

Abstract
Fibrosis is the major determinant of morbidity and mortality in patients with nonalcoholic steatohepatitis (NASH) but has no approved pharmacotherapy in part because of incomplete understanding of its pathogenic mechanisms. Here, we report that hepatocyte Notch activity tracks with disease severity and treatment response in patients with NASH and is similarly increased in a mouse model of diet-induced NASH and liver fibrosis. Hepatocyte-specific Notch loss-of-function mouse models showed attenuated NASH-associated liver fibrosis, demonstrating causality to obesity-induced liver pathology. Conversely, forced activation of hepatocyte Notch induced fibrosis in both chow- and NASH diet–fed mice by increasing Sox9-dependent Osteopontin (Opn) expression and secretion from hepatocytes, which activate resident hepatic stellate cells. In a cross-sectional study, we found that OPN explains the positive correlation between liver Notch activity and fibrosis stage in patients. Further, we developed a Notch inhibitor [Nicastrin antisense oligonucleotide (Ncst ASO)] that reduced fibrosis in NASH diet–fed mice. In summary, these studies demonstrate the pathological role and therapeutic accessibility of the maladaptive hepatocyte Notch response in NASH-associated liver fibrosis.
INTRODUCTION
Obesity and its metabolic consequences are among the most pressing public health challenges (1, 2). As the obese population at risk increases (3), the prevalence of obesity-related comorbidities such as nonalcoholic fatty liver disease (NAFLD), already the most common chronic liver disease (4), grows in parallel. NAFLD ranges in severity from simple steatosis (SS) to hepatocellular injury and necroinflammatory changes that define nonalcoholic steatohepatitis (NASH), which predisposes patients to fibrosis and hepatocellular carcinoma (5). Whereas SS is considered a predisease state and NASH is potentially reversible (6), fibrosis severity is the main determinant of mortality in patients with NAFLD (7–10). Because organ availability for transplantation is already limited, new pharmaceutical targets to address NASH-associated fibrosis are a large unmet need for an increasingly obese population.
The Notch family of transmembrane receptors (Notch1–4) determines cell fate during development (11) through ligand binding and γ-secretase–mediated cleavage that generates Notch intracellular domain (NICD) (12), which binds Rbp-Jκ and Mastermind (MAM) to activate transcription of canonical Notch targets including the Hairy enhancer of split (Hes) and Hes-related (Hey) family genes (13). In the liver, Notch activation pushes hepatic progenitor cells’ differentiation to cholangiocytes and formation of the biliary tree, whereas Notch-inactive progenitors commit to hepatocyte lineage (14), which sets the quiescent baseline of Notch signaling in hepatocytes in normal liver. However, we found that liver Notch activity is increased in obese rodents (15) and in patients with SS or NASH (16), although the relative contributions of hepatocyte and nonparenchymal cell (NPC) Notch activity were unclear.
Here, we found that the number of HES1+ hepatocytes, but not of NPCs, is increased in patients with NASH. In longitudinal analysis using paired baseline and end-of-treatment biopsy specimens from the Pioglitazone versus Vitamin E versus Placebo for the Treatment of Nondiabetic Patients with Nonalcoholic Steatohepatitis (PIVENS) trial ( 17, 18), we found that Notch activity, specifically in hepatocytes, tracks with NASH severity. These data suggested that hepatocyte Notch activation may be a biomarker or a causal determinant of NASH severity. To distinguish these possibilities, we used a palmitate/cholesterol-rich diet coupled with fructose-containing drinking water (19) fed to wild-type (WT) mice to induce NASH/fibrosis, which increased hepatocyte but not NPC Notch activation. Two genetic hepatocyte-specific Notch loss-of-function mouse models showed lower hepatic stellate cell (HSC) activity and liver collagen deposition without changes in hepatocellular injury or inflammation, whereas forced activation of hepatocyte Notch induced fibrosis in chow-fed mice because of Sox9 (sex determining region Y-box 9)–dependent expression of Spp1 (encoding the secreted fibrogenic factor Opn). Treating NASH diet–fed WT mice with a Notch antagonist [Nicastrin antisense oligonucleotide (Ncst ASO)] decreased liver fibrosis. In sum, these data support the necessity and therapeutic tractability of hepatocyte Notch signaling for the development of NASH-associated liver fibrosis.
RESULTS
Hepatocyte Notch activity tracks with NASH severity in patients
Steady-state liver Notch activation is associated with higher serum alanine aminotransferase and a NAFLD activity score (NAS), which are biochemical and pathologic markers of NASH, independent of steatosis or insulin resistance (16). To test relative hepatocyte and NPC contribution, we stained liver biopsy specimens taken at the time of bariatric surgery from patients with pathologically normal livers, SS, or NASH/fibrosis. We observed detectable basal NPC expression of HES1, which, although modestly increased in SS, did not further change in NASH/fibrosis (Fig. 1A and fig. S1). Hepatocyte HES1, however, was nearly absent in normal livers, mildly elevated in SS, but highly increased in patients with NASH/fibrosis (Fig. 1A).
Fig. 1. Hepatocyte Notch activation in NASH.

(A) Representative images of HES1 (red) and hepatocyte nuclear factor 4α (HNF4α; green) immunofluorescence in liver biopsies from patients with histologically normal liver, SS, or NASH/fibrosis and quantification of the percentage of HES1+ cells among HNF4α+ hepatocytes and HNF4α− nonhepatocytes (n = 3 to 4 per group). (B) Expression of Notch target genes HES1 and HEYL after 96 weeks of treatment in nonresponders (n = 49) or responders (n = 69) from the PIVENS trial. (C) Liver HES1 and (D) HEYL expression in paired baseline and 96-week end-of-treatment liver biopsy specimens from PIVENS subjects (n = 10 to 11 per group) and (E) percentage change of HES1+/HNF4α+ hepatocytes and HES1+/HNF4− NPCs in nonresponders (NR) and responders (R) from baseline to end of treatment (n = 7 per group). (F) Hes1 expression in liver, fractionated hepatocytes, and NPCs. (G) Representative images of Hes1 and HNF4α immunofluorescence in chow- and NASH diet–fed WT mouse livers and quantification of the percentage of hepatocytes with nuclear Hes1 staining (n = 9 per group). (H) Expression of Notch receptors in hepatocytes and (I) Western blots of Notch1 and Notch2 intracellular domains (ICDs) in livers from chow- and NASH diet–fed WT mice. (J) Quantification of Venus+ hepatocytes in livers from chow- and NASH diet–fed Notch reporter mice (n = 5 per group). *P < 0.05, **P < 0.01, and ***P < 0.001 as compared to the indicated controls by two-tailed t tests (two groups) or one-way analysis of variance (ANOVA), followed by post hoc t tests (three groups). All data are shown as the means ± SEM. NS, not significant; AU, arbitrary units.
Next, to determine whether liver Notch activity may covary with NASH severity and treatment response, we performed parallel cross-sectional and longitudinal analyses in samples derived from the PIVENS trial. PIVENS investigators randomized adult, nondiabetic patients with NASH to placebo, vitamin E (800 IU daily), or pioglitazone (30 mg daily) arms, taking liver biopsies at enrollment and end-of-treatment (96 weeks) visits (20). A “responder” was defined as an NAS response of at least 2 points or resolution of NASH. In a cross-sectional analysis of 118 samples (table S1), responders showed lower liver Notch activity than nonresponders at the end of the trial (Fig. 1B). Next, in a longitudinal analysis from a subset of PIVENS subjects that had paired baseline and end-of-treatment complementary DNA (table S1), we found a reduction from baseline HES1 and HEYL expression in responders but not in nonresponders (Fig. 1, C and D). Last, we found reduced hepatocyte, but not NPC, HES1 staining in liver sections from PIVENS responders from baseline and as compared to nonresponders (Fig. 1E). In sum, these data suggest that liver Notch activity, specifically in hepatocytes, tracks with NASH disease severity.
