

Abstract
Non-alcoholic steatosis and non-alcoholic steatohepatitis (NASH) are liver pathologies characterized by severe metabolic alterations due to fat accumulation that lead to liver damage, inflammation, and fibrosis. We demonstrate that the voltage-dependent anion channel 1 (VDAC1)-based peptide R-Tf-D-LP4 arrested steatosis and NASH progression, as produced by a high-fat diet (HFD-32) in a mouse model, and reversed liver pathology to a normal-like state. VDAC1, a multi-functional mitochondrial protein, regulates cellular metabolic and energetic functions and apoptosis and interacts with many proteins. R-Tf-D-LP4 treatment eliminated hepatocyte ballooning degeneration, inflammation, and liver fibrosis associated with steatosis, NASH, and hepatocarcinoma, and it restored liver pathology-associated enzyme and glucose levels. Peptide treatment affected carbohydrate and lipid metabolism, increasing the expression of enzymes and factors associated with fatty acid transport to mitochondria, enhancing β-oxidation and thermogenic processes, yet decreasing the expression of enzymes and regulators of fatty acid synthesis. The VDAC1-based peptide thus offers a promising therapeutic approach for steatosis and NASH.
Keywords: liver, metabolism, mitochondria, NAFLD, NASH, peptide, steatosis, VDAC1
Graphical Abstract
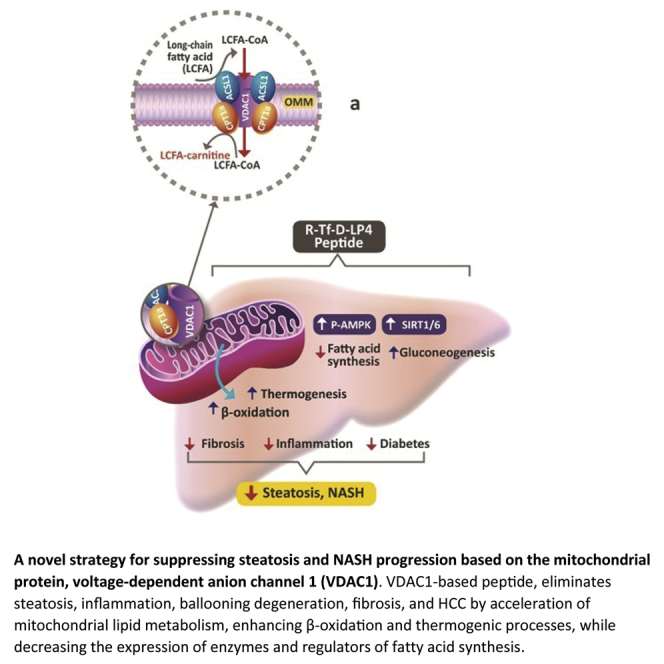
A VDAC1-derived peptide arrested steatosis and NASH in a high-fat diet mouse model, eliminating steatosis- and NASH-associated liver pathogenesis, including inflammation, fibrosis, and hepatocarcinoma. The peptide increased fatty acid transport to mitochondria, β-oxidation, and thermogenesis while decreasing fatty acid synthesis. The peptide thus offers a therapeutic approach for steatosis and NASH.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is characterized by excessive abnormal accumulation of fatty acids and triglycerides within the hepatocytes of non-alcohol users. NAFLD and the progressive state, non-alcoholic steatohepatitis (NASH), are the most common causes of abnormal liver function.1 About 20%–30% of the general population in the western world suffers from NAFLD.2 Prevalence is further increased in patients with type 2 diabetes (T2D),3 and it is commonly associated with obesity and cardiovascular disease.4 NASH represents a spectrum of disorders, including inflammation, hepatic fibrosis, a severe form of liver fibrosis-cirrhosis, and hepatocellular carcinoma (HCC).4 Various NASH mouse models have been reported, yet most do not replicate the full spectrum of human NASH.5 One mouse model that depicts liver pathology from steatosis to NASH to HCC with a pre-diabetic background is the steatosis-NASH high-fat diet (HFD)-32/Stelic animal model (STAM).6
Various studies of hepatosteatosis have shown that mitochondria play a prominent role in disease pathogenesis.7 Mitochondria can influence cell fate at the levels of energy production, lipid metabolism, production and detoxification of reactive oxygen species (ROS), and apoptotic and necrotic hepatocyte cell death induction. The voltage-dependent anion channel 1 (VDAC1), found in the outer mitochondria membrane (OMM), has been identified as a dynamic regulator of global mitochondrial function under normal physiological conditions and disease states.8, 9, 10 VDAC1 regulates the flux of metabolites and ions between mitochondria and cytoplasm, linking energy and signaling pathways in mitochondria and other cell compartments, and it also participates in apoptosis. Accordingly, VDAC1 function or dysfunction is associated with various diseases.8, 9, 10
VDAC1 is involved in cholesterol transport and was proposed to be part of a complex mediating the transport of fatty acids across the OMM.11, 12 Specifically, VDAC1 serves as an anchoring site for long-chain acyl-CoA synthetase (ACSL), associated with the outer surface of the OMM, and for carnitine palmitoyltransferase 1a (CPT1a), which faces the intermembrane space (IMS). It is thought that the long-chain fatty acyl-CoA formed by ACSL is transferred across the OMM by VDAC1 to the IMS, where CPT1a converts the acyl CoA into long-chain fatty acyl-carnitine.11, 12 Acyl-carnitine crosses the inner mitochondria membrane (IMM) by carnitine or acylcarnitine translocase (CACT), converts back into acyl-CoA by IMM-associated CPT2, and subsequently undergoes β-oxidation in the matrix. Furthermore, the GSK3-mediated VDAC1 phosphorylation state is correlated with steatosis severity, with that status controlling OMM permeability in hepatosteatosis.13
Interacting with many proteins, VDAC1 is considered a hub protein.10, 14 The VDAC1 interactome includes metabolism-, apoptosis-, signal transduction-, DNA-, RNA-, and anti-oxidation related proteins.10, 14 Given the roles played by VDAC1 in cell metabolism and apoptosis and its regulation by many interacting partners, modulating such interactions by VDAC1-based peptides represents a potential therapeutic for treating liver metabolic disorders, such as NAFLD. Accordingly, we have designed VDAC1-based peptides targeting these interactions.15, 16 One of these peptides, R-Tf-D-LP4, was selected for this study.
R-Tf-D-LP4 comprises a VDAC1-derived sequence defined as LP4 fused to a cell-penetrating peptide, the human transferrin receptor (hTfR) recognition sequence HAIYPRH (Tf).17 Transferrin receptor 2 (TfR2) is highly expressed in hepatocytes.18 The amino acids of the VDAC1-derived sequence are in the D-configuration,16 allowing higher bioavailability, and they are less immunogenic than the corresponding L-peptides.17 In the retro-inverso peptide R-Tf-D-LP4, the direction of the peptide bond is reversed, allowing side-chain topology similar to that of the original L-amino acid peptide.17
Here, using the HFD-32-fed steatosis-NASH-HCC/STAM mouse model, we demonstrated that the cell-penetrating VDAC1-based peptide R-Tf-D-LP4 largely eliminated all steatosis- and NASH-associated pathogenesis, including ballooning degeneration (cell injury), inflammation, hepatic fibrosis, as well as HCC.19 These effects resulted from the peptide affecting carbohydrate and lipid metabolism via alterated expression of the liver transcriptional program and moduation of mitochondrial proteins mediating import of long-chain fatty acyl-CoAs.
Results
In this study, we employed the steatosis-NASH-HCC/HFD-32-STAM mouse model6 to study the effects of the VDAC1-based peptide R-Tf-D-LP4 on steatosis and NASH pathology. This model sequentially depicts the liver pathology of NAFLD to NASH to HCC against a pre-diabetic background.20
R-Tf-D-LP4 Inhibits Steatosis
To study the effects of the peptide on steatosis, mice were fed a high-fat diet (HFD-32) from weeks 5–8 after the STZ injection. In the last 3 weeks, the mice were treated with Hank’s balanced salt solution (HBSS) (control) or R-Tf-D-LP4 peptide (14 mg/kg) (Figure 1A). Mice provided with a regular chow diet (normal diet [ND]) served as controls. Body weight of the HFD-32-fed, peptide-treated mice was slightly decreased (Figure S1A). Once sacrificed, the removed livers showed an increase of ∼43% in liver weight in the HFD-32-fed mice that was attenuated upon peptide treatment by 86% (Figures 1B and 1C), which was also reflected in the liver:body weight ratio (Figure S1B). Abdominal visceral fat mass in the HFD-32-fed mice was increased 3-fold relative to ND-fed mice. This value was decreased by 50% in the peptide-treated group (Figure S1C). Half of each liver was then fixed in formalin and the second half was frozen for further histopathological, imunoblotting, or qPCR analysis.
Figure 1.

