

Abstract
Objective
The long-acting glucagon-like peptide-1 receptor (GLP-1R) agonist, liraglutide, stimulates insulin secretion and efficiently suppresses food intake to reduce body weight. As such, liraglutide is growing in popularity in the treatment of diabetes and chronic weight management. Within the brain, liraglutide has been shown to alter the activity of hypothalamic proopiomelanocortin (POMC) and Neuropeptide Y/Agouti-related peptide (NPY/AgRP) neurons. Moreover, the acute activities of POMC and NPY neurons have been directly linked to feeding behavior, body weight, and glucose metabolism. Despite the increased usage of liraglutide and other GLP-1 analogues as diabetic and obesity interventions, the cellular mechanisms by which liraglutide alters the activity of metabolically relevant neuronal populations are poorly understood.
Methods
In order to resolve this issue, we utilized neuron-specific transgenic mouse models to identify POMC and NPY neurons for patch-clamp electrophysiology experiments.
Results
We found that liraglutide directly activated arcuate POMC neurons via TrpC5 channels, sharing a similar mechanistic pathway to the adipose-derived peptide leptin. Liraglutide also indirectly increases excitatory tone to POMC neurons. In contrast, liraglutide inhibited NPY/AgRP neurons through post-synaptic GABAA receptors and enhanced activity of pre-synaptic GABAergic neurons, which required both TrpC5 subunits and K-ATP channels. In support of an additive role of leptin and liraglutide in suppressing food intake, leptin potentiated the acute effects of liraglutide to activate POMC neurons. TrpC5 subunits in POMC neurons were also required for the intact pharmacological effects of liraglutide on food intake and body weight. Thus, the current study adds to recent work from our group and others, which highlight potential mechanisms to amplify the effects of GLP-1 agonists in vivo. Moreover, these data highlight multiple sites of action (both pre- and post-synaptic) for GLP-1 agonists on this circuit.
Conclusions
Taken together, our results identify critical molecular mechanisms linking GLP-1 analogues in arcuate POMC and NPY/AgRP neurons with metabolism.
Keywords: Melanocortin, POMC, NPY/AgRP, Liraglutide, Patch-clamp, Electrophysiology
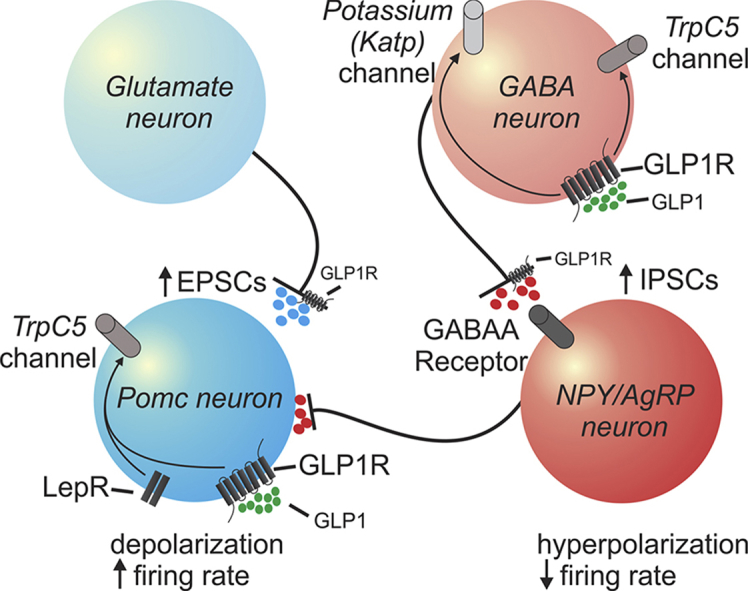
Graphical abstract
Model for direct and indirect effects of GLP-1 and its agonist (liraglutide) on arcuate POMC and NPY/AgRP neurons.
Highlights
-
•
Liraglutide directly activates arcuate POMC neurons, while also increasing pre-synaptic excitatory inputs to POMC neurons.
-
•
Leptin potentiates the acute effects of liraglutide to activate POMC neurons.
-
•
Liraglutide indirectly inhibits arcuate NPY/AgRP neurons via presynaptic TrpC 5 subunits and KATP channels.
-
•
TrpC5 subunits in POMC neurons are required for the intact pharmacological effects of liraglutide.
1. Introduction
Type 2 diabetes mellitus (T2DM) is a global health problem closely linked to obesity [1], [2], [3]. Glucagon-like peptide 1 (GLP-1) is a gut derived hormone that plays a key role in regulation of glucose metabolism by acting in the periphery to stimulate insulin secretion as well as suppress glucagon secretion in a glucose dependent manner [4], [5]. In addition, GLP-1 suppresses feeding by relaying meal-related information on nutritional status to the brain [6], [7], [8], [9]. As a consequence, compounds that activate GLP-1 receptors (e.g. liraglutide) are widely used for the treatment of type 2 diabetes and chronic weight management [10], [11], [12], [13], [14], [15].
The extensive distribution of GLP-1 receptors in the central nervous system (CNS), and findings from rodents and mammalian studies, indicate that the liraglutide-induced satiety and weight effects require activity within the brain [16], [17], [18], [19], [20]. GLP-1 receptors expressed in CNS glutamatergic neurons are essential to the liraglutide induced physiological effects [16]. Within the hypothalamus, arcuate pro-opiomelanocortin (POMC) and Neuropeptide Y (NPY)/Agouti gene-related peptide (AgRP) neurons play an important role in regulating energy balance and glucose homeostasis [21], [22], [23], [24], [25]. Notably, arcuate POMC neurons are glutamatergic and POMC expression in glutamatergic neurons is also essential for proper energy balance [26], [27], [28], [29]. Moreover, GLP-1 receptors in POMC neurons are required for weight regulation on a High fat diet (HFD) [30]. Both peripheral and central administration of liraglutide alter arcuate POMC and NPY/AgRP neuronal activity and contribute to changes in food intake and body weight [31], [32]. These data support an important regulation of metabolism by GLP-1 which requires activity within the central nervous system including melanocortin neurons.
There is increasing evidence that highlights a potential melanocortin-dependent compensatory/additive role for GLP-1Rs in the absence/presence of leptin. In particular, the acute effects of GLP-1 receptor activation in melanocortin neurons mirrors that of the description of leptin [33], [34], [35], [36], [37]. GLP-1 may also be beneficial in the absence of leptin [38], [39]. Moreover, leptin potentiates the effects of GLP-1 on food intake and body weight [32], [40]. Thus, a better understanding of GLP-1 receptors mechanism of action in melanocortin neurons may advance our knowledge of how GLP-1 contributes to food intake and body weight control with or without leptin.
In the current study, we hypothesized that GLP-1 and leptin may activate melanocortin neurons in an additive manner. To test this hypothesis, we utilized transgenic and Cre-Lox technology to identify NPY/AgRP and POMC neurons which express LepRs. We found that leptin and GLP-1 directly modulate neuronal excitability of melanocortin neurons in an additive manner, an activity that requires TrpC5 signaling. We also describe a melanocortin pre-synaptic network altered in response to GLP-1, which requires TrpC5 and K-ATP signaling.
2. Methods
2.1. Animals
Male (6- to 18-week-old) pathogen-free mice were used for all experiments. All mice were housed under standard laboratory conditions (12 h on/off; lights on at 7:00 a.m.) and temperature-controlled environment with food and water available ad libitum. All experiments were performed in accordance with the guidelines established by the National Institute of Health Guide for the Care and Use of Laboratory Animals and approved by the University of Texas Institutional Animal Care and Use Committee.
To identify POMC and NPY neurons with or without LepR, we generated POMC-hrGFP::LepR-cre::tdTomato (PLT) and NPY-hrGFP::LepR-cre::tdTomato (NLT) mice as previously described [41], [42], [43], [44]. Briefly, LepR reporter mice were made by mating LepR-cre mice [45] with the tdTomato reporter mouse (#007908; The Jackson Laboratory). LepR-cre::tdTomato reporter mice were subsequently mated with either POMC- or NPY-humanized Renilla green fluorescent protein (hrGFP) mice [46], [47]. TrpC5 KO mice [48], [49] were subsequently mated with PLT or NLT mice to identify POMC or NPY neurons which express LepR on a TrpC5 KO background. Separately, POMC-creERT2 mice [50] were mated with TrpC5-flox mice to generate mice with selective deficiency of TrpC5 in all POMC neurons (POMC-creER::TrpC5-flox mice).
Tamoxifen treatment to induce adult-onset ablation of TrpC5 in POMC neurons:
Tamoxifen (Sigma, 20 mg/ml) dissolved in corn oil (Sigma) was administered i.p. for 2 consecutive days (80 μl/day) to 5- to 7-week-old male POMC-creER::TrpC5-flox mice. Moist food was provided after the injection of tamoxifen. After 2 weeks of injection, mice were used for physiology studies.
2.2. Electrophysiology studies
2.2.1. Slice preparation
Brain slices were prepared from young adult male mice (6–10 weeks old) as previously described. Briefly, male mice were deeply anesthetized with i.p. injection of 7% chloral hydrate and transcardially perfused with a modified ice-cold artificial CSF (ACSF) (described below). The mice were then decapitated, and the entire brain was removed and immediately submerged in ice-cold, carbogen-saturated (95% O2 and 5% CO2) ACSF (126 mM NaCl, 2.8 mM KCl, 1.2 mM MgCl2, 2.5 mM CaCl2, 1.25 mM NaH2PO4, 26 mM NaHCO3, and 5 mM glucose). For some experiments, slices were perfused with ACSF containing 0.5 mM or 3 mM glucose by replacing glucose with equimolar amounts of sucrose [42], [51]. Coronal sections (250 μm) were cut with a Leica VT1000S Vibratome and then incubated in oxygenated ACSF (32 °C–34 °C) for at least 1 h before recording. The slices were bathed in oxygenated ACSF (32 °C–34 °C) at a flow rate of ∼2 ml/min. All electrophysiology recordings were performed at room temperature.