Hepatocyte Notch activity is increased in a mouse model of diet-induced NASH
As a first step toward determining whether a causal relationship exists between Notch activity and NASH, we fed WT C57BL/6J mice a NASH-provoking diet for 16 weeks, which induced hepatic steatosis, inflammation, and fibrosis as compared to chow-fed mice (fig. S2) (19). As in patients with NASH/fibrosis, we found higher liver and hepatocyte, but not NPC, Hes1 in NASH diet–fed mice (Fig. 1F), reflecting an increased number of Hes1+ hepatocytes and increased Hes1 staining intensity per cell (Fig. 1G), consistent with increased expression and activation of Notch1 and Notch2 (Fig. 1, H and I). In parallel, using a Notch reporter mouse that expresses the fluorescent protein Venus under the control of four Rbp-jκ binding sites (21), we found a large increase in Venus+ hepatocytes in NASH diet–fed mice (Fig. 1J).
Hepatocyte Notch mediates NASH diet–induced liver fibrosis
The positive correlations between hepatocyte Notch activation and NASH severity provided the impetus to generate hepatocyte-specific Notch loss-of-function mice to test causality to the phenotype. Because the MAM transcriptional coactivator is downstream of all four Notch receptors, we transduced mice with a dominant-negative MAM (DNMAM) allele (22) in the Rosa26 locus (RosaDNMAM) with AAV8-Tbg-Cre to take advantage of liver tropism of AAV8 and the hepatocyte-specific thyroxine-binding globulin (Tbg) promoter (23, 24). This approach generated mice with postdevelopment hepatocyte-specific expression of DNMAM (L-DNMAM mice; Fig. 2A). After 16 weeks of NASH diet, L-DNMAM mice showed similar body weight and adiposity (fig. S3, A and B) as compared with control RosaDNMAM mice transduced with AAV8-TBG-LacZ but lower liver Hes1 expression (Fig. 2B), fewer Hes1+ hepatocytes, and weaker Hes1 staining intensity per cell (Fig. 2C and fig. S3C). Despite unchanged liver lipid accumulation, terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) staining, inflammatory gene expression, CD45+ immune cell infiltrate, and serum transaminases (Fig. 2D and fig. S3, D to I), markers of HSC activation (Col1a1 , Acta2, and Timp1) were decreased (Fig. 2E), which translated to less collagen deposition in L-DNMAM livers (Fig. 2F).
Fig. 2. Hepatocyte Notch blockade ameliorates NASH-associated fibrosis.
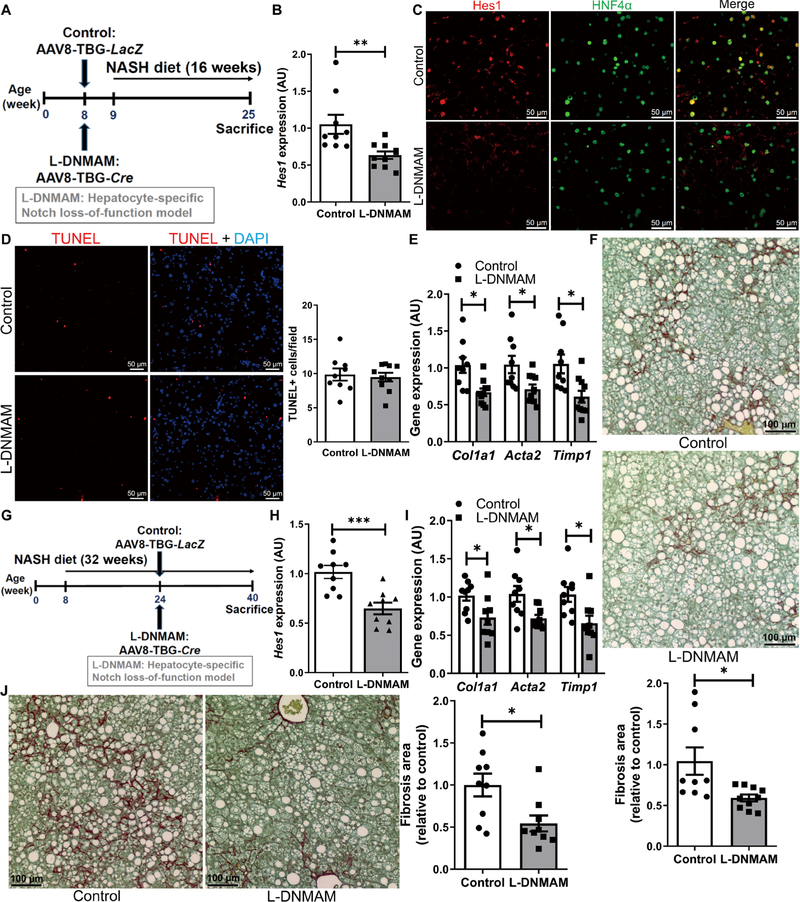
(A) RosaDNMAM mice (8 weeks old) were transduced with AAV8-TBG-LacZ (control) or AAV8-TBG-Cre to generate L-DNMAM mice and then fed for 16 weeks with NASH diet (n = 9 to 10 per group). (B) Hes1 expression and (C) representative images of Hes1 (red) and HFN4α (green) staining, (D) TUNEL (red) staining and quantification, (E) expression of fibrogenic genes, and (F) liver collagen staining and quantification in livers from Cre− control and L-DNMAM mice. DAPI, 4′,6-diamidino-2-phenylindole. (G) RosaDNMAM mice (8 weeks old) were fed with NASH diet for 32 weeks with AAV8-TBG-LacZ or AAV8-TBG-Cre transduction in the 16th week (n = 9 per group). (H) Hes1 and (I) fibrogenic gene expression, and (J) collagen staining and quantification in livers from Cre− controls and mice with delayed expression of DNMAM. *P < 0.05, **P < 0.01, and ***P < 0.001 as compared to the indicated controls by two-tailed t tests (two groups). All data are shown as the means ± SEM.
Next, to ensure reproducibility of these results, we ablated hepatocyte Nicastrin (25), the γ-secretase targeting subunit necessary for ligand-dependent Notch activation (L-Ncst mice) (26). Similar to L-DNMAM mice, NASH diet–fed L-Ncst mice showed normal body weight and adiposity (fig. S4, A and B), but reduced hepatic Hes1 (fig. S4C) and fibrogenic gene expression (fig. S4D), and a tendency toward less fibrosis (fig. S4E) as compared to Cre− controls, despite unchanged hepatic TUNEL staining or serum transaminases, liver inflammation, and only minor changes in liver lipid content (fig. S4, F to L).
These data suggest that hepatocyte Notch signaling is necessary for the full development of NASH diet–induced fibrosis. To test whether a late inhibition of Notch in hepatocytes may ameliorate NASH-associated fibrosis, we fed adult RosaDNMAM mice the NASH diet for 16 weeks before AAV8-Tbg-Cre transduction (Fig. 2G). In this experimental paradigm, we again observed decreased liver Hes1 (Fig. 2H) and a parallel reduction in HSC-mediated liver fibrosis (Fig. 2, I and J) despite unchanged body weight, adiposity, liver steatosis, inflammation, TUNEL staining, or serum transaminases (fig. S5, A to I). These data indicate that inhibition of hepatocyte Notch signaling can interrupt NASH-associated fibrosis even after fibrosis has begun to develop.
Hepatocyte Notch is not required for MCD diet–induced fibrosis
Acute toxin exposure increases Notch activity in HSCs and immune cells (27–29), contrary to what we find in fluorescence-activated cell sorter (FACS)–purified HSCs and myeloid cells isolated from NASH diet–fed mice (fig. S6A). Similarly, we observed that a methionine-choline deficient (MCD) diet, which induces severe liver injury, inflammation, and fibrosis in addition to anorexia and rapid weight loss (30, 31), led to increased liver Hes1 expression relative to chow-fed controls (fig. S6B). This increased Hes1 expression was predominantly seen in NPCs (fig. S6C), unlike in NASH diet feeding (fig. S6D). These data are consistent with a recent study showing un-changed expression of Notch targets in hepatocytes in MCD diet-fed mice (32) and with our finding that MCD diet–fed L-DNMAM mice show no difference in liver Hes1 and fibrogenic gene expression or collagen deposition (fig. S6, E to H), suggesting specificity of the hepatocyte Notch response in NASH.