R-Tf-D-LP4 Peptide-Mediated Inhibition of Steatotic Liver Pathology in an HFD-32 Mouse Model
(A) Schematic presentation of the course of steatosis development induced by an HFD-32 diet and peptide treatment initiation. (B–D) Livers from mice fed chow (normal diet [ND]), HFD-32, or HFD-32 and treated with R-Tf-D-LP4 (14 mg/kg) from weeks 6–8 by intravenous (i.v.) injection of 100 μL 0.8% DMSO in HBSS (control) or with R-Tf-D-LP4 in HBSS every 2 days. The final blood DMSO concentration was 0.07% in control and peptide-treated mice. Mice were then sacrificed, and livers were removed, photographed (B), and weighed (C). Results are means ± SEM (n = 10; **p ≤ 0.01, ***p ≤ 0.001). Representative liver sections were stained with H&E (D) or oil red O (E).
Morphological changes and fat accumulation were evaluated on formaldehyde-fixed, paraffin-embedded liver sections using H&E and Sirius red or oil red O staining of sections obtained from freshly frozen liver.
H&E-stained liver sections from HFD-32-fed mice displayed signs of steatosis, characterized by fat droplet accumulation that was highly reduced in peptide-treated mice (Figure 1D). Liver sections from HFD-32-fed mice, but not those treated with the peptide or from regular ND-fed mice, stained with oil red O showed high staining of fat droplets that occupied liver cells (Figure 1E).
Livers from HFD-32-fed mice displayed ballooning cell accumulation with a wispy clear cytoplasm, characteristic of steatosis. Peptide treatment greatly reduced the number of ballooning cells (Figure 2A). Ballooning-related hepatocyte degeneration is associated with hepatocyte senescence and cell death.21 H&E staining also showed inflamed regions in HFD-32-fed mouse livers that were highly reduced in HFD-32-fed, peptide-treated mice (Figure 2A).
Figure 2.

R-Tf-D-LP4 Peptide-Mediated Inhibition of Inflammation and Fibrosis Activation in Steatosis in an HFD-32-Fed Mouse Model
Representative sections of livers from ND, HFD-32, and HFD-32-fed mice treated with R-Tf-D-LP4 (14 mg/kg) at steatosis, stained with H&E for ballooning degeneration and inflammation (A) and for the macrophage F4/80 marker by IF (B) and showing quantification of the percentage of the F4/80-stained area (C). (D) qRT-PCR analysis of mRNA levels of inflammation-related molecule in livers from ND-fed (black bars); HFD-32-fed (dark gray bars); and HFD-32-fed, peptide-treated (light gray bars) mice. Results are means ± SEM (n = 3–5; **p ≤ 0.01, ***p ≤ 0.001). Immunoblot (E) and quantitative (F) analysis of TGF-β in livers from ND-fed (black bars); HFD-32-fed (dark gray bars); and HFD-32-fed, peptide-treated (light gray bars) mice. β-actin served as an internal loading control. (G and H) Representative liver sections from HFD-32-fed and HFD-32-fed, peptide-treated mice stained with Sirius red (G) and anti-α-SMA antibodies (H). (I) and (J) show quantification of the percentages of the α-SMA- (I) and Sirius red-stained areas (J). Arrows point to major results presented in the image.
Immunofluorescent (IF) staining of liver sections for F4/80 demonstrated reduced numbers of macrophages in liver sections derived from HFD-32-fed, peptide-treated mice (Figure 2B), as also shown by quantification of the percentage of the F4/80-stained area (Figure 2C). Moreover, in HFD-32-fed mice, inflammation in the liver, as reflected in the increased (4- to 5-fold) mRNA levels of pro-inflammatory genes such as interleukin (IL)-1β and IL-6, was reduced in peptide-treated mice to the same levels as seen in ND-fed mouse livers.
Levels of tumor necrosis factor alpha (TNF-α) were similar in the three groups, while protein and mRNA levels of transforming growth factor β (TGF-β), essential for fibrotic activity, were increased (2- to 3-fold) in HFD-32-fed mice; these were reduced upon peptide treatment (Figures 2D–2F).
To analyze fibrosis, liver sections from HFD-32-fed mice with or without peptide treatment were stained for collagen with Sirius red or with antibodies against α-smooth muscle actin (SMA), a fibrosis marker (Figures 2G and 2H). The HFD-32 group showed Sirius red staining, pointing to the presence of collagen, reflecting fibrosis. In the livers of peptide-treated mice, Sirius red staining was decreased and restricted to the perisinusoidal space, as in healthy livers (Figure 2G). Quiescent hepatic stellate cells (HSCs), when activated by pathological conditions, differentiate into fibrotic cells expressing α-SMA, collagen 1, and desmin, and they secrete collagen, resulting in cirrhosis.22 In the HFD-32-fed mice, HSCs expressed large amounts of α-SMA (Figure 2H) that was highly reduced in R-Tf-D-LP4-treated mice, as also shown by quantification of the percentage of the α-SMA- or Sirius red-stained areas (Figures 2I and 2J), pointing to attenuated fibrotic progression.
The NAFLD activity score (NAS), performed as previously described23 for both the steatosis and NASH stages, analyzing steatosis, ballooning, inflammation, and fibrosis, is presented in Table S1.
The results show that peptide treatment decreased the NAS from 7.3 ± 0.16 to 3.1 ± 0.1 in the steatosis stage and from 9.6 ± 0.15 to 4.4 ± 0.17 in the NASH stage, which also included fibrosis. Thus, peptide treatment decreased the NAS by 58% and 55% in steatosis and NASH, respectively.
We compared intravenous and intramuscular peptide administration to HFD-32-fed mice, and we found similar effects on steatosis, ballooning degeneration of hepatocytes, inflammation, and fibrosis (Figures S1D and S1E).
Finally, peptide treatment of HFD-32-fed mice reduced liver damage, reflected in blood functional markers and cholesterol and triglyceride levels, all reduced to levels similar to those of ND-fed mice (Figure S2).
Effects of R-Tf-D-LP4 Treatment on Lipid Metabolism in HFD-32-Fed Mice
The effects of R-Tf-D-LP4 on the expression levels of proteins associated with lipid transport and metabolism, synthesis, and β-oxidation were revealed by immunohistochemistry (IHC), immunoblotting, and qPCR (Figure 3). Peptide treatment increased the expressions of the OMM anchored CPT1a, transferring the fatty acyl groups of acyl-CoA to carnitine to form acyl-carnitine24 (Figures 3A–3D); PPAR-γ (peroxisome proliferator-activated receptor γ) (Figures 3A, 3C, and 3E), involved in regulating fatty acid β-oxidation; and PPAR-α (Figure 3E). Expression levels of the uncoupler proteins UCP1 and UCP2, associated with thermogenic respiration, were also increased in livers from HFD-32-fed, peptide-treated mice (2.65- and 4.8-fold at the protein level and 26.5- and 3-fold at the mRNA level, relative to their levels in ND-fed mice, and 1.8- and 2.4-fold at the protein level and 24.1- and 1.3-fold at the mRNA level, when comparing to the values seen in HFD-32; Figures 3A, 3C, and 3F).
Figure 3.

R-Tf-D-LP4 Peptide Treatment of HFD-32-Fed Mice Altered Liver Lipid Metabolism and Steatosis Pathology
(A) Immunoblots of several proteins in livers from ND-fed; HFD-32-fed; and HFD-32-fed, peptide-treated mice. (B) IHC of CPT1 with the relative intensity (n = 3 liver sections) is presented in parentheses. (C) Immunoblot quantitative analysis. (D) qRT-PCR analyses of mRNA of proteins associated with lipid metabolism in livers from ND-fed (black bars); HFD-32-fed (dark gray bars); and HFD-32-fed, peptide-treated (light gray bars) mice. Transcription factors and co-activators involved in lipid metabolism regulation (E) and mRNA levels of UCP1 and UCP2, associated with thermogenic respiration (F), were analyzed in the same groups of mice as above. (G) qRT-PCR analysis of mRNA levels of proteins associated with lipid biosynthesis in livers from ND-fed (black bars); HFD-32-fed (dark gray bars); and HFD-32-fed, peptide-treated (light gray bars) mice. Results are means ± SEM (n = 3–5; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001). Immunoblot (H) and quantitative analysis (I) of ACC1 and FAS. β-actin served as the loading control.
The expression of the transcription activator of PPAR-γ, PPARγ coactivator (PGC1α), was also increased over 5.6-fold (Figure 3E). PGC1α is a transcription factor involved in mitochondrial biogenesis and fatty acid oxidation that acts by regulating other transcription factors, and it is an essential protein in the regulation of lipid metabolic processes, such as high oxidative capacity.25 mRNA levels of ACSL1 and ACSL5, present in the OMM and catalyzing the formation of acyl-CoAs from fatty acids, were also increased 5- to 6-fold in livers of HFD-32-fed, peptide-treated mice (Figure 3D). mRNA levels of acyl-CoA dehydrogenase long chain (ACADL) and 3-hydroxyacyl-CoA dehydrogenase (HADH), involved in β-oxidation, were unchanged (Figure 3D).