2.2.2. Whole-cell recordings
The pipette solution for whole-cell recording was modified to include an intracellular dye (Alexa Fluor 350 hydrazide dye) for whole-cell recording: 120 mM K-gluconate or KCl, 10 mM KCl, 10 mM HEPES, 5 mM EGTA, 1 mM CaCl2, 1 mM MgCl2, and 2 mM MgATP, 0.03 mM Alexa Fluor 350 hydrazide dye (pH 7.3). Epifluorescence was briefly used to target fluorescent cells, at which time the light source was switched to infrared differential interference contrast imaging to obtain the whole-cell recording (Zeiss Axioskop FS2 Plus equipped with a fixed stage and a QuantEM:512SC electron-multiplying charge-coupled device camera). Electrophysiological signals were recorded using an Axopatch 700B amplifier (Molecular Devices), low-pass filtered at 2–5 kHz, and analyzed offline on a PC with pCLAMP programs (Molecular Devices). Membrane potential and firing rate were measured by whole-cell current clamp recordings from POMC and NPY neurons in brain slices. Recording electrodes had resistances of 2.5–5 MΩ when filled with the K-gluconate internal solution. Input resistance was assessed by measuring voltage deflection at the end of the response to a hyperpolarizing rectangular current pulse steps (500 ms of −10 to −50 pA).
Neurons were voltage-clamped at −70 mV (for excitatory postsynaptic currents) and −15 mV (for inhibitory postsynaptic currents). Frequency and peak amplitude were measured by using the Mini Analysis program (Synaptosoft, Inc.).
2.3. Drugs
Drug working concentrations (WC) and stock preparation (SP) were as follows: liraglutide (6 mg/ml, Novo Nordisk), GLP-1 (WC = .001-1 μM, SP = 1 mg dissolved in 1 ml deionized water, Tocris), tetrodotoxin (TTX, WC = 2 μM, SP = 1 mg dissolved in 1 ml deionized water, Tocris), picrotoxin (WC = 50 μM, SP = 1 mg dissolved in 30 μl dimethyl sulfoxide, Sigma–Aldrich), 6-cyano-7-nitroquinoxaline-2, 3-dione (CNQX; WC = 10 μM, SP = 1 mg dissolved in 400 μl dimethyl sulfoxide, Sigma–Aldrich), AP5 (WC = 50 μM, SP = 1 mg dissolved in 200 μl deionized water, Sigma–Aldrich), Leptin (WC = 100 nM, SP = 1 mg dissolved in 1 ml Dulbecco's PBS from Gibco, provided by A.F. Parlow, through the National Hormone and Peptide Program), THIP (WC = 10 μM, SP = 1 mg dissolved in 100 μl deionized water, Tocris), tolbutamide (WC = 200 μM, SP = 25 mg dissolved in 500 μl Ethanol alcohol, Sigma–Aldrich). The final concentration of dimethyl sulfoxide applied to the slice was <0.1%.
TTX + Synaptic blockers (SB): picrotoxin (GABAA receptor antagonist), CNQX (Potent and selective non-NMDA iGluR antagonist), and AP5 (Potent and selective NMDA antagonist) were combined with TTX (Sodium Channel blocker) in the ACSF and resulted in the final concentration as described above.
Solutions containing drug were typically perfused for 5 min. A drug effect was required to be associated temporally with compound application, and the response had to be stable within a few minutes. A change in membrane potential was required to be at least 2 mV in amplitude, the onset was required to be associated temporally with the peptide application (i.e. usually beginning at about 1–2 min after changing solutions, the time it took for compound to arrive at the recording chamber), and the response had to be saturated and stable within a few minutes (i.e. did not continually change). The value of the membrane potential was measured at a specific time after compound application (i.e. 3–4 min after the compound arrived in the chamber and no continual changes).
2.4. Animal studies
For the liraglutide daily injection study, age matched male POMC-creER::TrpC5-flox mice (WT and deletion mice are all homozygous of TrpC5-flox, while KO or WT is defined as with either creER expression or not, n = ≥5 per genotype) were single housed and fed with chow diet. Daily food intake, body weight, and blood glucose were measured at 9:00 am for 5 days as baseline. Starting from day 6, liraglutide (300 μg/kg) and vehicle (sterile saline, 10 ml/kg) were administered i.p. in a counterbalanced manner to both controls and POMC-creER::TrpC5-flox mice at 9:00 am for 5 days. Body weight, food intake and blood glucose were measured daily.
2.5. Analysis and statistics
A value of twice the mean peak-to-peak noise level for a given recording in control solutions was used as the detection limit for minimal PSC amplitude (i.e. typically 5–10 pA). For spontaneous EPSCs and IPSCs (sEPSCs and sIPSCs), at least 2 min of activity was examined to identify effects on amplitude and frequency distributions. Membrane potential values were not compensated to account for junction potential (−8 mV). For animal studies, data were normalized with the average of 5 days baseline. Food intake was also normalized with body weight from the same day. All graphs were carried out using Graphpad Prism 7.0 software. All figures were carried out using CorelDraw C8 (64 Bit). Data from responding cells were analyzed using a paired t test. Proportions of responding cells from different groups were analyzed using unpaired a two-tailed Student's t test. Results are reported as the mean ± SEM unless indicated otherwise; where n represents the number of cells studied. Significance was set at *p < 0.05 for all statistical measures.
3. Results
3.1. Liraglutide depolarizes LepR expressing POMC neurons in arcuate nucleus of hypothalamus
In order to analyze the effects of liraglutide on arcuate POMC neurons, whole-cell patch-clamp recordings were performed on both leptin receptor expressing and non-leptin receptor expressing POMC neurons from POMC-hrGFP::LepR-cre::tdtomato (PLT) mice. Alexa Fluor 350 hydrazide dye was added to the intracellular pipette solution for real-time confirmation that hrGFP-positive neurons were targeted for recording (Figure 1A–E). Recordings were made in 37 POMC-hrGFP neurons (both LepR expressing and non LepR expressing, see Figure S1). Bath application of liraglutide (1 μM) depolarized 10 of 27 LepR expressing POMC neurons (37%, 1 μM, n = 10, change of resting membrane potential: +10.2 ± 1.8 mV. Figure 1F and M, Figure S2C). Within a recording, the depolarization was at least partially reversible within 15 min in 7 out of 10 neurons (Figure 1M). Moreover, application of liraglutide induced dose-dependent depolarization in LepR expressing POMC neurons (EC50 = 80 nM, Figure 1G). Rectangular current steps (500 ms; ±50 pA) were applied to the membrane in order to obtain a current–voltage (I–V) plot. The liraglutide induced depolarization of POMC neurons was concomitant with a decrease in input resistance (29%, 1.0 ± 0.1 GΩ for ACSF control; 0.7 ± 0.1 GΩ for liraglutide administration, n = 5, Figure 1H, I). Linear extrapolation of current–voltage relation revealed the reversal potential of the liraglutide-induced depolarization to be −9.3 mV ± 3.8 mV (n = 5, Figure 1I), suggesting an activation of a mixed-cation conductance contributing to the liraglutide induced depolarization of arcuate POMC neurons.
Figure 1.

Liraglutide depolarizes LepR expressing POMC neurons in arcuate nucleus of hypothalamus. (A) Brightfield illumination of POMC-hrGFP::Lepr-cre::tdtomato neuron from PLT mice. (B) and (C) The same neuron under FITC (hrGFP) and Alexa Fluor 594 (tdtomato) illumination. (D) Complete dialysis of Alexa Fluor 350 from the intracellular pipette. (E) Merge image illustrates colocalization of hr-GFP, tdtomato, and Alexa Fluor 350 indicative of LepR expressing (white arrows) and non LepR expressing (blue arrows) POMC neurons. (F) Electrophysiological study demonstrates a POMC-hrGFP::Lepr-cre::tdtomato (green/red) neuron that is depolarized in response to liraglutide (1 μM). (G) Dosage response of liraglutide on LepR-expressing POMC neurons. (H) Traces showing decreased voltage deflection and increased action potential frequency after liraglutide application. (I) Current versus voltage (I–V) plot illustrating a characteristic decrease in input resistance subsequent to liraglutide application. Shown are responses before (ACSF) and during liraglutide application. (J) Representative trace showing pretreatment with TTX (2 μM) and synaptic blockers (SB) do not abrogate liraglutide-induced depolarization of LepR-expressing POMC neurons. (K) Representative trace showing that GLP 1 depolarizes LepR-expressing POMC neurons. (L) Representative trace showing that pretreatment with TTX (2 μM) and synaptic blockers do not abrogate liraglutide-induced depolarization of LepR-expressing POMC neurons. (M) Histogram summarizing the acute effect of liraglutide on the membrane potential of POMC neurons which express LepR (G&R: LepR expressing POMC neurons). Data are expressed as mean ± SEM.
Pretreatment with TTX and synaptic blockers (picrotoxin, CNQX and AP5) failed to abrogate the liraglutide-induced depolarization on LepR expressing POMC neurons (1 μM, n = 5, change of resting membrane potential: +9.3 ± 1.2 mV, Figure 1J, M). The remaining LepR expressing POMC neurons were unresponsive to liraglutide treatment (63%, n = 17, 1 μM, change of resting membrane potential: +0.1 ± 0.3 mV, Figure 1M, Figure S1). Similarly, liraglutide (1 μM) failed to alter the membrane potential of non LepR expressing POMC neurons (1 μM, n = 10, change of resting membrane potential: −0.1 ± 0.3 mV, Figures S1 and S2A–B).