Forced hepatocyte Notch activation exacerbates diet-induced NASH and fibrosis
We next evaluated whether forced hepatocyte Notch activation by AAV8-TBG-Cre transduction of mice that carry a Cre-inducible constitutively active (RosaNICD) Notch1 (33) may exacerbate NASH/fibrosis (L-NICD-16wks, Fig. 3A). In parallel, because 8 weeks of NASH diet provoke steatosis but no apparent fibrosis (19), we transduced RosaNICD mice with AAV8-TBG-Cre halfway through the diet study (L-NICD-8wks, Fig. 3A) to determine whether hepatocyte Notch activity promotes the transition from steatosis to NASH/fibrosis. As compared to AAV8-TBG-LacZ–transduced RosaNICD controls, both L-NICD groups showed a mild (~2-fold) increase in liver Hes1 due to increased Hes1+ hepatocyte number and cellular Hes1 content (Fig. 3, B and C, and fig. S7A), as well as increased HSC activation and pericellular collagen deposition (Fig. 3, D and E), even with a mild decrease of liver triglyceride (fig. S7, B and C). Hepatocyte Notch gain of function also did not change hepatocellular death (Fig. 3F), underscoring the predominant effects of hepatocyte Notch activation on liver fibrosis development but did show enhanced liver inflammation and CD45+ immune cell infiltration (fig. S7, D and E).
Fig. 3. Hepatocyte Notch activation exacerbates NASH and fibrosis.
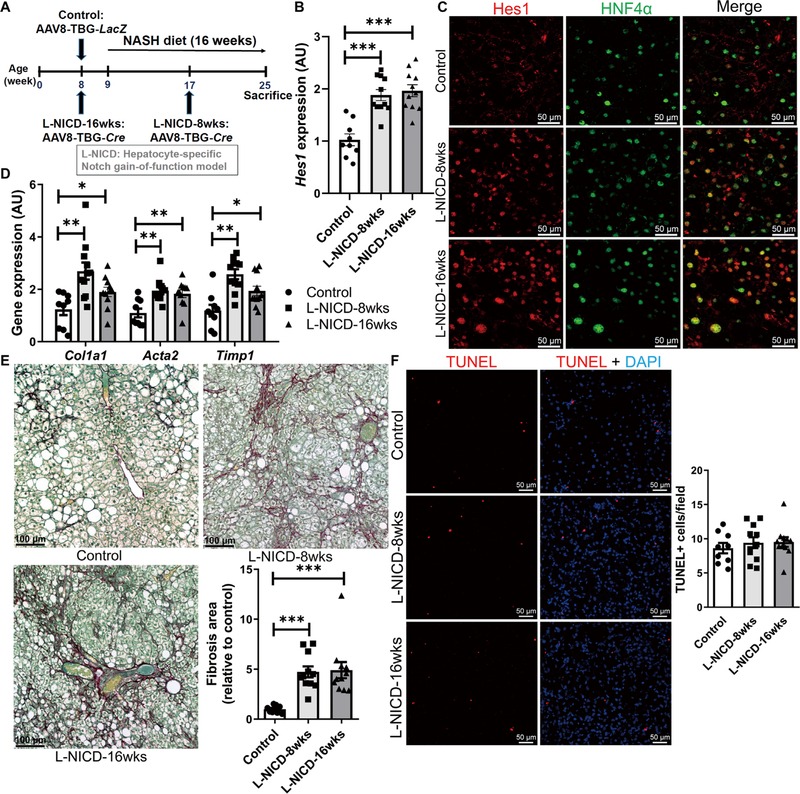
(A) RosaNICD mice (8 weeks old) were transduced with AAV8-TBG-LacZ (control) or AAV8-TBG-Cre to generate hepatocyte-specific Notch gain-of-function (L-NICD-16wks) mice before 16 weeks of NASH diet feeding or halfway through NASH diet feeding (L-NICD-8wks) (n = 9 to 11 per group). (B) Hes1 expression and (C) representative images of Hes1 (red) and HFN4α (green) staining, (D) expression of fibrogenic genes, (E) collagen staining and quantification, and (F) TUNEL (red) staining in livers from control and L-NICD mice. *P < 0.05, **P < 0.01, and ***P < 0.001 as compared to Cre− control mice by one-way ANOVA, followed by post hoc t tests (three groups). All data are shown as the means ± SEM.
Although NASH is prevalent in both male and female humans (34), female mice tend to be protected from diet-induced obesity and consequent metabolic complications. Consistent with their male counterparts, despite unchanged body weight and adiposity (fig. S7, F and G), female L-NICD mice showed increased liver Hes1, inflammatory and fibrogenic gene expression (fig. S7, H to J), and exacerbated liver inflammation and fibrosis as compared to AAV8-TBG-LacZ–transduced controls (fig. S7, K and L).
Hepatocyte Notch activation causes liver fibrosis in the absence of steatohepatitis
NASH is associated with simultaneous pathologic insults: lipotoxicity, hepatocellular injury, inflammation, and fibrosis. Each hit may affect development and/or progression of the others (35, 36); for example, lipotoxicity-induced hepatocellular injury contributes to HSC activation (37, 38). Although we observed unchanged hepato-cellular death in Notch loss- or gain-of-function mice, it is difficult to disentangle the effects of hepatocyte Notch on liver fibrosis from other changes associated with an obesogenic diet. However, even lean, chow-fed L-NICD male mice (Fig. 4A and fig. S8, A to C), without apparent hepatocellular injury (Fig. 4B and fig. S8, D and E), showed increased liver inflammation (fig. S8, F and G), fibrogenic gene expression (Fig. 4C), and greater collagen deposition (Fig. 4D) than Cre− controls. This phenotype was recapitulated in L-NICD female mice (fig. S9). These data suggest that hepatocyte Notch activation is sufficient to trigger HSC activation and liver fibrosis even in the absence of liver steatosis or hepatocyte injury.
Fig. 4. Hepatocyte Notch activation in chow-fed mice induces Sox9-dependent Spp1 expression and liver fibrosis.

(A) Male normal chow-fed RosaNICD mice (8 weeks old) were transduced with AAV8-TBG-LacZ (control) or AAV-TBG-Cre (L-NICD ) and maintained on normal chow diet for 16 more weeks (n = 6 to 8 per group). (B) Quantification of TUNEL staining, (C) fibrogenic gene expression, and (D) collagen staining and quantification in livers from control and L-NICD mice. (E) Fibrogenic gene expression in plated HSCs isolated form WT mice, exposed to control or Ad-NICD–transduced hepatocyte conditioned medium (CM; n = 3 per group). (F) Hes1 and Spp1 in control and Notch-activated primary hepatocytes (n = 6 per group). (G to J) Spp1 expression in (G) livers and isolated hepatocytes of NASH diet–fed mice (n = 9 per group), (H) hepatocytes sorted on the basis of Notch activity from Notch reporter mice (n = 13 per group), (I) livers and FACS-separated hepatocytes from NASH diet–fed L-DNMAM mice (n = 8 per group), and (J) NASH diet–fed L-NICD mice (n = 7 per group). (K) Spp1 expression in siSox9- or siHes1-transfected primary hepatocytes (n = 3 per group). *P < 0.05, **P < 0.01, and ***P < 0.001 as compared to the indicated controls by two-tailed t tests (two groups) or one-way ANOVA, followed by post hoc t tests (more than two groups). All data are shown as the means ± SEM.
Loss of hepatocyte Notch activity does not affect DRs
Notch activation in hepatocytes may promote transdifferentiation to a cholangiocyte lineage (23, 39), contributing to ductular reaction (DR). Because DR and liver fibrosis frequently coexist (40–43), we hypothesized that Notch-induced liver fibrosis may depend on Notch-induced DR. NASH diet induced ductular proliferation, as evidenced by increased liver Krt19 expression and number of cytokeratin 19-positive (CK19+) cells (fig. S10, A and B), which was exacerbated in L-NICD mice (fig. S10, C and D). Further, as previously observed (23), a subset of L-NICD hepatocytes adopted a biliary fate (fig. S10E). However, NASH diet–fed Notch loss-of-function mice showed comparable DR to controls (fig. S10, F and G). These data suggest that constitutive Notch overexpression can induce hepatocyte trans-differentiation, but hepatocyte Notch activation is not necessary for NASH diet–induced DR that likely derives from cholangiocytes.