In contrast to the peptide-induced increased expression of fatty acid oxidation-related genes, those associated with lipid synthesis were increased in the HFD-32-fed mice and decreased below the levels seen in ND-fed mice in HFD-32-fed, peptide-treated mice (Figures 3G–3I). These included sterol regulatory element-binding protein-1A (SREBP1A) and SREBP1C, acetyl-CoA carboxylase1 (ACC1), ACC2, acyl-CoA desaturase1 (SCD1), elongation of very long-chain fatty acid protein 6 (ELOVL6), and fatty acid synthase (FAS) (Figures 3G–3I). These findings suggest that peptide treatment of HFD-32-fed mice at the steatosis stage increased mitochondrial fatty acid uptake, β-oxidation, and thermogenic respiration while inhibiting fatty acid synthesis.
Effects of R-Tf-D-LP4 on Carbohydate Metabolism in HFD-32-Fed Mice
NAFLD pathogenesis is associated with metabolic changes involving increased glucose uptake by liver cells, which is utilized for glycogen and lipid synthesis.26 The liver carbohydrate metabolic features of ND-fed; HFD-32-fed; and HFD-32-fed, peptide-treated mice were analyzed by IHC, qPCR, and immunoblotting (Figure 4).
Figure 4.

R-Tf-D-LP4 Peptide Treatment of HFD-32-Fed Mice at the Steatosis Stage Altered the Expression of Metabolism-Related Enzymes
(A) Representative IHC staining of proteins mediating glucose transport (Glut-1), glycolysis (GAPDH, LDH-A), mitochondria metabolite transport (VDAC1), and OXPHOS (ATP synthase-5a) in liver sections from ND-fed mice and HFD-32-fed mice with and without peptide treatment. (B) Quantification of IHC staining intensity. (C) qRT-PCR analysis of the glucose transporters Glut-1, Glut-2, and Glut-4 in the 3 mouse groups. Immunoblot (D) and quantitative analysis (E) of metabolism-related enzymes and SIRT1 and SIRT6. Results are means ± SEM (n = 3–5; **p ≤ 0.01, ***p ≤ 0.001). (F) Immunoblot and quantitative analysis of phosphorylated AMPK (p-AMPK). RU represents relative unit. (G) Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining of liver sections from HFD-32-fed and HFD-32-fed, peptide-treated mice. β-actin served as a loading control.
HFD-32-fed mice showed high expression levels of glucose transporter 1 (Glut-1) that were reduced upon peptide treatment (Figures 4A and 4B). However, mRNA levels of Glut-2 and Glut-4 were highly increased (3.8- and 19.3-fold, respectively) in HFD-32-fed, peptide-treated mice (Figure 4C). Expression levels of the glycolytic enzymes glyceraldehyde dehydrogenase (GAPDH) and lactate dehydrogenase-A (LDH-A), the Kreb’s cycle enzyme citrate synthase (CS), VDAC1, and ATP synthase-5a, as analyzed by IHC, qPCR, and immunoblotting, were also increased upon peptide treatment of HFD-32-fed mice (Figures 4A–4D). Sirtuin1 and 6 (SIRT1 and SIRT6) expressions were highly increased in HFD-32-fed, peptide-treated mice (Figures 4D and 4E). SIRT1 targets metabolic processes via deacetylating regulatory transcription factors.27 The level of phosphorylated AMP-activated protein kinase (p-AMPK) was decreased in HFD-32-fed mice, but it highly increased when mice were treated with the peptide (Figure 4F). AMPK is an energy-sensor28 and a key regulator of fatty acid and cholesterol biosynthesis and lipid metabolism, acting as an upstream kinase of ACC and 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGR).29
R-Tf-D-LP4 peptide was found to induce apoptotic cell death in liver19 and other cancers.30 No cell death was found in liver sections derived from the HFD-32-fed mice with or without peptide treatment (Figure 4G).
R-Tf-D-LP4 Inhibits NASH-Associated Pathologies
NASH is characterized by fat droplets occupying hepatocytes, ballooning degeneration of hepatocytes, and expanded fibrosis in the liver parenchyma.20 For NASH, mice were HFD-32 fed in weeks 5–11, and, in the last 3 weeks, they were untreated or treated with R-Tf-D-LP4 (14 mg/kg) (Figure 5A). At the end of week 11, the mice were sacrificed, and livers were photographed (Figure 5B), weighed (Figure 5C), and then fixed or frozen for further proccesing. Livers from HFD-32-fed mice were yellow and weighed 1.6-fold more than livers from chow-fed mice or those when peptide treated (Figures 5B and 5C). Similarly, abdominal visceral fat mass in HFD-32-fed mice was 2.6-fold higher than in ND-fed mice. This value was decreased by ∼50% in HFD-fed, peptide-treated mice (Figure S3C).
Figure 5.

R-Tf-D-LP4 Peptide Treatment of HFD-32-Fed Mice Inhibits NASH Liver Pathology
(A) Schematic presentation of NASH development as post-steatosis induced by an HFD-32 diet and peptide treatment. The experiment was carried out as in Figure 1 except that mice were fed with HFD-32 for 8 weeks (weeks 5–12) and treatment with the peptide was started at the beginning of week 9 for 3 weeks. Control mice were injected i.v. every 2 days with 100 μL HBSS containing 0.8% DMSO or R-Tf-D-LP4 (14 mg/kg). The final blood DMSO concentration was 0.07% in control and peptide-treated mice. At the end of week 12, the mice were sacrificed, and livers were photographed (B) and weighed (C). Results are means ± SEM (n = 10; *p ≤ 0.05, ***p ≤ 0.001). (D) Representative H&E-stained liver sections from ND-fed; HFD-32-fed; and HFD-32-fed, peptide-treated mice. (E) Liver sections as in (D) were stained with oil red O. (F) TUNEL staining of liver sections from HFD-32-fed and HFD-32-fed, peptide-treated mice.
Liver sections from HFD-32-fed mice showed accumulated fat droplets. These were highly decreased in the peptide-treated group, as visualized by H&E (Figure 5D) or oil red O staining (Figure 5E). No cell death was seen in peptide-treated or untreated, HFD-32-fed mouse livers (Figure 5F). As in steatosis, serum biochemical analyses of HFD-32-fed mice at the NASH stage showed increased fatty liver marker levels, with peptide treatment bringing them close to those of chow-fed mice (Figures S3D–S3L).
In liver sections from HFD-32-fed mice, ballooned hepatocyte morphology and inflammation were clearly observed, but they were highly reduced in HFD-32-fed, peptide-treated mice (Figure 6A). Macrophages were mobilized to inflammation sites in HFD-32-fed mice, as identified by F4/80 staining (Figure 6B) and quantification of the percentage of the F4/80-stained area (Figure 6C). mRNA levels for pro-inflammatory genes, IL-1β, TNF-α, and IL-6 (Figure 6D), and anti-inflammatory pro-fibrotic TGF-β (Figure 6E) were increased in HFD-32-fed mice, yet they were reduced by the peptide to levels close to those in chow-fed mice.
Figure 6.

R-Tf-D-LP4 Peptide-Mediated Inhibition of Inflammation and Fibrosis Activities and Enhanced Metabolism in a NASH Mouse Model
(A) Representative H&E staining of liver sections from HFD-32-fed mice at the NASH stage, untreated or treated with R-Tf-D-LP4 (14 mg/kg), showing hepatocyte ballooning degeneration and inflammation. (B and C) Staining of the macrophage marker F4/80 in liver sections from HFD-32-fed and HFD-32-fed, peptide-treated mice. (B) IF staining shows arrows pointing to major results presented in the image. (C) Quantification of the percentage of the F4/80-stained area. (D) qRT-PCR analysis of mRNA levels of inflammation-related molecule in livers from chow-fed mice (black bars) and HFD-32-fed mice untreated (dark gray bars) or treated with R-Tf-D-LP4 peptide (light gray bars). (E) Immunoblot analysis of TGF-β with the relative band intensity presented in relative units (RUs). (F and G) Representative liver sections from HFD-32-fed mice untreated or peptide treated and stained with anti-α-SMA antibodies (F) or Sirius red (G), with arrows pointing to major results presented in the image. Quantification of the α-SMA (H) and Sirius red-stained areas (I). Results are means ± SEM (n = 10; *p ≤ 0.05, ***p ≤ 0.001).
Livers from HFD-32-fed mice, but not when treated with the peptide, showed high levels of collagen, a sign of fibrosis, as revealed by Sirius red (Figures 6G–6I) and Masson’s trichrome (Figure S4) staining. Similarly, staining of liver sections from HFD-32-fed mice with anti-α-SMA antibodies revealed high staining of HSCs, while in HFD-32-fed, peptide-treated mice, α-SMA was detected solely in the perisinusoidal space, as in healthy liver (Figures 6F and 6H).
R-Tf-D-LP4-Mediated Effects on Carbohydrate and Fat Metabolism in NASH
As in steatosis, the metabolic features of HFD-32-fed mice at the NASH stage showed high expression levels of Glut-1 that were decreased in HFD-32-fed, peptide-treated mice (Figure 7A). Peptide treatment increased Glut-4 mRNA levels from an ∼2-fold decrease in HFD-32-fed mice to an about 3-fold increase (Figure 7A). The expression levels of VDAC1, GAPDH, LDH-A, and ATP synthase-5a were slightly increased in the HFD-32-fed mice, and they were further increased upon peptide treatment (Figures 7B, 7C, and S5A–S5D). Interestingly, IHC staining of liver sections for LDH-A revealed a nuclear localization (Figures S5A and S5B). Hexokinase (HK)-I and HK-II levels were found to increase in HSCs (Figures S5E and S5F), reported to adopt aerobic glycolysis during activation, including higher glucose consumption and increased HK activity.31 Finally, SIRT1 and SIRT6 expression levels were increased 3- to 4-fold (Figures 7B and 7C), while that of p-AMPK rose ∼3-fold (Figure 7D) in HFD-32-fed, peptide-treated mice.