Similar to liraglutide, GLP-1 depolarized arcuate LepR expressing POMC neurons (1 μM, n = 5, change of resting membrane potential: +9.8 ± 2.3 mV, Figure 1K, M). Moreover, analogous results were obtained by using GLP-1 (1 μM) following the pretreatment of TTX (2 μM) and synaptic blockers (1 μM, n = 6, change of resting membrane potential: +8.8 ± 1.2 mV, Figure 1L, M). Together, these data support a GLP-1/liraglutide direct membrane depolarization/activation of LepR expressing POMC neurons via a mixed cation conductance, independent of pre-synaptic inputs.
3.2. Additive effect of liraglutide and leptin on LepR expressing POMC neurons
GLP-1R agonism rescues many of the metabolic deficits observed in ob/ob mice [39]. Moreover, GLP-1R agonists potentiate leptin's effect to suppress food intake and body weight [32], [40]. In the current study, we observed a selective activation of LepR expressing POMC neurons by GLP-1R agonists. Thus, we hypothesized that liraglutide and leptin may have additive effects on the excitability of arcuate POMC neurons.
Consistent with previous reports [52], [53], [54], [55], leptin depolarized LepR expressing POMC neurons (100 nM, n = 5, change of resting membrane potential: +6.1 ± 0.8, n = 5, Figure 2A, E). Liraglutide (100 nM) combined with leptin (100 nM) resulted in a larger depolarization of the resting membrane potential when compared with the changes observed of either liraglutide or leptin, alone (100 nM liraglutide: n = 10, change of resting membrane: +6.6 ± 0.8, Figure 2B, E; 100 nM liraglutide + 100 nM leptin: n = 4, change of resting membrane potential: +10.1 ± 1.5, Figure 2C, E, *p < 0.05). Interestingly, higher doses of liraglutide (1 μM) combined with leptin (100 nM) resulted in a similar depolarization of the resting membrane potential that was observed with the sub-maximal dose of liraglutide (100 nM) and leptin (100 nM) co-treatment (1 μM liraglutide + 100 nM leptin, n = 5, change of resting membrane potential: +10.4 ± 1.7, Figure 2D, E, *p < 0.05). Moreover, the change of resting membrane potential of liraglutide (100 nM) and leptin co-treatment is analogous to the change in membrane potential observed with maximal concentrations of liraglutide (1 μM) or GLP-1 (1 μM) application alone (Figure 1, Figure 2C). These data suggest that both liraglutide and leptin have additive effects on LepR expressing POMC neurons and support a potential shared cellular mechanism to depolarize LepR expressing POMC neurons.
Figure 2.

Synergetic effect of liraglutide and leptin on LepR expressing POMC neurons. (A) Current-clamp record depicting the characteristic depolarization of arcuate LepR-expressing POMC neurons by leptin (100 nM). (B) Representative trace showing that low dose of liraglutide (100 nM) depolarizes LepR-expressing POMC neurons. (C) Representative electrophysiological trace demonstrating that additive administration of liraglutide (100 nM) with leptin (100 nM) induced larger depolarized effect of LepR-expressing POMC neurons. (D) Representative trace showing that additive administration of liraglutide (1 μM) with leptin (100 nM) induced depolarization of LepR-expressing POMC neurons. (E) Histogram summarizing the acute effect of liraglutide and leptin on the membrane potential of LepR expressing POMC neurons. Data are expressed as mean ± SEM. *p < 0.05, unpaired t-test was used to compare.
3.3. Similar to leptin, liraglutide directly depolarizes LepR expressing POMC neurons via TrpC5 subunits
Transient receptor potential cation 5 (TrpC5) subunits are required for the acute effects of leptin on POMC neurons [49]. In the current study, the conductance activated by liraglutide in LepR-expressing POMC neurons (Erev = −9.3 ± 3.8 mV) is in agreement with that described for activation of TrpC5 subunits [56], [57]. In order to examine the requirement of TrpC5 subunits in the liraglutide induced activation of POMC neurons, LepR expressing POMC neurons deficient for TRPC5 subunits were targeted for electrophysiological recordings from POMC-hrGFP::LepR-cre-tdtomato::TRPC5KO (PLT5KO) mice. All POMC neurons deficient for TRPC5 subunits failed to depolarize in response to liraglutide (1 μM, n = 17, change of resting membrane potential: −0.1 ± 0.3 mV, Figure 3A–G). Next, we investigated the requirement of TrpC5 subunits in adult POMC neurons (POMC-creERT2-cre allows temporal control of cre-recombinase activity in POMC neurons) in regulating the pharmacological effects of liraglutide to inhibit food intake, decrease body weight, and lower blood glucose. Male age matched litter mates control and POMC-creER::TrpC5-flox mice were treated daily with liraglutide (300 μg/kg; I.P.; once daily.; for 5 days; Figure S3A). Within 24 h after the first liraglutide injection, both WT mice and mice deficient for TrpC5 subunits in POMC neurons exhibited a decrease in body weight (∼8%), food intake (∼40%), and blood glucose levels (∼45%) (Figure 3I–K). Importantly, mice deficient for TrpC5 subunits in POMC neurons exhibited a rapid reversal of the liraglutide effects on food intake, bodyweight and blood glucose (Figure 3H–K, Figure S3B). Together, these data support a distributed network of GLP-1 receptors to mediate the pleiotropic effects of liraglutide. Moreover, TrpC5 subunits in POMC neurons are required for the sustained intact pharmacological effects of liraglutide.
Figure 3.

Liraglutide depolarizes LepR expressing POMC neurons via TrpC 5 subunits. (A) Brightfield illumination of POMC-hrGFP::Lepr-cre::tdtomato neuron from POMC-hrGFP::Lepr-cre::tdtomato::TrpC5 KO mice. (B) and (C) The same neuron under FITC (hrGFP) and Alexa Fluor 594 (tdtomato) illumination. (D) Complete dialysis of Alexa Fluor 350 from the intracellular pipette. (E) Merge image illustrates colocalization of hr-GFP, tdtomato, and Alexa Fluor 350 indicative of LepR expressing (white arrows) POMC neurons. (F) Representative trace showing that liraglutide fails to induce a depolarization in a POMC-hrGFP::Lepr-cre::tdtomato::TrpC5 KO (green/red) neuron. (G) Histogram summarizing the acute effect of liraglutide on the membrane potential of LepR expressing POMC neurons which deleted of TrpC5. Data are expressed as mean ± SEM. Plots show the changes in cumulative food intake (H), body weight (I), daily food intake (J), and blood glucose (K) with daily injection of liraglutide on both litter mates control and POMC-creER::TrpC5-flox mice. Data are from male chow-fed mice. Number of mice studied for each group equals or above 5. Unpaired t-test compared to controls. *p < 0.05, **p < 0.01.
3.4. Liraglutide indirectly hyperpolarizes NPY neurons in arcuate nucleus of hypothalamus
To better understand the effect of liraglutide on arcuate NPY/AgRP neurons, whole-cell patch-clamp recording was performed on leptin receptor expressing and non-expressing NPY neurons from NPY-hrGFP::LepR-cre::tdtomato (NLT) mice (Figure 4A–E). In total, 32 NPY-hrGFP neurons were targeted in control artificial cerebrospinal fluid (ACSF) bath solutions. Opposite to the effects on arcuate POMC neurons, liraglutide hyperpolarized arcuate NPY neurons (53%, 1 μM, n = 17, change of resting membrane potential: −10.1 ± 0.9 mV, Figure 4F, J, Figure S4). The liraglutide induced hyperpolarization occurred in both LepR expressing (1 μM, n = 8, change of resting membrane potential: −8.7 ± 1.3 mV, Figure 4J) and non LepR expressing NPY neurons (1 μM, n = 9, change of resting membrane potential: −11.4 ± 1.1 mV, Figure 4J), suggesting liraglutide effects a broader cell population in NPY neurons when compared to POMC neurons. Within a recording, the hyperpolarization was at least partially reversible within 15 min in 4 out of 8 LepR expressing NPY neurons and 5 out of 9 non LepR expressing NPY neurons (Figure 4J). The membrane potential of the remaining neurons remained unchanged.
Figure 4.

Liraglutide indirectly hyperpolarizes NPY neurons in arcuate nucleus of hypothalamus. (A) Brightfield illumination of NPY-hrGFP::Lepr-cre::tdtomato neuron from NLT mice. (B) and (C) The same neuron under FITC (hrGFP) and Alexa Fluor 594 (tdtomato) illumination. (D) Complete dialysis of Alexa Fluor 350 from the intracellular pipette. (E) Merge image illustrates colocalization of hr-GFP, tdtomato, and Alexa Fluor 350 indicative of LepR expressing (white arrows) and non LepR expressing (blue arrows) NPY neurons. (F) Electrophysiological study demonstrates a NPY-hrGFP neuron that is hyperpolarized in response to liraglutide (1 μM). (G) Traces showing decreased voltage deflection and decreased action potential frequency after liraglutide application. (H) Current versus voltage (I–V) plot illustrating a characteristic decrease in input resistance subsequent to liraglutide application. Shown are responses before (ACSF) and during liraglutide application. (I) Electrophysiological study demonstrates that pretreatment with TTX (2 μM) and synaptic blockers (SB) abrogate liraglutide (1 μM)-induced hyperpolarization of NPY neurons. (J) Histogram summarizing the acute effect of liraglutide (1 μM) on the membrane potential of NPY neurons with or without pretreatment of TTX and synaptic blockers. (G: non LepR expressing NPY neurons. G&R: LepR expressing NPY neurons). Data are expressed as mean ± SEM.