Notch-induced Sox9 activates hepatocyte Spp1 expression
We next considered alternative explanations for the robust effect of hepatocyte Notch on liver fibrosis and hypothesized that Notch-active hepatocytes may secrete fibrogenic factors that activate HSCs (44). CM collected from primary hepatocytes transduced with Ad-NICD stimulated greater activation of primary HSCs as compared to Ad-GFP (green fluorescent protein)–transduced hepatocyte CM (Fig. 4E). Notch activation did not globally affect the secretome but elevated secretion of the fibrogenic factor Opn (fig. S11A), likely because of increased Spp1 expression (Fig. 4F). Although Opn is known to activate HSCs (45–48), hepatocyte-derived Opn has not been well described. We found that Spp1 was increased in hepatocytes derived from NASH diet–fed WT mice (Fig. 4G), with a preferential increase in Venus+ hepatocytes in Notch reporter mice (Fig. 4H). Thus, NASH diet–fed L-DNMAM mice showed lower Spp1 as compared to Cre− controls (Fig. 4I), whereas Spp1 expression was markedly increased in chow- or NASH diet–fed L-NICD mice (Fig. 4J and fig. S11, B and C). This regulation appeared fairly specific: Similar to results from the cytokine array, expression of other secreted fibrogenic factors (Hedgehog, transforming growth factor–β , and platelet-derived growth factors) was not Notch-activated in hepatocytes (fig. S11D) and/or does not show reciprocal reduction with Notch loss of function (fig. S11E).
These data suggest a cell-autonomous effect of hepatocyte Notch activation on Spp1 transcription. Because the Spp1 promoter does not have a consensus Rbp-Jk binding sequence, we identified additional candidates—Sox9, a direct Notch transcriptional target (49), which we observed to track with genetic manipulations of Notch signaling (fig. S11, F to I), and Hes1, which has been suggested to regulate Spp1 expression in osteoblasts (50). To distinguish between these candidate mechanisms, we used small interfering RNA (siRNA) to knock down Hes1 or Sox9 expression and found that only siRNA directed to Sox9 blunted NICD-induced Spp1 (Fig. 4K), suggesting that Sox9 is the transcriptional mediator of Notch-induced Spp1 ex-pression in hepatocytes.
Notch-mediated Opn secretion from hepatocytes activates HSCs to induce liver fibrosis
To determine whether increased Opn secretion may explain HSC activation induced by hepatocyte Notch activation, we transfected hepatocytes with siRNA to Spp1 (Fig. 5, A and B, and fig. S11J), which abrogated HSC activation induced by Ad-NICD hepatocyte CM (Fig. 5C). In a parallel approach, we pretreated CM with an Opn-neutralizing antibody before addition to HSCs, which similarly negated Notch-induced fibrogenic gene expression (Fig. 5D).
Fig. 5. Notch-induced hepatocyte Opn activates HSCs and induces liver fibrosis.
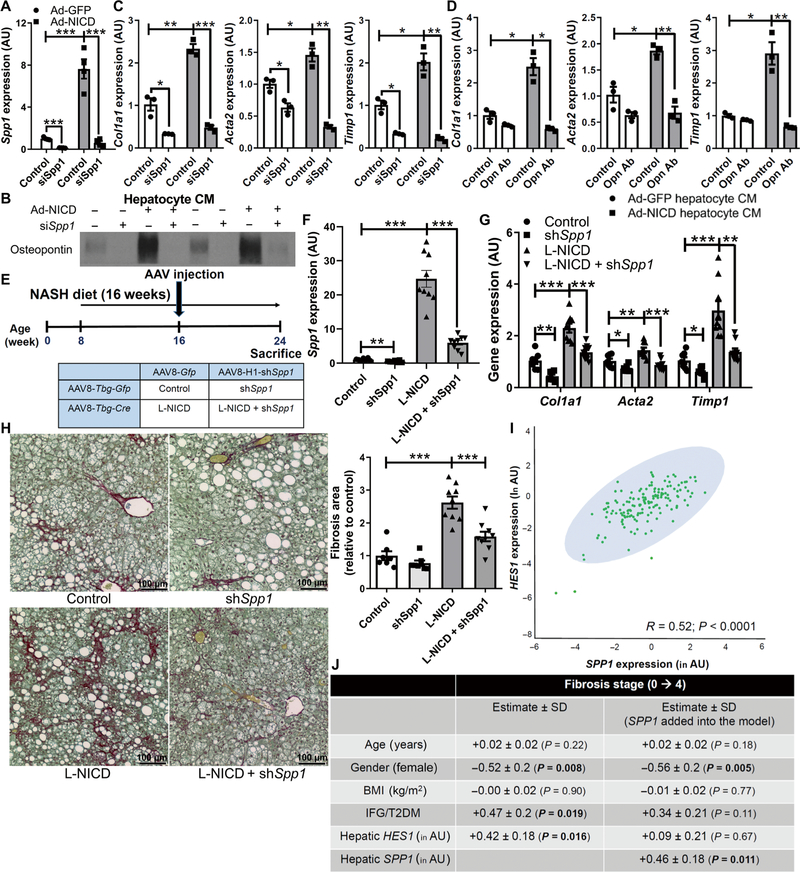
(A) Hepatocyte Spp1 expression (n = 4 per group) and (B) Opn secreted in hepatocyte CM from control or Notch-activated hepatocytes transfected with siRNA directed against Spp1 (siSpp1) or scrambled control. Expression of fibrogenic genes in HSCs exposed to CM (C) from control or Notch-activated siSpp1-transfected hepatocytes (n = 3 per group) or (D) CM pretreated with an Opn-neutralizing antibody (Opn Ab; n = 3 per group). (E) RosaNICD mice (8 weeks old) were fed with NASH diet for 8 weeks and then transduced with AAV8-TBG-Gfp or AAV8-TBG-Cre (to generate control and L-NICD mice, respectively) and simultaneously with AAV8-H1-Gfp or AAV8-H1-shSpp1 (n = 7 to 9 per group). (F) Spp1 and (G) fibrogenic gene expression and (H) collagen staining and quantification in livers from control and L-NICD mice transduced with shSpp1 (short hairpin–mediated RNA–transfected Spp1). *P < 0.05, **P < 0.01, and ***P < 0.001 as compared to the indicated controls by one-way ANOVA, followed by post hoc t tests (more than two groups). All data are shown as the means ± SEM. (I) Correlation between HES1 and SPP1 expression in liver biopsies from patients at risk of NASH (n = 159). (J) Table of association between HES1 expression and liver fibrosis stage after adjustment for key demographic variables (left column) or when additionally adjusted for SPP1 expression (right column). Expression of HES1 and SPP1 was log transformed to ensure the assumption of normal distribution. All data in the table are shown as the regression estimates ± SD and P values, which were generated by multivariate ordinal regression analyses. BMI, body mass index; IFG, impaired fasting glucose; T2DM, type 2 diabetes mellitus.
Next, we generated AAV8-H1-shSpp1 to silence hepatocyte Spp1 and transduced adult NASH diet–fed RosaNICD mice with AAV8-TBG-Gfp or AAV8-TBG-Cre, with or without simultaneous Spp1 knockdown (Fig. 5E). AAV8-H1-shSpp1 transduction did not affect body weight, adiposity, Hes1 expression, liver injury, or inflammatory gene expression (fig. S12, A to F) but successfully attenuated Notch-induced hepatocyte Spp1 without affecting cholangiocyte Opn (Fig. 5F and fig. S12, G and H). Consistent with in vitro experiments, preventing Notch-induced hepatocyte Spp1 expression nearly normalized increased fibrogenic gene expression (Fig. 5G) and excess collagen deposition seen in NASH diet–fed L-NICD mice (Fig. 5H).
To test the clinical SPP1 and HES1 in 159 patients undergoing percutaneous liver biopsy for suspected NASH (table S2). We observed a strong concordance between HES1 and SPP1 (Fig. 5I). Moreover, when we performed multivariate regression analysis to adjust for potential demographic, metabolic, and genetic confounders, HES1 was significantly (P = 0.016) and positively correlated with fibrosis stage, but not when SPP1 was added into the regression model (Fig. 5J). In combination with our mechanistic experiments in vitro and associated mouse modeling, these data suggest that Notch-induced HSC activation and liver fibrosis are likely Opn dependent.