Figure 7.

R-Tf-D-LP4 Peptide Treatment Increased Carbohydrate Metabolism in HFD-32 Mice at the NASH Stage
(A) qRT-PCR analysis of mRNA levels of Glut-1, Glut-2, and Glut-4 in chow-fed mice (black bars) and HFD-32-fed mice untreated (dark gray bars) or treated with R-Tf-D-LP4 (light gray bars). Immunoblot (B) and quantitative analysis (C) of metabolism-related proteins and SIRT1 and SIRT6. (D) Immunoblot of phosphorylated AMPK (p-AMPK), with the relative band intensity presented in relative units (RUs) (n = 3). β-actin served as an internal loading control. Results are means ± SEM (n = 3–5; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001).
The peptide also affected fatty acid and cholesterol metabolism at the NASH stage. Immunoblotting of proteins associated with lipid metabolism, such as CPT1a and PPARγ, and their quantitative analysis demonstrated increased expression in the livers of HFD-32-fed mice that was further increased upon peptide treatment, as in steatosis (Figures 8A, 8B, 8D, and 8E).
Figure 8.

R-Tf-D-LP4 Peptide-Mediated Lipid Metabolism Reprogramming in HFD-32 Mice at the NASH Stage
Proteins extracted from liquid nitrogen-frozen liver obtained from HFD-32-fed mice untreated or peptide treated (14 mg/kg) at the NASH stage were subjected to immunoblotting (A) and quantitative protein analysis (B). (C) UCP1 and UCP2 mRNA levels were analyzed in mRNA isolated from frozen livers by qRT-PCR of chow-fed mice (black bars) and untreated (dark gray bars) or peptide-treated HFD-32-fed mice (light gray bars). (D) IHC of CPT1a in liver sections from HFD-32-fed mice untreated (control) or treated with R-Tf-D-L4 peptide, with the relative intensity (n = 3 liver sections) presented in parentheses. (E and F) qRT-PCR analysis of mRNA levels of proteins, factors, and co-activators involved in lipid metabolism and regulation in chow-fed mice (black bars) and HFD-32-fed mice untreated (dark gray bars) or treated with R-Tf-D-LP4 peptide (light gray bars). Proteins associated with lipid metabolism (E) or involved in fatty acid transport (F). Immunoblot (G) and quantitative analysis (H) of proteins related to lipid biosynthesis in livers from HFD-32-fed mice untreated (dark gray bars) or treated with R-Tf-D-LP4 peptide (light gray bars). β-actin served as a loading control. (I) mRNA levels of lipid synthesis-associated proteins in chow-fed mice (black bars) and HFD-32-fed mice untreated (dark gray bars) or treated with R-Tf-D-LP4 peptide (light gray bars). Results are mean ± SEM (n = 3–5; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001).
The expression levels of UCP1 and UCP2, associated with thermogenic respiration, were dramaticaly increased in the peptide-treated mice, 3- and 5-fold at the protein level and 26.5- and 3.0-fold at the mRNA level, relative to ND-fed mice, and 2- and 3.7-fold at the protein level and 11- and 1.8-fold at the mRNA level, relative to the levels seen in HFD-32-fed mice (Figures 8A–8C). Expression levels of PPARγ and PGC1α, involved in regulating lipid metabolic processes, were increased (Figures 8A, 8B, and 8E). ACSL1 and ACSL5 levels were also increased 3- to 4-fold in the livers of HFD-32-fed, peptide-treated mice (Figure 8F). mRNA levels of ACADL and HADHa (hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase/enoyl-CoA hydratase, alpha subunit), involved in β-oxidation, were slightly increased in livers from HFD-32-fed mice, and they were further increased upon peptide treatment (Figure 8F).
In contrast to the increased expression of proteins associated with fatty acid oxidation elicited by peptide treatment, those associated with lipid synthesis were decreased (Figures 8G–8I). SREBP1A, SREBP1C, ACC1, ACC2, SCD1, and ELOVL6 levels were all increased 2- to 3-fold in the HFD-32-fed mice, and they decreased to the levels seen in the ND-fed mice following peptide treatment (Figures 8G–8I). The expression of FAS was also decreased upon peptide treatment (Figures 8G and 8H).
Finally, the mRNA levels of the nuclear bile acid receptor, farnesoid X receptor (FXR), were highly increased in HFD-32-fed mice at the NASH stage, with peptide treatment restoring FXR levels to those of ND-fed mice (Figure 8I). FXR regulates glucose, lipid, and energy metabolism,32 while activated FXR negatively regulates liver triglycerides and cholesterol.33
R-Tf-D-LP4-Mediated Effects on Glycogen Levels and Metabolism
As glucose stored as glycogen may further contribute to hepatic steatosis by diverting excess carbohydrates into fatty acids by the de novo lipogenesis pathway (DNL),34 we analyzed the levels of glycogen and glycogen metabolism-related enzymes in the HFD-32-fed mice without and with peptide treatment (Figure S6). The results showed that, at both the steatosis and NASH stages, glycogen levels were decreased in the HFD-32-fed mice, while the peptide increased them to the levels in ND-fed mice (Figures S6A–S6D). Enzymes associated with glycogen metabolism, namely glucose-6-phosphate phosphatase (G6Pase) and phosphoenolpyruvate carboxykinase (PEP-CK), were increased in the HFD-32-fed, peptide-treated mice over their levels in ND- or HFD-fed mice. On the contrary, the expression levels of pyruvate carboxylase (PCX) and fructose bisphosphatase (FBPase), both supporting gluconeogenesis, were increased in HFD-32-fed mice, but they decreased in such mice treated with the peptide (Figures S6E and S6F). These results suggest that, in HFD-32-fed mice, glycogen is synthesized and used in the DNL and that this process was inhibited by peptide treatment, in which case carbohydrates and fat were oxidized.
R-Tf-D-LP4-Mediated Effects on Physiological Parameters of the HFD-32/STAM Mouse Model and Metabolic Cages
The effects of the peptide on the metabolic parameters of HFD-32-fed mice at steatosis were also evaluated by indirect calorimetry using metabolic cages. Heat production (EE [energy expenditure]), respiratory exchange ratio (RER), oxygen consumption ratio, and carbon dioxide production (CDP) were evaluated (Figures S7A–S7E). Rates of EE were 10%–20% higher in HFD-32-fed, peptide-treated mice, as compared to HFD-32-fed mice (Figure S7A).
The RER value for HFD-32-fed mice was ∼0.7 and decreased to about 0.6 in peptide-treated mice (Figure S7B), indicating a higher use of fat in the HFD-32-fed mice. Oxygen consumption rate (OCR) and CDP levels were also increased in HFD-32-fed, peptide-treated mice (Figures S7C and S7D), reflecting respiratory-based metabolism. Peptide treatment had no effect on locomotor activity (Figure S7E), led to a slight increase in food intake (Figures S8A and S8B), and increased water consumption (Figure S8C). The results suggest that HFD-32-fed mice, when treated with the peptide, utilized fat and produced more heat.
VDAC1 Interactions with CPT1a and ACSL1 Suggest a Mechanism for Increased β-Oxidation
CPT1a and CPT2 contribute to fatty acid oxidation by mediating the transport of acyl-CoA formed by ACSL1 via its conversion into acyl-carnitine by CPT1a and then by CPT2 to acyl-CoA for oxidation in the matrix. CPT1a, ACSL1, and VDAC1 were reported to form a complex.11 To confirm the presence of such a complex, rat liver-isolated mitochondria were solubilized under gentle conditions using β-octyl glucoside (OG), and they were subjected to two-step column chromatography in the presence of OG (Figures 9A and 9B). CPT1a, ACSL1, and VDAC1 were identified in the same fractions, along with CPT2, localized in the IMM24 (Figure 9A). Size-exclusion chromatography revealed a complex with an estimated molecular mass of ∼420 kDa (Figure 9B). As the expected mass of the complex is 272 kDa, it would appear that some proteins are present in more than a single copy. The presence of CPT2 in the complex suggests that the complex is enriched at OMM-IMM contact sites (Figure 9E).
Figure 9.