Using rectangular current steps (500 ms; ±50 pA), we found that the liraglutide induced hyperpolarization on NPY neurons was concomitant with a decrease in input resistance (39.5%, 1.2 ± 0.1 GΩ for control ACSF, 0.7 ± 0.1 GΩ for liraglutide administration, n = 5, Figure 4G, H). Linear extrapolation of current–voltage relation revealed the reversal potential of the liraglutide-induced hyperpolarization to be −76.6 ± 1.5 mV (n = 5, Figure 4H). The reversal potential of the liraglutide induced hyperpolarization of arcuate NPY neuron resembled the reversal potential of chloride (calculated ECl = −65 mV in our solutions), suggesting that liraglutide activates a chloride channel to hyperpolarize NPY neurons. Importantly, the liraglutide-induced hyperpolarization of NPY neurons was blocked in the presence of TTX and synaptic blockers (n = 14, change of resting membrane potential: −0.1 ± 0.3 mV. Figure 4I, J, Figure S4), supporting an indirect (dependent upon action potentials and/or fast neurotransmitter release) hyperpolarization of arcuate NPY neurons.
NPY neurons (n = 19) were voltage-clamped at −70 mV, and changes in whole cell current were monitored during bath application of liraglutide. Eleven of these neurons displayed a change in whole-cell current subsequent to liraglutide application. Superfusion of liraglutide (1 μM) resulted in an outward current in 10 out of 19 NPY neurons (Figure 5A, B). Most of the remaining cells were unchanged in response to liraglutide (n = 8), while 1 NPY neuron exhibited an inward current (change of current: −48.5 pA). In order to further investigate the chloride dependence of the liraglutide-induced inhibition of NPY neurons we utilized a high KCl (120 mM) intracellular solution. This resulted in a shift of the calculated reversal potential of Cl from −65 mV to 0 mV in our solution. Neurons were then voltage-clamped at −70 mV. In this configuration, we observed an inward current in 3 of 6 NPY neurons (Figure S5A). Importantly, when neurons were voltage-clamped at membrane potentials closer to the reversal potential of Cl (ECL = 0 mV; neurons held at −15 mV), we failed to observe a liraglutide (1 μM) induced change of holding current in all NPY neurons (n = 9, Figure S5B). These data are in agreement with a dependence of chloride for the effects of liraglutide on NPY cellular activity.
Figure 5.

Liraglutide enhances inhibitory synaptic inputs to arcuate NPY neurons, while no effect on excitatory synaptic inputs. (A) Voltage clamp recording demonstrates liraglutide (1 μM) induced outward current on NPY neurons (Vm = −15 mV). (B) Histogram summarizing the acute effect of liraglutide (1 μM) on the currents of NPY neurons (Vm = −15 mV). Data are expressed as mean ± SEM. (C) Voltage clamp recording of spontaneous inhibitory postsynaptic currents (sIPSCs) observed in NPY-hrGFP neuron from NLT mice before (upper) and during (lower) liraglutide administration (Vm = −15 mV). (D&E) Plots indicating increased sIPSC frequency (Hz), but not amplitude (pA) in arcuate NPY neurons with liraglutide (1 μM) administration. (F) Voltage clamp recording of spontaneous excitatory postsynaptic currents (sEPSCs) observed in NPY-hrGFP neuron from NLT mice before (upper) and during (lower) liraglutide administration (Vm = −70 mV). (G&H) Plots indicating no change of both sEPSC frequency (Hz) and amplitude (pA) in arcuate NPY neurons with liraglutide (1 μM) administration (black bar: ACSF; red bar: ACSF + liraglutide). Data are expressed as mean ± SEM. ****p < 0.0001, paired t-test compared to controls.
3.5. Liraglutide enhances inhibitory synaptic inputs to arcuate NPY neurons, while enhances excitatory synaptic inputs to POMC neurons
In order to characterize the effects of liraglutide on the synaptic inputs to arcuate NPY and POMC neurons in the absence of voltage fluctuations, we targeted 22 arcuate NPY neurons and 12 LepR expressing POMC neurons in voltage clamp configuration. With a similar time course to the liraglutide induced membrane hyperpolarization, liraglutide (1 μM) increased the spontaneous IPSCs frequency in NPY neurons (n = 19, control ACSF: 1.1 ± 0.1 Hz, liraglutide administration: 1.6 ± 0.15 Hz, ****p < 0.0001, paired t test, Figure 5C, D) independent of changes in amplitude (Figure 5C–E). Both the frequency and amplitude of spontaneous EPSCs were unaltered in NPY neurons following application of liraglutide (1 μM; Figure 5F–H). In contrast, liraglutide increased the spontaneous EPSCs frequency to arcuate LepR expressing POMC neurons independent of changes in amplitude (*p < 0.05, paired t test, Figure S6A–B). However, there was a trend toward a liraglutide induced decrease in spontaneous IPSC frequency of LepR expressing POMC neurons, this was not statistically significant (p = 0.051, paired t test, Figure S6C–D). Similar results were obtained in the presence of TTX (Figure S6E–H).
3.6. Liraglutide hyperpolarizes NPY neurons via both pre-synaptic inputs and post-synaptic GABAA receptor
Pretreatment of TTX and synaptic blockers abrogated the liraglutide induced hyperpolarization of NPY neurons, suggesting the requirement of presynaptic GLP-1Rs in the inhibition of NPY neurons. To investigate the possible location of GLP-1 receptors on the presynaptic neurons, we analyzed the effect of liraglutide with and without TTX, which blocks action potential-dependent synaptic activity. TTX reversed the liraglutide induced hyperpolarization in one-third of NPY neurons (3 of 9 NPY neurons; Figure 6B, E). The remaining 6 NPY neurons hyperpolarized by liraglutide were unaffected by TTX application (Figure 6A, E). In a separate experiment, application of the GABAA receptor antagonist (picrotoxin) reversed the liraglutide induced hyperpolarization in all 6 neurons recorded in control ACSF (Figure 6C, F). Picrotoxin also reversed the liraglutide induced inhibition of all NPY neurons in the presence of TTX (n = 5, Figure 6D, G). These data suggest liraglutide/GLP-1 may act at both terminal and soma/dendritic loci on afferent neurons. Moreover, presynaptic GLP-1 receptor expressing neurons release GABA, binding to GABAA receptors on NPY neurons, and leading to the inhibition of NPY neuronal excitability. Consistent with a liraglutide-induced hyperpolarization of arcuate NPY neurons via a putative chloride channel (GABAA receptors), administration of the GABAA receptor agonist (THIP) results in an analogous inhibition of NPY neurons (10 μM, n = 5, change of resting membrane potential: −10.0 ± 1.2 mV, Figure S7).
Figure 6.

Liraglutide hyperpolarizes NPY neurons via both pre-synaptic GLP1 receptor and post-synaptic GABAA receptor. (A) Representative trace showing that TTX (2 μM) does not prevent the liraglutide (1 μM)-induced hyperpolarization of NPY neurons. (B) Representative trace showing that TTX (2 μM) blocked the liraglutide (1 μM)-induced hyperpolarization of NPY neurons. (C) Representative trace showing that picrotoxin (50 μM) reversed the liraglutide (1 μM)-induced hyperpolarization of NPY neurons. (D) Representative trace showing that picrotoxin (50 μM) reversed the liraglutide (1 μM)-induced hyperpolarization of NPY neurons in which TTX (2 μM) does not blocked the liraglutide-induced effect. (E) Histogram summarizing the acute effect of liraglutide and followed by TTX (2 μM) administration on the membrane potential of arcuate NPY neurons (black: liraglutide, red: TTX does not block the liraglutide-induced effect, blue: TTX blocked the liraglutide-induced effect). (F) Histogram summarizing the acute effect of liraglutide and followed by picrotoxin (50 μM) administration on the membrane potential of arcuate NPY neurons (black: liraglutide, red: picrotoxin reversed the liraglutide effect). (G) Histogram summarizing the acute effect of liraglutide and followed by picrotoxin (50 μM) administration on the membrane potential of arcuate NPY neurons, in which TTX (2 μM) does not block the liraglutide-induced effect. Data are expressed as mean ± SEM. ***p < 0.001, ****p < 0.0001, unpaired & paired t-test were used to compare the significance.
3.7. ATP-sensitive potassium channel (K-ATP) and TrpC 5 subunits were involved in liraglutide effect in CNS
We found that TrpC5 subunits are involved in the liraglutide induced depolarization of arcuate POMC neurons. Previous work also suggest that the ATP-sensitive potassium channels (K-ATP channel) are essential for GLP-1 induced effects in peripheral tissues [58], [59], [60], [61], [62] In order to investigate if TrpC5 subunits and/or K-ATP channels are required for the indirect inhibition of NPY neurons (i.e. activate afferent GABAergic neurons to arcuate NPY neurons), we first examined the effects of liraglutide on NPY neurons from mice globally deficient for TrpC5 subunits (NLT::TrpC5 KO mice; Figure 7A–D). Liraglutide hyperpolarized 6 of the 15 NPY neurons from NLT::TrpC5 KO mice (40%, 1 μM, n = 6, change of resting membrane potential: −11.2 ± 0.6, Figure 7E, F). This represents a 13% reduction in the population of liraglutide responsive NPY neurons when compared to NPY neurons from wildtype mice; however, this did not reach statistical significance (53% on NLT mice, 40% on NLT::TrpC5 KO mice). Pretreatment with an ATP-sensitive potassium channel blocker (tolbutamide) followed by liraglutide administration prevented the liraglutide induced hyperpolarization in the majority of neurons examined (Figure 7G, H), such that 3 of 17 neurons were hyperpolarized (35% decrease in the population when compared to NPY neurons in control ACSF; tolbutamide + liraglutide: 18%, n = 3, change of resting membrane potential: −11.5 ± 1.2 mV, Figure 7H; liraglutide in control ACSF: 53%, n = 17, change of resting membrane potential: −10.1 ± 0.9 mV, Figure 4J). Importantly, summation of the percent of NPY neurons responsive in mice deficient for TrpC5 subunits (40%) and those responsive in the presence of tolbutamide (18%) is analogous to the 53% responsive NPY neurons from wildtype mice. Moreover, pretreatment of tolbutamide totally abrogated the liraglutide induced hyperpolarization on NPY neurons from NLT::TrpC5 KO mice (1 μM, n = 10, change of resting membrane potential: −0.1 ± 0.2, Figure 7I, J). Together, these data suggest that both K-ATP channels and TrpC5 subunits are required for the liraglutide induced inhibition on arcuate NPY neurons.
