367–74. [DOI] [PubMed] [Google Scholar]
- 52.Reilly PA, Lehr T, Haertter S et al. The effect of dabigatran plasma concentrations and patient characteristics on the frequency of ischemic stroke and major bleeding in atrial fibrillation patients: the RE-LY Trial (Randomized Evaluation of Long-Term Anticoagulation Therapy). J Am Coll Cardiol 2014;63:321–8. [DOI] [PubMed] [Google Scholar]
- 53.Porchet R, Probst A, Bouras C, Draberova E, Draber P, Riederer BM. Analysis of glial acidic fibrillary protein in the human entorhinal cortex during aging and in Alzheimer’s disease. Proteomics 2003;3:1476–85. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.