Purification of a VDAC1/CPT1a/ACSL1 Complex and Peptide Treatment of HFD-32-Fed Mice Increase Complex Levels and Association
(A) Immunoblot of complexed VDAC1, CPT1a, ACSL1, and CPT2 isolated from rat liver mitochondria using reactive red agarose (RRA). (B) Sephacryl S-200 chromatography with absorbance at 280 nm (presented as mAU) of elution fractions is shown, and the inset shows immunoblots of the peak fractions as functions of the elution volume (Vol). (C and D) Liver sections from HFD-32-fed mice untreated and peptide treated (14 mg/kg) at the steatosis (C) or NASH (D) stage were subjected to in situ PLA to test for interactions between VDAC1 and CPT1a or CPT1a and ACSL1 or VDAC1 and ACSL1 (see the Supplemental Materials and Methods). DAPI-stained nuclei are shown. (E) Schematic presentation of proposed mode of peptide-mediated increased β-oxidation in steatosis and NASH, as induced by an HFD-32 diet. The complex ACSL1/VDAC1/CPT1a and CPT2 is modified by the VDAC1-derived peptide. (a) VDAC1 serves as an anchoring site for ACSL1, associated with the outer surface of the OMM, and CPT1a, which faces the IMS. The acyl-CoA formed by ACSL1 is transferred across the OMM by VDAC1 to the IMS, where CPT1a converts it into acyl-carnitine that is transported by CACT into the matrix, where it is converted back into acyl-CoA via IMM-associated CPT2 and, subsequently, undergoes β-oxidation in the matrix. (b) The VDAC1-based peptide serves as a decoy, interacting with CPT1a and/or ACSL1 such that their interaction with VDAC1 is modified, leading to increased fatty acid translocation to the mitochondria, thereby increasing β-oxidation. The presence of CPT2 in the purified complex suggests the direct channeling of fatty acids from the cytosol to the matrix for β-oxidation.
Next, we used the in situ proximity ligation assay (PLA)35 to visualize endogenous protein-protein interactions between VDAC1-CPT1a, CPT1a-ACSL1, and VDAC1-ACSL1 (Figures 9C and 9D). These complexes were present in the liver sections of ND-fed mice, with a signal similar to that in HFD-32-fed mice. However, HFD-32-fed, peptide-treated mice showed a strong signal for each of the three protein pairs (i.e., VDAC1-CPT1a, CPT1a-ACSL1, and VDAC1-ACSL1) in both steatosis and NASH, suggesting an increase in the amounts of the complex. This is in agreement with the highly increased expression levels of VDAC1, CPT1a, and ACSL1 in the livers of HFD-32-fed, peptide-treated mice (Figures 3, 4, 7, 8, and S5). As a control, we performed PLA with VDAC1 and CS, located in the matrix. Although their expressions were increased in the HFD-32-fed, peptide-treated mice, no increased PLA signal was observed, in line with their localization in two different compartments (Figure S9).
Discussion
Chronic liver disease represents a significant public health problem worldwide, with viral hepatitis and NAFLD affecting about 20% of the general population.1 NAFLD can result in cirrhosis, leading to liver failure. Currently, there is no effective treatment, and many patients end up with a progressive form of the disease, eventually requiring a liver transplant.
Here we demonstrated that the VDAC1-based peptide R-Tf-D-LP4 dramatically reduced pathophysiological features related to steatohepatitis and/or NASH that developed in an HFD-32/STAM mouse model. This mouse model displayed the metabolic, histological, and clinical endpoints of human steatosis and/or NASH. HFD-32-fed mice developed steatohepatitis, with fat droplet accumulation in hepatocytes, ballooning degeneration, scattered inflammatory cell infiltration, fibrosis, and hyperglycemia, seen in HFD-32-fed mice and reduced in R-Tf-D-LP4-treated mice.
Fibrous scars in liver change the physiological architecture of hepatic tissues, affecting normal liver function.36 Stellate cell activation and differentiation into fibrotic cells, as reflected by α-SMA and TGF-β expressions, were reduced in HFD-32-fed, peptide-treated mice. The peptide also reduced inflammatory macrophages and the expressions of cytokines (IL-1β and IL-6) in livers from HFD-32-fed mice.
Remarkably, in mice continuously fed HFD-32, the peptide not only stopped the progression of the diseases when treatment started at the steatosis, NASH, or HCC stages19 but also reversed liver physiology and anatomy to a state similar to ND-fed mice. The VDAC1-based peptide eliminated or highly reduced numerous indicators of liver pathogenesis mediated via regulating fat and carbohydrate metabolism, leading to an altered transcriptional program (see below).
The VDAC1-Based Peptide Altered Carbohydrate and Lipid Metabolism in the Livers of HFD-32-Fed Mice
The peptide affected both carbohydrate and lipid metabolism in HFD-32-fed mouse livers. This was reflected in the peptide decreasing serum triglycerides, body fat, and hepatocyte fat droplets and increasing hepatocyte glycogen accumulation in HFD-32-fed mice. The peptide stimulated catabolic pathways, such as mitochondrial lipid transport and fatty acid oxidation, carbohydrate metabolism, and thermogenesis while inhibiting lipid synthesis. The increased fat metabolism and energy expenditure following peptide treatment was also reflected in the physiological parameters of the mice, as revealed in the metabolic cage studies and the altered expression of genes and proteins associated with lipid and carbohydrate metabolism. For example, the increased catabolic processes upon peptide treatment were reflected in the increased expression of UCP1 and UCP2 proteins associated with thermogenic respiration and high water consumption. In this respect, the capacity to increase energy expenditure in response to caloric excess, referred to as diet-induced thermogenesis (DIT), was proposed as a defense against diet-induced obesity.37 UCP1 is thought to drive DIT and is critical for thermal homeostasis, as mice lacking UCP1 show induced obesity and abolished DIT37 and are sensitive to acute cold exposure.38
Glycogen synthesis and degradation are highly regulated processes contributing to glucose homeostasis. Glycogen levels in HFD-32-fed mice were decreased, and peptide treatment increased the levels to that of ND-fed mice, in agreement with the peptide increasing Glut-2 and, dramatically so, Glut-4 mRNA levels. Generally, Glut-4, an insulin-dependent glucose transporter, is not expressed in liver, although glucose stimulates Glut-4 expression in rat hepatocytes.39 Elevated Glut-4 expression in liver cells should increase glucose uptake into hepatocytes and, thus, glycolysis. Indeed, the expressions of GAPDH and LDH-A were increased in the livers of HFD-32-fed, peptide-treated mice. Moreover, the nuclear location of LDH-A is associated with the regulation of SIRT1 activity (see below).
Taken together, our results suggest that the peptide increases glucose and fat metabolism.
Peptide-Altered Lipid and Carbohydrate Metabolism Is Mediated via Alterations in the Liver Transcriptional Program
Cell metabolic state and gene expression are tightly linked, with the dynamic interface of cellular metabolism and gene expression mediated by bioenergetic pathways and enzymes providing metabolic cofactors for epigenetic regulation of gene expression.40 Our results suggest that the global effects of the peptide on the liver pathology, as induced by HFD-32, are mediated via reprogrammed metabolism, inducing alterations in the liver transcriptional program. Peptide treatment of HFD-32-fed mice increased liver expression of genes and proteins involved in fatty acid transfer to mitochondria, fatty acid oxidation, and thermogenic respiration, at both the steatosis and NASH stages. Moreover, the levels of genes and proteins engaged in fatty acid and glycogen synthesis were reduced.
The levels of SIRT1 and SIRT6, key enzymes in transcriptional regulation, were highly increased. SIRT1 activity is linked with metabolic homeostasis through its ability to deacetylate target proteins.27 In response to low nutritional availability, SIRT1 functions as a master switch to maintain lipid and glucose homeostasis, insulin secretion, and energy balance by regulating important metabolic regulators, such as SREBP1C, PGC1α, and forkhead box protein O1 (Foxo-1).41 Thus, peptide-induced SIRT1 overexpression is in line with the downregulation of SIRT1 in mouse liver, increasing the expressions of lipogenic genes and decreasing the expressions of genes involved in fatty acid β-oxidation and bile acid biosynthesis.42
SIRT1-mediated deacetylation of SREBP1C inhibits its activity.41 SREBP1C is a key transcription factor that regulates genes in the de novo lipogenesis and glycolysis pathways and contributes to liver steatosis and insulin resistance.43 The expressions of SREBP1C and SREBP1A (dominantly expressed in liver) were increased in livers from HFD-32-fed mice, while peptide treatment decreased their expressions, in line with the inhibition of their pro-lipogenesis effects.43
SIRT1-signaling activation has been shown to protect mice from high saturated fat diet-induced steatosis and inflammation through inhibiting fatty acid biosynthesis while promoting fatty acid β-oxidation44 and activating AMPK.45 Similar effects were induced by the peptide in HFD-32-fed mice. AMPK is a central regulator of cellular metabolism, transcription factors and their co-activators, and histones to regulate gene expression, leading to metabolic reprogramming and cell survival.46 p-AMPK levels were decreased by the peptide in HFD-32-fed mice but increased 2-fold over the level in ND-fed mice. AMPK, via phosphorylation, converts ACC into the inactive form that promotes fatty acid biosynthesis and steatosis while inhibiting fatty acid β-oxidation.47 Both ACC1 and ACC2 levels were increased in HFD-32-fed mice and decreased by the peptide below their levels in ND-fed mice. This, together with their possible inactivation via increased p-AMPK, suggests that the peptide inhibits fatty acid biosynthesis and steatosis and stimulates fatty acid β-oxidation.