Figure 7.

ATP-sensitive potassium channel (K-ATP) and TrpC 5 subunits were involved in liraglutide effect. (A) Brightfield illumination of NPY-hrGFP neuron from NLT::TRPC5 KO mice. (B) The same neuron under FITC (hrGFP) illumination. (C) Complete dialysis of Alexa Fluor 350 from the intracellular pipette. (D) Merge image illustrates colocalization of hr-GFP and Alexa Fluor 350 indicative of NPY neuron which was deleted of TRPC5. (E) Electrophysiological study demonstrates a TRPC5 deleted NPY-hrGFP neuron that is hyperpolarized in response to liraglutide (1 μM). (F) Histogram summarizing the acute effect of liraglutide (1 μM) on the membrane potential of TRPC5 deleted NPY neurons. (G) Electrophysiological study demonstrates that liraglutide fails to hyperpolarize NPY-hrGFP neuron with the pretreatment of tolbutamide(200 μM). (H) Histogram summarizing the acute effect of liraglutide (1 μM) on the membrane potential of NPY neurons, with the pretreatment of tolbutamide(200 μM). (I) Representative trace showing that liraglutide fails to hyperpolarize NPY-hrGFP neuron from NLT:TrpC5 KO mice with the pretreatment of tolbutamide (200 μM). (J) Histogram summarizing the acute effect of liraglutide (1 μM) on the membrane potential of NPY neurons from NLT:TrpC5 KO mice, with the pretreatment of tolbutamide (200 μM). Data are expressed as mean ± SEM.
4. Discussion
The predominant effect of liraglutide on leptin receptor-expressing arcuate POMC neurons was excitatory, acting to depolarize the neurons directly and increase the excitatory synaptic inputs they receive. Leptin potentiated these effects on POMC cellular activity such that leptin and liraglutide concomitantly resulted in an enhanced depolarization when compared to either peptide alone. In contrast, the overall effect of liraglutide was inhibitory in adjacent NPY/AgRP neurons. However, the effects on NPY/AgRP neurons were indirect, requiring increased inhibitory synaptic inputs to suppress NPY/AgRP neuronal activity. These effects in combination with those from previous work support a liraglutide dependent enhancement of melanocortin neural circuitry within the arcuate nucleus.
4.1. Liraglutide directly actives arcuate POMC neurons of hypothalamus
The GLP-1 agonist, liraglutide, dose-dependently activates arcuate POMC neurons. Pretreatment with the voltage-gated sodium channel blocker (TTX) and fast ionotropic receptors antagonists failed to abrogate the liraglutide and GLP-1 induced depolarization on POMC neurons, suggesting these effects require direct action within POMC neurons and are independent of pre-synaptic inputs. These data are consistent with studies showing that GLP-1 receptors are widely expressed in the arcuate nucleus of hypothalamus and co-express with POMC neurons [17], [31]. Similarly, GLP-1 receptors on glutamatergic neurons are critical for liraglutide-induced effects on appetite and body weight within the brain, but not on GABAergic neurons [16]. Importantly, Jones and colleagues also recently demonstrated that glutamatergic POMC neurons are essential for body weight control [27]. Furthermore, targeted delivery of Exendin-4 to the arcuate nucleus reduces food intake, while deficiency of GLP-1R in POMC neurons results in increased HFD induced weight gain [30]. Together, these data highlight acute mechanisms by which liraglutide alters the activity of arcuate POMC neurons that contribute to regulating food intake and energy balance.
4.2. Additive effects of liraglutide and leptin on LepR expressing POMC neuronal activity
The liraglutide-induced activation of arcuate POMC neurons occurred almost exclusively in leptin receptor expressing neurons. Moreover, leptin potentiated the effects of low concentrations of liraglutide on LepR expressing POMC neurons when compared with treatment of either compound alone. However, high doses of liraglutide with co-administration of leptin resulted in similar maximum change in resting membrane potential compared with the high doses of either liraglutide or GLP-1 alone. This is in agreement with the ability of low dose GLP-1R agonists to potentiate the effects of leptin to suppress food intake and body weight [63], [64]. Moreover, these data suggest a common intracellular mechanism may be triggered by leptin and GLP-1 receptor agonists [64]. In particular, combined administration of liraglutide and leptin elevated pSTAT3 in hypothalamic tissue and reduced the expression of PTP1B. As a negative regulator of leptin receptor signaling and a key promoter of insulin and leptin resistance, PTP1B could be a potential mechanism for the enhanced pSTAT3 response observed after liraglutide-leptin co-administration [64]. However, it is important to note that leptin-induced pSTAT3 has been dissociated from the acute ability of leptin to reduce food intake and activate arcuate POMC neurons [51], [65]. Thus the changes in cellular activity observed in the current study likely involve mechanisms independent of STAT3 and PTP1B (discussed in more detail below).
Plasma concentrations of liraglutide measured during liraglutide therapy have been suggested to be 10–40 nM [66], [67]. In the current study, we utilized doses of liraglutide within this therapeutic range and observed that liraglutide effects on POMC neurons occurred in a dose-dependent manner (from 1 nM to 1 μM). Importantly, the EC50 of liraglutide on POMC neurons is ∼80 nM, which is ∼2–8 folds higher than plasma concentrations of liraglutide during therapy [66], [67]. This illustrates that current dosing of liraglutide for T2DM treatment is far from a maximal dose for this circuit. Given the observed adverse side effects of liraglutide (e.g., nausea), combinatorial therapy to boost GLP-1 efficacy might be advantageous [68]. The current study adds to recent work from our group and others, which highlight potential mechanisms to amplify the effects of GLP-1 agonists in vivo [68], [69], [70], [71]. Together, these data highlight potential mechanisms of combinatorial treatment for the liraglutide in treatment of T2DM.
4.3. Liraglutide indirectly inhibits arcuate NPY/AgRP neurons
In contrast with effects observed in POMC neurons, liraglutide inhibited both LepR expressing and non LepR expressing NPY/AgRP neurons. The hyperpolarization was not observed in the presence of TTX and synaptic blockers, and thus required synaptic transmission in afferent neurons, implying an indirect effect on the soma/dendritic membrane potential. These observations are consistent with previous reports showing liraglutide fails to bind directly to NPY neurons due to low or absent GLP-1 receptor expression [31], [72]. It is important to note that there may be species variability to these results given that GLP-1 receptors colocalize with both NPY and AgRP neurons in T2DM patients [73]. However, liraglutide's direct action on POMC neurons (putative glutamatergic neurons) [74], [75], [76] and its indirect effect on NPY neurons (putative GABAergic neurons) [77] highlighted in the current study are in agreement with the hypothesis that GLP-1 receptors on glutamatergic neurons are key factors for liraglutide effects on appetite and body weight within the brain, independent of GLP-1 receptors on GABAergic neurons [16].
4.4. Synaptic effects of liraglutide on both NPY/AgRP and POMC neurons
Liraglutide enhanced the frequency of spontaneous GABAergic input to NPY neurons. There are two classes of GABA receptors (GABAA and GABAB) [78], [79]. GABAA receptors are ionotropic receptors, whereas GABAB receptors are G protein-coupled receptors. While GABAB receptors are typically linked to activation of potassium conductance, GABAA receptors facilitate chloride entry into the cell [80], [81]. Importantly, the involvement of GABAA receptors is consistent with the I–V relationship in current study which accompanied the liraglutide-induced hyperpolarization of arcuate NPY neurons. Moreover, treatment of the GABAA receptor antagonist (picrotoxin) reversed the liraglutide induced hyperpolarization on NPY neurons. While GABAA receptors are largely recognized for their phasic action, recent work suggests an ability of GABAA receptors to alter membrane potential and cellular activity via tonic activity [82]. The tonic activity of GABAA receptors has been attributed to possible spillover of GABA from the synaptic cleft and/or possible inefficient removal of GABA from the synapse. While these are both intriguing hypotheses in this model, this requires further investigation. Although the GABAA receptor antagonist reversed the liraglutide-induced hyperpolarization in every NPY cell examined, TTX alone reversed the hyperpolarization in only one-third of neurons targeted. The hyperpolarization persisted in the remaining two-thirds of neurons with following treatment of TTX, suggesting liraglutide may act at terminal and soma/dendritic loci on afferent neurons. Collectively, these data detail a mechanism by which liraglutide activates pre-synaptic GLP-1 receptors, resulting in an increased release of GABA and enhanced binding to GABAA receptors on post-synaptic NPY/AgRP neurons, ultimately inhibiting cellular activity.
Interestingly, we also found liraglutide increased excitatory input to LepR expressing POMC neurons, independent of changing inhibitory input. Pretreatment of TTX failed to alter the liraglutide induced increase of excitatory input to LepR expressing POMC neurons suggesting liraglutide may act at terminal loci on afferent neurons. Unlike the effects observed on arcuate NPY neurons, synaptic blockade failed to blunt the liraglutide induced excitation of arcuate POMC neurons. Thus, while liraglutide enhanced excitatory synaptic activity to LepR expressing POMC neurons, this presynaptic effect was not required for the changes in POMC membrane potential.