Pharmacologic Notch inhibition ameliorates NASH-associated liver fibrosis
Notch inhibitors are in clinical trials for cancer (51)—the most commonly studied of these are γ-secretase inhibitors (GSIs) (52) that block endogenous production of NICD. We have previously shown that dibenzazepine, a well-characterized and bioavailable GSI (53), can potently inhibit Notch signaling in vivo (15, 54). To test whether acute Notch inhibition can ameliorate NASH-associated liver fibrosis, we treated WT mice with GSI or vehicle once daily for 1 week, initiating treatment after nearly 4 months of NASH diet. Acute GSI treatment did not alter body weight, adiposity, or liver inflammation (fig. S13, A to E) but, as expected from our genetic models, lowered liver Notch activity, Spp1 (fig. S13F), and fibrogenic gene expression (fig. S13G), with only a trend toward less fibrosis (fig. S13H). These data imply that 1 week of GSI exposure is insufficient to reverse liver fibrosis, so next, we used a chronic, intermittent dosing strategy at a lower GSI dose to mitigate potential toxicity (Fig. 6A). As previously observed (55), chronic GSI treatment lowered body weight, adiposity, and liver Notch activity (fig. S14, A and B, and Fig. 6B) and was sufficient to reduce HSC activation and liver fibrosis (Fig. 6, C and D), without effects on inflammation (fig. S14, C and D).
Fig. 6. Pharmacologic Notch inhibitors ameliorate NASH diet–induced fibrosis.
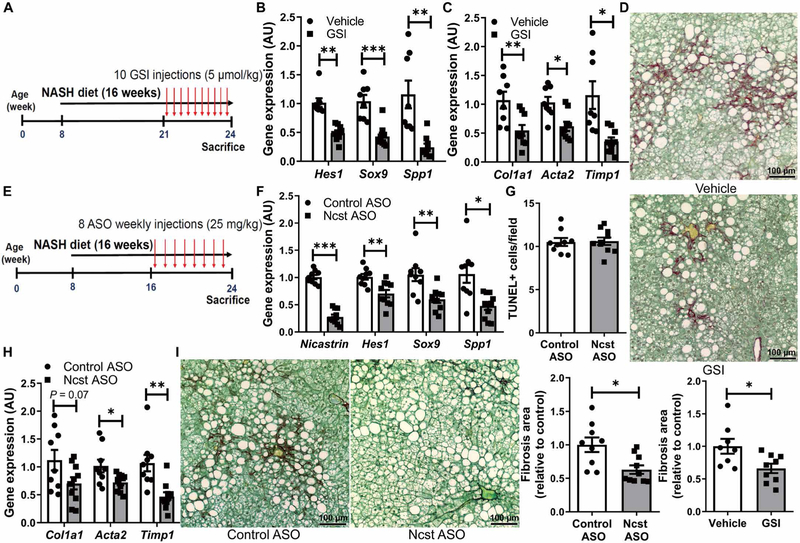
(A) WT mice received vehicle or GSI (5μmol/kg of body weight) every other day for the last 3 weeks of NASH diet feeding (n = 8 to 9 per group). (B) Expression of Notch targets and (C) fibrogenic genes and (D) collagen staining and quantification in livers from vehicle or GSI-treated mice. (E) WT mice received weekly injections of control or Ncst ASO (25 mg/kg of body weight) for the last 8 weeks of NASH diet feeding (n = 9 to 10 per group). (F) Ncst and Notch target gene expression, (G) quantification of TUNEL staining, (H) expression of fibrogenic genes, and (I) collagen staining and quantification in livers from Control ASO– and Ncst ASO–treated mice. *P < 0.05, **P < 0.01, and ***P < 0.001 as compared to the indicated controls by two-tailed t tests (two groups). All data are shown as the means ± SEM.
Unfortunately, chronic GSI treatment induces goblet cell metaplasia (53), even at the lower dose we used here (fig. S14E). Resultant intestinal toxicity likely precludes repurposing GSIs for NASH. Thus, we developed a liver-selective γ-secretase antagonist to bypass intestinal distribution—Ncst ASO (26). On the basis of preclinical data with ASOs of similar chemistry (56–58), we predicted that weekly administration of Ncst ASO would be well tolerated and would effectively block liver Notch signaling as compared to control ASO. Weekly Ncst ASO dosing of NASH diet–fed WT mice (Fig. 6E) reduced liver Ncst, Notch activity, and Spp1 expression (Fig. 6F) as compared to control ASO-treated mice. Ncst ASO did not affect liver TUNEL staining (Fig. 6G), serum transaminases, or liver inflammation (fig. S14, F to I) but effectively reduced expression of HSC markers and collagen deposition (Fig. 6, H and I). Ncst ASO lowered body weight and adiposity (fig. S14, J and K), likely because of adipose ASO up-take (59), but showed no intestinal toxicity (fig. S14L), in contrast to chronic GSI dosing. Control ASO had no effect (fig. S15). These proof-of-principle studies echo data from Notch loss-of-function mice, suggesting that hepatocyte Notch activation is central to the development of obesity-induced liver fibrosis and may be therapeutically targeted in NASH (fig. S16).
DISCUSSION
Fibrosis, although not a diagnostic determinant of NASH, predisposes to cirrhosis and liver malignancy and is the major determinant of long-term mortality of patients with NASH (8–10). NASH is projected to be the leading cause of liver transplantation because of the lack of approved therapeutics (60, 61), underscoring the urgency to uncover mechanisms of NASH-induced fibrosis to find targetable pathways. Our study demonstrates that hepatocyte Notch signaling is virtually absent in normal liver, but tracks with NASH severity in mouse and human, and is maladaptive, leading ultimately to Opn secretion and liver fibrosis. Opn is a known regulator of HSC activation, but the contribution of hepatocyte-derived Opn in NASH suggests that Notch-activated hepatocytes are not simple bystanders but rather alter the liver microenvironment to potentiate fibrosis.
To place our work in context, although several studies have suggested increased Notch activity in NPCs with CCl4-induced acute liver injury (27, 29), none has linked hepatocyte Notch to fibrosis in the setting of NASH. We hypothesize that CCl4 treatment or MCD diet feeding provokes rapid hepatocellular death (27, 32), which induces Notch activation in inflammatory cells and HSCs as part of a proliferative injury response. However, these stimuli provoke minimal and/or nonsustained hepatocyte Notch activation; thus, hepatocyte-specific Notch inhibition has no impact on liver fibrosis in MCD diet–fed mice. In contrast, in the low-grade but long-term injuries associated with obesity/insulin resistance seen in NASH diet–fed mice (and in human NASH), hepatocyte Notch activity is specifically increased to cope with the chronic insult. Our proof-of-principle experiments with Ncst ASO highlight the potential of Notch-directed therapeutics to address NASH-associated fibrosis without the intestinal toxicity associated with systemic Notch inhibition (GSIs). However, because liver-targeted Notch antagonism may be fraught with side effects including disruption of hepatic vascular integrity (62), N-acetylgalactosamine–modified siRNA (63, 64) or nanoparticle strategies (65) retain great potential to address the inappropriate hepatocyte Notch activity in patients with NASH without affecting normal Notch activity in liver NPCs and other tissues.
Our data show that hepatocyte Notch–mediated Opn secretion can directly activate HSCs, independent of hepatocellular injury, leading to excessive collagen deposition. However, our data also point to other possible paths of increased fibrosis with hepatocyte Notch activity. For instance, a subset of L-NICD hepatocytes adopted a biliary fate, consistent with Notch’s role in bile duct development (14) and ability to promote transdifferentiation to a biliary fate (23). Although DR arises primarily from cholangiocytes in liver injury (66), because DR is associated with liver fibrosis ( 40–43), Notch-induced hepatocyte cell fate change may augment fibrosis in L-NICD mice. Hepatocyte secretion of Opn, normally a biliary marker, in response to Notch activation may suggest an incomplete transition of hepatocytes toward biliary fate. Notch loss-of-function mice, however, show unchanged DR as assessed by the CK19+ cell number but still less fibrosis than controls; further, a specific knockdown of hepatocyte Opn reduces fibrosis in L-NICD mice. Thus, our data support the conclusion that hepatocyte Notch activity facilitates liver fibrosis in NASH by both direct (Opn-mediated HSC activation) and possibly indirect (inflammation and DR) pathways.