Metabolic reprogramming in response to glucose and fatty acid availabilities is also controlled by PGC1α. PGC1α is indirectly involved in lipid metabolism, with hepatic overexpression of PGC1α leading to increased fatty acid β-oxidation and reduced triacylglycerol accumulation.25 Moreover, fatty liver is associated with impaired activity of PGC1α levels.48 Thus, our finding of increased PGC1α mRNA levels in the livers of peptide-treated mice agrees with the above and with our findings of fatty acid oxidation and reduced fatty acid accumulation in the liver.
SIRT1 deacetylation of PGC1α increases its activity, which enhances the expressions of metabolic genes involved in hepatic gluconeogenesis, β-oxidation, and mitochondrial functions.49 The highly increased levels of PGC1α (6-fold) and of SIRT1 induced by the peptide are expected to increase PGC1α deacetylation, thereby stimulating fatty acid oxidation.
Another target of SIRT1, the FXR, negatively regulates liver triglycerides and cholesterol,33 thereby regulating hepatic lipid metabolism.50 FXR levels were increased in the livers of HFD-32-fed mice, and they were reduced by the peptide to levels seen in chow-fed mice.
Peptide treatment of HFD-32-fed mice increased the expression levels of PPARγ and the PPARγ co-activator PGC1α, which is also activated by SIRT1-mediated deacetylation. This, in turn, activates PPARγ. Peptide activation of PPARγ is in agreement with the findings that, in patients with NASH, the PPARγ agonist pioglitazone improves steatosis and reduces liver inflammation and fibrosis51 and that overexpression of PPARγ attenuated steatohepatitis.52 Indeed, many PPARγ-activating drugs have been developed.53
SIRT6 expression levels were also increased in HFD-32-fed, peptide-treated mice. SIRT6, like SIRT1, regulates liver metabolism, reflected in the finding that hepatic Sirt6-knockout (KO) mice develop liver steatosis.54 In addition, ablation of SIRT6 in adipocytes leads to hyperglycemia, insulin resistance, obesity, hepatic steatosis, and reduced thermogenesis in mice.55 At the same time, transgenic mice overexpressing SIRT6 and fed an HFD are less sensitive to diet-induced obesity and insulin resistance.56 SIRT6 expression is positively correlated with the expressions of genes associated with β-oxidation, such as CPT1a, and negatively correlated with the expressions of genes associated with lipogenesis, such as ACC1, FAS, and SCD1,54 or involved in cholesterol synthesis, such as SREBP.57 These observations correlate with our results showing that the peptide reduced the expression levels of the above genes in HFD-32-fed mice.
Finally, the presence of metabolic enzymes in the nucleus has been recognized as an important mechanism linking metabolic flux to the regulation of gene expression.58 LDH-A is expressed in the liver and was detected in the nucleus,59 where it generates NAD+ required by the NAD+-dependent deacetylase SIRT1.60 Peptide treatment of HFD-32-fed mice highly increased LDH-A expression levels and also induced its translocation into the nucleus, where SIRT1 is located, thus indirectly activating SIRT1.
These findings further demonstrate the link between cell metabolic alteration induced by the peptide in HFD-32-fed mice and gene expression associated with inhibited fatty acid biosynthesis and steatosis and stimulated fatty acid β-oxidation.
VDAC1, Steatosis, NASH, and Their Inhibition by the VDAC1-Based Peptide: Proposed Mode of Peptide Action
A lack of VDAC1 led to mitochondria that no longer oxidize fatty acids, and a VDAC1 inhibitor inhibited the oxidation of palmitate.61 In addition, it is proposed that VDAC closure produces steatosis both in alcoholic steatohepatitis (ASH) and NASH.62, 63 Here we showed that VDAC1-based peptide treatment led to increased β-oxidation.
Lipids are oxidized primarily in the mitochondria via β-oxidation, which begins with mitochondrial import of long-chain fatty acyl-CoAs. It was proposed that the long-chain fatty acyl-CoA formed by ACSL at the outer surface of the OMM is transferred via VDAC1 to the IMS, where CPT1a, also at the OMM but facing the IMS, converts the molecule into long-chain fatty acyl-carnitine that then crosses the IMM to reach the matrix, with such transport being mediated by CACT. Once in the matrix, the long-chain fatty acyl-carnitine is converted back into a long-chain fatty acyl-CoA by CPT2.11, 12 Once restored to acyl-CoA in the matrix, β-oxidation occurs (Figure 9E).
Here we purified large complexes containing CPT1a, ACSL1/5, and VDAC1 located in the OMM/IMS and CPT2 at the IMM with a molecular mass of 415 kDa, higher than the expected 272 kDa for a complex containing a single copy of each protein. The presence of CPT2 in the complex suggests that the complex provides a shuttle for fatty acids from crossing the OMM-IMM contact site and reaching the matrix (Figure 9E).
The PLA reporting protein-protein interactions at single-molecule resolution64 confirmed the close proximity of CPT1a, ACSL1, and VDAC1. Moreover, in peptide-treated mice, the levels of these proteins were increased, leading to enhanced complex formation (as reflected in the PLA), which subsequently increases fatty acid transport to the mitochondria. We propose that the peptide serves as a decoy, interacting with CPT1a and/or ACSL1 such that their interaction with VDAC1 is modified, leading to their partial detachment from VDAC1, thus rendering the channel being more accessible to fatty acid transport (Figure 9E), and in agreement with the proposal that VDAC closure produces steatosis.62, 63 This, in turn, leads to increased fatty acid translocation to the mitochondria, thereby increasing β-oxidation (Figure 9E), as manifested by decreased hepatocyte fat levels, increased expression of enzymes related to lipid oxidation and thermogenesis, and the metabolic cage results.
To conclude, a cell-penetrating VDAC1-based peptide was found to be an effective treatment for liver NAFLD pathology, as developed in the HFD-32/STAM, recapitulating the various phenotypes of NAFLD. Given the dramatic effects of the peptide on fat accumulation in these mice, with increased expressions of fatty acid oxidation-related proteins and reduced levels of fatty acid synthesis-related proteins, together with the metabolic cage results, we suggest that the R-Tf-D-LP4 peptide inhibits steatosis and NASH by increasing fatty acid oxidation (Figure 9E) via alterations in the liver transcriptional program. In addition, the decreased glycogen levels in the HFD-fed mice, relative to peptide-treated or ND-fed mice, suggest that these mice perform de novo lipogenesis,34 which was decreased upon peptide treatment. The results point to the VDAC1-based peptide addressed here as a potential novel strategy for treating steatosis and NASH.
Materials and Methods
See the Supplemental Materials and Methods for details.
Peptide Synthesis and Preparation
R-Tf-D-LP4 peptide (KWTWK-216-NSNGATWALNVATELKK-199-EWTWSHRPYIAH), comprising 34 residues in D-configuration (except the underlined Tf sequence),16 was prepared (Supplemental Materials and Methods).
Steatosis-NASH-HCC STAM Mouse Model and Dietary Interventions
Steatosis-NASH-HCC model mice were obtained as described previously6 and detailed in the Supplemental Materials and Methods. Mice were intravenously treated three times a week with R-Tf-D-LP4 (14 mg/kg) for 3 weeks, from weeks 6–8 for steatosis and from weeks 9–11 for NASH. Livers were removed, and part was frozen in liquid nitrogen for immunoblotting and qPCR analysis, while the other part was fixed and used for immunostaining (Supplemental Materials and Methods). Experimental protocols used were approved by the Institutional Animal Care and Use Committee.
Biochemical Analysis
Serum biochemical liver function markers, fat metabolism status, and blood glucose levels were measured (Supplemental Materials and Methods).
Fat Staining with Oil Red O, IHC, Immunofluorescence, and Immunoblotting Analyses of Liver Tissue
These were carried out as described in the Supplemental Materials and Methods. Images were captured using a confocal microscope (Olympus 1X81) or a light microscope (Leica DM2500).
Glycogen Level Measurement
Glycogen amounts in livers derived from ND-fed; HFD-32-fed; and HFD-32-fed, peptide-treated mice were determined using two different methods (Supplemental Materials and Methods).
RNA Preparation and Real-Time PCR
These were carried out as described in the Supplemental Materials and Methods.
CPT1-ACSL1-VDAC1 Complex Purification
CPT1 was purified from freshly isolated rat liver mitochondria by column chromatography using reactive red-agarose and Sephacryl S-200 columns (Supplemental Materials and Methods).
Proximity ligation assay (PLA)
VDAC1-CPT1a, VDAC1-ACSL1, and CPT1a-ACSL1 interactions were analyzed using a PLA35 (Supplemental Materials and Methods).
Assessing Metabolic Parameters Using Metabolic Cages
Metabolic parameters such as O2 consumption, CO2 production, energy expenditure, and locomotor activity of HFD-32-fed mice with and without peptide treatment were assessed using metabolic cages (Supplemental Materials and Methods).
Statistics and Data Analysis
The mean ± SEM of results obtained from at least three independent experiments is presented. The significance of differences was calculated by a two-tailed Student’s t test and is reported as *p < 0.05, **p < 0.01, or ***p < 0.001. Significance was also analyzed using a non-parametric Mann-Whitney U test to compare control and experimental groups, using GraphPad Prism 7.04 software.