4.5. Cellular mechanisms of liraglutide effects in arcuate POMC and NPY neurons – a link to metabolism
The effects of liraglutide on arcuate POMC neuronal activity mirror those described in response to leptin [52], [53], [54], [55]. This includes activation of a mixed-cation conductance, which in a recent report on the effects of leptin in POMC neurons has been identified as a TrpC5 channel [49]. This led to the hypothesis that GLP-1 receptors may activate TrpC5 channels to regulate the cellular signaling resulting in a similar increased cellular activity of arcuate POMC neurons. Similar to the effects of leptin, liraglutide failed to activate POMC neurons from mice deficient for TrpC5 channels. Moreover, pharmacological effects of liraglutide to inhibit food intake and decrease body weight was blunted in mice selectively deficient for TrpC5 channels in POMC neurons. We also observed an abrogation of the liraglutide induced lowering of blood glucose levels. It is important to note that the liraglutide induced decrease in blood glucose levels mirrored the time-course for the observed decrease in food intake and body weight. It is difficult to directly dissociate the glucose effects from that of secondary effects of altered energy balance. However, if the glucose effects are secondary, the fall in glucose levels would be expected to be slower or lag behind the change in energy balance. Therefore, it is likely that the liraglutide-induced effects on blood glucose levels in the current study are due to acute effects on glucose metabolism.
In our current model, liraglutide activates POMC neurons through both direct membrane depolarization and indirect enhancement of excitatory inputs from presynaptic glutamatergic neurons. Liraglutide also indirectly inhibits NPY/AgRP neurons through increased inhibitory input from presynaptic GABAergic neurons. Recent work suggests that arcuate NPY neurons acutely regulate feeding behavior (within minutes to hours), while arcuate POMC neurons alter feeding on a longer timescale (hours to days) [83], [84]. In mice with the deficiency of TrpC5 in POMC neurons, we demonstrate an inability of POMC neurons to be activated in response to liraglutide, while the acute effects of liraglutide remain intact in NPY neurons. While currently unclear, the acute responses observed might be due to indirect inhibition of arcuate NPY neurons or other cell populations. However, the differences observed over the subsequent days of liraglutide treatment might have more relevance to activity of POMC neurons.
It is important to note that the liraglutide-induced changes in synaptic input to POMC reinforce the post synaptic effect, providing an increased excitatory tone. Deficiency of TrpC5 in POMC neurons blocks only the post synaptic effects of liraglutide, while the changes in pre synaptic input remains intact. This is an inherent caveat of all previous POMC specific manipulations of GLP-1R expression [18], [30], as we and others cannot exclude the contribution/compensatory synaptic regulation of POMC neurons on the metabolic phenotypes described.
Opposite to effects observed in arcuate POMC neurons, global deficiency of TrpC5 channels resulted in a ∼13% reduction of NPY neurons which were responsive to liraglutide. In particular, 53% of NPY neurons from wild-type mice were inhibited in response to liraglutide, compared to 40% of NPY neurons from mice with global TrpC5 deficiency. Consistent with previous reports of K-ATP channels is essential for GLP-1 induced effects through GLP-1 receptors in peripheral tissues [58], [59], [60], [61], [62], pretreating with the K-ATP potassium channel blocker (tolbutamide) resulted in a 35% decrease of NPY neurons that responded to liraglutide (liraglutide: 53%, tolbutamide + liraglutide: 18%). Importantly, with the pretreatment of tolbutamide, liraglutide failed to alter the activity of NPY neurons form global deficient of TrpC5 subunits. These data support the conclusion that the liraglutide induced inhibition of NPY activity requires both TrpC5 and K-ATP channels in pre-synaptic neurons.
5. Conclusions
In summary, liraglutide and leptin activate arcuate LepR expressing POMC neurons via TrpC5 subunits. Liraglutide inhibits arcuate NPY neurons by indirectly enhancing inhibitory tone via presynaptic TrpC5 subunits and K-ATP channels on GABAergic neurons. The effects of GLP-1 analogues in the arcuate nucleus of hypothalamus are likely play a role in understanding the pathway by which GLP-1R agonists regulate energy balance and metabolism in the CNS.
Acknowledgment
We thank Dr. Joel K. Elmquist (of the Division of Hypothalamic Research, Department of Internal Medicine, UT Southwestern Medical Center, Dallas, Texas) for kindly providing us with the NPY-hrGFP and POMC-hrGFP mice. We also thank the help from The Guangdong Provincial Clinical Medical Centre for Neurosurgery, No. 2013B020400005. This work was supported by grants to K.W.W. (R01 DK100699, R01 DK119169, R01 DK123185 and P01 DK119130).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.molmet.2019.07.008.
Conflicts of interest
We wish to confirm that there are no known conflicts of interest.
Appendix A. Supplementary data
The following is the supplementary data to this article:
References
- 1.DeFronzo R.A., Ferrannini E., Groop L., Henry R.R., Herman W.H., Holst J.J. Type 2 diabetes mellitus. Nature Review Disease Primers. 2015;1:15019. doi: 10.1038/nrdp.2015.19. [DOI] [PubMed] [Google Scholar]
- 2.Whiting D.R., Guariguata L., Weil C., Shaw J. IDF diabetes atlas: global estimates of the prevalence of diabetes for 2011 and 2030. Diabetes Research and Clinical Practice. 2011;94(3):311–321. doi: 10.1016/j.diabres.2011.10.029. [DOI] [PubMed] [Google Scholar]
- 3.Flegal K.M., Carroll M.D., Kit B.K., Ogden C.L. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999–2010. Journal of the American Medical Association. 2012;307(5):491–497. doi: 10.1001/jama.2012.39. [DOI] [PubMed] [Google Scholar]
- 4.Armstrong M.J., Gaunt P., Aithal G.P., Barton D., Hull D., Parker R. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet. 2016;387(10019):679–690. doi: 10.1016/S0140-6736(15)00803-X. [DOI] [PubMed] [Google Scholar]
- 5.Halawi H., Khemani D., Eckert D., O'Neill J., Kadouh H., Grothe K. Effects of liraglutide on weight, satiation, and gastric functions in obesity: a randomised, placebo-controlled pilot trial. The Lancet. Gastroenterology & Hepatology. 2017;2(12):890–899. doi: 10.1016/S2468-1253(17)30285-6. [DOI] [PubMed] [Google Scholar]
- 6.van Bloemendaal L., RG I.J., Ten Kulve J.S., Barkhof F., Konrad R.J., Drent M.L. GLP-1 receptor activation modulates appetite- and reward-related brain areas in humans. Diabetes. 2014;63(12):4186–4196. doi: 10.2337/db14-0849. [DOI] [PubMed] [Google Scholar]
- 7.Holst J.J. The physiology of glucagon-like peptide 1. Physiological Reviews. 2007;87(4):1409–1439. doi: 10.1152/physrev.00034.2006. [DOI] [PubMed] [Google Scholar]
- 8.Fineman M.S., Cirincione B.B., Maggs D., Diamant M. GLP-1 based therapies: differential effects on fasting and postprandial glucose. Diabetes, Obesity and Metabolism. 2012;14(8):675–688. doi: 10.1111/j.1463-1326.2012.01560.x. [DOI] [PubMed] [Google Scholar]
- 9.Drucker D.J., Habener J.F., Holst J.J. Discovery, characterization, and clinical development of the glucagon-like peptides. Journal of Clinical Investigation. 2017;127(12):4217–4227. doi: 10.1172/JCI97233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O'Neil P.M., Birkenfeld A.L., McGowan B., Mosenzon O., Pedersen S.D., Wharton S. Efficacy and safety of semaglutide compared with liraglutide and placebo for weight loss in patients with obesity: a randomised, double-blind, placebo and active controlled, dose-ranging, phase 2 trial. Lancet. 2018;392(10148):637–649. doi: 10.1016/S0140-6736(18)31773-2. [DOI] [PubMed] [Google Scholar]
- 11.le Roux C.W., Astrup A., Fujioka K., Greenway F., Lau D.C.W., Van Gaal L. 3 years of liraglutide versus placebo for type 2 diabetes risk reduction and weight management in individuals with prediabetes: a randomised, double-blind trial. Lancet. 2017;389(10077):1399–1409. doi: 10.1016/S0140-6736(17)30069-7. [DOI] [PubMed] [Google Scholar]
- 12.Garber A.J. Long-acting glucagon-like peptide 1 receptor agonists: a review of their efficacy and tolerability. Diabetes Care. 2011;34(Suppl 2):S279–S284. doi: 10.2337/dc11-s231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Astrup A., Rossner S., Van Gaal L., Rissanen A., Niskanen L., Al Hakim M. Effects of liraglutide in the treatment of obesity: a randomised, double-blind, placebo-controlled study. Lancet. 2009;374(9701):1606–1616. doi: 10.1016/S0140-6736(09)61375-1. [DOI] [PubMed] [Google Scholar]
- 14.Drucker D.J., Nauck M.A. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368(9548):1696–1705. doi: 10.1016/S0140-6736(06)69705-5. [DOI] [PubMed] [Google Scholar]
- 15.Drucker D.J. Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metabolism. 2018;27(4):740–756. doi: 10.1016/j.cmet.2018.03.001. [DOI] [PubMed] [Google Scholar]