There are several limitations to our study. For instance, our conclusions were derived primarily with the use of a NASH-provoking diet. Although we saw robust and reproducible diet-induced obesity, liver inflammation, and fibrosis across cohorts, additional studies are necessary to determine whether this diet accurately models NASH pathogenesis. Further, there may be additional Notch-regulated factors involved in the pathogenesis of NASH/fibrosis yet to be identified, and future work is necessary to isolate the upstream signal leading to hepatocyte Notch activation in NASH. Last, although we find an association between hepatocyte Notch activation and NASH severity and therapeutic outcome in patients, it is important to recognize that all intervention studies were performed in mouse models, and the potential translational relevance of Notch inhibitors in human NASH and fibrosis requires formal testing. Nevertheless, we believe that these data provide strong impetus to develop hepatocyte-specific Notch inhibitors to treat the burgeoning health crisis of NASH-associated fibrosis.
MATERIALS AND METHODS
Study design
The objectives of this study were to (i) determine what cell type in liver is responsible for increased Notch activation seen in patients with NASH, (ii) define what role Notch activation plays in NASH-associated fibrosis development by combining a dietary mouse model of NASH with Notch loss- (L-DNMAM and L-Ncst) and gain-of-function (L-NICD) transgenic mice, (iii) define the fibrogenic machinery downstream of hepatocyte Notch activation by coculture of primary hepatocytes and stellate cells in vitro and the AAV-mediated knockdown approach in vivo with validation from translational studies in patients with NASH, and (iv) determine whether pharmacologic Notch inhibition can ameliorate NASH-induced fibrosis in mice.
All data presented here have been replicated in independent cohorts of mice or in at least three biological replicates for in vitro experiments, with all staining data quantitatively analyzed by Zen software. Statistical significance was performed by Student’s t test or ANOVA, followed by post hoc t tests for multiple comparisons. On the basis of predicted effects of Notch activation on various pathologic metrics in mice, with a power of 0.8 and P < 0.05, we calculated a sample size necessary of between 6 and 11 mice per group. Animals were randomly allocated into control and experimental groups, with the group assignment recorded in a master spreadsheet and only unmasked when all samples of the respective experiments were analyzed. Human samples were obtained from a completed random-ized, controlled trial (PIVENS) or a cross-sectional study for which randomization was not applicable—group assignments and other demographic information were blinded to the investigators until after all data were obtained. Data collection of each experiment was detailed in the respective figures, figure legends, and methods. No data were excluded from studies in this manuscript.
Human liver biopsies
Cross-sectional HES1 staining
Normal (n = 3), SS (n = 3), and NASH liver (n = 4) sections were obtained from morbidly obese individuals who underwent liver biopsy at the time of bariatric surgery to assess liver histopathology. The protocol was approved by the Ethical Committee of the Fonda-zione Institute for Research, Hospitalization and Health Care of Milan, and each patient signed a written informed consent.
PIVENS trial
We analyzed liver gene expression of HES1 and HEYL and performed immunostaining of HES1 and HNF4α using liver biopsy samples from PIVENS trial. PIVENS is a NASH treatment trial sponsored by the National Institute of Diabetes and Digestive and Kidney Diseases and conducted by the NASH Clinical Research Network (CRN). The PIVENS trial study design has been described, and the ClinicalTrials.gov identifier is NCT00063622.
Cross-sectional gene expression analysis in patients with suspected NASH
We analyzed liver gene expression of HES1 and SPP1 in individuals who underwent liver biopsy for suspected NASH because of the presence of persistent elevations in liver enzymes or because of severe obesity (67). The protocol was approved by the Ethical Committee of the Fondazione IRCCS of Milan, and each patient signed a written informed consent. For statistical analysis, comparisons were made by fitting data to generalized linear models, unadjusted (univariate analyses), or considering the following as independent variables: age, gender, body mass index, presence of impaired fasting glucose, or type 2 diabetes. Hepatic HES1 and SPP1 mRNA expression was normalized to ACTB expression and natural log transformed before analyses to ensure a normal distribution.
Animal studies
Homozygous RosaDNMAM (22), RosaNICD (33), and Notch-Venus (21) male mice were crossed with female C57BL/6J (strain no. 000664, The Jackson Laboratory) mice to generate heterozygous trans-genic mice for experiments. Ncstflox/flox mice (25) were crossed with albumin-Cre mice (68) to generate L-Ncst mice. All strains were maintained on C57BL/6J genetic background. Mice were weaned to standard chow (PicoLab rodent diet 20, no. 5053) for all experiments and then started on NASH diet (TD.160785, Teklad) with fructose-containing drinking water (23.1 g of fructose and 18.9 g of glucose dissolved in 1 liter of water and then filter sterilized) or MCD diet (no. 518810GI, Dyets) at 8 weeks of age, unless otherwise noted. Animals were housed in standard cages at 22°C in a 12-hour light/12-hour dark cycle and monitored for overall well-being and signs of distress with body weight measured weekly. Upon completion of each study, all mice were weighed and euthanized. Blood was collected via cardiac puncture. Perigonadal adipose tissues were removed and weighed. Livers were weighed, excised, and split for fixation, RNA, and protein isolation and were frozen for future analyses. The Columbia University Institutional Animal Care and Use Committee approved all animal procedures.
Statistical analysis
Results are shown as means ± SEM unless indicated otherwise. Statistical analysis was performed using Prism software (version 6, GraphPad Software). Differences between two groups were calculated using a two-tailed Student’s t test. Analysis involving multiple groups was performed using one-way ANOVA, followed by post hoc t tests. Risk factors for fibrosis severity (stages 0 to 4) in patients were evaluated by multivariate ordinal regression analysis. HES1 and SPP1 expression in the cross-sectional study was log transformed to ensure the assumption of normal distribution. P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments:
We thank A. Flete, T. Kolar, and J. Weber for excellent technical support; W. Wang, L. Lu, S. Shah, and S. Ho for assistance with cell sorting; and members of the Pajvani, Tabas, and Schwabe laboratories for insightful discussion. We also acknowledge J. Clarke and N. Cherrington (Arizona) and N. Tanaka and F. Gonzalez (NIH) for sharing samples related to this work, H. Grajal and J. Kitajewski (UIC) for sharing mouse strains, and the Ancillary Studies Committee and Publications and Presentations Committee of the NASH CRN, who reviewed and approved this manuscript.
Funding:
This work was supported by NIH DK103818 (to U.B.P.), NIH DK105303 (to U.B.P.), MyFIRST AIRC Grant no. 16888 (to L.V.), ALF Liver Scholar Award (to X.W.), and an AHA Predoctoral fellowship 17PRE33120000 (to C.Z.). Cell sorting experiments were performed in the CCTI Flow Cytometry Core, supported by NIH S10OD020056, and the Diabetes and Endocrinology Research Center Flow Core Facility funded in part through NIH 5P30DK063608. The PIVENS trial was supported by NIH U01DK061734 and U01DK061730.
Footnotes
Competing interests:
The authors declare that they have no competing financial interests.
Data and materials availability:
All data associated with this study are present in the manuscript and the Supplementary Materials.