Author Contributions
S.P. performed the research and together with Y. Krelin analyzed the data. Y. Kuperman helped with the metabolic cage experiment and analyzed the data. V.S.-B. assessed the results and wrote the paper.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This research was supported by Phil and Sima Needleman and Yafa and Ezra Yerucham research funds and by a grant from the Israel Science Foundation (307/13). We thank professor Ruth Birk for her advice.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2019.06.017.
Supplemental Information
References
- 1.Law K., Brunt E.M. Nonalcoholic fatty liver disease. Clin. Liver Dis. 2010;14:591–604. doi: 10.1016/j.cld.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 2.Friedman S.L., Neuschwander-Tetri B.A., Rinella M., Sanyal A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018;24:908–922. doi: 10.1038/s41591-018-0104-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leite N.C., Salles G.F., Araujo A.L., Villela-Nogueira C.A., Cardoso C.R. Prevalence and associated factors of non-alcoholic fatty liver disease in patients with type-2 diabetes mellitus. Liver Int. 2009;29:113–119. doi: 10.1111/j.1478-3231.2008.01718.x. [DOI] [PubMed] [Google Scholar]
- 4.Lomonaco R., Sunny N.E., Bril F., Cusi K. Nonalcoholic fatty liver disease: current issues and novel treatment approaches. Drugs. 2013;73:1–14. doi: 10.1007/s40265-012-0004-0. [DOI] [PubMed] [Google Scholar]
- 5.Takahashi Y., Soejima Y., Fukusato T. Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J. Gastroenterol. 2012;18:2300–2308. doi: 10.3748/wjg.v18.i19.2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fujii M., Shibazaki Y., Wakamatsu K., Honda Y., Kawauchi Y., Suzuki K., Arumugam S., Watanabe K., Ichida T., Asakura H., Yoneyama H. A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med. Mol. Morphol. 2013;46:141–152. doi: 10.1007/s00795-013-0016-1. [DOI] [PubMed] [Google Scholar]
- 7.Pessayre D. Role of mitochondria in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2007;22(Suppl 1):S20–S27. doi: 10.1111/j.1440-1746.2006.04640.x. [DOI] [PubMed] [Google Scholar]
- 8.Lemasters J.J., Holmuhamedov E. Voltage-dependent anion channel (VDAC) as mitochondrial governator--thinking outside the box. Biochim. Biophys. Acta. 2006;1762:181–190. doi: 10.1016/j.bbadis.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 9.Shoshan-Barmatz V., De Pinto V., Zweckstetter M., Raviv Z., Keinan N., Arbel N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Aspects Med. 2010;31:227–285. doi: 10.1016/j.mam.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 10.Shoshan-Barmatz V., Krelin Y., Shteinfer-Kuzmine A., Arif T. Voltage-Dependent Anion Channel 1 As an Emerging Drug Target for Novel Anti-Cancer Therapeutics. Front. Oncol. 2017;7:154. doi: 10.3389/fonc.2017.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee K., Kerner J., Hoppel C.L. Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex. J. Biol. Chem. 2011;286:25655–25662. doi: 10.1074/jbc.M111.228692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tonazzi A., Giangregorio N., Console L., Indiveri C. Mitochondrial carnitine/acylcarnitine translocase: insights in structure/ function relationships. Basis for drug therapy and side effects prediction. Mini Rev. Med. Chem. 2015;15:396–405. doi: 10.2174/138955751505150408142032. [DOI] [PubMed] [Google Scholar]
- 13.Martel C., Allouche M., Esposti D.D., Fanelli E., Boursier C., Henry C., Chopineau J., Calamita G., Kroemer G., Lemoine A., Brenner C. Glycogen synthase kinase 3-mediated voltage-dependent anion channel phosphorylation controls outer mitochondrial membrane permeability during lipid accumulation. Hepatology. 2013;57:93–102. doi: 10.1002/hep.25967. [DOI] [PubMed] [Google Scholar]
- 14.Shoshan-Barmatz V., Maldonado E.N., Krelin Y. VDAC1 at the crossroads of cell metabolism, apoptosis and cell stress. Cell Stress. 2017;1:11–36. doi: 10.15698/cst2017.10.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arzoine L., Zilberberg N., Ben-Romano R., Shoshan-Barmatz V. Voltage-dependent anion channel 1-based peptides interact with hexokinase to prevent its anti-apoptotic activity. J. Biol. Chem. 2009;284:3946–3955. doi: 10.1074/jbc.M803614200. [DOI] [PubMed] [Google Scholar]
- 16.Prezma T., Shteinfer A., Admoni L., Raviv Z., Sela I., Levi I., Shoshan-Barmatz V. VDAC1-based peptides: novel pro-apoptotic agents and potential therapeutics for B-cell chronic lymphocytic leukemia. Cell Death Dis. 2013;4:e809. doi: 10.1038/cddis.2013.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shteinfer-Kuzmine A., Amsalem Z., Arif T., Zooravlov A., Shoshan-Barmatz V. Selective induction of cancer cell death by VDAC1-based peptides and their potential use in cancer therapy. Mol. Oncol. 2018;12:1077–1103. doi: 10.1002/1878-0261.12313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anderson E.R., Shah Y.M. Iron homeostasis in the liver. Compr. Physiol. 2013;3:315–330. doi: 10.1002/cphy.c120016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pittala S., Krelin Y., Shoshan-Barmatz V. Targeting Liver Cancer and Associated Pathologies in Mice with a Mitochondrial VDAC1-Based Peptide. Neoplasia. 2018;20:594–609. doi: 10.1016/j.neo.2018.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kleiner D.E., Brunt E.M., Van Natta M., Behling C., Contos M.J., Cummings O.W., Ferrell L.D., Liu Y.C., Torbenson M.S., Unalp-Arida A., Nonalcoholic Steatohepatitis Clinical Research Network Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 21.Liangpunsakul S., Chalasani N. Treatment of Nonalcoholic Fatty Liver Disease. Curr. Treat. Options Gastroenterol. 2003;6:455–463. doi: 10.1007/s11938-003-0047-0. [DOI] [PubMed] [Google Scholar]
- 22.Tsuchida T., Friedman S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017;14:397–411. doi: 10.1038/nrgastro.2017.38. [DOI] [PubMed] [Google Scholar]
- 23.Liang W., Menke A.L., Driessen A., Koek G.H., Lindeman J.H., Stoop R., Havekes L.M., Kleemann R., van den Hoek A.M. Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PLoS One. 2014;9:e115922. doi: 10.1371/journal.pone.0115922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fraser F., Corstorphine C.G., Zammit V.A. Topology of carnitine palmitoyltransferase I in the mitochondrial outer membrane. Biochem. J. 1997;323:711–718. doi: 10.1042/bj3230711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morris E.M., Meers G.M., Booth F.W., Fritsche K.L., Hardin C.D., Thyfault J.P., Ibdah J.A. PGC-1α overexpression results in increased hepatic fatty acid oxidation with reduced triacylglycerol accumulation and secretion. Am. J. Physiol. Gastrointest. Liver Physiol. 2012;303:G979–G992. doi: 10.1152/ajpgi.00169.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Towle H.C., Kaytor E.N., Shih H.M. Regulation of the expression of lipogenic enzyme genes by carbohydrate. Annu. Rev. Nutr. 1997;17:405–433. doi: 10.1146/annurev.nutr.17.1.405. [DOI] [PubMed] [Google Scholar]
- 27.Knight J.R., Milner J. SIRT1, metabolism and cancer. Curr. Opin. Oncol. 2012;24:68–75. doi: 10.1097/CCO.0b013e32834d813b. [DOI] [PubMed] [Google Scholar]
- 28.Hardie D.G., Ross F.A., Hawley S.A. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carling D., Zammit V.A., Hardie D.G. A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 1987;223:217–222. doi: 10.1016/0014-5793(87)80292-2. [DOI] [PubMed] [Google Scholar]
- 30.Shteinfer-Kuzmine A., Arif T., Krelin Y., Tripathi S.S., Paul A., Shoshan-Barmatz V. Mitochondrial VDAC1-based peptides: Attacking oncogenic properties in glioblastoma. Oncotarget. 2017;8:31329–31346. doi: 10.18632/oncotarget.15455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chandrashekaran V., Das S., Seth R.K., Dattaroy D., Alhasson F., Michelotti G., Nagarkatti M., Nagarkatti P., Diehl A.M., Chatterjee S. Purinergic receptor X7 mediates leptin induced GLUT4 function in stellate cells in nonalcoholic steatohepatitis. Biochim. Biophys. Acta. 2016;1862:32–45. doi: 10.1016/j.bbadis.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiang J.Y.L. Bile acid metabolism and signaling in liver disease and therapy. Liver Res. 2017;1:3–9. doi: 10.1016/j.livres.2017.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sinal C.J., Tohkin M., Miyata M., Ward J.M., Lambert G., Gonzalez F.J. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102:731–744. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- 34.Schwarz J.-M., Linfoot P., Dare D., Aghajanian K. Hepatic de novo lipogenesis in normoinsulinemic and hyperinsulinemic subjects consuming high-fat, low-carbohydrate and low-fat, high-carbohydrate isoenergetic diets. Am. J. Clin. Nutr. 2003;77:43–50. doi: 10.1093/ajcn/77.1.43. [DOI] [PubMed] [Google Scholar]