- 16.Adams J.M., Pei H., Sandoval D.A., Seeley R.J., Chang R.B., Liberles S.D. Liraglutide modulates appetite and body weight through glucagon-like peptide 1 receptor-expressing glutamatergic neurons. Diabetes. 2018;67(8):1538–1548. doi: 10.2337/db17-1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cork S.C., Richards J.E., Holt M.K., Gribble F.M., Reimann F., Trapp S. Distribution and characterisation of glucagon-like peptide-1 receptor expressing cells in the mouse brain. Molecular Metabolism. 2015;4(10):718–731. doi: 10.1016/j.molmet.2015.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sisley S., Gutierrez-Aguilar R., Scott M., D'Alessio D.A., Sandoval D.A., Seeley R.J. Neuronal GLP1R mediates liraglutide's anorectic but not glucose-lowering effect. Journal of Clinical Investigation. 2014;124(6):2456–2463. doi: 10.1172/JCI72434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richards P., Parker H.E., Adriaenssens A.E., Hodgson J.M., Cork S.C., Trapp S. Identification and characterization of GLP-1 receptor-expressing cells using a new transgenic mouse model. Diabetes. 2014;63(4):1224–1233. doi: 10.2337/db13-1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tornehave D., Kristensen P., Romer J., Knudsen L.B., Heller R.S. Expression of the GLP-1 receptor in mouse, rat, and human pancreas. Journal of Histochemistry and Cytochemistry. 2008;56(9):841–851. doi: 10.1369/jhc.2008.951319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caron A., Dungan Lemko H.M., Castorena C.M., Fujikawa T., Lee S., Lord C.C. POMC neurons expressing leptin receptors coordinate metabolic responses to fasting via suppression of leptin levels. Elife. 2018;7 doi: 10.7554/eLife.33710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gautron L., Elmquist J.K., Williams K.W. Neural control of energy balance: translating circuits to therapies. Cell. 2015;161(1):133–145. doi: 10.1016/j.cell.2015.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cone R.D. Anatomy and regulation of the central melanocortin system. Nature Neuroscience. 2005;8(5):571–578. doi: 10.1038/nn1455. [DOI] [PubMed] [Google Scholar]
- 24.Schwartz M.W., Porte D., Jr. Diabetes, obesity, and the brain. Science. 2005;307(5708):375–379. doi: 10.1126/science.1104344. [DOI] [PubMed] [Google Scholar]
- 25.Rossi M., Kim M.S., Morgan D.G., Small C.J., Edwards C.M., Sunter D. A C-terminal fragment of Agouti-related protein increases feeding and antagonizes the effect of alpha-melanocyte stimulating hormone in vivo. Endocrinology. 1998;139(10):4428–4431. doi: 10.1210/endo.139.10.6332. [DOI] [PubMed] [Google Scholar]
- 26.Mercer A.J., Hentges S.T., Meshul C.K., Low M.J. Unraveling the central proopiomelanocortin neural circuits. Frontiers in Neuroscience. 2013;7:19. doi: 10.3389/fnins.2013.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones G.L., Wittmann G., Yokosawa E.B., Yu H., Mercer A.J., Lechan R.M. Selective restoration of Pomc expression in glutamatergic POMC neurons: evidence for a dynamic hypothalamic neurotransmitter network. eNeuro. 2019;6(2) doi: 10.1523/ENEURO.0400-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wittmann G., Hrabovszky E., Lechan R.M. Distinct glutamatergic and GABAergic subsets of hypothalamic pro-opiomelanocortin neurons revealed by in situ hybridization in male rats and mice. Journal of Comparative Neurology. 2013;521(14):3287–3302. doi: 10.1002/cne.23350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dicken M.S., Tooker R.E., Hentges S.T. Regulation of GABA and glutamate release from proopiomelanocortin neuron terminals in intact hypothalamic networks. Journal of Neuroscience. 2012;32(12):4042–4048. doi: 10.1523/JNEUROSCI.6032-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burmeister M.A., Ayala J.E., Smouse H., Landivar-Rocha A., Brown J.D., Drucker D.J. The hypothalamic glucagon-like peptide 1 receptor is sufficient but not necessary for the regulation of energy balance and glucose homeostasis in mice. Diabetes. 2017;66(2):372–384. doi: 10.2337/db16-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Secher A., Jelsing J., Baquero A.F., Hecksher-Sorensen J., Cowley M.A., Dalboge L.S. The arcuate nucleus mediates GLP-1 receptor agonist liraglutide-dependent weight loss. Journal of Clinical Investigation. 2014;124(10):4473–4488. doi: 10.1172/JCI75276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kanoski S.E., Rupprecht L.E., Fortin S.M., De Jonghe B.C., Hayes M.R. The role of nausea in food intake and body weight suppression by peripheral GLP-1 receptor agonists, exendin-4 and liraglutide. Neuropharmacology. 2012;62(5–6):1916–1927. doi: 10.1016/j.neuropharm.2011.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oswal A., Yeo G.S. The leptin melanocortin pathway and the control of body weight: lessons from human and murine genetics. Obesity Reviews. 2007;8(4):293–306. doi: 10.1111/j.1467-789X.2007.00378.x. [DOI] [PubMed] [Google Scholar]
- 34.Beckers S., Zegers D., Van Gaal L.F., Van Hul W. The role of the leptin-melanocortin signalling pathway in the control of food intake. Critical Reviews in Eukaryotic Gene Expression. 2009;19(4):267–287. doi: 10.1615/critreveukargeneexpr.v19.i4.20. [DOI] [PubMed] [Google Scholar]
- 35.Bjorbaek C., Hollenberg A.N. Leptin and melanocortin signaling in the hypothalamus. Vitamins & Hormones. 2002;65:281–311. doi: 10.1016/s0083-6729(02)65068-x. [DOI] [PubMed] [Google Scholar]
- 36.Gautron L., Elmquist J.K. Sixteen years and counting: an update on leptin in energy balance. Journal of Clinical Investigation. 2011;121(6):2087–2093. doi: 10.1172/JCI45888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Williams K.W., Scott M.M., Elmquist J.K. Modulation of the central melanocortin system by leptin, insulin, and serotonin: co-ordinated actions in a dispersed neuronal network. European Journal of Pharmacology. 2011;660(1):2–12. doi: 10.1016/j.ejphar.2010.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clemmensen C., Chabenne J., Finan B., Sullivan L., Fischer K., Kuchler D. GLP-1/glucagon coagonism restores leptin responsiveness in obese mice chronically maintained on an obesogenic diet. Diabetes. 2014;63(4):1422–1427. doi: 10.2337/db13-1609. [DOI] [PubMed] [Google Scholar]
- 39.Ding X., Saxena N.K., Lin S., Gupta N.A., Anania F.A. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology. 2006;43(1):173–181. doi: 10.1002/hep.21006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Williams D.L., Baskin D.G., Schwartz M.W. Leptin regulation of the anorexic response to glucagon-like peptide-1 receptor stimulation. Diabetes. 2006;55(12):3387–3393. doi: 10.2337/db06-0558. [DOI] [PubMed] [Google Scholar]
- 41.He Z., Gao Y., Lieu L., Afrin S., Guo H., Williams K.W. Acute effects of zinc and insulin on arcuate anorexigenic proopiomelanocortin neurons. British Journal of Pharmacology. 2019;176(5):725–736. doi: 10.1111/bph.14559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.He Z., Gao Y., Alhadeff A.L., Castorena C.M., Huang Y., Lieu L. Cellular and synaptic reorganization of arcuate NPY/AgRP and POMC neurons after exercise. Molecular Metabolism. 2018;18:107–119. doi: 10.1016/j.molmet.2018.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang Y., He Z., Gao Y., Lieu L., Yao T., Sun J. Phosphoinositide 3-kinase is integral for the acute activity of leptin and insulin in male arcuate NPY/AgRP neurons. Journal of the Endocrine Society. 2018;2(6):518–532. doi: 10.1210/js.2018-00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yao T., Deng Z., Gao Y., Sun J., Kong X., Huang Y. Ire1alpha in Pomc neurons is required for thermogenesis and glycemia. Diabetes. 2017;66(3):663–673. doi: 10.2337/db16-0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scott M.M., Lachey J.L., Sternson S.M., Lee C.E., Elias C.F., Friedman J.M. Leptin targets in the mouse brain. Journal of Comparative Neurology. 2009;514(5):518–532. doi: 10.1002/cne.22025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van den Pol A.N., Yao Y., Fu L.Y., Foo K., Huang H., Coppari R. Neuromedin B and gastrin-releasing peptide excite arcuate nucleus neuropeptide Y neurons in a novel transgenic mouse expressing strong Renilla green fluorescent protein in NPY neurons. Journal of Neuroscience. 2009;29(14):4622–4639. doi: 10.1523/JNEUROSCI.3249-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parton L.E., Ye C.P., Coppari R., Enriori P.J., Choi B., Zhang C.Y. Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature. 2007;449(7159):228–232. doi: 10.1038/nature06098. [DOI] [PubMed] [Google Scholar]