REFERENCES AND NOTES
- 1.Hossain P, Kawar B, El Nahas M, Obesity and diabetes in the developing world—A growing challenge. N. Engl. J. Med 356, 213–215 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Lazo M, Clark JM, The epidemiology of nonalcoholic fatty liver disease: A global perspective. Semin. Liver Dis 28, 339–350 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Ogden CL, Carroll MD, Fryar CD, Flegal KM, Prevalence of obesity among adults and youth: United States, 2011–2014. NCHS data brief 28, 339–350 (2015). [PubMed] [Google Scholar]
- 4.Younossi ZM, Stepanova M, Afendy M, Fang Y, Younossi Y, Mir H, Srishord M, Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin. Gastroenterol. Hepatol 9, 524–530.e1 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Brunt EM, Pathology of nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol 7, 195–203 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Gawrieh S, Chalasani N, Pharmacotherapy for nonalcoholic fatty liver disease. Semin. Liver Dis 35, 338–348 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim D, Kim WR, Kim HJ, Therneau TM, Association between noninvasive fibrosis markers and mortality among adults with nonalcoholic fatty liver disease in the United States. Hepatology 57, 1357–1365 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dulai PS, Singh S, Patel J, Soni M, Prokop LJ, Younossi Z, Sebastiani G, Ekstedt M, Hagstrom H, Nasr P, Stal P, Wong VW, Kechagias S, Hultcrantz R, Loomba R, Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology 65, 1557–1565 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Angulo P, Kleiner DE, Dam-Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, Mills PR, Keach JC, Lafferty HD, Stahler A, Haflidadottir S, Bendtsen F, Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 149, 389–397.e10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ekstedt M, Hagström H, Nasr P, Fredrikson M, Stål P, Kechagias S, Hultcrantz R, Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 61, 1547–1554 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Fortini ME, Bilder D, Endocytic regulation of Notch signaling. Hepatology 19, 323–328 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Strooper B, Nicastrin: Gatekeeper of the gamma-secretase complex. Cell 122, 318–320 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Bolós V, Grego-Bessa J, de la Pompa JL, Notch signaling in development and cancer. Endocr. Rev 28, 339–363 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Zong Y, Stanger BZ, Molecular mechanisms of liver and bile duct development. Wiley Interdiscip. Rev. Dev. Biol 1, 643–655 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Pajvani UB, Shawber CJ, Samuel VT, Birkenfeld AL, Shulman GI, Kitajewski J, Accili D, Inhibition of Notch signaling ameliorates insulin resistance in a FoxO1-dependent manner. Nat. Med 17, 961–967 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valenti L, Mendoza RM, Rametta R, Maggioni M, Kitajewski C, Shawber CJ, Pajvani UB, Hepatic notch signaling correlates with insulin resistance and nonalcoholic fatty liver disease. Diabetes 62, 4052–4062 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, Neuschwander-Tetri BA, Lavine JE, Tonascia J, Unalp A, Van Natta M, Clark J, Brunt EM, Kleiner DE, Hoofnagle JH, Robuck PR; Nash CRN, Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med 362, 1675–1685 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chalasani NP, Sanyal AJ, Kowdley KV, Robuck PR, Hoofnagle J, Kleiner DE, Unalp A, Tonascia J; NASH CRN Research Group, Pioglitazone versus vitamin E versus placebo for the treatment of non-diabetic patients with non-alcoholic steatohepatitis: PIVENS trial design. Contemp. Clin. Trials 30, 88–96 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang X, Zheng Z, Caviglia JM, Corey KE, Herfel TM, Cai B, Masia R, Chung RT, Lefkowitch JH, Schwabe RF, Tabas I, Hepatocyte TAZ/WWTR1 promotes inflammation and fibrosis in nonalcoholic steatohepatitis. Cell Metab 24, 848–862 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, Yeh M, McCullough AJ, Sanyal AJ; Nonalcoholic Steatohepatitis Clinical Research Network, Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41, 1313–1321 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Nowotschin S, Xenopoulos P, Schrode N, Hadjantonakis A-K, A bright single-cell resolution live imaging reporter of Notch signaling in the mouse. BMC Dev. Biol 13, 15 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tu L, Fang TC, Artis D, Shestova O, Pross SE, Maillard I, Pear WS, Notch signaling is an important regulator of type 2 immunity. J. Exp. Med 202, 1037–1042 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yanger K, Zong Y, Maggs LR, Shapira SN, Maddipati R, Aiello NM, Thung SN, Wells RG, Greenbaum LE, Stanger BZ, Robust cellular reprogramming occurs spontaneously during liver regeneration. Genes Dev 27, 719–724 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mu X, Español-Suñer R, Mederacke I, Affò S, Manco R, Sempoux C, Lemaigre FP, Adili A, Yuan D, Weber A, Unger K, Heikenwälder M, Leclercq IA, Schwabe RF, Hepatocellular carcinoma originates from hepatocytes and not from the progenitor/ biliary compartment. J. Clin. Invest 125, 3891–3903 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tabuchi K, Chen G, Südhof TC, Shen J, Conditional forebrain inactivation of nicastrin causes progressive memory impairment and age-related neurodegeneration. J. Neurosci 29, 7290–7301 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim K, Goldberg IJ, Graham MJ, Sundaram M, Bertaggia E, Lee SX, Qiang L, Haeusler RA, Metzger D, Chambon P, Yao Z, Ginsberg HN, Pajvani UB, γ-secretase inhibition lowers plasma triglyceride-rich lipoproteins by stabilizing the LDL receptor. Cell Metab 27, 816–827.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Y, Zheng S, Qi D, Zheng S, Guo J, Zhang S, Weng Z, Inhibition of notch signaling by a γ-secretase inhibitor attenuates hepatic fibrosis in rats. PLOS ONE 7, e46512 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bansal R, van Baarlen J, Storm G, Prakash J, The interplay of the Notch signaling in hepatic stellate cells and macrophages determines the fate of liver fibrogenesis. Sci. Rep 5, 18272 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He F, Guo FC, Li Z, Yu HC, Ma PF, Zhao JL, Feng L, Li WN, Liu XW, Qin HY, Dou KF, Han H, Myeloid-specific disruption of recombination signal binding protein Jκ ameliorates hepatic fibrosis by attenuating inflammation through cylindromatosis in mice. Hepatology 61, 303–314 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Hebbard L, George J, Animal models of nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol 8, 35–44 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Rinella ME, Green RM, The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J. Hepatol 40, 47–51 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Morell CM, Fiorotto R, Meroni M, Raizner A, Torsello B, Cadamuro M, Spagnuolo G, Kaffe E, Sutti S, Albano E, Strazzabosco M, Notch signaling and progenitor/ductular reaction in steatohepatitis. PLOS One 12, e0187384 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murtaugh LC, Stanger BZ, Kwan KM, Melton DA, Notch signaling controls multiple steps of pancreatic differentiation. Proc. Natl. Acad. Sci. U.S.A. 100, 14920–14925 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pan JJ, Fallon MB, Gender and racial differences in nonalcoholic fatty liver disease. World J. Hepatol 6, 274–283 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Machado MV, Diehl AM, Pathogenesis of nonalcoholic steatohepatitis. Gastroenterology 150, 1769–1777 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hardy T, Oakley F, Anstee QM, Day CP, Nonalcoholic fatty liver disease: Pathogenesis and disease spectrum. Annu. Rev. Pathol 11, 451–496 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Iredale JP, Hepatic stellate cell behavior during resolution of liver injury. Semin. Liver Dis 21, 427–436 (2001). [DOI] [PubMed] [Google Scholar]