- 35.Gustafsdottir S.M., Schallmeiner E., Fredriksson S., Gullberg M., Söderberg O., Jarvius M., Jarvius J., Howell M., Landegren U. Proximity ligation assays for sensitive and specific protein analyses. Anal. Biochem. 2005;345:2–9. doi: 10.1016/j.ab.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 36.Friedman S.L. Evolving challenges in hepatic fibrosis. Nat. Rev. Gastroenterol. Hepatol. 2010;7:425–436. doi: 10.1038/nrgastro.2010.97. [DOI] [PubMed] [Google Scholar]
- 37.Feldmann H.M., Golozoubova V., Cannon B., Nedergaard J. UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab. 2009;9:203–209. doi: 10.1016/j.cmet.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 38.Enerbäck S., Jacobsson A., Simpson E.M., Guerra C., Yamashita H., Harper M.E., Kozak L.P. Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature. 1997;387:90–94. doi: 10.1038/387090a0. [DOI] [PubMed] [Google Scholar]
- 39.Bhattacharya S., Ghosh R., Maiti S., Khan G.A., Sinha A.K. The activation by glucose of liver membrane nitric oxide synthase in the synthesis and translocation of glucose transporter-4 in the production of insulin in the mice hepatocytes. PLoS ONE. 2013;8:e81935. doi: 10.1371/journal.pone.0081935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Etchegaray J.-P., Mostoslavsky R. Interplay between metabolism and epigenetics: a nuclear adaptation to environmental changes. Mol. Cell. 2016;62:695–711. doi: 10.1016/j.molcel.2016.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ponugoti B., Kim D.H., Xiao Z., Smith Z., Miao J., Zang M., Wu S.Y., Chiang C.M., Veenstra T.D., Kemper J.K. SIRT1 deacetylates and inhibits SREBP-1C activity in regulation of hepatic lipid metabolism. J. Biol. Chem. 2010;285:33959–33970. doi: 10.1074/jbc.M110.122978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rodgers J.T., Puigserver P. Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proc. Natl. Acad. Sci. USA. 2007;104:12861–12866. doi: 10.1073/pnas.0702509104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pettinelli P., Del Pozo T., Araya J., Rodrigo R., Araya A.V., Smok G., Csendes A., Gutierrez L., Rojas J., Korn O. Enhancement in liver SREBP-1c/PPAR-alpha ratio and steatosis in obese patients: correlations with insulin resistance and n-3 long-chain polyunsaturated fatty acid depletion. Biochim. Biophys. Acta. 2009;1792:1080–1086. doi: 10.1016/j.bbadis.2009.08.015. [DOI] [PubMed] [Google Scholar]
- 44.Purushotham A., Schug T.T., Xu Q., Surapureddi S., Guo X., Li X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009;9:327–338. doi: 10.1016/j.cmet.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hou X., Xu S., Maitland-Toolan K.A., Sato K., Jiang B., Ido Y., Lan F., Walsh K., Wierzbicki M., Verbeuren T.J. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J. Biol. Chem. 2008;283:20015–20026. doi: 10.1074/jbc.M802187200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sanli T., Steinberg G.R., Singh G., Tsakiridis T. AMP-activated protein kinase (AMPK) beyond metabolism: a novel genomic stress sensor participating in the DNA damage response pathway. Cancer Biol. Ther. 2014;15:156–169. doi: 10.4161/cbt.26726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fullerton M.D., Galic S., Marcinko K., Sikkema S., Pulinilkunnil T., Chen Z.P., O’Neill H.M., Ford R.J., Palanivel R., O’Brien M. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med. 2013;19:1649–1654. doi: 10.1038/nm.3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aharoni-Simon M., Hann-Obercyger M., Pen S., Madar Z., Tirosh O. Fatty liver is associated with impaired activity of PPARγ-coactivator 1α (PGC1α) and mitochondrial biogenesis in mice. Lab. Invest. 2011;91:1018–1028. doi: 10.1038/labinvest.2011.55. [DOI] [PubMed] [Google Scholar]
- 49.Rodgers J.T., Lerin C., Gerhart-Hines Z., Puigserver P. Metabolic adaptations through the PGC-1 alpha and SIRT1 pathways. FEBS Lett. 2008;582:46–53. doi: 10.1016/j.febslet.2007.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kemper J.K., Xiao Z., Ponugoti B., Miao J., Fang S., Kanamaluru D., Tsang S., Wu S.Y., Chiang C.M., Veenstra T.D. FXR acetylation is normally dynamically regulated by p300 and SIRT1 but constitutively elevated in metabolic disease states. Cell Metab. 2009;10:392–404. doi: 10.1016/j.cmet.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boettcher E., Csako G., Pucino F., Wesley R., Loomba R. Meta-analysis: pioglitazone improves liver histology and fibrosis in patients with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2012;35:66–75. doi: 10.1111/j.1365-2036.2011.04912.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu C.W., Chu E.S., Lam C.N., Cheng A.S., Lee C.W., Wong V.W., Sung J.J., Yu J. PPARgamma is essential for protection against nonalcoholic steatohepatitis. Gene Ther. 2010;17:790–798. doi: 10.1038/gt.2010.41. [DOI] [PubMed] [Google Scholar]
- 53.Sun X., Zhang Y., Xie M. Review. The role of peroxisome proliferator-activated receptor in the treatment of non-alcoholic fatty liver disease. Acta Pharm. 2017;67:1–13. doi: 10.1515/acph-2017-0007. [DOI] [PubMed] [Google Scholar]
- 54.Kim H.S., Xiao C., Wang R.H., Lahusen T., Xu X., Vassilopoulos A., Vazquez-Ortiz G., Jeong W.I., Park O., Ki S.H. Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab. 2010;12:224–236. doi: 10.1016/j.cmet.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kuang J., Zhang Y., Liu Q., Shen J., Pu S., Cheng S., Chen L., Li H., Wu T., Li R. Fat-Specific Sirt6 Ablation Sensitizes Mice to High-Fat Diet-Induced Obesity and Insulin Resistance by Inhibiting Lipolysis. Diabetes. 2017;66:1159–1171. doi: 10.2337/db16-1225. [DOI] [PubMed] [Google Scholar]
- 56.Kanfi Y., Peshti V., Gil R., Naiman S., Nahum L., Levin E., Kronfeld-Schor N., Cohen H.Y. SIRT6 protects against pathological damage caused by diet-induced obesity. Aging Cell. 2010;9:162–173. doi: 10.1111/j.1474-9726.2009.00544.x. [DOI] [PubMed] [Google Scholar]
- 57.Elhanati S., Kanfi Y., Varvak A., Roichman A., Carmel-Gross I., Barth S., Gibor G., Cohen H.Y. Multiple regulatory layers of SREBP1/2 by SIRT6. Cell Rep. 2013;4:905–912. doi: 10.1016/j.celrep.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 58.Boukouris A.E., Zervopoulos S.D., Michelakis E.D. Metabolic Enzymes Moonlighting in the Nucleus: Metabolic Regulation of Gene Transcription. Trends Biochem. Sci. 2016;41:712–730. doi: 10.1016/j.tibs.2016.05.013. [DOI] [PubMed] [Google Scholar]
- 59.Ferriero R., Nusco E., De Cegli R., Carissimo A., Manco G., Brunetti-Pierri N. Pyruvate dehydrogenase complex and lactate dehydrogenase are targets for therapy of acute liver failure. J. Hepatol. 2018;69:325–335. doi: 10.1016/j.jhep.2018.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Castonguay Z., Auger C., Thomas S.C., Chahma M., Appanna V.D. Nuclear lactate dehydrogenase modulates histone modification in human hepatocytes. Biochem. Biophys. Res. Commun. 2014;454:172–177. doi: 10.1016/j.bbrc.2014.10.071. [DOI] [PubMed] [Google Scholar]
- 61.Turkaly P., Kerner J., Hoppel C. A 22 kDa polyanion inhibits carnitine-dependent fatty acid oxidation in rat liver mitochondria. FEBS Lett. 1999;460:241–245. doi: 10.1016/s0014-5793(99)01354-x. [DOI] [PubMed] [Google Scholar]
- 62.Zhong Z., Lemasters J.J. A Unifying Hypothesis Linking Hepatic Adaptations for Ethanol Metabolism to the Proinflammatory and Profibrotic Events of Alcoholic Liver Disease. Alcohol. Clin. Exp. Res. 2018;42:2072–2089. doi: 10.1111/acer.13877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Holmuhamedov E.L., Czerny C., Beeson C.C., Lemasters J.J. Ethanol suppresses ureagenesis in rat hepatocytes: role of acetaldehyde. J. Biol. Chem. 2012;287:7692–7700. doi: 10.1074/jbc.M111.293399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Söderberg O., Gullberg M., Jarvius M., Ridderstråle K., Leuchowius K.J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L.G., Landegren U. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.