- 48.Riccio A., Li Y., Moon J., Kim K.S., Smith K.S., Rudolph U. Essential role for TRPC5 in amygdala function and fear-related behavior. Cell. 2009;137(4):761–772. doi: 10.1016/j.cell.2009.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gao Y., Yao T., Deng Z., Sohn J.W., Sun J., Huang Y. TrpC5 mediates acute leptin and serotonin effects via Pomc neurons. Cell Reports. 2017;18(3):583–592. doi: 10.1016/j.celrep.2016.12.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berglund E.D., Liu C., Sohn J.W., Liu T., Kim M.H., Lee C.E. Serotonin 2C receptors in pro-opiomelanocortin neurons regulate energy and glucose homeostasis. Journal of Clinical Investigation. 2013;123(12):5061–5070. doi: 10.1172/JCI70338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hill J.W., Williams K.W., Ye C., Luo J., Balthasar N., Coppari R. Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. Journal of Clinical Investigation. 2008;118(5):1796–1805. doi: 10.1172/JCI32964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cowley M.A., Smart J.L., Rubinstein M., Cerdan M.G., Diano S., Horvath T.L. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411(6836):480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- 53.Hill J.W., Elias C.F., Fukuda M., Williams K.W., Berglund E.D., Holland W.L. Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metabolism. 2010;11(4):286–297. doi: 10.1016/j.cmet.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Williams K.W., Margatho L.O., Lee C.E., Choi M., Lee S., Scott M.M. Segregation of acute leptin and insulin effects in distinct populations of arcuate proopiomelanocortin neurons. Journal of Neuroscience. 2010;30(7):2472–2479. doi: 10.1523/JNEUROSCI.3118-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang Y., van der Klaauw A.A., Zhu L., Cacciottolo T.M., He Y., Stadler L.K.J. Steroid receptor coactivator-1 modulates the function of Pomc neurons and energy homeostasis. Nature Communications. 2019;10(1):1718. doi: 10.1038/s41467-019-08737-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Blair N.T., Kaczmarek J.S., Clapham D.E. Intracellular calcium strongly potentiates agonist-activated TRPC5 channels. The Journal of General Physiology. 2009;133(5):525–546. doi: 10.1085/jgp.200810153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jung S., Muhle A., Schaefer M., Strotmann R., Schultz G., Plant T.D. Lanthanides potentiate TRPC5 currents by an action at extracellular sites close to the pore mouth. Journal of Biological Chemistry. 2003;278(6):3562–3571. doi: 10.1074/jbc.M211484200. [DOI] [PubMed] [Google Scholar]
- 58.Light P.E., Manning Fox J.E., Riedel M.J., Wheeler M.B. Glucagon-like peptide-1 inhibits pancreatic ATP-sensitive potassium channels via a protein kinase A- and ADP-dependent mechanism. Molecular Endocrinology. 2002;16(9):2135–2144. doi: 10.1210/me.2002-0084. [DOI] [PubMed] [Google Scholar]
- 59.Aizawa T., Komatsu M., Asanuma N., Sato Y., Sharp G.W. Glucose action 'beyond ionic events' in the pancreatic beta cell. Trends in Pharmacological Sciences. 1998;19(12):496–499. doi: 10.1016/s0165-6147(98)01273-5. [DOI] [PubMed] [Google Scholar]
- 60.McClenaghan N.H., Flatt P.R., Ball A.J. Actions of glucagon-like peptide-1 on KATP channel-dependent and -independent effects of glucose, sulphonylureas and nateglinide. Journal of Endocrinology. 2006;190(3):889–896. doi: 10.1677/joe.1.06949. [DOI] [PubMed] [Google Scholar]
- 61.Kwon H.J., Park H.S., Park S.H., Park J.H., Shin S.K., Song S.E. Evidence for glucagon-like peptide-1 receptor signaling to activate ATP-sensitive potassium channels in pancreatic beta cells. Biochemical and Biophysical Research Communications. 2016;469(2):216–221. doi: 10.1016/j.bbrc.2015.11.127. [DOI] [PubMed] [Google Scholar]
- 62.MacDonald P.E., Salapatek A.M., Wheeler M.B. Glucagon-like peptide-1 receptor activation antagonizes voltage-dependent repolarizing K(+) currents in beta-cells: a possible glucose-dependent insulinotropic mechanism. Diabetes. 2002;51(Suppl 3):S443–S447. doi: 10.2337/diabetes.51.2007.s443. [DOI] [PubMed] [Google Scholar]
- 63.Zhao S., Kanoski S.E., Yan J., Grill H.J., Hayes M.R. Hindbrain leptin and glucagon-like-peptide-1 receptor signaling interact to suppress food intake in an additive manner. International Journal of Obesity. 2012;36(12):1522–1528. doi: 10.1038/ijo.2011.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kanoski S.E., Ong Z.Y., Fortin S.M., Schlessinger E.S., Grill H.J. Liraglutide, leptin and their combined effects on feeding: additive intake reduction through common intracellular signalling mechanisms. Diabetes, Obesity and Metabolism. 2015;17(3):285–293. doi: 10.1111/dom.12423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ernst M.B., Wunderlich C.M., Hess S., Paehler M., Mesaros A., Koralov S.B. Enhanced Stat3 activation in POMC neurons provokes negative feedback inhibition of leptin and insulin signaling in obesity. Journal of Neuroscience. 2009;29(37):11582–11593. doi: 10.1523/JNEUROSCI.5712-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Idorn T., Knop F.K., Jorgensen M.B., Jensen T., Resuli M., Hansen P.M. Safety and efficacy of liraglutide in patients with type 2 diabetes and end-stage renal disease: an investigator-initiated, placebo-controlled, double-blind, Parallel-group, randomized trial. Diabetes Care. 2016;39(2):206–213. doi: 10.2337/dc15-1025. [DOI] [PubMed] [Google Scholar]
- 67.Jacobsen L.V., Flint A., Olsen A.K., Ingwersen S.H. Liraglutide in type 2 diabetes mellitus: clinical pharmacokinetics and pharmacodynamics. Clinical Pharmacokinetics. 2016;55(6):657–672. doi: 10.1007/s40262-015-0343-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ratner C., He Z., Grunddal K.V., Skov L.J., Hartmann B., Zhang F. Long acting neurotensin synergizes with liraglutide to reverse obesity through a melanocortin-dependent pathway. Diabetes. 2019 Jun;68(6):1329–1340. doi: 10.2337/db18-1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Finan B., Clemmensen C., Muller T.D. Emerging opportunities for the treatment of metabolic diseases: glucagon-like peptide-1 based multi-agonists. Molecular and Cellular Endocrinology. 2015;418(Pt 1):42–54. doi: 10.1016/j.mce.2015.07.003. [DOI] [PubMed] [Google Scholar]
- 70.Muller T.D., Sullivan L.M., Habegger K., Yi C.X., Kabra D., Grant E. Restoration of leptin responsiveness in diet-induced obese mice using an optimized leptin analog in combination with exendin-4 or FGF21. Journal of Peptide Science. 2012;18(6):383–393. doi: 10.1002/psc.2408. [DOI] [PubMed] [Google Scholar]
- 71.Trevaskis J.L., Sun C., Athanacio J., D'Souza L., Samant M., Tatarkiewicz K. Synergistic metabolic benefits of an exenatide analogue and cholecystokinin in diet-induced obese and leptin-deficient rodents. Diabetes, Obesity and Metabolism. 2015;17(1):61–73. doi: 10.1111/dom.12390. [DOI] [PubMed] [Google Scholar]
- 72.Lee S.J., Sanchez-Watts G., Krieger J.P., Pignalosa A., Norell P.N., Cortella A. Loss of dorsomedial hypothalamic GLP-1 signaling reduces BAT thermogenesis and increases adiposity. Molecular Metabolism. 2018;11:33–46. doi: 10.1016/j.molmet.2018.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ten Kulve J.S., van Bloemendaal L., Balesar R., RG I.J., Swaab D.F., Diamant M. Decreased hypothalamic glucagon-like peptide-1 receptor expression in type 2 diabetes patients. Journal of Clinical Endocrinology & Metabolism. 2016;101(5):2122–2129. doi: 10.1210/jc.2015-3291. [DOI] [PubMed] [Google Scholar]
- 74.Collin M., Backberg M., Ovesjo M.L., Fisone G., Edwards R.H., Fujiyama F. Plasma membrane and vesicular glutamate transporter mRNAs/proteins in hypothalamic neurons that regulate body weight. European Journal of Neuroscience. 2003;18(5):1265–1278. doi: 10.1046/j.1460-9568.2003.02840.x. [DOI] [PubMed] [Google Scholar]
- 75.Kiss J., Csaba Z., Csaki A., Halasz B. Glutamatergic innervation of neuropeptide Y and pro-opiomelanocortin-containing neurons in the hypothalamic arcuate nucleus of the rat. European Journal of Neuroscience. 2005;21(8):2111–2119. doi: 10.1111/j.1460-9568.2005.04012.x. [DOI] [PubMed] [Google Scholar]
- 76.Hentges S.T., Otero-Corchon V., Pennock R.L., King C.M., Low M.J. Proopiomelanocortin expression in both GABA and glutamate neurons. Journal of Neuroscience. 2009;29(43):13684–13690. doi: 10.1523/JNEUROSCI.3770-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wu Q., Palmiter R.D. GABAergic signaling by AgRP neurons prevents anorexia via a melanocortin-independent mechanism. European Journal of Pharmacology. 2011;660(1):21–27. doi: 10.1016/j.ejphar.2010.10.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sigel E., Steinmann M.E. Structure, function, and modulation of GABA(A) receptors. Journal of Biological Chemistry. 2012;287(48):40224–40231. doi: 10.1074/jbc.R112.386664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Newland C.F., Colquhoun D., Cull-Candy S.G. Single channels activated by high concentrations of GABA in superior cervical ganglion neurones of the rat. Journal of Physiology. 1991;432:203–233. doi: 10.1113/jphysiol.1991.sp018382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Maconochie D.J., Zempel J.M., Steinbach J.H. How quickly can GABAA receptors open? Neuron. 1994;12(1):61–71. doi: 10.1016/0896-6273(94)90152-x. [DOI] [PubMed] [Google Scholar]
- 81.Kalueff A.V. Mapping convulsants' binding to the GABA-A receptor chloride ionophore: a proposed model for channel binding sites. Neurochemistry International. 2007;50(1):61–68. doi: 10.1016/j.neuint.2006.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pal B. Astrocytic actions on extrasynaptic neuronal currents. Frontiers in Cellular Neuroscience. 2015;9:474. doi: 10.3389/fncel.2015.00474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhan C., Zhou J., Feng Q., Zhang J.E., Lin S., Bao J. Acute and long-term suppression of feeding behavior by POMC neurons in the brainstem and hypothalamus, respectively. Journal of Neuroscience. 2013;33(8):3624–3632. doi: 10.1523/JNEUROSCI.2742-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Aponte Y., Atasoy D., Sternson S.M. AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nature Neuroscience. 2011;14(3):351–355. doi: 10.1038/nn.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.