- 38.Friedman SL, Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev 88, 125–172 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yimlamai D, Christodoulou C, Galli GG, Yanger K, Pepe-Mooney B, Gurung B, Shrestha K, Cahan P, Stanger BZ, Camargo FD, Hippo pathway activity influences liver cell fate. Cell 157, 1324–1338 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang X, Lopategi A, Ge X, Lu Y, Kitamura N, Urtasun R, Leung TM, Fiel MI, Nieto N, Osteopontin induces ductular reaction contributing to liver fibrosis. Gut 63, 1805–1818 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Machado MV, Michelotti GA, Pereira TA, Xie G, Premont R, Cortez-Pinto H, Diehl AM, Accumulation of duct cells with activated YAP parallels fibrosis progression in non-alcoholic fatty liver disease. J. Hepatol 63, 962–970 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Williams MJ, Clouston AD, Forbes SJ, Links between hepatic fibrosis, ductular reaction, and progenitor cell expansion. Gastroenterology 146, 349–356 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Richardson MM, Jonsson JR, Powell EE, Brunt EM, Neuschwander-Tetri BA, Bhathal PS, Dixon JB, Weltman MD, Tilg H, Moschen AR, Purdie DM, Demetris AJ, Clouston AD, Progressive fibrosis in nonalcoholic steatohepatitis: Association with altered regeneration and a ductular reaction. Gastroenterology 133, 80–90 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, Pradere JP, Schwabe RF, Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun 4, 2823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Syn WK, Choi SS, Liaskou E, Karaca GF, Agboola KM, Oo YH, Mi Z, Pereira TA, Zdanowicz M, Malladi P, Chen Y, Moylan C, Jung Y, Bhattacharya SD, Teaberry V, Omenetti A, Abdelmalek MF, Guy CD, Adams DH, Kuo PC, Michelotti GA, Whitington PF, Diehl AM, Osteopontin is induced by hedgehog pathway activation and promotes fibrosis progression in nonalcoholic steatohepatitis. Hepatology 53, 106–115 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Urtasun R, Lopategi A, George J, Leung T-M, Lu Y, Wang X, Ge X, Fiel MI, Nieto N, Osteopontin, an oxidant stress sensitive cytokine, up-regulates collagen-I via integrin αγβ3 engagement and PI3K/pAkt/NFκB signaling. Hepatology 55, 594–608 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Coombes JD, Swiderska-Syn M, Dollé L, Reid D, Eksteen B, Claridge L, Briones-Orta MA, Shetty S, Oo YH, Riva A, Chokshi S, Papa S, Mi Z, Kuo PC, Williams R, Canbay A, Adams DH, Diehl AM, van Grunsven LA, Choi SS, Syn WK, Osteopontin neutralisation abrogates the liver progenitor cell response and fibrogenesis in mice. Gut 64, 1120–1131 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sahai A, Malladi P, Melin-Aldana H, Green RM, Whitington PF, Upregulation of osteopontin expression is involved in the development of nonalcoholic steatohepatitis in a dietary murine model. Am. J. Physiol. Gastrointest. Liver Physiol 287, G264–G273 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Zong Y, Panikkar A, Xu J, Antoniou A, Raynaud P, Lemaigre F, Stanger BZ, Notch signaling controls liver development by regulating biliary differentiation. Development 136, 1727–1739 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shen Q, Christakos S, The vitamin D receptor, Runx2, and the Notch signaling pathway cooperate in the transcriptional regulation of osteopontin. J. Biol. Chem 280, 40589–40598 (2005). [DOI] [PubMed] [Google Scholar]
- 51.Espinoza I, Miele L, Notch inhibitors for cancer treatment. Pharmacol. Ther 139, 95–110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Takebe N, Nguyen D, Yang SX, Targeting notch signaling pathway in cancer: Clinical development advances and challenges. Pharmacol. Ther 141, 140–149 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van Es JH, van Gijn ME, Riccio O, van den Born M, Vooijs M, Begthel H, Cozijnsen M, Robine S, Winton DJ, Radtke F, Clevers H, Notch/γ-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 435, 959–963 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Sparling DP, Yu J, Kim K, Zhu C, Brachs S, Birkenfeld AL, Pajvani UB, Adipocyte-specific blockade of gamma-secretase, but not inhibition of Notch activity, reduces adipose insulin sensitivity. Mol. Metab 5, 113–121 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bi P, Shan T, Liu W, Yue F, Yang X, Liang XR, Wang J, Li J, Carlesso N, Liu X, Kuang S, Inhibition of Notch signaling promotes browning of white adipose tissue and ameliorates obesity. Nat. Med 20, 911–918 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gaudet D, Brisson D, Tremblay K, Alexander VJ, Singleton W, Hughes SG, Geary RS, Baker BF, Graham MJ, Crooke RM, Witztum JL, Targeting APOC3 in the familial chylomicronemia syndrome. N. Engl. J. Med 371, 2200–2206 (2014). [DOI] [PubMed] [Google Scholar]
- 57.Graham MJ, Lee RG, Bell TA III, Fu W, Mullick AE, Alexander VJ, Singleton W, Viney N, Geary R, Su J, Baker BF, Burkey J, Crooke ST, Crooke RM, Antisense oligonucleotide inhibition of apolipoprotein C-III reduces plasma triglycerides in rodents, nonhuman primates, and humans. Circ. Res 112, 1479–1490 (2013). [DOI] [PubMed] [Google Scholar]
- 58.Gaudet D, Alexander VJ, Baker BF, Brisson D, Tremblay K, Singleton W, Geary RS, Hughes SG, Viney NJ, Graham MJ, Crooke RM, Witztum JL, Brunzell JD, Kastelein JJ, Antisense inhibition of apolipoprotein C-III in patients with hypertriglyceridemia. N. Engl. J. Med 373, 438–447 (2015). [DOI] [PubMed] [Google Scholar]
- 59.Geary RS, Norris D, Yu R, Bennett CF, Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev 87, 46–51 (2015). [DOI] [PubMed] [Google Scholar]
- 60.Suzuki A, Diehl AM, Nonalcoholic Steatohepatitis. Annu. Rev. Med 68, 85–98 (2017). [DOI] [PubMed] [Google Scholar]
- 61.Rinella ME, Nonalcoholic fatty liver disease: A systematic review. JAMA 313, 2263–2273 (2015). [DOI] [PubMed] [Google Scholar]
- 62.Cuervo H, Nielsen CM, Simonetto DA, Ferrell L, Shah VH, Wang RA, Endothelial notch signaling is essential to prevent hepatic vascular malformations in mice. Hepatology 64, 1302–1316 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wittrup A, Lieberman J, Knocking down disease: A progress report on siRNA therapeutics. Nat. Rev. Genet 16, 543–552 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lorenzer C, Dirin M, Winkler AM, Baumann V, Winkler J, Going beyond the liver: Progress and challenges of targeted delivery of siRNA therapeutics. J. Control. Release 203, 1–15 (2015). [DOI] [PubMed] [Google Scholar]
- 65.Dehaini D, Fang RH, Zhang L, Biomimetic strategies for targeted nanoparticle delivery. Bioeng. Transl. Med 1, 30–46 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jörs S, Jeliazkova P, Ringelhan M, Thalhammer J, Dürl S, Ferrer J, Sander M, Heikenwalder M, Schmid RM, Siveke JT, Geisler F, Lineage fate of ductular reactions in liver injury and carcinogenesis. J. Clin. Invest 125, 2445–2457 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mancina RM, Dongiovanni P, Petta S, Pingitore P, Meroni M, Rametta R, Borén J, Montalcini T, Pujia A, Wiklund O, Hindy G, Spagnuolo R, Motta BM, Pipitone RM, Craxì A, Fargion S, Nobili V, Käkelä P, Kärjä V, Männistö V, Pihlajamaki J, Reilly DF, Castro-Perez J, Kozlitina J, Valenti L, Romeo S, The MBOAT7-TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of European descent. Gastroenterology 150, 1219–1230.e6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Postic C, Magnuson MA, DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis 26, 149–150 (2000). [DOI] [PubMed] [Google Scholar]
- 69.Pajvani UB, Qiang L, Kangsamaksin T, Kitajewski J, Ginsberg HN, Accili D, Inhibition of Notch uncouples Akt activation from hepatic lipid accumulation by decreasing mTorc1 stability. Nat. Med 19, 1055–1060 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Miller CM, Tanowitz M, Donner AJ, Prakash TP, Swayze EE, Harris EN, Seth PP, Receptor-mediated uptake of phosphorothioate antisense oligonucleotides in different cell types of the liver. Nucleic Acid Ther 28, 119–127 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lisowski L, Dane AP, Chu K, Zhang Y, Cunningham SC, Wilson EM, Nygaard S, Grompe M, Alexander IE, Kay MA, Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature 506, 382–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim K, Qiang L, Hayden MS, Sparling DP, Purcell NH, Pajvani UB, mTORC1-independent Raptor prevents hepatic steatosis by stabilizing PHLPP2. Nat. Commun 7, 10255 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Font-Burgada J, Shalapour S, Ramaswamy S, Hsueh B, Rossell D, Umemura A, Taniguchi K, Nakagawa H, Valasek MA, Ye L, Kopp JL, Sander M, Carter H, Deisseroth K, Verma IM, Karin M, Hybrid periportal hepatocytes regenerate the injured liver without giving rise to cancer. Cell 162, 766–779 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mederacke I, Dapito DH, Affò S, Uchinami H, Schwabe RF, High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat. Protoc 10, 305–315 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Folch J, Lees M, Sloane Stanley GH, A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem 226, 497–509 (1957). [PubMed] [Google Scholar]
- 76.Pradere JP, Kluwe J, De Minicis S, Jiao JJ, Gwak GY, Dapito DH, Jang MK, Guenther ND, Mederacke I, Friedman R, Dragomir AC, Aloman C, Schwabe RF, Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 58, 1461–1473 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.